
Analysis of SLC26A4, FOXI1, and KCNJ10 Gene Variants in Patients with Incomplete Partition of the Cochlea and Enlarged Vestibular Aqueduct (EVA) Anomalies

 , and
, and
Abstract
1. Introduction
2. Results
2.1. Allelic Frequency and Clinical Significance of the Identified Variants in the SLC26A4, FOXI1, and KCNJ10 Genes
2.2. In Silico Assessment of the Pathogenic Effect of the c.441G>A p.(Met147Ile) Missense Variant of the SLC26A4 Gene on the Function and/or Structure of Pendrin (SLC26A4)
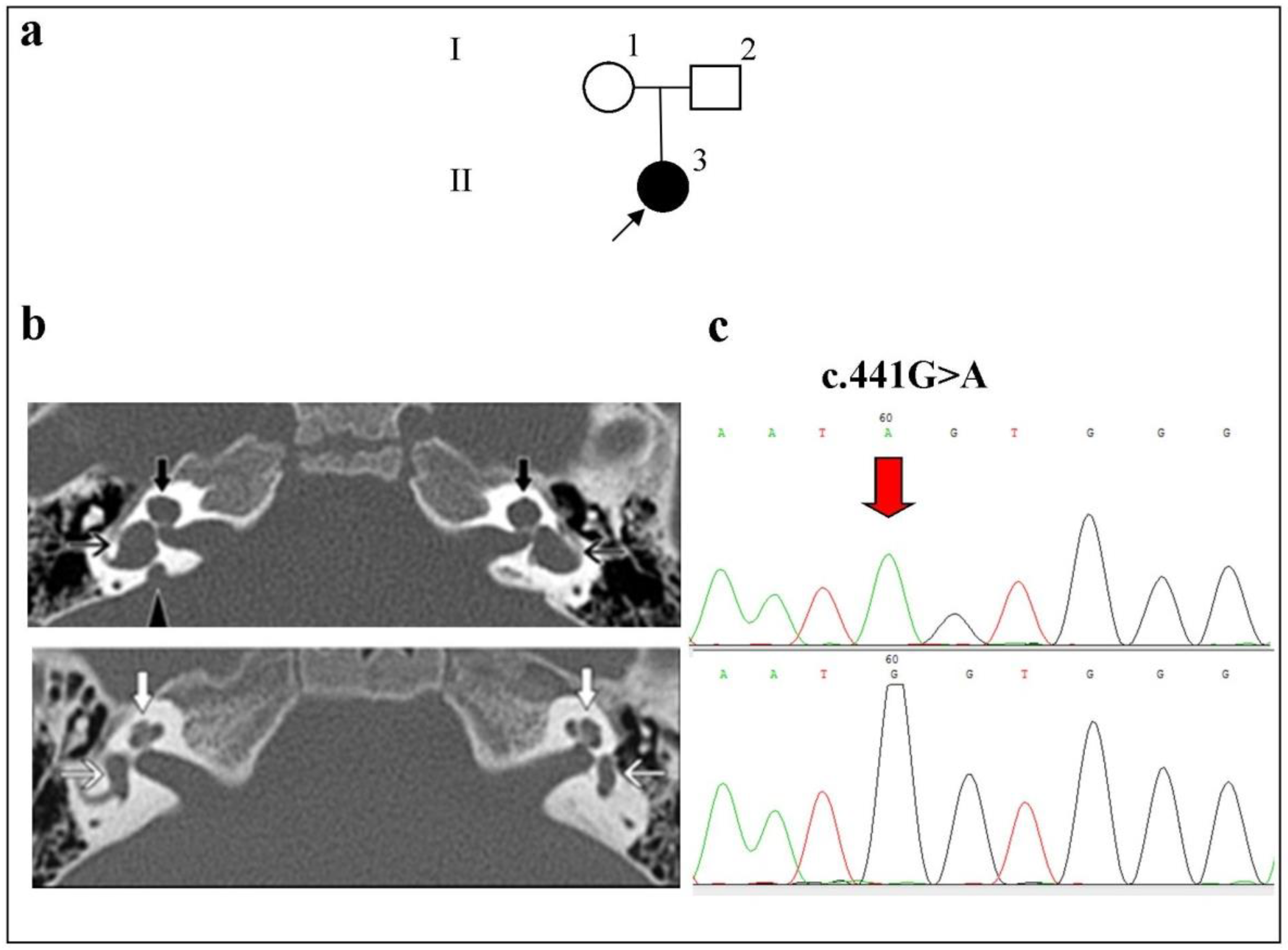
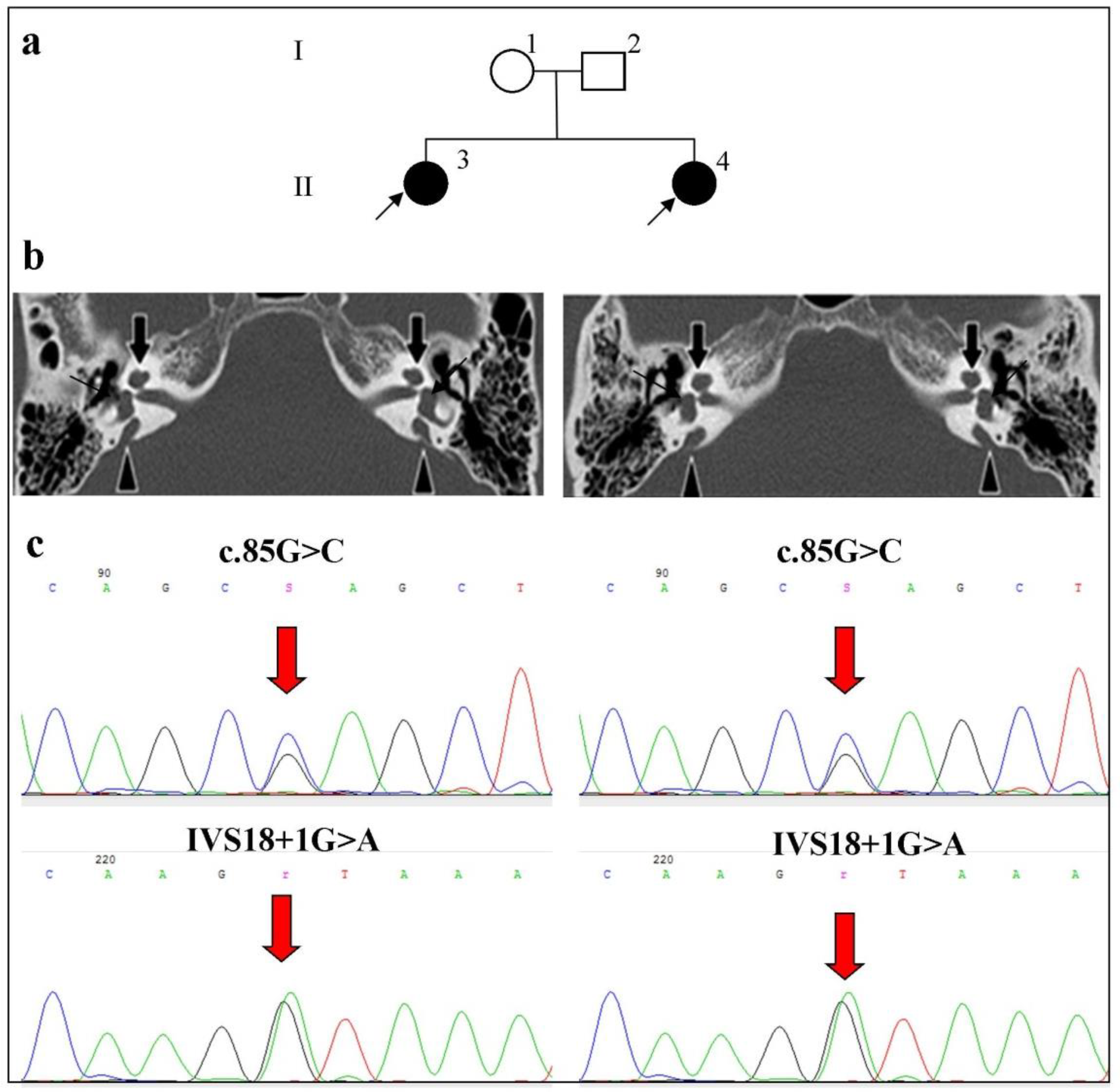
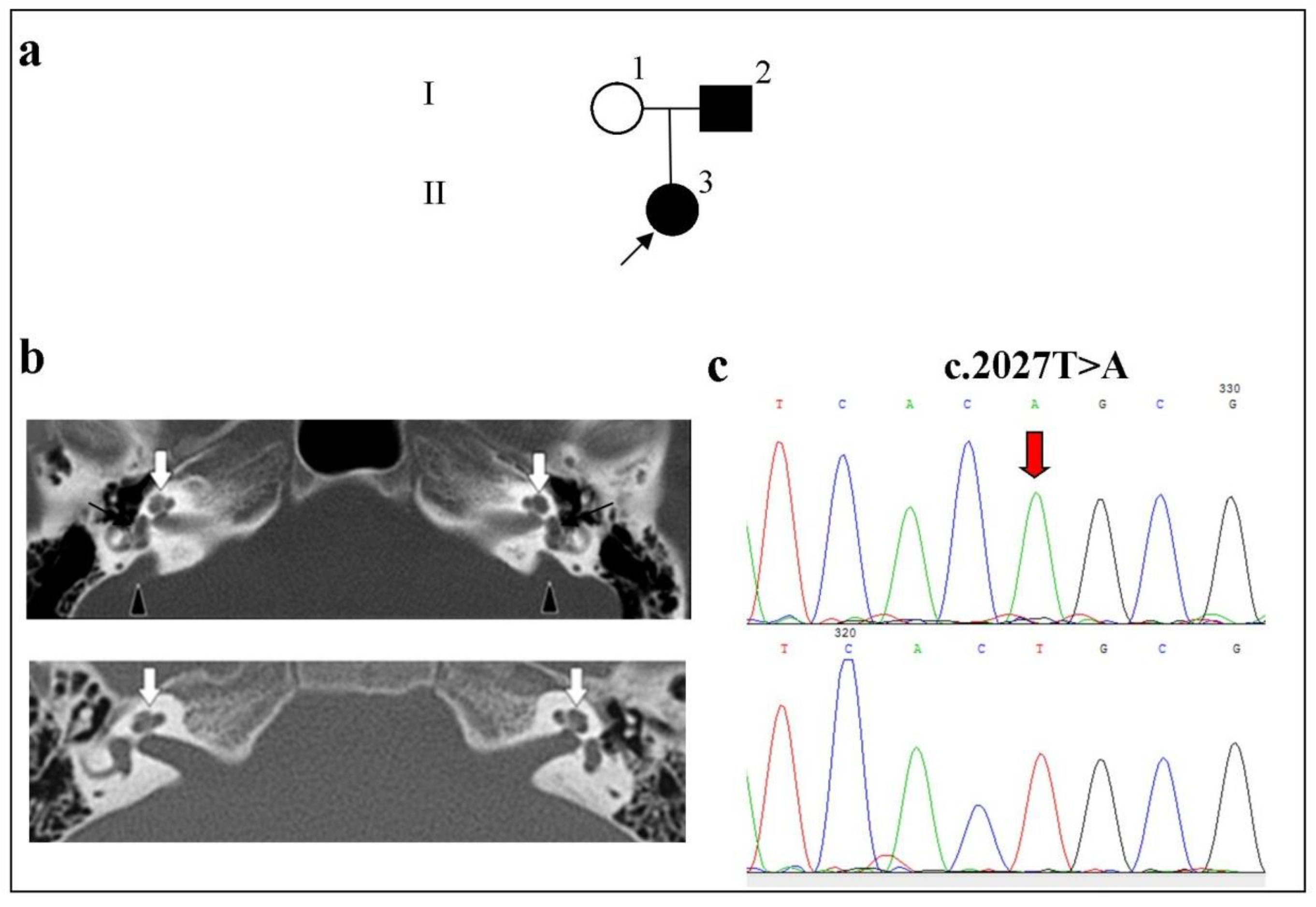
2.3. Phenotypes of Families with Biallelic Variants in the SLC26A4 Gene
- Family 1
- Family 2
- Family 3
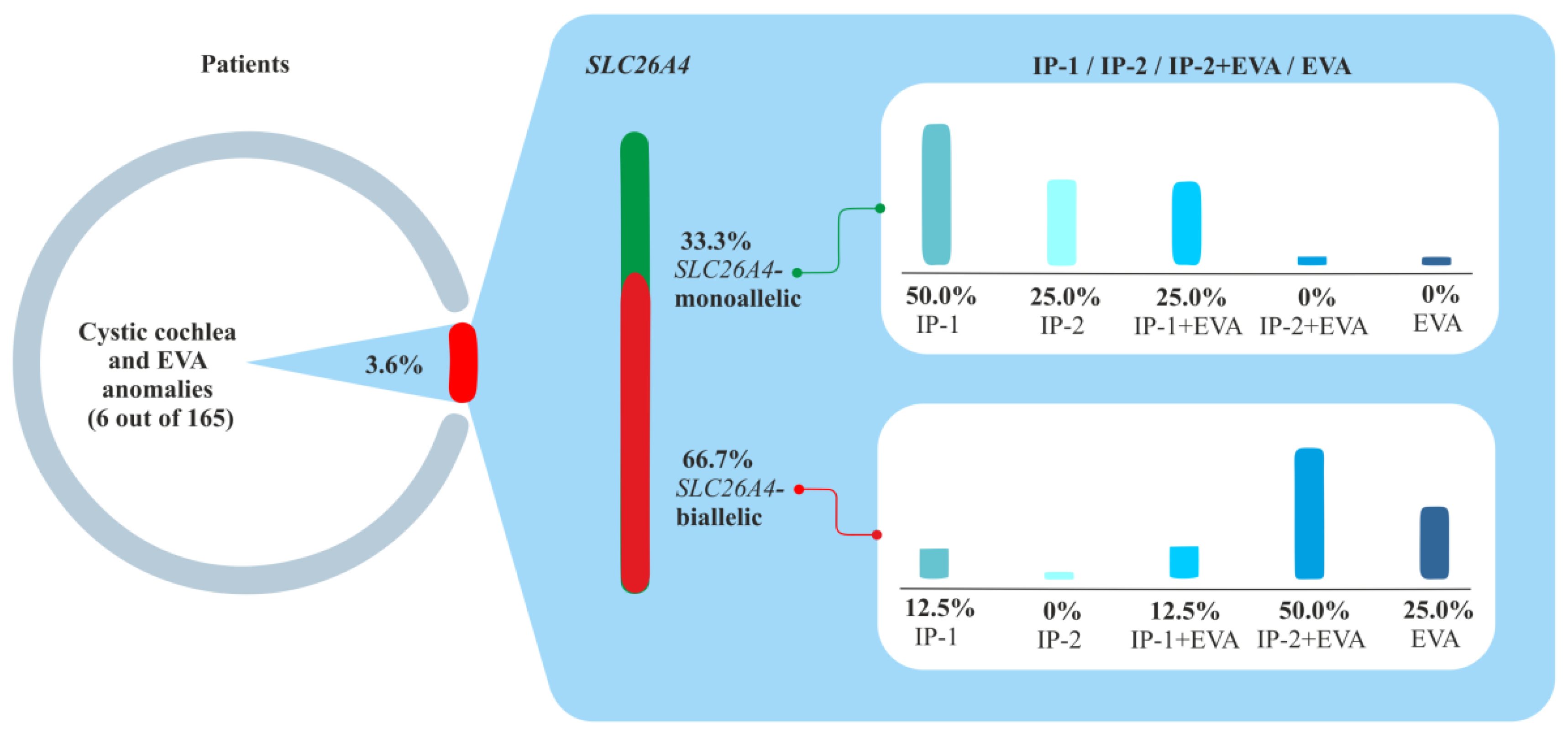
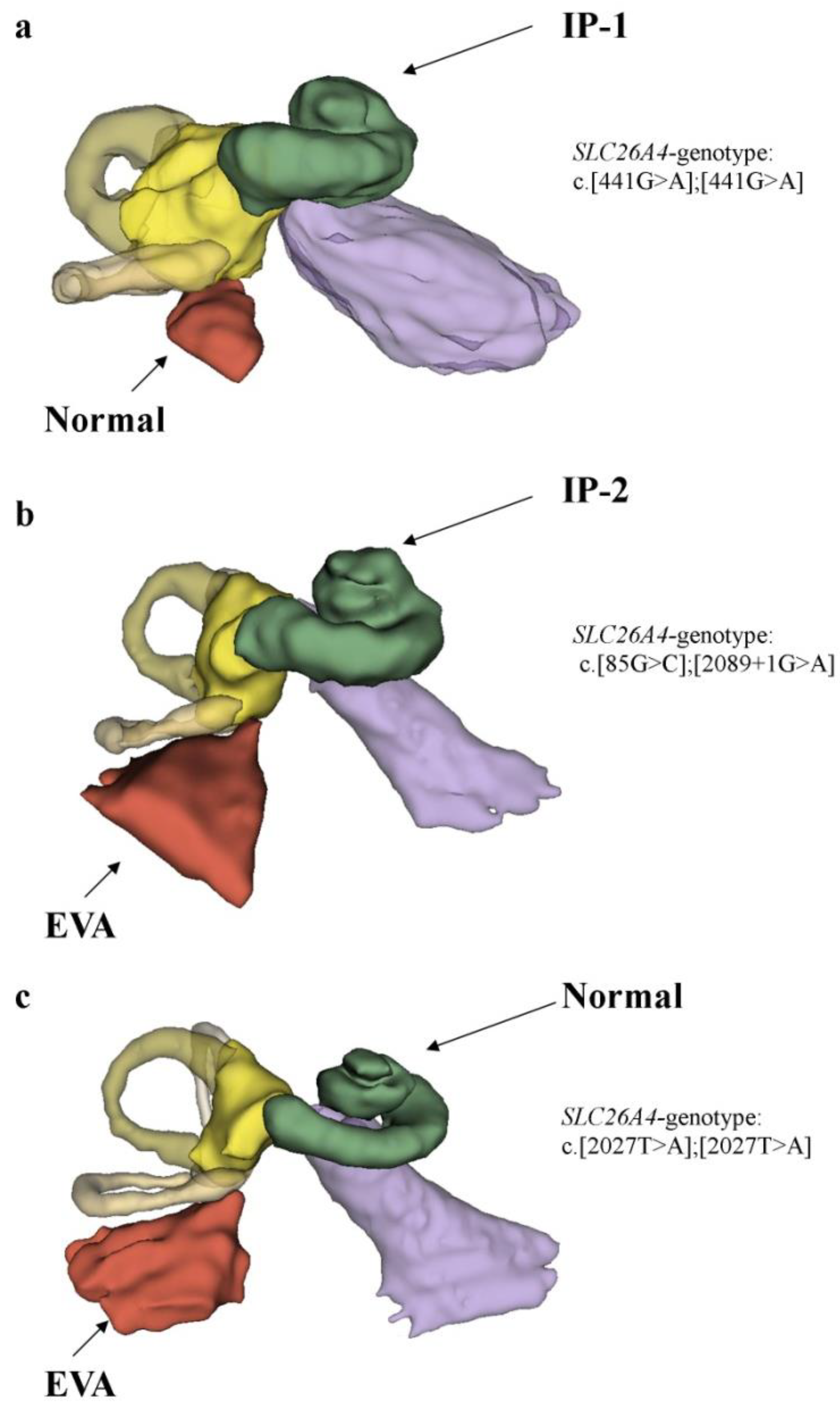
2.4. Clinical and Molecular Genetic Characteristics of Patients with the Studied Inner Ear Malformations
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Audiological Examination
4.3. Computed Tomography of the Temporal Bone
4.4. Pathogenic Variant Analysis
4.5. Database Searches
4.6. In Silico Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baldwin, C.T.; Weiss, S.; Farrer, L.A.; De Stefano, A.L.; Adair, R.; Franklyn, B.; Kidd, K.K.; Korostishevsky, M.; Bonné-Tamir, B. Linkage of congenital, recessive deafness (DFNB4) to chromosome 7q31 and evidence for genetic heterogeneity in the Middle Eastern Druze population. Hum. Mol. Genet. 1995, 4, 1637–1642. [Google Scholar] [CrossRef] [PubMed]
- Everett, L.A.; Glaser, B.; Beck, J.C.; Idol, J.R.; Buchs, A.; Heyman, M.; Adawi, F.; Hazani, E.; Nassir, E.; Baxevanis, A.D.; et al. Pendred Syndrome Is Caused by Pathogenic variants in a Putative Sulphate Transporter Gene (PDS). Nat. Genet. 1997, 17, 411–422. [Google Scholar] [CrossRef]
- Abe, S.; Usami, S.; Hoover, D.M.; Cohn, E.; Shinkawa, H.; Kimberling, W.J. Fluctuating sensorineural hearing loss associated with enlarged vestibular aqueduct maps to 7q31, the region containing the Pendred gene. Am. J. Med. Genet. 1999, 82, 322–328. [Google Scholar] [CrossRef]
- Li, X.C.; Everett, L.A.; Lalwani, A.K.; Desmukh, D.; Friedman, T.B.; Green, E.D.; Wilcox, E.R. A pathogenic variant in PDS causes non-syndromic recessive deafness. Nat. Genet. 1998, 18, 215–217. [Google Scholar] [CrossRef] [PubMed]
- Usami, S.; Abe, S.; Weston, M.D.; Shinkawa, H.; van Camp, G.; Kimberling, W.J. Non-syndromic hearing loss associated with enlarged vestibular aqueduct is caused by PDS pathogenic variants. Hum. Genet. 1999, 104, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.; Cucci, R.A.; Prasad, S.; Green, G.E.; Edeal, J.B.; Galer, C.E.; Karniski, L.P.; Sheffield, V.C.; Smith, R.J. Pendred syndrome, DFNB4, and PDS/SLC26A4 identification of eight novel pathogenic variants and possible genotype-phenotype correlations. Hum. Mutat. 2001, 17, 403–411. [Google Scholar] [CrossRef]
- Jackler, R.K.; Luxford, W.M.; House, W.F. Congenital Malformations of the Inner Ear: A Classification Based on Embryogenesis. Laryngoscope 1987, 97, 2–14. [Google Scholar] [CrossRef]
- Valvassori, G.E.; Clemis, J.D. The large vestibular aqueduct syndrome. Laryngoscope 1978, 88, 723–728. [Google Scholar] [CrossRef]
- Sennaroglu, L.; Saatci, I. A New Classification for Cochleovestibular Malformations. Laryngoscope 2002, 112, 2230–2241. [Google Scholar] [CrossRef]
- Sennaroğlu, L.; Bajin, M.D. Classification and Current Management of Inner Ear Malformations. Balkan Med. J. 2017, 34, 397–411. [Google Scholar] [CrossRef]
- Scott, D.A.; Wang, R.; Kreman, T.M.; Sheffield, V.C.; Karniski, L.P. The Pendred Syndrome Gene Encodes a Chloride-Iodide Transport Protein. Nat. Genet. 1999, 21, 440–443. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.A.; Karniski, L.P. Human Pendrin Expressed in Xenopus Laevis Oocytes Mediates Chloride/Formate Exchange. Am. J. Physiol. Cell Physiol. 2000, 278, C207–C211. [Google Scholar] [CrossRef] [PubMed]
- Soleimani, M. Molecular physiology of the renal chloride-formate exchanger. Curr. Opin. Nephrol. Hypertens. 2001, 10, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Royaux, I.E.; Suzuki, K.; Mori, A.; Katoh, R.; Everett, L.A.; Kohn, L.D.; Green, E.D. Pendrin, the Protein Encoded by the Pendred Syndrome Gene (PDS), Is an Apical Porter of Iodide in the Thyroid and Is Regulated by Thyroglobulin in FRTL-5 Cells. Endocrinology 2000, 141, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Royaux, I.E.; Wall, S.M.; Karniski, L.P.; Everett, L.A.; Suzuki, K.; Knepper, M.A.; Green, E.D. Pendrin, Encoded by the Pendred Syndrome Gene, Resides in the Apical Region of Renal Intercalated Cells and Mediates Bicarbonate Secretion. Proc. Natl. Acad. Sci. USA 2001, 98, 4221–4226. [Google Scholar] [CrossRef] [PubMed]
- Dossena, S.; Bernardinelli, E.; Sharma, A.K.; Alper, S.L.; Paulmichlet, M. The Pendrin Polypeptide. In The Role of Pendrin in Health and Disease; Dossena, S., Paulmichl, M., Eds.; Springer: Cham, Switzerland, 2017; pp. 187–220. [Google Scholar] [CrossRef]
- Tsirkin, V.I.; Trukhina, S.I.; Trukhin, A.N. Neurophysiology: Physiology of Sensory Systems: A Textbook for Universities, 2nd ed.; Corrected and Additional; Yurayt Publishing House: Moscow, Russia, 2020; 459p, ISBN 978-5-534-12590-0. Available online: https://urait.ru/bcode/447840 (accessed on 6 June 2022). (In Russian)
- Tsukada, K.; Nishio, S.Y.; Hattori, M.; Usami, S. Ethnic-specific spectrum of GJB2 and SLC26A4 pathogenic variants: Their origin and a literature review. Ann. Otol. Rhinol. Laryngol. 2015, 124 (Suppl. 1), S61–S76. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.; Chai, Y.; Chen, P.; He, L.; Wang, X.; Wu, H.; Yang, T. Mono-allelic pathogenic variants of SLC26A4 is over-presented in deaf patients with non-syndromic enlarged vestibular aqueduct. Int. J. Pediatr. Otorhinolaryngol. 2015, 79, 1351–1353. [Google Scholar] [CrossRef]
- Tsukamoto, K.; Suzuki, H.; Harada, D.; Namba, A.; Abe, S.; Usami, S.-I. Distribution and frequencies of PDS (SLC26A4) pathogenic variants in Pendred Syndrome and nonsyndromic hearing loss associated with enlarged vestibular aqueduct: A unique spectrum of pathogenic variants in Japanese. Eur. J. Hum. Genet. 2003, 11, 916–922. [Google Scholar] [CrossRef]
- Park, H.-J.; Shaukat, S.; Liu, X.-Z.; Hahn, S.H.; Naz, S.; Ghosh, M.; Kim, H.-N.; Moon, S.-K.; Abe, S.; Tukamoto, K.; et al. Origins and Frequencies of SLC26A4 (PDS) Pathogenic variants in East and South Asians: Global Implications for the Epidemiology of Deafness. J. Med. Genet. 2003, 40, 242–248. [Google Scholar] [CrossRef]
- Han, J.J.; Nguyen, P.D.; Oh, D.Y.; Han, J.H.; Kim, A.R.; Kim, M.Y.; Park, H.R.; Tran, L.H.; Dung, N.H.; Koo, J.W.; et al. Elucidation of the unique pathogenic variant spectrum of severe hearing loss in a Vietnamese pediatric population. Sci. Rep. 2019, 9, 1604. [Google Scholar] [CrossRef]
- Guo, Y.F.; Liu, X.W.; Guan, J.; Han, M.K.; Wang, D.Y.; Zhao, Y.L.; Rao, S.Q.; Wang, Q.J. GJB2, SLC26A4 and mitochondrial DNA A1555G pathogenic variants in prelingual deafness in Northern Chinese subjects. Acta Otolaryngol. 2008, 128, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Miyagawa, M.; Nishio, S.Y.; Usami, S. Deafness Gene Study Consortium. Pathogenic variant spectrum and genotype-phenotype correlation of hearing loss patients caused by SLC26A4 pathogenic variants in the Japanese: A large cohort study. J. Hum. Genet. 2014, 5, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Chai, Y.; Huang, Z.; Tao, Z.; Li, X.; Li, L.; Li, Y.; Wu, H.; Yang, T. Molecular etiology of hearing impairment associated with nonsyndromic enlarged vestibular aqueduct in East China. Am. J. Med. Genet. Part A 2013, 161, 2226–2233. [Google Scholar] [CrossRef]
- Xiang, Y.B.; Tang, S.H.; Li, H.Z.; Xu, C.Y.; Chen, C.; Xu, Y.Z.; Ding, L.R.; Xu, X.Q. Pathogenic variant analysis of common deafness-causing genes among 506 patients with nonsyndromic hearing loss from Wenzhou city, China. Int. J. Pediatr. Otorhinolaryngol. 2019, 122, 185–190. [Google Scholar] [CrossRef]
- Zhang, M.; Han, Y.; Zhang, F.; Bai, X.; Wang, H. Pathogenic variant spectrum and hotspots of the common deafness genes in 314 patients with nonsyndromic hearing loss in Heze area, China. Acta Otolaryngol. 2019, 139, 612–617. [Google Scholar] [CrossRef]
- Erdenechuluun, J.; Lin, Y.-H.; Ganbat, K.; Bataakhuu, D.; Makhbal, Z.; Tsai, C.-Y.; Lin, Y.-H.; Chan, Y.-H.; Hsu, C.-J.; Hsu, W.-C.; et al. Unique spectra of deafness-associated pathogenic variants in Mongolians provide insights into the genetic relationships among Eurasian populations. PLoS ONE 2018, 13, e0209797. [Google Scholar] [CrossRef] [PubMed]
- Blons, H.; Feldmann, D.; Duval, V.; Messaz, O.; Denoyelle, F.; Loundon, N.; Sergout-Allaoui, A.; Houang, M.; Duriez, F.; Lacombe., D.; et al. Screening of SLC26A4 (PDS) gene in Pendred’s syndrome: A large spectrum of pathogenic variants in France and phenotypic heterogeneity. Clin. Genet. 2004, 66, 333–340. [Google Scholar] [CrossRef]
- Albert, S.; Blons, H.; Jonard, L.; Feldmann, D.; Chauvin, P.; Loundon, N.; Sergent-Allaoui, A.; Houang, M.; Joannard, A.; Schmerber, S.; et al. SLC26A4 gene is frequently involved in nonsyndromic hearing impairment with enlarged vestibular aqueduct in Caucasian populations. Eur. J. Hum. Genet. 2006, 14, 773–779. [Google Scholar] [CrossRef]
- Hutchin, T.; Coy, N.N.; Conlon, H.; Telford, E.; Bromelow, K.; Blaydon, D.; Taylor, G.; Coghill, E.; Brown, S.; Trembath, R.; et al. Assessment of the genetic causes of recessive childhood non-syndromic deafness in the UK—Implications for genetic testing. Clin. Genet. 2005, 68, 506–512. [Google Scholar] [CrossRef]
- Pourová, R.; Janousek, P.; Jurovcík, M.; Dvoráková, M.; Malíková, M.; Rasková, D.; Bendová, O.; Leonardi, E.; Murgia, A.; Kabelka, Z.; et al. Spectrum and frequency of SLC26A4 pathogenic variants among Czech patients with early hearing loss with and without Enlarged Vestibular Aqueduct (EVA). Ann. Hum. Genet. 2010, 74, 299–307. [Google Scholar] [CrossRef]
- Koohiyan, M. A systematic review of SLC26A4 pathogenic variants causing hearing loss in the Iranian population. Int. J. Pediatr. Otorhinolaryngol. 2019, 125, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Pryor, S.P.; Demmler, G.J.; Madeo, A.C.; Yang, Y.; Zalewski, C.K.; Brewer, C.C.; Butman, J.A.; Fowler, K.B.; Griffith, A.J. Investigation of the Role of Congenital Cytomegalovirus Infection in the Etiology of Enlarged Vestibular Aqueducts. Arch. Otolaryngol. Head Neck Surg. 2005, 131, 388–392. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tian, Y.; Xu, H.; Liu, D.; Zhang, J.; Yang, Z.; Zhang, S.; Liu, H.; Li, R.; Tian, Y.; Zeng, B.; et al. Increased diagnosis of enlarged vestibular aqueduct by multiplex PCR enrichment and next-generation sequencing of the SLC26A4 gene. Mol. Genet. Genom. Med. 2021, 9, e1734. [Google Scholar] [CrossRef] [PubMed]
- Roesch, S.; Rasp, G.; Sarikas, A.; Dossena, S. Genetic Determinants of Non-Syndromic Enlarged Vestibular Aqueduct: A Review. Audiol. Res. 2021, 11, 40. [Google Scholar] [CrossRef]
- Chattaraj, P.; Munjal, T.; Honda, K.; Rendtorff, N.D.; Ratay, J.S.; Muskett, J.A.; Risso, D.S.; Roux, I.; Gertz, E.M.; Schäffer, A.A.; et al. A common SLC26A4-linked haplotype underlying non-syndromic hearing loss with enlargement of the vestibular aqueduct. J. Med. Genet. 2017, 10, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Vidarsson, H.; Rodrigo-Blomqvist, S.; Rosengren, S.S.; Enerback, S.; Smith, R.J. Transcriptional control of SLC26A4 is involved in Pendred syndrome and nonsyndromic enlargement of vestibular aqueduct (DFNB4). Am J. Hum. Genet. 2007, 80, 1055–1063. [Google Scholar] [CrossRef] [PubMed]
- Mercer, S.; Mutton, P.; Dahl, H.H. Identification of SLC26A4 pathogenic variants in patients with hearing loss and enlarged vestibular aqueduct using high-resolution melting curve analysis. Genet. Test. Mol. Biomark. 2011, 15, 365–368. [Google Scholar] [CrossRef]
- Wu, C.C.; Lu, Y.C.; Chen, P.J.; Yeh, P.L.; Su, Y.N.; Hwu, W.L.; Hsu, C.J. Phenotypic analyses and pathogenic variant screening of the SLC26A4 and FOXI1 genes in 101 Taiwanese families with bilateral nonsyndromic enlarged vestibular aqueduct (DFNB4) or Pendred syndrome. Audiol. Neurootol. 2010, 15, 57–66. [Google Scholar] [CrossRef]
- Lai, R.; Hu, P.; Zhu, F.; Zhu, G.; Vivero, R.; Peng, A.; Wu, W.; Xiao, Z.; Liu, X.; Xie, D. Genetic diagnosis and cochlear implantation for patients with nonsyndromic hearing loss and enlarged vestibular aqueduct. J. Laryngol. Otol. 2012, 126, 349–355. [Google Scholar] [CrossRef]
- Tada, Y.; Horio, Y.; Takumi, T.; Terayama, M.; Tsuji, L.; Copeland, N.G.; Jenkins, N.A.; Kurachi, Y. Assignment of the glial inwardly rectifying potassium channel K(AB)-2/Kir4.1 (Kcnj10) gene to the distal region of mouse chromosome 1. Genomics 1997, 45, 629–630. [Google Scholar] [CrossRef]
- Scholl, U.I.; Choi, M.; Liu, T.; Ramaekers, V.T.; Hausler, M.G.; Grimmer, J.; Tobe, S.W.; Farhi, A.; Nelson-Williams, C.; Lifton, R.P. Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by pathogenic variants in KCNJ10. Proc. Nat. Acad. Sci. USA 2009, 106, 5842–5847. [Google Scholar] [CrossRef] [PubMed]
- Reichold, M.; Zdebik, A.A.; Lieberer, E.; Rapedius, M.; Schmidt, K.; Bandulik, S.; Sterner, C.; Tegtmeier, I.; Penton, D.; Baukrowitz, T.; et al. KCNJ10 gene pathogenic variants causing EAST syndrome (epilepsy, ataxia, sensorineural deafness, and tubulopathy) disrupt channel function. Proc. Natl. Acad. Sci. USA 2010, 107, 14490–14495. [Google Scholar] [CrossRef] [PubMed]
- Larsson, C.; Hellqvist, M.; Pierrou, S.; White, I.; Enerback, S.; Carlsson, P. Chromosomal localization of six human forkhead genes, freac-1 (FKHL5), -3 (FKHL7), -4 (FKHL8), -5 (FKHL9), -6 (FKHL10), and -8 (FKHL12). Genomics 1995, 30, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Hulander, M.; Wurst, W.; Carlsson, P.; Enerback, S. The winged helix transcription factor Fkh10 is required for normal development of the inner ear. Nat. Genet. 1998, 20, 374–376. [Google Scholar] [CrossRef]
- Yang, T.; Gurrola, J.G.; Wu, H.; Chiu, S.M.; Wangemann, P.; Snyder, P.M.; Richard, J.H. Pathogenic variants of KCNJ10 together with pathogenic variants of SLC26A4 cause digenic nonsyndromic hearing loss associated with enlarged vestibular aqueduct syndrome. Am. J. Hum. Genet. 2009, 84, 651–657. [Google Scholar] [CrossRef]
- Chen, K.; Wang, X.; Sun, L.; Jiang, H. Screening of SLC26A4, FOXI1, KCNJ10, and GJB2 in bilateral deafness patients with inner ear malformation. Otolaryngol. Head Neck Surg. 2012, 146, 972–978. [Google Scholar] [CrossRef]
- Pique, L.M.; Brennan, M.; Davidson, C.J.; Schaefer, F.; Greinwald, J., Jr.; Schrijver, I. Pathogenic variant analysis of the SLC26A4, FOXI1 and KCNJ10 genes in individuals with congenital hearing loss. PeerJ 2014, 2, e384. [Google Scholar] [CrossRef]
- Lobov, S.L. Structural Features of the Pendrina (SLC26A4) and Prestin (SLC26A5) Genes in Patients with Hereditary Non-Syndromic Sensorineural Deafness; Institute of Biochemistry and Genetics, USC RAS: Moscow, Russia, 2013. (In Russian) [Google Scholar]
- Mironovich, O.L.; Bliznetz, E.A.; Polyakov, A.V.; Markova, T.G.; Geptner, E.N.; Lalayants, M.R.; Zelikovich, E.I.; Tavartkiladze, G.A. Results of molecular genetic testing in Russian patients with Pendred syndrome and allelic disorders. Russ. J. Genet. 2017, 53, 128–138. [Google Scholar] [CrossRef]
- Danilchenko, V.Y.; Zytsar, M.V.; Maslova, E.A.; Bady-Khoo, M.S.; Barashkov, N.A.; Morozov, I.V.; Bondar, A.A.; Posukh, O.L. Different Rates of the SLC26A4-Related Hearing Loss in Two Indigenous Peoples of Southern Siberia (Russia). Diagnostics 2021, 11, 2378. [Google Scholar] [CrossRef]
- Jonard, L.; Niasme-Grare, M.; Bonnet, C.; Feldmann, D.; Rouillon, I.; Loundon, N.; Calais, C.; Catros, H.; David, A.; Dollfus, H.; et al. Screening of SLC26A4, FOXI1 and KCNJ10 Genes in Unilateral Hearing Impairment with Ipsilateral Enlarged Vestibular Aqueduct. Int. J. Pediatr. Otorhinolaryngol. 2010, 74, 1049–1053. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; ACMG Laboratory Quality Assurance Committee; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Oza, A.M.; DiStefano, M.T.; Hemphill, S.E.; Cushman, B.J.; Grant, A.R.; Siegert, R.K.; Shen, J.; Chapin, A.; Boczek, N.J.; Schimmenti, L.A.; et al. ClinGen Hearing Loss Clinical Domain Working Group. Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum. Mutat. 2018, 39, 1593–1613. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Mooers, B.H.M.; Brown, M.E. Templates for writing PyMOL scripts. Protein Sci. 2021, 30, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Reardon, W.; OMahoney, C.F.; Trembath, R.; Jan, H.; Phelps, P.D. Enlarged vestibular aqueduct: A radiological marker of pendred syndrome, and pathogenic variant of the PDS gene. QJM 2000, 93, 99–104. [Google Scholar] [CrossRef]
- Honda, K.; Griffith, A.J. Genetic architecture and phenotypic landscape of SLC26A4-related hearing loss. Hum. Genet. 2022, 141, 455–464. [Google Scholar] [CrossRef]
- Fu, C.; Zheng, H.; Zhang, S.; Chen, Y.; Su, J.; Wang, J.; Xie, B.; Hu, X.; Fan, X.; Luo, J.; et al. Pathogenic variant screening of the SLC26A4 gene in a cohort of 192 Chinese patients with congenital hypothyroidism. Arch. Endocrinol. Metab. 2016, 60, 323–327. [Google Scholar] [CrossRef][Green Version]
- Mey, K.; Muhamad, A.A.; Tranebjaerg, L.; Rendtorff, N.D.; Rasmussen, S.H.; Bille, M.; Cayé-Thomasen, P. Association of SLC26A4 Pathogenic variants, Morphology, and Hearing in Pendred Syndrome and NSEVA. Laryngoscope 2019, 129, 2574–2579. [Google Scholar] [CrossRef]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the Effects of Coding Non-Synonymous Variants on Protein Function Using the SIFT Algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense pathogenic variants. Nat. Methods 2010, 4, 248–249. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. Pathogenic variantTaster2: Pathogenic variant prediction for the deep-sequencing age. Nat. Methods 2014, 4, 361–362. [Google Scholar] [CrossRef]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef] [PubMed]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Žídek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly accurate protein structure prediction for the human proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.-H.G.; Tsai, C.-J.; Ma, B.; Nussinov, R. In Silico Protein Design by Combinatorial Assembly of Protein Building Blocks. Protein Sci. 2004, 13, 2753–2765. [Google Scholar] [CrossRef] [PubMed]
- Bordogna, A.; Pandini, A.; Bonati, L. Predicting the accuracy of protein-ligand docking on homology models. J. Comput. Chem. 2011, 32, 81–98. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Exon | Variant | dbSNP | Molecular Consequence | ClinVar Interpretation | Allelic Frequency ** |
|---|---|---|---|---|---|---|
| SLC26A4 | 2 | c.85G>C p.(Glu29Gln) | rs111033205 | Missense | Pathogenic | 10% (1/10) |
| 5 | c.441G>A p.(Met147Ile) | rs201905280 | Missense | Conflicting interpretations of pathogenicity: uncertain significance(6), likely benign(1) | 20% (2/10) | |
| 6 | c.757A>G p.(Ile253Val) | rs773657545 | Missense | Likely pathogenic * | 20% (2/10) | |
| FOXI1 | 17 | c.2027T>A p.(Leu676Gln) | rs111033318 | Missense | Pathogenic/likely pathogenic | 20% (2/10) |
| Intron 18 | c.2089+1G>A (IVS18+1G>A) | rs727503430 | Splice donor site | Pathogenic | 10% (1/10) | |
| 1 | c.279G>A p.(Arg93=) | rs2277944 | Synonymous | Benign | 30% (3/10) | |
| 2 | c.1044T>C p.(Tyr348=) | rs10063424 | Synonymous | Benign | 90% (9/10) | |
| KCNJ10 | 1 | c.811C>T p.(Arg271Cys) | rs1130183 | Missense | Benign/ likely benign | 10% (1/10) |
| Patient Code | Age | Cipher | Sex | Ethnicity | Degree of HL | VD | Goiter/PDS | Side | Type of IP | EVA | DV | MM | SLC26A4 Genotypes | FOXI1 Genotypes | KCNJ10 Genotypes | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Allele 1 | Allele 2 | Allele 1 | Allele 2 | Allele 1 | Allele 2 | |||||||||||||
| 1095 | 2 | - | M | Yakut | Severe | - | -/- | R | IP-1 | - | + | - | c.757A>G p.(Ile253Val) | wt | c.279G>A p.(Arg93=) | c.1044T>C p.(Tyr348=) | wt | wt |
| L | IP-1 | + (2.4 mm) | + | - | ||||||||||||||
| 18 * | 52 | - | F | Yakut | Profound | Dizziness | -/- | R | IP-1 | - | + | - | c.757A>G p.(Ile253Val) | wt | c.279G>A p.(Arg93=) c.1044T>C p.(Tyr348=) | c.1044T>C p.(Tyr348=) | wt | wt |
| L | IP-2 | - | + | - | ||||||||||||||
| 16 * | 38 | II:3 | F | Russian | Profound | Dizziness | -/- | R | IP-2 | + (3.5 mm) | + | + | c.2089+1G>A (IVS18+1G>A) | c.85G>C p.(Glu29Gln) | wt | wt | c.811C>T p.(Arg271Cys) | wt |
| L | IP-2 | + (3.6 mm) | + | + | ||||||||||||||
| 17 * | 32 | II:4 | F | Russian | Profound | - | -/- | R | IP-2 | + (2.9 mm) | + | + | c.2089+1G>A (IVS18+1G>A) | c.85G>C p.(Glu29Gln) | c.1044T>C p.(Tyr348=) | c.1044T>C p.(Tyr348=) | wt | wt |
| L | IP-2 | + (2.7 mm) | + | + | ||||||||||||||
| 192 | 5 | II:3 | F | Russian | Severe | - | -/- | R | IP-1 | + (2.5 mm) | + | - | c.441G>A p.(Met147Ile) | c.441G>A p.(Met147Ile) | c.1044T>C p.(Tyr348=) | c.1044T>C p.(Tyr348=) | ||
| L | IP-1 | - | + | - | ||||||||||||||
| 1091 | 33 | II:3 | F | Buryat | Moderate | - | Nodular goiter/PDS | R | Normal | + (5.4 mm) | + | - | c.2027T>A p.(Leu676Gln) | c.2027T>A p.(Leu676Gln) | c.279G>A p.(Arg93=) c.1044T>C p.(Tyr348=) | c.1044T>C p.(Tyr348=) | wt | wt |
| L | Normal | + (5.7 mm) | + | - | ||||||||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klarov, L.A.; Pshennikova, V.G.; Romanov, G.P.; Cherdonova, A.M.; Solovyev, A.V.; Teryutin, F.M.; Luginov, N.V.; Kotlyarov, P.M.; Barashkov, N.A. Analysis of SLC26A4, FOXI1, and KCNJ10 Gene Variants in Patients with Incomplete Partition of the Cochlea and Enlarged Vestibular Aqueduct (EVA) Anomalies. Int. J. Mol. Sci. 2022, 23, 15372. https://doi.org/10.3390/ijms232315372
Klarov LA, Pshennikova VG, Romanov GP, Cherdonova AM, Solovyev AV, Teryutin FM, Luginov NV, Kotlyarov PM, Barashkov NA. Analysis of SLC26A4, FOXI1, and KCNJ10 Gene Variants in Patients with Incomplete Partition of the Cochlea and Enlarged Vestibular Aqueduct (EVA) Anomalies. International Journal of Molecular Sciences. 2022; 23(23):15372. https://doi.org/10.3390/ijms232315372
Chicago/Turabian StyleKlarov, Leonid A., Vera G. Pshennikova, Georgii P. Romanov, Aleksandra M. Cherdonova, Aisen V. Solovyev, Fedor M. Teryutin, Nikolay V. Luginov, Petr M. Kotlyarov, and Nikolay A. Barashkov. 2022. "Analysis of SLC26A4, FOXI1, and KCNJ10 Gene Variants in Patients with Incomplete Partition of the Cochlea and Enlarged Vestibular Aqueduct (EVA) Anomalies" International Journal of Molecular Sciences 23, no. 23: 15372. https://doi.org/10.3390/ijms232315372
APA StyleKlarov, L. A., Pshennikova, V. G., Romanov, G. P., Cherdonova, A. M., Solovyev, A. V., Teryutin, F. M., Luginov, N. V., Kotlyarov, P. M., & Barashkov, N. A. (2022). Analysis of SLC26A4, FOXI1, and KCNJ10 Gene Variants in Patients with Incomplete Partition of the Cochlea and Enlarged Vestibular Aqueduct (EVA) Anomalies. International Journal of Molecular Sciences, 23(23), 15372. https://doi.org/10.3390/ijms232315372