
Integrated Transcriptome and Metabolome Analysis Reveals Phenylpropanoid Biosynthesis and Phytohormone Signaling Contribute to “Candidatus Liberibacter asiaticus” Accumulation in Citrus Fruit Piths (Fluffy Albedo)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
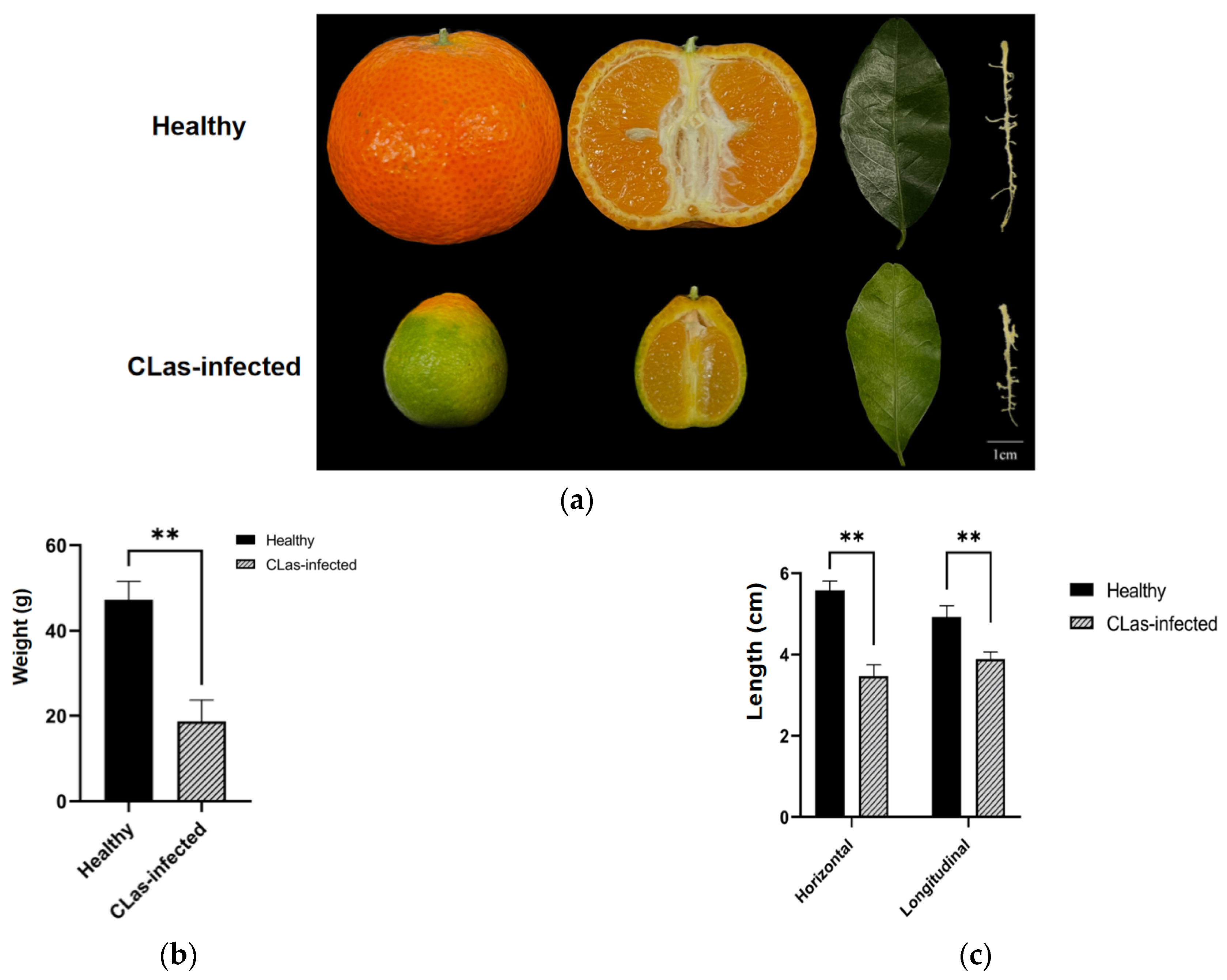
2.1. Phenotype of the CLas-Infected and Uninfected Fruits and Leaves
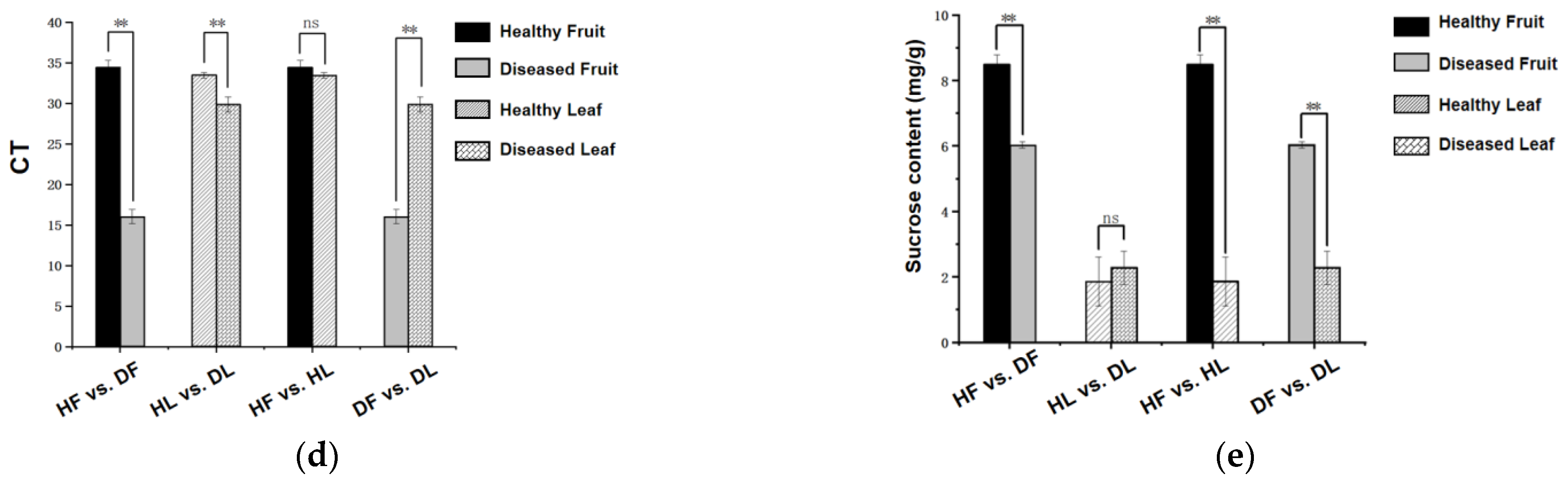
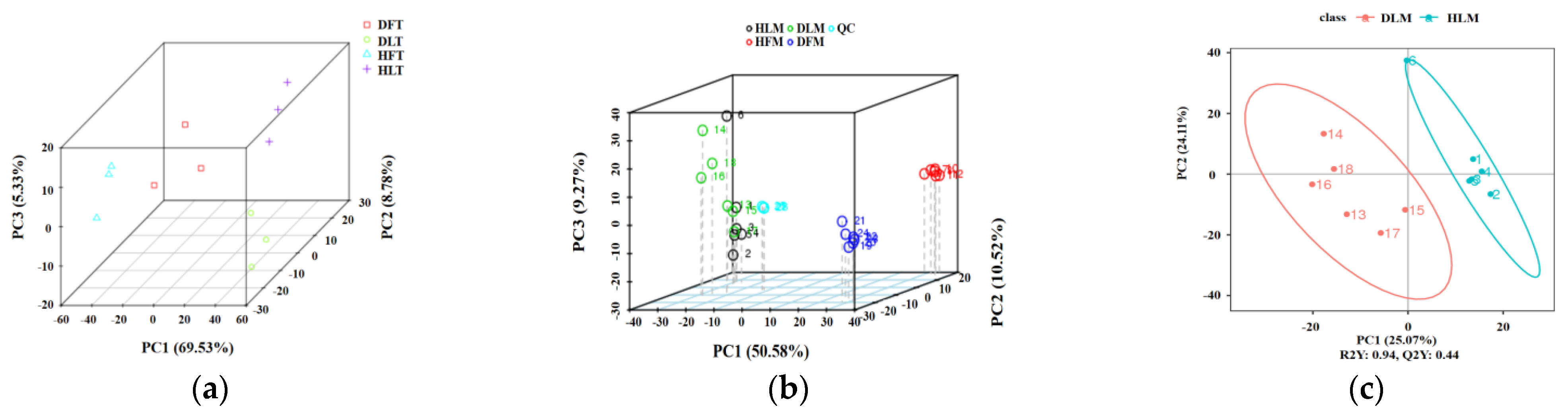
2.2. Quality Control of RNA-seq and UHPLC–MS/MS
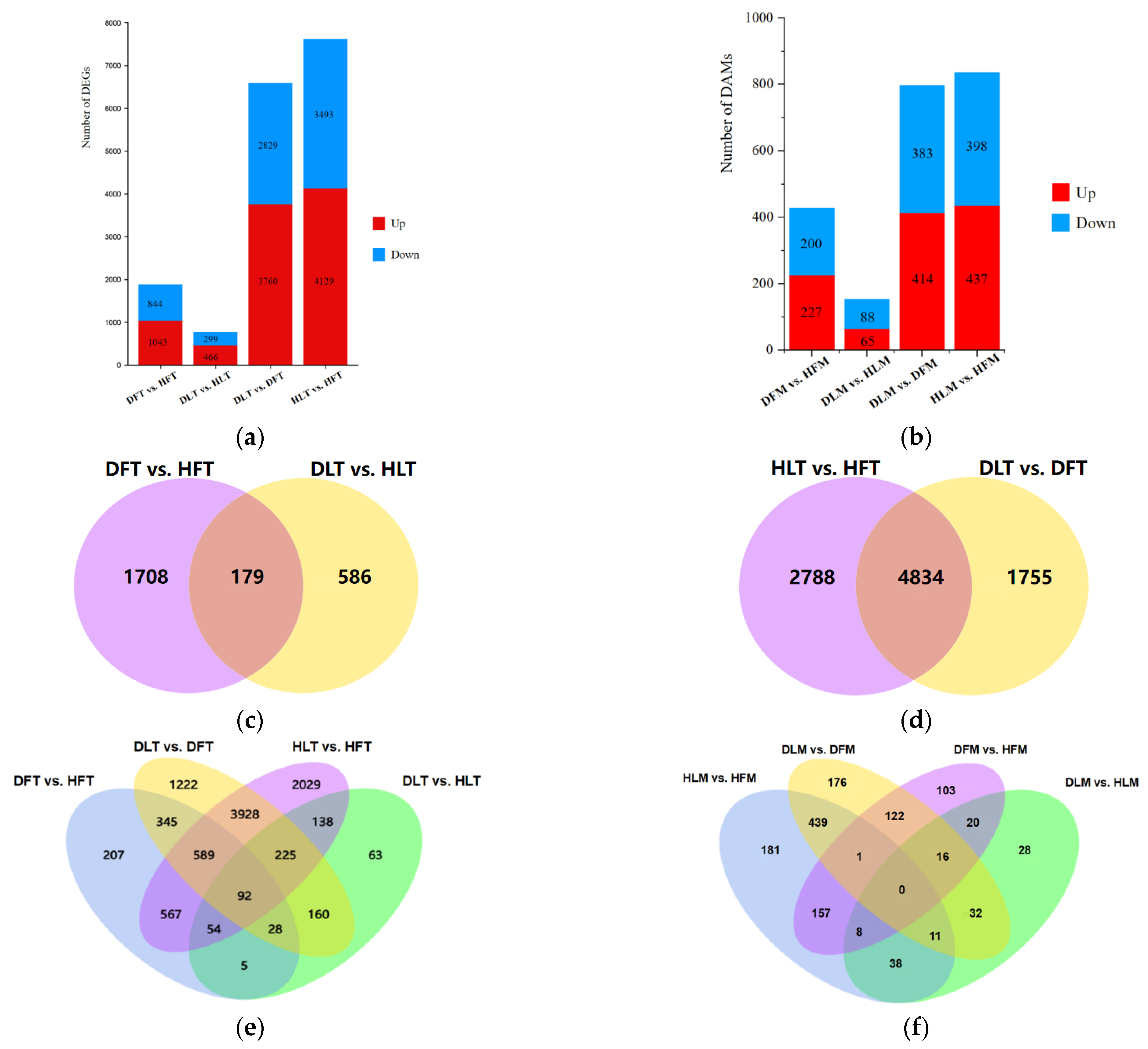
2.3. General Description of the Differentially Expressed Genes and Differential Metabolites
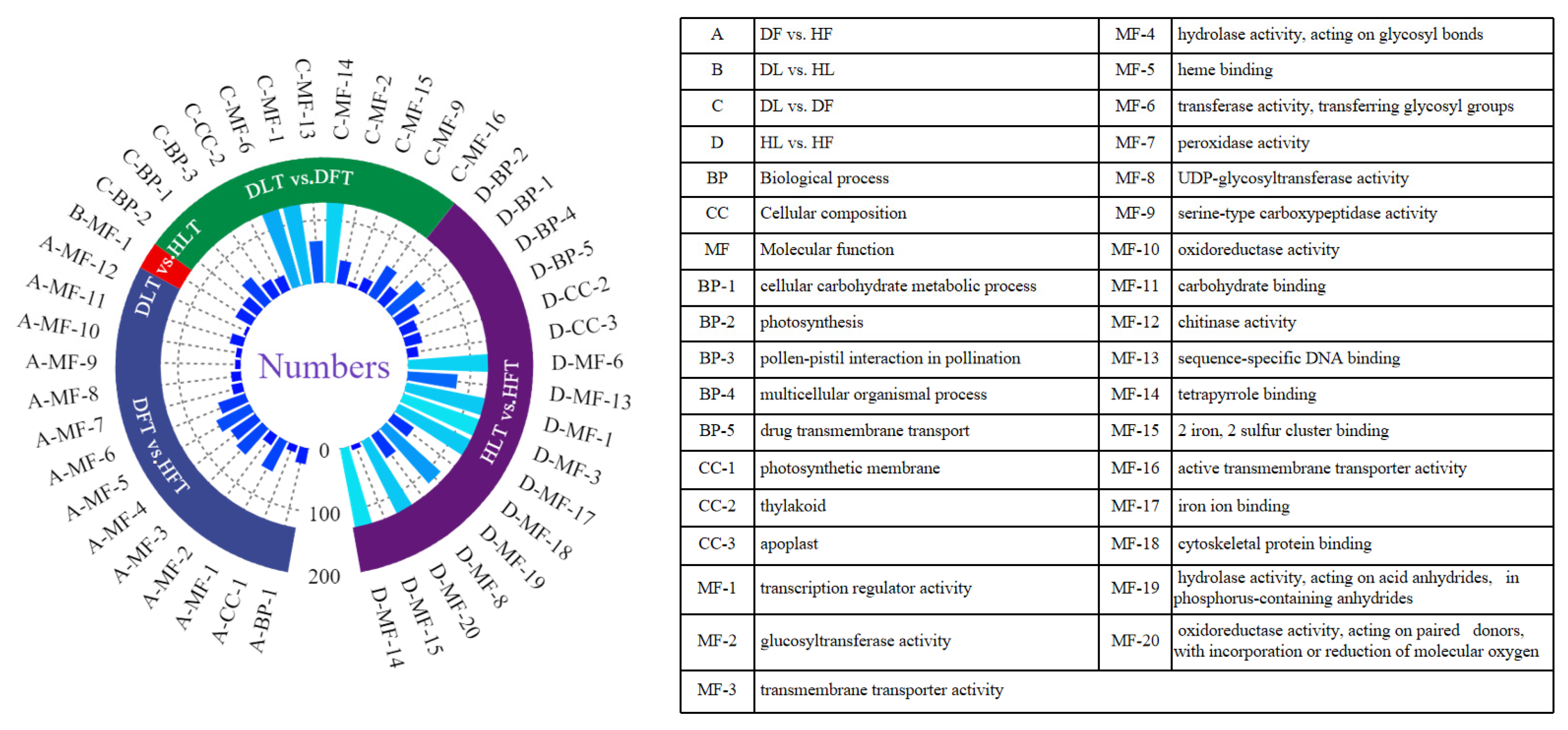
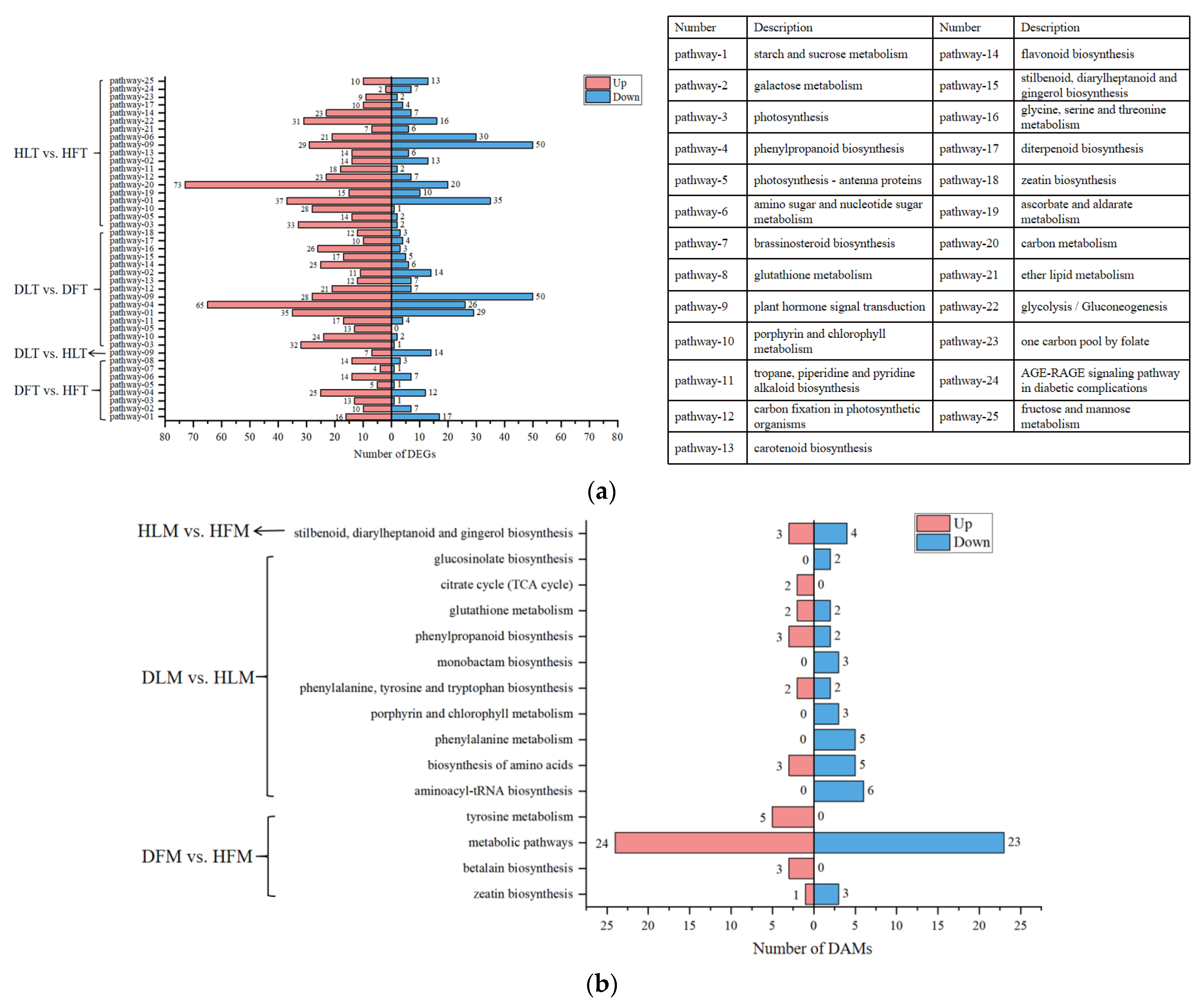
2.4. GO and KEGG Enrichment Analyses of Differentially Expressed Genes and Differential Accumulated Metabolites
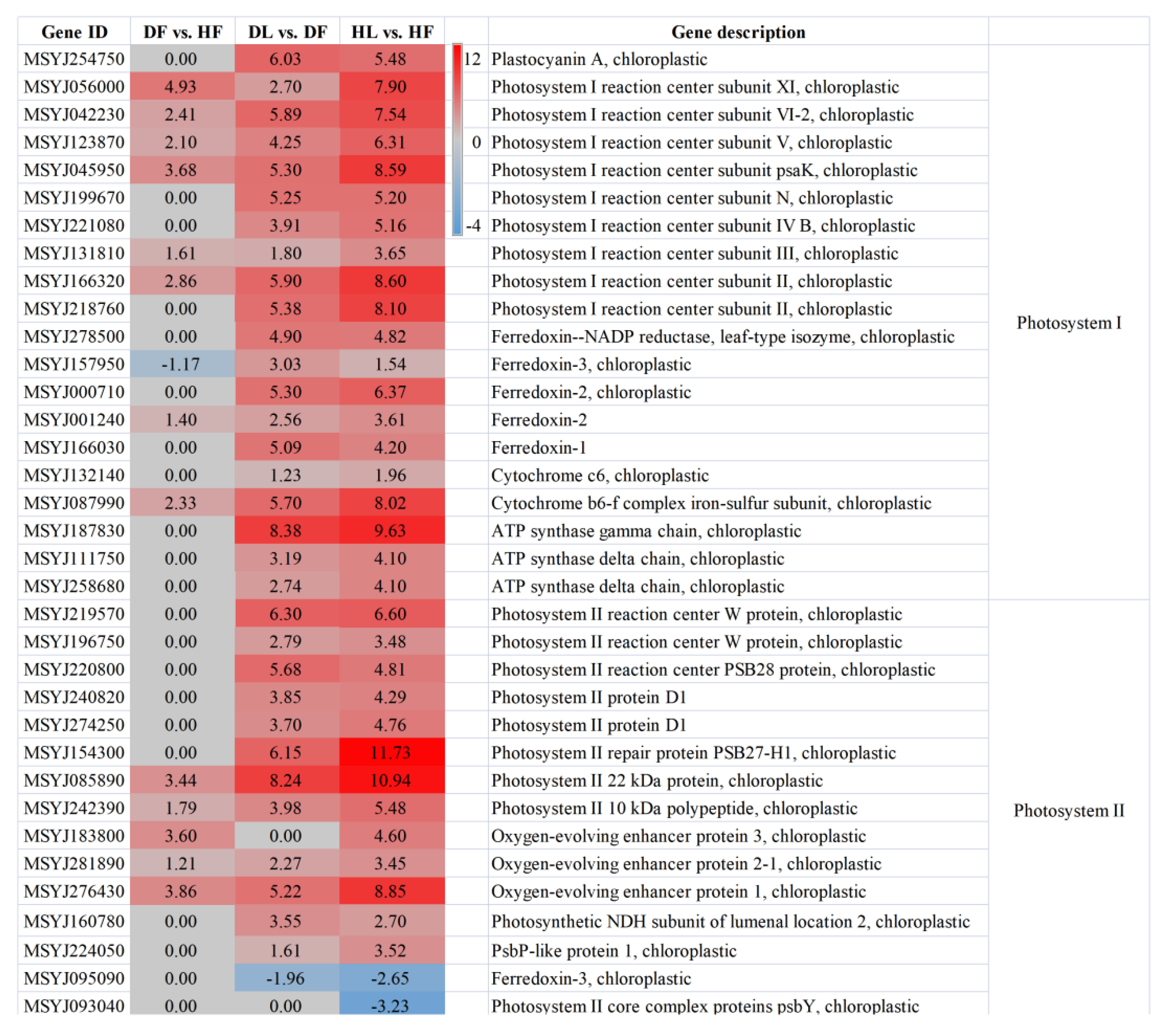
2.5. Photosynthesis and Starch and Sucrose Metabolism Were Related to the Citrus Response to CLas
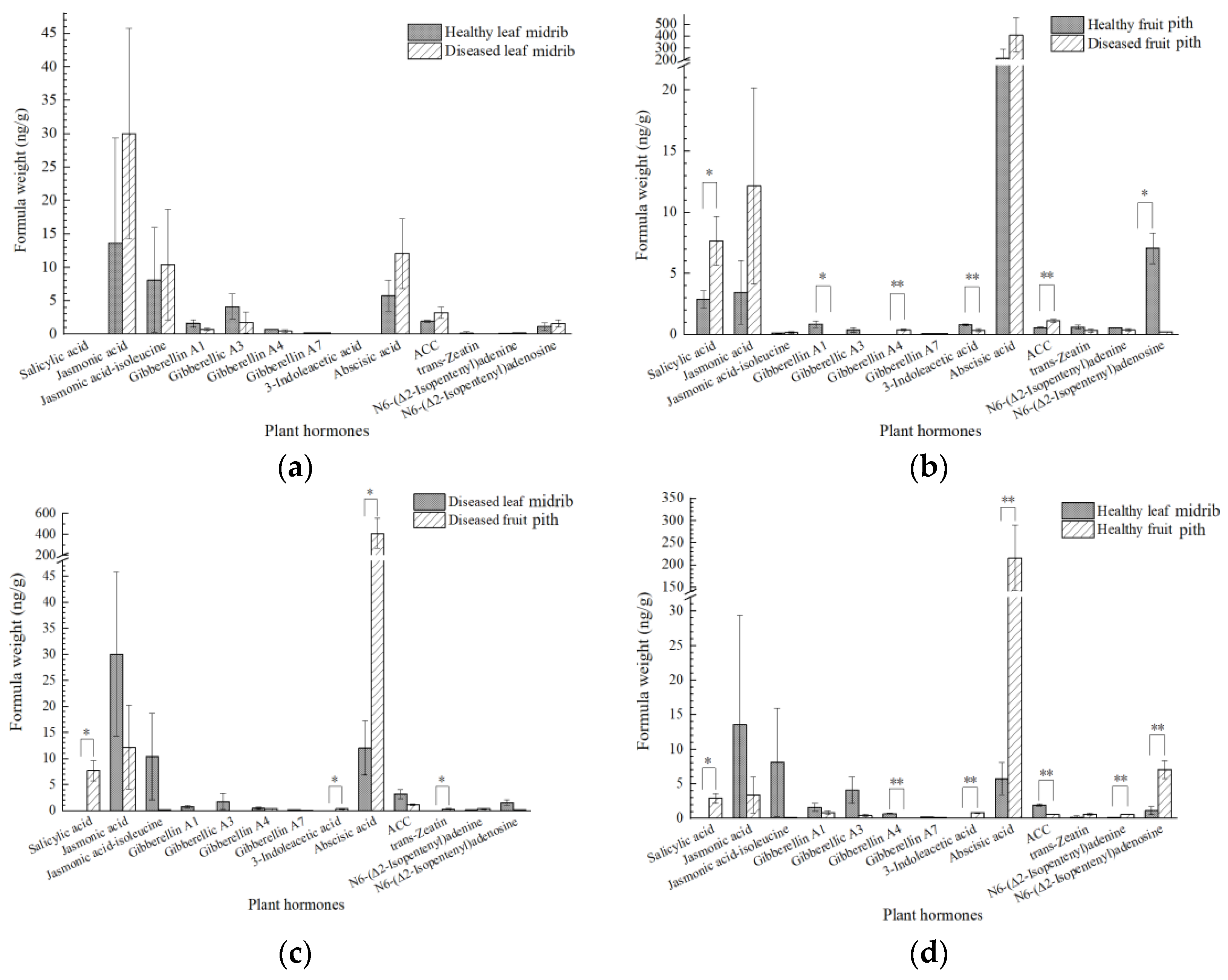
2.6. Plant Hormone Signal Transduction Was Induced upon CLas Infection in the Leaves
2.7. Phenylpropanoid Biosynthesis Was Induced Specifically in the CLas-Infected Leaves
3. Discussion
3.1. HLB Symptoms Are Closely Related to the Photosynthetic Reaction of Leaves and Fruits
3.2. CLas Infection Mainly Influences Starch Metabolism in Leaves or Sucrose Metabolism in Fruits
3.3. The Induction of IAA and Depression of JA Are Closely Related to CLas Accumulation in Fruit Piths
3.4. Depression of Phenylpropanoid Biosynthesis in the Fruit Pith Decreased Defense Responses
4. Materials and Methods
4.1. Plant Material
4.2. Determination of Phenotypes and Measurement of Carbohydrate Contents
4.3. DNA Extraction and PCR Detection of Ca. L. asiaticus
4.4. RNA Extraction, RNA-seq, and Differential Expression Analysis
4.5. Metabolite Analysis by UHPLC–MS
4.6. Phytohormone Profiling
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Deng, X.L.; Tang, W.W. The studies on detection of citrus huanglongbing pathogen by polymerase chain reaction. J. South China Agric. Univ. 1996, 3, 122–123. [Google Scholar]
- Da Graça, J.V.; Douhan, G.W.; Halbert, S.E.; Keremane, M.L.; Lee, R.F.; Vidalakis, G.; Zhao, H.W. Huanglongbing: An overview of a complex pathosystem ravaging the world’s citrus. J. Integr. Plant Biol. 2016, 58, 373–387. [Google Scholar] [CrossRef] [PubMed]
- Bové, J.M. Huanglongbing: A destructive, newly-emerging, century-old disease of citrus. J. Plant Pathol. 2006, 88, 7–37. [Google Scholar]
- Lin, K.X. Observations on yellow shoot of citrus. Acta Phytopathol. Sin. 1956, 2, 12–42. [Google Scholar] [CrossRef]
- Ding, F.; Duan, Y.P.; Paul, C.; Brlansky, R.H.; Hartung, J.S. Localization and distribution of “Candidatus Liberibacter asiaticus” in citrus and periwinkle by direct tissue blot immuno assay with an Anti-OmpA polyclonal antibody. PLoS ONE 2015, 10, e0123939. [Google Scholar] [CrossRef] [PubMed]
- Tatineni, S.; Sagaram, U.S.; Gowda, S.; Robertson, C.J.; Dawson, W.O.; Iwanami, T.; Wang, N. In planta distribution of “Candidatus Liberibacter asiaticus” as revealed by polymerase chain reaction (PCR) and real-time PCR. Phytopathology 2008, 98, 592–599. [Google Scholar] [CrossRef]
- Chen, Y.L.; Tang, R.; Liu, Y.; Deng, X.L.; Xu, M.R. Distribution and genetic diversity of “Candidatus Liberibacter asiaticus” in citrus canopy shoots. Acta Phytopathol. Sin. 2018, 48, 728–737. [Google Scholar] [CrossRef]
- Fang, F.; Guo, H.Y.; Zhao, A.M.; Li, T.; Liao, H.H.; Deng, X.L.; Xu, M.R.; Zheng, Z. A significantly high abundance of “Candidatus Liberibacter asiaticus” in citrus fruit pith: In planta transcriptome and anatomical analyses. Front. Microbiol. 2021, 12, 681251. [Google Scholar] [CrossRef]
- Huang, Z.H.; Wang, J.; Gao, J.; Zheng, Z.; Deng, X.L.; Xu, M.R. Distribution of “Candidatus Liberibacter asiaticus” in the fruit branch of four citrus cultivars. J. South China Agric. Univ. 2022, 43, 35–42. [Google Scholar] [CrossRef]
- Teng, C.L.; Zhong, X.; Wu, H.D.; Hu, Y.; Zhou, C.Y.; Wang, X.F. Biologic and transcriptomic analysis of Citrus hystrix responses to “Candidatus Liberibacter asiaticus” at different infection stages. Sci. Agric. Sin. 2020, 53, 1368–1380. [Google Scholar] [CrossRef]
- Zhao, W.; Baldwin, E.A.; Bai, J.; Plotto, A.; Irey, M. Comparative analysis of the transcriptomes of the calyx abscission zone of sweet orange insights into the huanglongbing-associated fruit abscission. Hort. Res. 2019, 6, 71. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, U.; Bowman, K.D. Transcriptional response of susceptible and tolerant citrus to infection with Candidatus Liberibacter asiaticus. Plant Sci. 2012, 185–186, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Zhong, X.; Liu, X.L.; Lou, B.H.; Zhou, C.Y.; Wang, X.F. Comparative transcriptome analysis unveils the tolerance mechanisms of Citrus hystrix in response to “Candidatus Liberibacter asiaticus” infection. PLoS ONE 2017, 12, e0189229. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.B.; Chen, C.X.; Du, D.L.; Huang, M.; Yao, J.Q.; Yu, F.H.; Brlansky, R.H.; Gmitter, F.G., Jr. Reprogramming of a defense signaling pathway in rough lemon and sweet orange is a critical element of the early response to “Candidatus Liberibacter asiaticus”. Hort. Res. 2017, 4, 17063. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Chen, C.X.; Yu, Q.B.; Brlansky, R.H.; Li, Z.G.; Gmitter, F.G., Jr. Comparative iTRAQ proteome and transcriptome analyses of sweet orange infected by “Candidatus Liberibacter asiaticus”. Physiol. Plant. 2011, 143, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.R.; Li, Y.; Zheng, Z.; Dai, Z.H.; Tao, Y.; Deng, X.L. Transcriptional analyses of mandarins seriously infected by “Candidatus Liberibacter asiaticus”. PLoS ONE 2015, 10, e0133652. [Google Scholar] [CrossRef]
- Balan, B.; Ibáñez, A.M.; Dandekar, A.M.; Caruso, T.; Martinelli, F. Identifying host molecular features strongly linked with responses to huanglongbing disease in citrus leaves. Front. Plant Sci. 2018, 9, 277. [Google Scholar] [CrossRef]
- Liao, H.L.; Burns, J.K. Gene expression in Citrus sinensis fruit tissues harvested from huanglongbing-infected trees: Comparison with girdled fruit. J. Exp. Bot. 2012, 63, 3307–3319. [Google Scholar] [CrossRef]
- Martinelli, F.; Uratsu, S.L.; Albrecht, U.; Reagan, R.L.; Phu, M.L.; Britton, M.; Buffalo, V.; Fass, J.; Leicht, E.; Zhao, W.X.; et al. Transcriptome profiling of citrus fruit response to huanglongbing disease. PLoS ONE 2012, 7, e38039. [Google Scholar] [CrossRef]
- Hung, W.L.; Wang, Y. A targeted mass spectrometry-based metabolomics approach toward the understanding of host responses to huanglongbing disease. J. Agric. Food Chem. 2018, 66, 10651–10661. [Google Scholar] [CrossRef]
- Cevallos-Cevallos, J.M.; Futch, D.B.; Shilts, T.; Folimonova, S.Y.; Reyes-De-Corcuera, J.I. GC-MS metabolomic differentiation of selected citrus varieties with different sensitivity to citrus huanglongbing. Plant Physiol. Biochem. 2012, 53, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Killiny, N.; Nehela, Y. Metabolomic response to Huanglongbing: Role of carboxylic compounds in citrus sinensis response to “Candidatus Liberibacter asiaticus” and its vector, Diaphorina citri. Mol. Plant-Microbe Interact. 2017, 30, 666–678. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.H.; Tang, X.X.; Zhang, Y.; Gmitter, F.G., Jr.; Wang, Y. Metabolomic analysis provides new insight into tolerance of Huanglongbing in citrus. Front. Plant Sci. 2021, 12, 710598. [Google Scholar] [CrossRef]
- Chin, E.L.; Mishchuk, D.O.; Breksa, A.P.; Slupsky, C.M. Metabolite signature of Candidatus Liberibacter asiaticus infection in two citrus varieties. J. Agric. Food Chem. 2014, 62, 6585–6591. [Google Scholar] [CrossRef] [PubMed]
- Slisz, A.M.; Breksa, A.P.; Mishchuk, D.O.; McCollum, G.; Slupsky, C.M. Metabolomic analysis of citrus infection by “Candidatus Liberibacter” reveals insight into pathogenicity. J. Proteome Res. 2012, 11, 4223–4230. [Google Scholar] [CrossRef]
- Freitas, D.D.S.; Carlos, E.F.; Gil, M.C.; Vieira, L.G.; Alcantara, G.B. NMR-based metabolomic analysis of Huanglongbing-asymptomatic and symptomatic citrus trees. J. Agric. Food Chem. 2015, 63, 7582–7588. [Google Scholar] [CrossRef]
- Yao, L.X.; Yu, Q.B.; Huang, M.; Hung, W.L.; Grosser, J.; Chen, S.C.; Wang, Y.; Gmitter, F.G., Jr. Proteomic and metabolomic analyses provide insight into the off-flavour of fruits from citrus trees infected with “Candidatus Liberibacter asiaticus”. Hort. Res. 2019, 6, 31. [Google Scholar] [CrossRef]
- Wang, F.Y.; Wu, Y.L.; Wu, W.; Huang, Y.J.; Zhu, C.Y.; Zhang, R.M.; Chen, J.Z.; Zeng, J.W. Integrative analysis of metabolome and transcriptome profiles provides insight into the fruit pericarp pigmentation disorder caused by “Candidatus Liberibacter asiaticus” infection. BMC Plant Biol. 2021, 21, 397. [Google Scholar] [CrossRef]
- Chin, E.L.; Ramsey, J.S.; Mishchuk, D.O.; Saha, S.; Foster, E.; Chavez, J.D.; Howe, K.; Zhong, X.F.; Polek, M.; Godfrey, K.E.; et al. Longitudinal transcriptomic, proteomic, and metabolomic analyses of Citrus sinensis (L.) Osbeck graft-inoculated with “Candidatus Liberibacter asiaticus”. J. Proteome Res. 2020, 19, 719–732. [Google Scholar] [CrossRef]
- Peng, T.; Kang, J.L.; Xiong, X.T.; Cheng, F.T.; Zhou, X.J.; Dai, W.S.; Wang, M.; Li, Z.Y.; Su, H.N.; Zhong, B.L. Integrated transcriptomics and metabolomics analyses provide insights into the response of Chongyi wild mandarin to Candidatus Liberibacter asiaticus infection. Front. Plant Sci. 2021, 12, 748209. [Google Scholar] [CrossRef]
- Robert-Seilaniantz, A.; Navarro, L.; Bari, R.; Jones, J.D. Pathological hormone imbalances. Curr. Opin. Plant Biol. 2007, 10, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Robert-Seilaniantz, A.; Grant, M.; Jones, J.D. Hormone crosstalk in plant disease and defense: More than just jasmonate-salicylate antagonism. Annu. Rev. Phytopathol. 2011, 49, 317–343. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.D.; Xu, M.R.; Guan, L.; Zheng, Z.; Zhu, Q.; Deng, X.L. Affects of blotchy-type Huanglongbing on fruit production and quality of Shatangju (Citrus reticulata Blanco). South China Fruits 2013, 42, 44–46. [Google Scholar] [CrossRef]
- Zhong, Y.; Cheng, C.Z.; Jiang, N.H.; Jiang, B.; Zhang, Y.Y.; Wu, B.; Hu, M.L.; Zeng, J.W.; Yan, H.X.; Yi, G.J.; et al. Comparative transcriptome and iTRAQ proteome analyses of citrus root responses to Candidatus Liberibacter asiaticus infection. PLoS ONE 2015, 10, e0126973. [Google Scholar] [CrossRef] [PubMed]
- Caspy, I.; Nelson, N. Structure of the plant photosystem I. Biochem. Soc. Trans. 2018, 46, 285–294. [Google Scholar] [CrossRef]
- Huang, W.; Cai, Y.F.; Wang, J.H.; Zhang, S.B. Chloroplastic ATP synthase plays an important role in the regulation of proton motive force in fluctuating light. J. Plant Physiol. 2018, 226, 40–47. [Google Scholar] [CrossRef]
- Kurisu, G.; Zhang, H.M.; Smith, J.L.; Cramer, W.A. Structure of the cytochrome b6f complex of oxygenic photosynthesis: Tuning the cavity. Science 2003, 302, 1009–1014. [Google Scholar] [CrossRef]
- Zhang, H.H.; Xu, Z.S.; Guo, K.W.; Huo, Y.Z.; He, G.Q.; Sun, H.W.; Guan, Y.P.; Xu, N.; Yang, W.; Sun, G.Y. Toxic effects of heavy metal Cd and Zn on chlorophyll, carotenoid metabolism and photosynthetic function in tobacco leaves revealed by physiological and proteomics analysis. Ecotoxicol. Environ. Saf. 2020, 202, 110856. [Google Scholar] [CrossRef]
- Warburg, O. Photosynthesis. Science 1958, 128, 68–73. [Google Scholar] [CrossRef]
- Asgher, M.; Sehar, Z.; Rehaman, A.; Rashid, S.; Ahmed, S.; Per, T.S.; Alyemeni, M.N.; Khan, N.A. Exogenously-applied L-glutamic acid protects photosynthetic functions and enhances arsenic tolerance through increased nitrogen assimilation and antioxidant capacity in rice (Oryza sativa L.). Environ. Pollut. 2022, 301, 119008. [Google Scholar] [CrossRef]
- Sugishima, M.; Wada, K.; Fukuyama, K.; Yamamoto, K. Crystal structure of phytochromobilin synthase in complex with biliverdin IXα, a key enzyme in the biosynthesis of phytochrome. J. Biol. Chem. 2020, 295, 771–782. [Google Scholar] [CrossRef] [PubMed]
- Adams, H.D.; Zeppel, M.; Anderegg, W.R.L.; Hartmann, H.; Landhäusser, S.M.; Tissue, D.T.; Huxman, T.E.; Hudson, P.J.; Franz, T.E.; Allen, C.D.; et al. A multi-speciessy nthesis of physiological mechanisms in drought-induced tree mortality. Nat. Ecol. Evol. 2017, 1, 1285–1291. [Google Scholar] [CrossRef]
- Webb, A.A.; Satake, A. Understanding circadian regulation of carbohydrate metabolism in arabidopsis using mathematical models. Plant Cell Physiol. 2015, 56, 586–593. [Google Scholar] [CrossRef] [PubMed]
- Etxeberria, E.; Gonzalez, P.; Achor, D.; Albrigo, G. Anatomical distribution of abnormally high levels of starch in HLB-affected Valencia orange trees. Physiol. Mol. Plant Pathol. 2009, 74, 76–83. [Google Scholar] [CrossRef]
- Fan, J.; Chen, C.; Brlansky, R.H.; Gmitter, F.G., Jr.; Li, Z.G. Changes in carbohydrate metabolism in Citrus sinensis infected with “Candidatus Liberibacter asiaticus”. Plant Pathol. 2010, 59, 1037–1043. [Google Scholar] [CrossRef]
- Guo, L.X.; Hussain, S.B.; Fernie, A.R.; Liu, Y.Z.; Yan, M.; Chen, H.; Alam, S.M. Multiomic analysis elucidates the reasons underlying the differential metabolite accumulation in citrus mature leaves and fruit juice sacs. J. Agric. Food Chem. 2020, 68, 11863–11874. [Google Scholar] [CrossRef]
- Wu, Y.; Su, H.N.; Huang, A.J.; Zhou, Y.; Li, Z.A.; Liu, J.X.; Zhou, C.Y. Effect of Candidatus Liberibacter asiaticus infection on carbohydrate metabolism in Citrus sinensis. Sci. Agric. Sin. 2015, 48, 63–72. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Yang, Z.L.; Liu, G.G.; Wu, Y.W.; Ouyang, J. Inhibitory effect of chestnut (Castanea mollissima Blume) inner skin extract on the activity of α-amylase, α-glucosidase, dipeptidyl peptidase IV and in vitro digestibility of starches. Food Chem. 2020, 324, 126847. [Google Scholar] [CrossRef]
- Akiyama, T.; Kaku, H.; Shibuya, N. A cell wall-bound beta-glucosidase from germinated rice: Purification and properties. Phytochemistry 1998, 48, 49–54. [Google Scholar] [CrossRef]
- Eklöf, S.; Åstot, C.; Sitbon, F.; Moritz, T.; Olsson, O.; Sandberg, G. Transgenic tobacco plants co-expressing Agrobacterium iaa and ipt genes have wild-type hormone levels but display both auxin- and cytokinin-overproducing phenotypes. Plant J. 2000, 23, 279–284. [Google Scholar] [CrossRef]
- Wallmeroth, N.; Jeschke, D.; Slane, D.; Nägele, J.; Veerabagu, M.; Mira-Rodado, V.; Berendzen, K.W. ARR22 overexpression can suppress plant Two-Component Regulatory Systems. PLoS ONE 2019, 14, e0212056. [Google Scholar] [CrossRef]
- He, L.; Zhang, F.; Wu, X.; Hu, Y.; Dong, L.; Dewitte, W.; Wen, B. Genome-wide characterization and expression of two-component system genes in cytokinin-regulated gall formation in Zizania latifolia. Plants 2020, 9, 1409. [Google Scholar] [CrossRef] [PubMed]
- Spaepen, S.; Vanderleyden, J.; Remans, R. Indole-3-acetic acid in microbial and microorganism-plant signaling. FEMS Microbiol. Rev. 2007, 31, 425–448. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mostafa, S.; Zeng, W.; Jin, B. Function and mechanism of jasmonic acid in plant responses to abiotic and biotic stresses. Int. J. Mol. Sci. 2021, 22, 8568. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Kano, A.; Tamaoki, D.; Miyamoto, A.; Shishido, H.; Miyoshi, S.; Taniguchi, S.; Akimitsu, K.; Gomi, K. Involvement of OsJAZ8 in jasmonate-induced resistance to bacterial blight in rice. Plant Cell Physiol. 2012, 53, 2060–2072. [Google Scholar] [CrossRef]
- Delfin, J.C.; Kanno, Y.; Seo, M.; Kitaoka, N.; Matsuura, H.; Tohge, T.; Shimizu, T. AtGH3.10 is another jasmonic acid-amido synthetase in Arabidopsis thaliana. Plant J. 2022, 110, 1082–1096. [Google Scholar] [CrossRef]
- Yang, Y.X.; Ahammed, G.J.; Wan, C.P.; Liu, H.J.; Chen, R.R.; Zhou, Y. Comprehensive analysis of TIFY transcription factors and their expression profiles under jasmonic acid and abiotic stresses in watermelon. Int. J. Genomics. 2019, 2019, 6813086. [Google Scholar] [CrossRef]
- Fan, J.; Hill, L.; Crooks, C.; Doerner, P.; Lamb, C. Abscisic acid has a key role in modulating diverse plant-pathogen interactions. Plant Physiol. 2009, 150, 1750–1761. [Google Scholar] [CrossRef]
- Zhou, J.M.; Zhang, Y.L. Plant immunity: Danger perception and signaling. Cell 2020, 181, 978–989. [Google Scholar] [CrossRef]
- Yadav, V.; Wang, Z.Y.; Wei, C.H.; Amo, A.; Ahmed, B.; Yang, X.Z.; Zhang, X. Phenylpropanoid pathway engineering: An emerging approach towards plant defense. Pathogens 2020, 9, 312. [Google Scholar] [CrossRef]
- Jadhav, P.R.; Mahatma, M.K.; Mahatma, L.; Jha, S.; Parekh, V.B.; Khandelwal, V. Expression analysis of key genes of phenylpropanoid pathway and phenol profiling during Ricinus communis-Fusarium oxysporum f. sp. ricini interaction. Ind. Crop. Prod. 2013, 50, 456–461. [Google Scholar] [CrossRef]
- Xue, A.H.; Liu, Y.Q.; Li, H.X.; Cui, M.; Huang, X.Y.; Wang, W.J.; Wu, D.; Guo, X.L.; Hao, Y.B.; Luo, L.P. Early detection of Huanglongbing with EESI-MS indicates a role of phenylpropanoid pathway in citrus. Anal. Biochem. 2022, 639, 114511. [Google Scholar] [CrossRef] [PubMed]
- Aghaei, K.; Pirbalouti, A.G.; Mousavi, A.; Badi, H.N.; Mehnatkesh, A. Effects of foliar spraying of l-phenylalanine and application of bio-fertilizers on growth, yield, and essential oil of hyssop [Hyssopus officinalis l. subsp. Angustifolius (Bieb.)]. Biocatal. Agric. Biotechnol. 2019, 21, 101318. [Google Scholar] [CrossRef]
- Miao, M.; Xiang, L. Pharmacological action and potential targets of chlorogenic acid. Adv. Pharmacol. 2020, 87, 71–88. [Google Scholar] [CrossRef]
- Zandalinas, S.I.; Sales, C.; Beltrán, J.; Gómez-Cadenas, A.; Arbona, A. Activation of secondary metabolism in citrus plants is associated to sensitivity to combined drought and high temperatures. Front. Plant Sci. 2017, 7, 1954. [Google Scholar] [CrossRef]
- Lafuente, M.T.; Zacarias, L.; Martínez-Téllez, M.A.; Sanchez-Ballesta, M.T.; Granell, A. Phenylalanine ammonia-lyase and ethylene in relation to chilling injury as affected by fruit age in citrus. Postharvest Biol. Technol. 2003, 29, 308–317. [Google Scholar] [CrossRef]
- Cui, K.; Wang, H.Y.; Liao, S.X.; Tang, Q.; Li, L.; Cui, Y.Z.; Yuan, H. Transcriptome sequencing and analysis for culm elongation of the world’s largest bamboo (Dendrocalamus sinicus). PLoS ONE 2016, 11, e0157362. [Google Scholar] [CrossRef]
- Hijaz, F.; Al-Rimawi, F.; Manthey, J.A.; Killiny, N. Phenolics, flavonoids and antioxidant capacities in Citrus species with different degree of tolerance to Huanglongbing. Plant Signal. Behav. 2020, 15, 1752447. [Google Scholar] [CrossRef]
- Bao, M.L.; Zheng, Z.; Sun, X.A.; Chen, J.C.; Deng, X.L. Enhancing PCR capacity to detect “Candidatus Liberibacter asiaticus” utilizing whole genome sequence information. Plant Dis. 2020, 104, 527–532. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Want, E.J.; Wilson, I.D.; Gika, H.; Theodoridis, G.; Plumb, R.S.; Shockcor, S.; Holmes, E.; Nicholson, J.K. Global metabolic profiling procedures for urine using UPLC–MS. Nat. Protoc. 2010, 5, 1005–1018. [Google Scholar] [CrossRef] [PubMed]
- Wen, B.; Mei, Z.; Zeng, C.; Liu, S. metaX: A flexible and comprehensive software for processing metabolomics data. BMC Bioinform. 2017, 18, 183. [Google Scholar] [CrossRef] [PubMed]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, X.; Zhan, X.; Liu, Y.; Huang, Z.; Deng, X.; Zheng, Z.; Xu, M. Integrated Transcriptome and Metabolome Analysis Reveals Phenylpropanoid Biosynthesis and Phytohormone Signaling Contribute to “Candidatus Liberibacter asiaticus” Accumulation in Citrus Fruit Piths (Fluffy Albedo). Int. J. Mol. Sci. 2022, 23, 15648. https://doi.org/10.3390/ijms232415648
Cui X, Zhan X, Liu Y, Huang Z, Deng X, Zheng Z, Xu M. Integrated Transcriptome and Metabolome Analysis Reveals Phenylpropanoid Biosynthesis and Phytohormone Signaling Contribute to “Candidatus Liberibacter asiaticus” Accumulation in Citrus Fruit Piths (Fluffy Albedo). International Journal of Molecular Sciences. 2022; 23(24):15648. https://doi.org/10.3390/ijms232415648
Chicago/Turabian StyleCui, Xiaoqing, Xuanlin Zhan, Yangyang Liu, Zhenhui Huang, Xiaoling Deng, Zheng Zheng, and Meirong Xu. 2022. "Integrated Transcriptome and Metabolome Analysis Reveals Phenylpropanoid Biosynthesis and Phytohormone Signaling Contribute to “Candidatus Liberibacter asiaticus” Accumulation in Citrus Fruit Piths (Fluffy Albedo)" International Journal of Molecular Sciences 23, no. 24: 15648. https://doi.org/10.3390/ijms232415648
APA StyleCui, X., Zhan, X., Liu, Y., Huang, Z., Deng, X., Zheng, Z., & Xu, M. (2022). Integrated Transcriptome and Metabolome Analysis Reveals Phenylpropanoid Biosynthesis and Phytohormone Signaling Contribute to “Candidatus Liberibacter asiaticus” Accumulation in Citrus Fruit Piths (Fluffy Albedo). International Journal of Molecular Sciences, 23(24), 15648. https://doi.org/10.3390/ijms232415648