

A Study on the Protective Effect of sRAGE-MSCs in a Rodent Reperfusion Model of Myocardial Infarction

Abstract
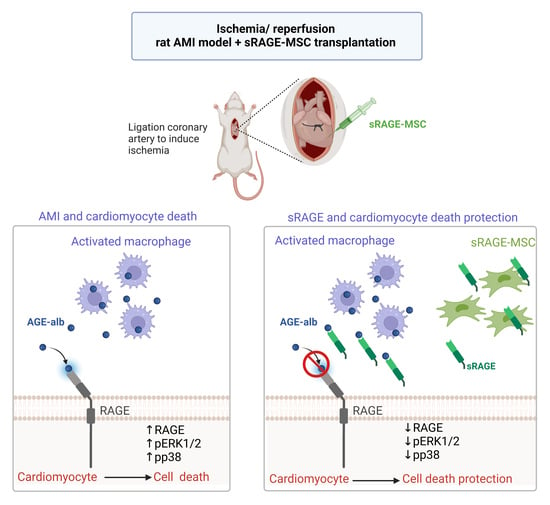
1. Introduction
2. Results
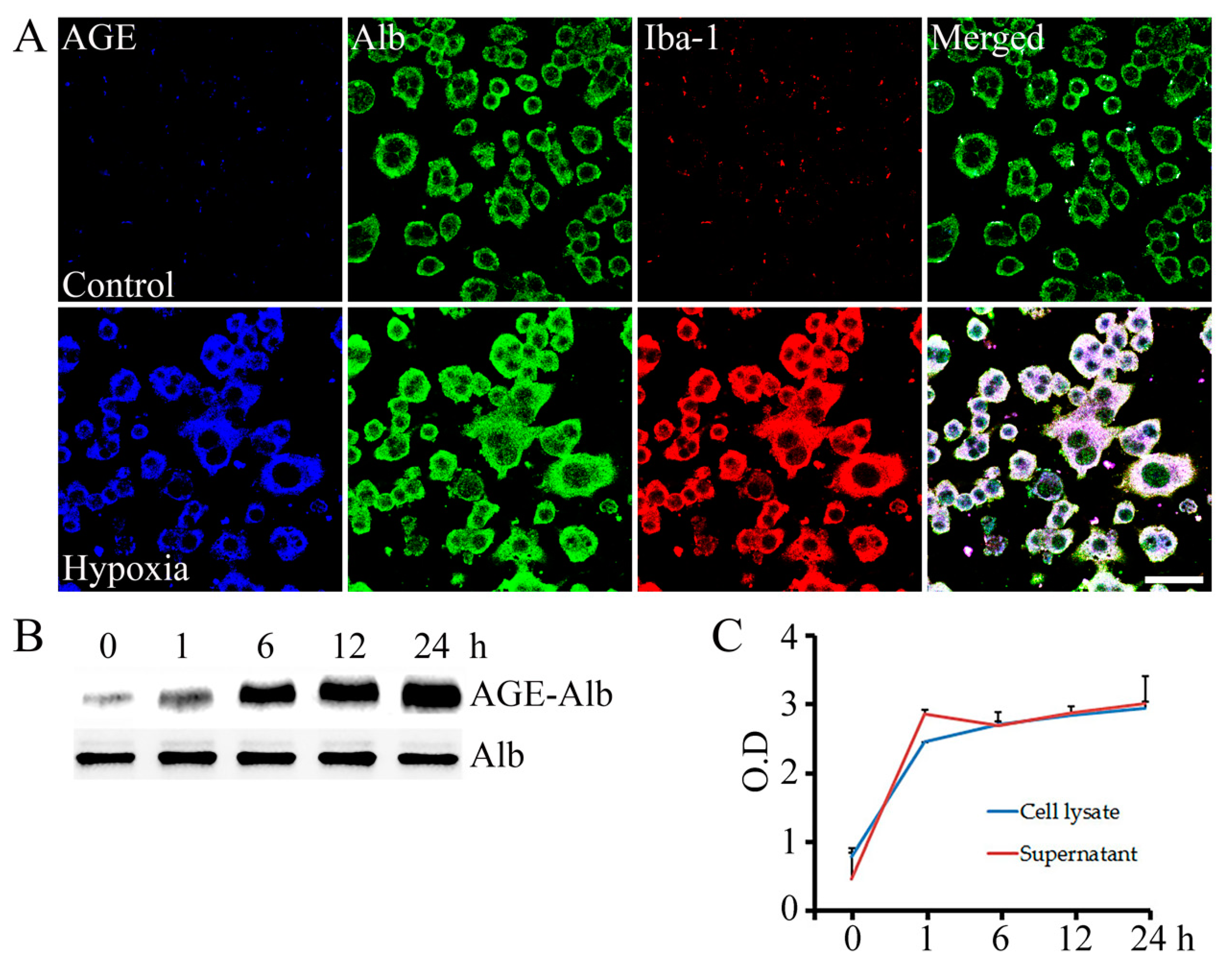
2.1. AGE-Albumin Synthesis and Secretion in AMI Hearts of Humans and Rats
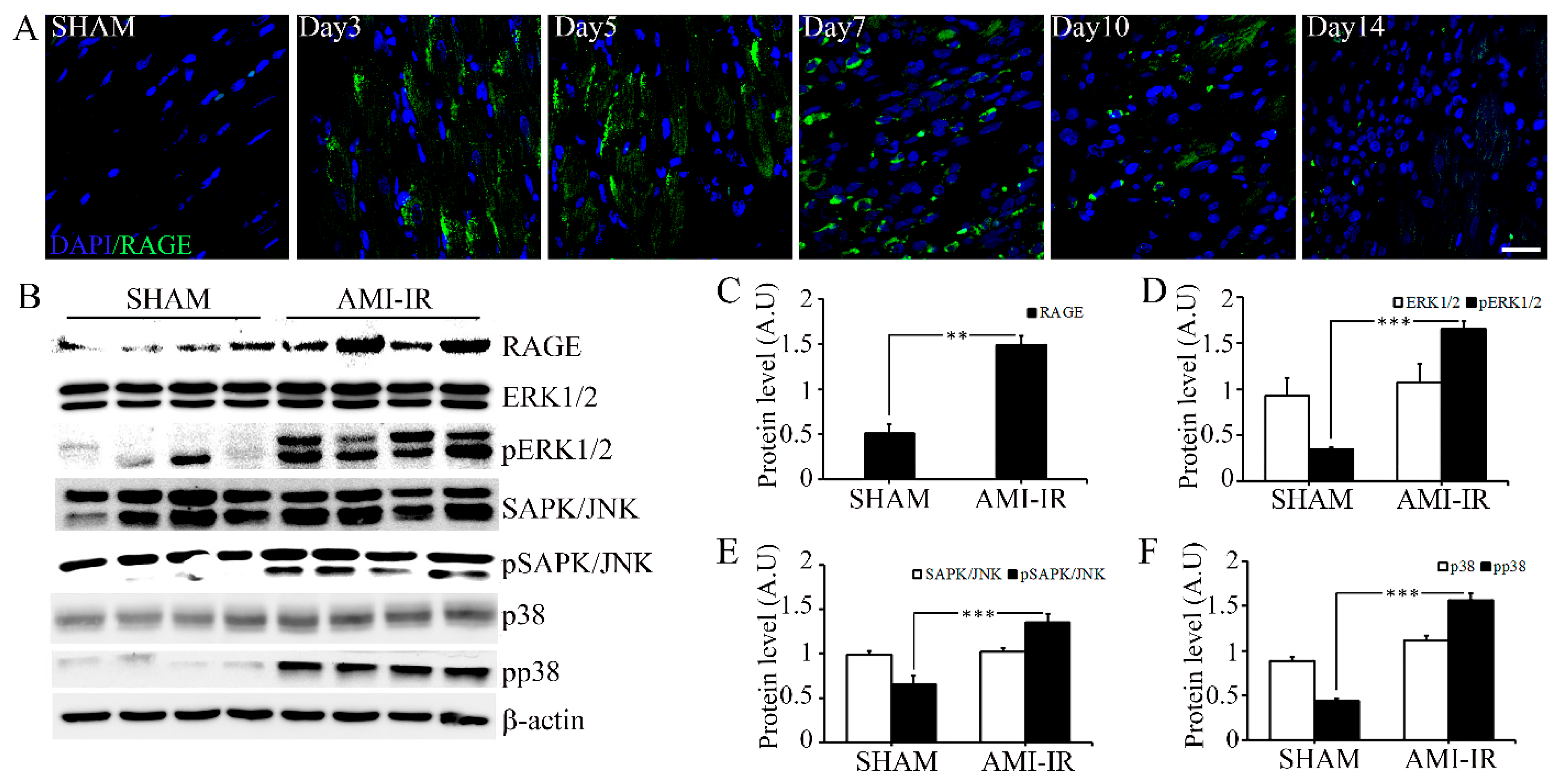
2.2. Post-Ischemic Reperfusion Injury Increases RAGE in AGE-Dependent Manner
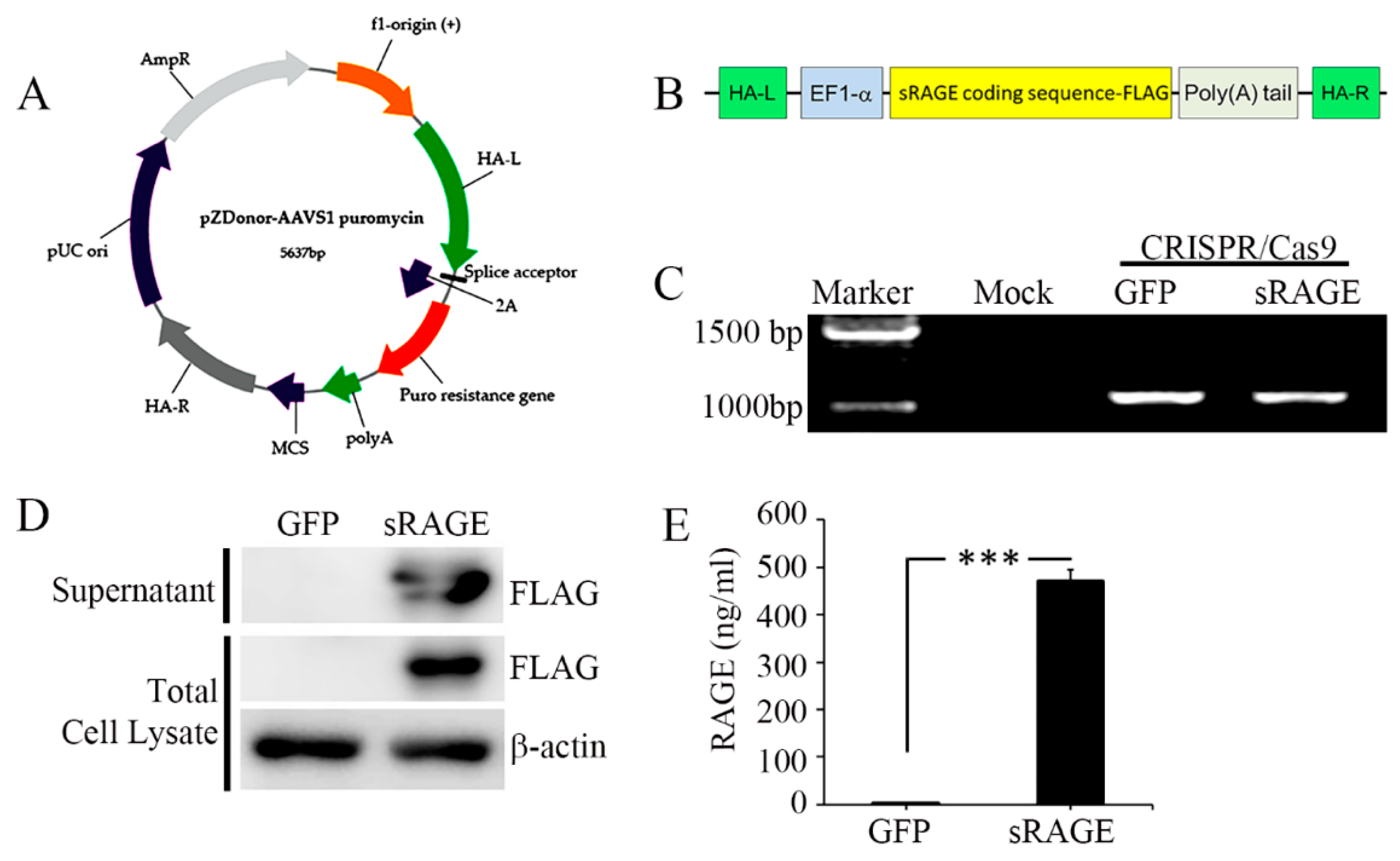
2.3. The sRAGE-Secreting MSC Is a Novel Therapy for AMI
3. Discussion
4. Materials and Methods
4.1. Human Heart Tissue
4.2. Cell Culture
4.3. Generation and Characterization of sRAGE-Secreting MSC
4.4. Myocardial Infarction Modeling and sRAGE-MSC Transplantation
4.5. Animal Heart Tissue Preparation
4.6. Evaluation of Infarct Size
4.7. Immunostaining
4.8. DAB (3,3′-Diaminobenzidine) Staining and Cell Counting
4.9. The Terminal Deoxynucleotidyl Transferase dUTP Nick End Labeling (TUNEL) Assay
4.10. Co-Immunoprecipitation
4.11. Enzyme-Linked Immunosorbent Assay (ELISA)
4.12. Densitometry and Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Bayarsaikhan, D.; Bayarsaikhan, G.; Lee, B. Recent advances in stem cells and gene editing: Drug discovery and therapeutics. Prog. Mol. Biol. Transl. Sci. 2021, 181, 231–269. [Google Scholar]
- Mc Namara, K.; Alzubaidi, H.; Jackson, J.K. Cardiovascular disease as a leading cause of death: How are pharmacists getting involved? Integr. Pharm. Res. Pract. 2019, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Buja, L.M. Myocardial ischemia and reperfusion injury. Cardiovasc Pathol. 2005, 14, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Frank, A.; Bonney, M.; Bonney, S.; Weitzel, L.; Koeppen, M.; Eckle, T. Myocardial ischemia reperfusion injury: From basic science to clinical bedside. Semin. Cardiothorac. Vasc. Anesth. 2012, 16, 123–132. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Yellon, D.M. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J. Clin. Investig. 2013, 123, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Dong, X.; Tian, M.; Liu, C.; Wang, K.; Li, L.; Liu, Z.; Liu, J. Stem cell-based therapies for ischemic stroke: A systematic review and meta-analysis of clinical trials. Stem. Cell Res. Ther. 2020, 11, 252. [Google Scholar] [CrossRef]
- Goradel, N.H.; Hour, F.G.; Negahdari, B.; Malekshahi, Z.V.; Hashemzehi, M.; Masoudifar, A.; Mirzaei, H. Stem Cell Therapy: A New Therapeutic Option for Cardiovascular Diseases. J. Cell Biochem. 2018, 119, 95–104. [Google Scholar] [CrossRef]
- Wernly, B.; Mirna, M.; Rezar, R.; Prodinger, C.; Jung, C.; Podesser, B.K.; Kiss, A.; Hoppe, U.C.; Lichtenauer, M. Regenerative Cardiovascular Therapies: Stem Cells and Beyond. Int. J. Mol. Sci. 2019, 20, 1420. [Google Scholar] [CrossRef]
- Müller, P.; Lemcke, H.; David, R. Stem Cell Therapy in Heart Diseases—Cell Types, Mechanisms and Improvement Strategies. Cell. Physiol. Biochem. 2018, 48, 2607–2655. [Google Scholar] [CrossRef]
- Nguyen, P.K.; Rhee, J.W.; Wu, J.C. Adult Stem Cell Therapy and Heart Failure, 2000 to 2016: A Systematic Review. JAMA Cardiol. 2016, 1, 831–841. [Google Scholar] [CrossRef]
- Kawaguchi, M.; Takahashi, M.; Hata, T.; Kashima, Y.; Usui, F.; Morimoto, H.; Izawa, A.; Takahashi, Y.; Masumoto, J.; Koyama, J.; et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation 2011, 123, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Weinberger, T.; Schulz, C. Myocardial infarction: A critical role of macrophages in cardiac remodeling. Front. Physiol. 2015, 6, 107. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G.; Smith, C.W.; Entman, M.L. The inflammatory response in myocardial infarction. Cardiovasc. Res. 2002, 53, 31–47. [Google Scholar] [CrossRef]
- Niu, J.; Kolattukudy, P.E. Role of MCP-1 in cardiovascular disease: Molecular mechanisms and clinical implications. Clin. Sci. 2009, 117, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, H.; Takahashi, M.; Izawa, A.; Ise, H.; Hongo, M.; Kolattukudy, P.E.; Ikeda, U. Cardiac overexpression of monocyte chemoattractant protein-1 in transgenic mice prevents cardiac dysfunction and remodeling after myocardial infarction. Circ. Res. 2006, 99, 891–899. [Google Scholar] [CrossRef]
- Kakio, T.; Matsumori, A.; Ono, K.; Ito, H.; Matsushima, K.; Sasayama, S. Roles and relationship of macrophages and monocyte chemotactic and activating factor/monocyte chemoattractant protein-1 in the ischemic and reperfused rat heart. Lab. Investig. 2000, 80, 1127–1136. [Google Scholar]
- Son, M.; Kang, W.C.; Oh, S.; Bayarsaikhan, D.; Ahn, H.; Lee, J.; Park, H.; Lee, S.; Choi, J.; Lee, H.S.; et al. Advanced glycation end-product (AGE)-albumin from activated macrophage is critical in human mesenchymal stem cells survival and post-ischemic reperfusion injury. Sci. Rep. 2017, 7, 11593. [Google Scholar] [CrossRef]
- Bayarsaikhan, G.; Bayarsaikhan, D.; Lee, J.; Lee, B. Targeting Scavenger Receptors in Inflammatory Disorders and Oxidative Stress. Antioxidants 2022, 11, 936. [Google Scholar] [CrossRef]
- Byun, K.; Bayarsaikhan, E.; Kim, D.; Kim, C.Y.; Mook-Jung, I.; Paek, S.H.; Kim, S.U.; Yamamoto, T.; Won, M.H.; Song, B.J.; et al. Induction of neuronal death by microglial AGE-albumin: Implications for Alzheimer’s disease. PLoS ONE 2012, 7, e37917. [Google Scholar] [CrossRef]
- Rungratanawanich, W.; Qu, Y.; Wang, X.; Essa, M.M.; Song, B.J. Advanced glycation end products (AGEs) and other adducts in aging-related diseases and alcohol-mediated tissue injury. Exp. Mol. Med. 2021, 53, 168–188. [Google Scholar] [CrossRef]
- Bayarsaikhan, G.; Bayarsaikhan, D.; Oh, P.C.; Kang, W.C.; Lee, B. CUPRAC-Reactive Advanced Glycation End Products as Prognostic Markers of Human Acute Myocardial Infarction. Antioxidants 2021, 10, 434. [Google Scholar] [CrossRef] [PubMed]
- Byun, K.; Yoo, Y.; Son, M.; Lee, J.; Jeong, G.B.; Park, Y.M.; Salekdeh, G.H.; Lee, B. Advanced glycation end-products produced systemically and by macrophages: A common contributor to inflammation and degenerative diseases. Pharmacol. Ther. 2017, 177, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Neviere, R.; Yu, Y.; Wang, L.; Tessier, F.; Boulanger, E. Implication of advanced glycation end products (Ages) and their receptor (Rage) on myocardial contractile and mitochondrial functions. Glycoconj. J. 2016, 33, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Zhao, Y.; Xu, S.; Ding, F.; Jin, C.; Fu, G.; Weng, S. Advanced Glycation End Product (AGE)-AGE Receptor (RAGE) System Upregulated Connexin43 Expression in Rat Cardiomyocytes via PKC and Erk MAPK Pathways. Int. J. Mol. Sci. 2013, 14, 2242–2257. [Google Scholar] [CrossRef]
- Cho, H.J.; Xie, C.; Cai, H. AGE-induced neuronal cell death is enhanced in G2019S LRRK2 mutation with increased RAGE expression. Transl. Neurodegener. 2018, 7, 1. [Google Scholar] [CrossRef]
- Aleshin, A.; Ananthakrishnan, R.; Li, Q.; Rosario, R.; Lu, Y.; Qu, W.; Song, F.; Bakr, S.; Szabolcs, M.; D’Agati, V.; et al. RAGE modulates myocardial injury consequent to LAD infarction via impact on JNK and STAT signaling in a murine model. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H1823–H1832. [Google Scholar] [CrossRef]
- Yan, S.F.; Ramasamy, R.; Schmidt, A.M. The RAGE axis: A fundamental mechanism signaling danger to the vulnerable vasculature. Circ. Res. 2010, 106, 842–853. [Google Scholar] [CrossRef]
- Xu, Y.; Toure, F.; Qu, W.; Lin, L.; Song, F.; Shen, X.; Rosario, R.; Garcia, J.; Schmidt, A.M.; Yan, S.F. Advanced glycation end product (AGE)-receptor for AGE (RAGE) signaling and up-regulation of Egr-1 in hypoxic macrophages. J. Biol. Chem. 2010, 285, 23233–23240. [Google Scholar] [CrossRef]
- Shang, L.; Ananthakrishnan, R.; Li, Q.; Quadri, N.; Abdillahi, M.; Zhu, Z.; Qu, W.; Rosario, R.; Touré, F.; Yan, S.F.; et al. RAGE modulates hypoxia/reoxygenation injury in adult murine cardiomyocytes via JNK and GSK-3beta signaling pathways. PLoS ONE 2010, 5, e10092. [Google Scholar] [CrossRef]
- Yue, J.; López, J.M. Understanding MAPK Signaling Pathways in Apoptosis. Int. J. Mol. Sci. 2020, 21, 2346. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, X.; Kubo, H.; Harris, D.M.; Mills, G.D.; Moyer, J.; Berretta, R.; Potts, S.T.; Marsh, J.D.; Houser, S.R. Ca2+ influx-induced sarcoplasmic reticulum Ca2+ overload causes mitochondrial-dependent apoptosis in ventricular myocytes. Circ. Res. 2005, 97, 1009–1017. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Xu, Q.; Yang, Q.; Cao, J.; Wu, C.; Peng, H.; Zhang, X.; Chen, J.; Cheng, G.; Wu, Y.; et al. Vascular peroxidase 1 is a novel regulator of cardiac fibrosis after myocardial infarction. Redox Biol. 2019, 22, 101151. [Google Scholar] [CrossRef]
- Byun, K.; Bayarsaikhan, E.; Kim, D.; Son, M.; Hong, J.; Jeong, G.B.; Paek, S.H.; Won, M.H.; Lee, B. Activated microglial cells synthesize and secrete AGE-albumin. Anat. Cell Biol. 2012, 45, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Byun, K.; Bayarsaikhan, D.; Bayarsaikhan, E.; Son, M.; Oh, S.; Lee, J.; Son, H.I.; Won, M.H.; Kim, S.U.; Song, B.J.; et al. Microglial AGE-albumin is critical in promoting alcohol-induced neurodegeneration in rats and humans. PLoS ONE 2014, 9, e104699. [Google Scholar] [CrossRef] [PubMed]
- Bayarsaikhan, E.; Bayarsaikhan, D.; Lee, J.; Son, M.; Oh, S.; Moon, J.; Park, H.J.; Roshini, A.; Kim, S.U.; Song, B.J.; et al. Microglial AGE-albumin is critical for neuronal death in Parkinson’s disease: A possible implication for theranostics. Int. J. Nanomed. 2016, 10, 281–292. [Google Scholar]
- Frangogiannis, N.G. Cardiac fibrosis: Cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol. Aspects Med. 2019, 65, 70–99. [Google Scholar] [CrossRef]
- Hu, S.; Yang, M.; Huang, S.; Zhong, S.; Zhang, Q.; Ding, H.; Xiong, X.; Hu, Z.; Yang, Y. Different Roles of Resident and Non-resident Macrophages in Cardiac Fibrosis. Front. Cardiovasc. Med. 2022, 9, 818188. [Google Scholar] [CrossRef]
- Yashima, H.; Terasaki, M.; Sotokawauchi, A.; Matsui, T.; Mori, Y.; Saito, T.; Osaka, N.; Kushima, H.; Hiromura, M.; Ohara, M.; et al. AGE-RAGE Axis Stimulates Oxidized LDL Uptake into Macrophages through Cyclin-Dependent Kinase 5-CD36 Pathway via Oxidative Stress Generation. Int. J. Mol. Sci. 2020, 21, 9263. [Google Scholar] [CrossRef]
- Zhang, L.; He, J.; Wang, J.; Liu, J.; Chen, Z.; Deng, B.; Wei, L.; Wu, H.; Liang, B.; Li, H.; et al. Knockout RAGE alleviates cardiac fibrosis through repressing endothelial-to-mesenchymal transition (EndMT) mediated by autophagy. Cell Death Dis. 2021, 12, 470. [Google Scholar] [CrossRef]
- Wang, Q.; Zhu, G.; Cao, X.; Dong, J.; Song, F.; Niu, Y. Blocking AGE-RAGE Signaling Improved Functional Disorders of Macrophages in Diabetic Wound. J. Diabetes Res. 2017, 2017, 1428537. [Google Scholar] [CrossRef]
- Senatus, L.M.; Schmidt, A.M. The AGE-RAGE Axis: Implications for Age-Associated Arterial Diseases. Front. Genet. 2017, 8, 187. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Su, X.; Huang, G.; Xin, X.F.; Cao, E.H.; Shi, Y.; Song, Y. sRAGE alleviates neutrophilic asthma by blocking HMGB1/RAGE signalling in airway dendritic cells. Sci. Rep. 2017, 7, 14268. [Google Scholar] [CrossRef]
- Jensen, L.J.; Lindberg, S.; Hoffmann, S.; Iversen, A.Z.; Pedersen, S.H.; Møgelvang, R.; Galatius, S.; Flyvbjerg, A.; Jensen, J.S.; Bjerre, M. Dynamic changes in sRAGE levels and relationship with cardiac function in STEMI patients. Clin. Biochem. 2015, 48, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Selejan, S.R.; Hewera, L.; Hohl, M.; Kazakov, A.; Ewen, S.; Kindermann, I.; Böhm, M.; Link, A. Suppressed MMP-9 Activity in Myocardial Infarction-Related Cardiogenic Shock Implies Diminished Rage Degradation. Shock 2017, 48, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Grauen Larsen, H.; Marinkovic, G.; Nilsson, P.M.; Nilsson, J.; Engström, G.; Melander, O.; Orho-Melander, M.; Schiopu, A. High Plasma sRAGE (Soluble Receptor for Advanced Glycation End Products) Is Associated with Slower Carotid Intima-Media Thickness Progression and Lower Risk for First-Time Coronary Events and Mortality. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 925–933. [Google Scholar] [CrossRef]
- Liu, Y.; Guo, X.; Zhang, J.; Han, X.; Wang, H.; Du, F.; Zeng, X.; Guo, C. Protective Effects of the Soluble Receptor for Advanced Glycation End-Products on Pyroptosis during Myocardial Ischemia-Reperfusion. Oxid. Med. Cell Longev. 2021, 2021, 9570971. [Google Scholar] [CrossRef]
- Zhang, X.; Xie, J.; Sun, H.; Wei, Q.; Nong, G. sRAGE Inhibits the Mucus Hypersecretion in a Mouse Model with Neutrophilic Asthma. Immunol. Investig. 2022, 51, 1243–1256. [Google Scholar] [CrossRef]
- Prasad, K. AGE-RAGE Stress in the Pathophysiology of Atrial Fibrillation and Its Treatment. Int. J. Angiol. 2020, 29, 72–80. [Google Scholar] [CrossRef]
- Quade-Lyssy, P.; Kanarek, A.M.; Baiersdörfer, M.; Postina, R.; Kojro, E. Statins stimulate the production of a soluble form of the receptor for advanced glycation end products. J. Lipid. Res. 2013, 54, 3052–3061. [Google Scholar] [CrossRef]
- Falcone, C.; Bozzini, S.; D’Angelo, A.; Matrone, B.; Colonna, A.; Benzi, A.; Paganini, E.M.; Falcone, R.; Pelissero, G. Plasma levels of soluble receptor for advanced glycation end products and coronary atherosclerosis: Possible correlation with clinical presentation. Dis. Markers 2013, 35, 135–140. [Google Scholar] [CrossRef]
- Lee, J.; Bayarsaikhan, D.; Arivazhagan, R.; Park, H.; Lim, B.; Gwak, P.; Jeong, G.B.; Lee, J.; Byun, K.; Lee, B. CRISPR/Cas9 Edited sRAGE-MSCs Protect Neuronal Death in Parkinson’s Disease Model. Int. J. Stem Cells 2019, 12, 114–124. [Google Scholar] [CrossRef]
- Park, M.J.; Lee, S.H.; Moon, S.J.; Lee, J.A.; Lee, E.J.; Kim, E.K.; Park, J.S.; Lee, J.; Min, J.K.; Kim, S.J.; et al. Overexpression of soluble RAGE in mesenchymal stem cells enhances their immunoregulatory potential for cellular therapy in autoimmune arthritis. Sci. Rep. 2016, 6, 35933. [Google Scholar] [CrossRef] [PubMed]
- Attar, A.; Monabati, A.; Montaseri, M.; Vosough, M.; Hosseini, S.A.; Kojouri, J.; Abdi-Ardekani, A.; Izadpanah, P.; Azarpira, N.; Pouladfar, G.; et al. Transplantation of mesenchymal stem cells for prevention of acute myocardial infarction induced heart failure: Study protocol of a phase III randomized clinical trial (Prevent-TAHA8). Trials 2022, 23, 632. [Google Scholar] [CrossRef] [PubMed]
- Attar, A.; Bahmanzadegan Jahromi, F.; Kavousi, S.; Monabati, A.; Kazemi, A. Mesenchymal stem cell transplantation after acute myocardial infarction: A meta-analysis of clinical trials. Stem. Cell Res. Ther. 2021, 12, 600. [Google Scholar] [CrossRef] [PubMed]
- van den Akker, F.; Deddens, J.C.; Doevendans, P.A.; Sluijter, J.P. Cardiac stem cell therapy to modulate inflammation upon myocardial infarction. Biochim. Biophys. Acta 2013, 1830, 2449–2458. [Google Scholar] [CrossRef]
- Suzuki, E.; Fujita, D.; Takahashi, M.; Oba, S.; Nishimatsu, H. Therapeutic Effects of Mesenchymal Stem Cell-Derived Exosomes in Cardiovascular Disease. Adv. Exp. Med. Biol. 2017, 998, 179–185. [Google Scholar]
- Milutinovic, P.S.; Englert, J.M.; Crum, L.T.; Mason, N.S.; Ramsgaard, L.; Enghild, J.J.; Sparvero, L.J.; Lotze, M.T.; Oury, T.D. Clearance kinetics and matrix binding partners of the receptor for advanced glycation end products. PLoS ONE 2014, 9, e88259. [Google Scholar] [CrossRef]
- Suchal, K.; Malik, S.; Khan, S.I.; Malhotra, R.K.; Goyal, S.N.; Bhatia, J.; Kumari, S.; Ojha, S.; Arya, D.S. Protective effect of mangiferin on myocardial ischemia-reperfusion injury in streptozotocin-induced diabetic rats: Role of AGE-RAGE/MAPK pathways. Sci. Rep. 2017, 7, 42027. [Google Scholar] [CrossRef]
- Takagawa, J.; Zhang, Y.; Wong, M.L.; Sievers, R.E.; Kapasi, N.K.; Wang, Y.; Yeghiazarians, Y.; Lee, R.J.; Grossman, W.; Springer, M.L. Myocardial infarct size measurement in the mouse chronic infarction model: Comparison of area- and length-based approaches. J. Appl. Physiol. 2007, 102, 2104–2111. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antigen | Host | Company (Cat No) | Working Dilution |
|---|---|---|---|
| Albumin | Mouse | Abcam (ab10241) | IF-1:100, WB-1:1000 |
| AGE | Rabbit | Abcam (ab23722) | IF-1:200, WB-1:3000 |
| Iba-1 | Goat | Abcam (ab5076) | IF-1:100 |
| RAGE | Goat | Abcam (ab7764) | IF-1:400, WB-1:4000 |
| FLAG | Rabbit | Sigma-aldrich (F7425) | WB-1:1000 |
| p38 | Rabbit | Cell signaling (9212L) | WB-1:1000 |
| pp38 | Rabbit | Cell signaling (9211S) | WB-1:1000 |
| ERK1/2 | Rabbit | Cell signaling (9102S) | WB-1:1000 |
| pERK1/2 | Rabbit | Cell signaling (4377S) | WB-1:1000 |
| SAPK/JNK | Rabbit | Cell signaling (9252S) | WB-1:1000 |
| pSAPK/JNK | Rabbit | Cell signaling (9251S) | WB-1:1000 |
| Β-Actin | Rabbit | Abcam (ab8227) | WB-1:1000 |
| Peroxidase-labeled anti-mouse IgG | Mouse | Vector (PI2000) | WB-1:5000 |
| Peroxidase labeled anti-rabbit IgG | Rabbit | Vector (PI 1000) | WB-1:5000 |
| Peroxidase-labeled anti-goat IgG | Goat | Vector (PI9500) | WB-1:5000 |
| Alexa Fluor 555 donkey anti-rabbit IgG | Rabbit | Invitrogen (A31572) | IF-1:500 |
| Alexa Fluor 633 goat anti-rabbit IgG | Rabbit | Invitrogen (A21070) | IF-1:500 |
| Alexa Fluor 555 donkey anti-goat IgG | Goat | Invitrogen (A21432) | IF-1:500 |
| Alexa Fluor 488 donkey anti mouse | Mouse | Invitrogen (A11001) | IF-1:500 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bayarsaikhan, D.; Bayarsaikhan, G.; Lee, J.; Lee, B. A Study on the Protective Effect of sRAGE-MSCs in a Rodent Reperfusion Model of Myocardial Infarction. Int. J. Mol. Sci. 2022, 23, 15630. https://doi.org/10.3390/ijms232415630
Bayarsaikhan D, Bayarsaikhan G, Lee J, Lee B. A Study on the Protective Effect of sRAGE-MSCs in a Rodent Reperfusion Model of Myocardial Infarction. International Journal of Molecular Sciences. 2022; 23(24):15630. https://doi.org/10.3390/ijms232415630
Chicago/Turabian StyleBayarsaikhan, Delger, Govigerel Bayarsaikhan, Jaewon Lee, and Bonghee Lee. 2022. "A Study on the Protective Effect of sRAGE-MSCs in a Rodent Reperfusion Model of Myocardial Infarction" International Journal of Molecular Sciences 23, no. 24: 15630. https://doi.org/10.3390/ijms232415630
APA StyleBayarsaikhan, D., Bayarsaikhan, G., Lee, J., & Lee, B. (2022). A Study on the Protective Effect of sRAGE-MSCs in a Rodent Reperfusion Model of Myocardial Infarction. International Journal of Molecular Sciences, 23(24), 15630. https://doi.org/10.3390/ijms232415630