
Cancer Stemness/Epithelial–Mesenchymal Transition Axis Influences Metastasis and Castration Resistance in Prostate Cancer: Potential Therapeutic Target



{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Epithelial–Mesenchymal Transition and Cancer Stem Cells in Prostate Cancer
3. Cancer Stem Cells and Epithelial–Mesenchymal Transition Control Prostate Cancer Progression
4. Role of Cancer Stem Cells in Metastatic Colonization and Progression
5. Different Malignant Cell Types Collaborate to Produce Distant Metastasis in Prostate Cancer
6. Cancer Stems Cells and Epithelial–Mesenchymal Transition in Relapse and Castration Resistance
7. Orthotopic Model for the Study of Human Prostate Cancer Metastasis
8. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Litwin, M.S.; Tan, H.J. The Diagnosis and Treatment of Prostate Cancer: A Review. JAMA 2017, 317, 2532–2542. [Google Scholar] [CrossRef] [PubMed]
- Luchini, C.; Fleischmann, A.; Boormans, J.L.; Fassan, M.; Nottegar, A.; Lucato, P.; Stubbs, B.; Solmi, M.; Porcaro, A.; Veronese, N.; et al. Extranodal extension of lymph node metastasis influences recurrence in prostate cancer: A systematic review and meta-analysis. Sci. Rep. 2017, 7, 2374. [Google Scholar] [CrossRef] [PubMed]
- Harris, K.S.; Kerr, B.A. Prostate Cancer Stem Cell Markers Drive Progression, Therapeutic Resistance, and Bone Metastasis. Stem. Cells Int. 2017, 2017, 8629234. [Google Scholar] [CrossRef] [Green Version]
- Mizokami, A.; Kadono, Y.; Kitagawa, Y.; Izumi, K.; Konaka, H. Therapies for castration-resistant prostate cancer in a new era: The indication of vintage hormonal therapy, chemotherapy and the new medicines. Int. J. Urol. 2017, 24, 566–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceder, Y.; Bjartell, A.; Culig, Z.; Rubin, M.A.; Tomlins, S.; Visakorpi, T. The Molecular Evolution of Castration-resistant Prostate Cancer. Eur. Urol. Focus. 2016, 2, 506–513. [Google Scholar] [CrossRef] [PubMed]
- Hoda, M.R.; Kramer, M.W.; Merseburger, A.S.; Cronauer, M.V. Androgen deprivation therapy with Leuprolide acetate for treatment of advanced prostate cancer. Expert Opin. Pharmacother. 2017, 18, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Einstein, D.J.; Arai, S.; Balk, S.P. Targeting the androgen receptor and overcoming resistance in prostate cancer. Curr. Opin. Oncol. 2019, 31, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Lorente, D.; Fizazi, K.; Sweeney, C.; de Bono, J.S. Optimal Treatment Sequence for Metastatic Castration-resistant Prostate Cancer. Eur. Urol. Focus. 2016, 2, 488–498. [Google Scholar] [CrossRef] [PubMed]
- Aurilio, G.; Cimadamore, A.; Mazzucchelli, R.; Lopez-Beltran, A.; Verri, E.; Scarpelli, M.; Massari, F.; Cheng, L.; Santoni, M.; Montironi, R. Androgen Receptor Signaling Pathway in Prostate Cancer: From Genetics to Clinical Applications. Cells 2020, 9, 2653. [Google Scholar] [CrossRef]
- Galletti, G.; Leach, B.I.; Lam, L.; Tagawa, S.T. Mechanisms of resistance to systemic therapy in metastatic castration-resistant prostate cancer. Cancer Treat. Rev. 2017, 57, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Bastos, D.A.; Antonarakis, E.S. CTC-derived AR-V7 detection as a prognostic and predictive biomarker in advanced prostate cancer. Expert Rev. Mol. Diagn. 2018, 18, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Sharp, A.; Coleman, I.; Yuan, W.; Sprenger, C.; Dolling, D.; Rodrigues, D.N.; Russo, J.W.; Figueiredo, I.; Bertan, C.; Seed, G.; et al. Androgen receptor splice variant-7 expression emerges with castration resistance in prostate cancer. J. Clin. Investig. 2019, 129, 192–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sciarra, A.; Gentilucci, A.; Silvestri, I.; Salciccia, S.; Cattarino, S.; Scarpa, S.; Gatto, A.; Frantellizzi, V.; Von Heland, M.; Ricciuti, G.P.; et al. Androgen receptor variant 7 (AR-V7) in sequencing therapeutic agents for castratrion resistant prostate cancer: A critical review. Medicine 2019, 98, e15608. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, P.; Zeigler-Johnson, C.M.; Spangler, E.; van der Merwe, A.; Jalloh, M.; Gueye, S.M.; Rebbeck, T.R. Androgen Metabolism Gene Polymorphisms, Associations with Prostate Cancer Risk and Pathological Characteristics: A Comparative Analysis between South African and Senegalese Men. Prostate Cancer 2012, 2012, 798634. [Google Scholar] [CrossRef] [Green Version]
- Bader, D.A.; McGuire, S.E. Tumour metabolism and its unique properties in prostate adenocarcinoma. Nat. Rev. Urol. 2020, 17, 214–231. [Google Scholar] [CrossRef] [PubMed]
- Benko, G.; Spajić, B.; Krušlin, B.; Tomas, D. Impact of the EpCAM expression on biochemical recurrence-free survival in clinically localized prostate cancer. Urol. Oncol. 2013, 31, 468–474. [Google Scholar] [CrossRef]
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular principles of metastasis: A hallmark of cancer revisited. Signal Transduct Target Ther. 2020, 5, 28. [Google Scholar] [CrossRef] [Green Version]
- Mollica, V.; Di Nunno, V.; Cimadamore, A.; Lopez-Beltran, A.; Cheng, L.; Santoni, M.; Scarpelli, M.; Montironi, R.; Massari, F. Molecular Mechanisms Related to Hormone Inhibition Resistance in Prostate Cancer. Cells 2019, 8, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, N.; Tiwari, V.K.; Waldmeier, L.; Balwierz, P.J.; Arnold, P.; Pachkov, M.; Meyer-Schaller, N.; Schübeler, D.; van Nimwegen, E.; Christofori, G. Sox4 is a master regulator of epithelial-mesenchymal transition by controlling Ezh2 expression and epigenetic reprogramming. Cancer Cell 2013, 23, 768–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, S.M.; Schaller, M.; Cieply, B. Mechanisms that link the oncogenic epithelial-mesenchymal transition to suppression of anoikis. J. Cell Sci. 2013, 126, 21–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.T.; Wu, K.J. Epigenetic regulation of epithelial-mesenchymal transition: Focusing on hypoxia and TGF-β signaling. J. Biomed. Sci. 2020, 27, 39. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brabletz, S.; Schuhwerk, H.; Brabletz, T.; Stemmler, M.P. Dynamic EMT: A multi-tool for tumor progression. EMBO J. 2021, 40, e108647. [Google Scholar] [CrossRef]
- Herrera, D.; Orellana-Serradell, O.; Villar, P.; Torres, M.J.; Paciucci, R.; Contreras, H.R. Silencing of the transcriptional factor ZEB1 alters the steroidogenic pathway, and increases the concentration of testosterone and DHT in DU145 cells. Oncol. Rep. 2019, 41, 1275–1283. [Google Scholar] [CrossRef] [Green Version]
- Haffner, M.C.; Zwart, W.; Roudier, M.P.; True, L.D.; Nelson, W.G.; Epstein, J.I.; De Marzo, A.M.; Nelson, P.S.; Yegnasubramanian, S. Genomic and phenotypic heterogeneity in prostate cancer. Nat. Rev. Urol. 2021, 18, 79–92. [Google Scholar] [CrossRef]
- Najafi, M.; Farhood, B.; Mortezaee, K. Cancer stem cells (CSCs) in cancer progression and therapy. J. Cell Physiol. 2019, 234, 8381–8395. [Google Scholar] [CrossRef]
- Lim, J.R.; Mouawad, J.; Gorton, O.K.; Bubb, W.A.; Kwan, A.H. Cancer stem cell characteristics and their potential as therapeutic targets. Med. Oncol. 2021, 38, 76. [Google Scholar] [CrossRef]
- Civenni, G.; Albino, D.; Shinde, D.; Vázquez, R.; Merulla, J.; Kokanovic, A.; Mapelli, S.N.; Carbone, G.M.; Catapano, C.V. Transcriptional Reprogramming and Novel Therapeutic Approaches for Targeting Prostate Cancer Stem Cells. Front. Oncol. 2019, 9, 385. [Google Scholar] [CrossRef]
- Castellón, E.A.; Valenzuela, R.; Lillo, J.; Castillo, V.; Contreras, H.R.; Gallegos, I.; Mercado, A.; Huidobro, C. Molecular signature of cancer stem cells isolated from prostate carcinoma and expression of stem markers in different Gleason grades and metastasis. Biol. Res. 2012, 45, 297–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castillo, V.; Valenzuela, R.; Huidobro, C.; Contreras, H.R.; Castellon, E.A. Functional characteristics of cancer stem cells and their role in drug resistance of prostate cancer. Int. J. Oncol. 2014, 45, 985–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez, C.A.; Andahur, E.I.; Valenzuela, R.; Castellón, E.A.; Fullá, J.A.; Ramos, C.G.; Triviño, J.C. Exosomes from bulk and stem cells from human prostate cancer have a differential microRNA content that contributes cooperatively over local and pre-metastatic niche. Oncotarget 2016, 7, 3993–4008. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.H.; Xing, T.; Yang, Z.; Dudek, R.; Lu, Q.; Chen, Y.-H. Epithelial Mesenchymal Transition in Embryonic Development, Tissue Repair and Cancer: A Comprehensive Overview. J. Clin. Med. 2017, 7, 1. [Google Scholar] [CrossRef] [Green Version]
- De Craene, B.; Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef]
- Plygawko, A.T.; Kan, S.; Campbell, K. Epithelial-mesenchymal plasticity: Emerging parallels between tissue morphogenesis and cancer metastasis. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2020, 375, 20200087. [Google Scholar] [CrossRef]
- Culig, Z. Epithelial mesenchymal transition and resistance in endocrine-related cancers. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1368–1375. [Google Scholar] [CrossRef] [PubMed]
- Montanari, M.; Rossetti, S.; Cavaliere, C.; D’Aniello, C.; Malzone, M.G.; Vanacore, D.; Di Franco, R.; La Mantia, E.; Iovane, G.; Piscitelli, R.; et al. Epithelial-mesenchymal transition in prostate cancer: An overview. Oncotarget 2017, 8, 35376–35389. [Google Scholar] [CrossRef] [Green Version]
- Osorio, L.A.; Farfán, N.M.; Castellón, E.A.; Contreras, H.R. SNAIL transcription factor increases the motility and invasive capacity of prostate cancer cells. Mol. Med. Rep. 2016, 13, 778–786. [Google Scholar] [CrossRef] [Green Version]
- Poblete, C.E.; Fulla, J.; Gallardo, M.; Muñoz, V.; Castellón, E.A.; Gallegos, I.; Contreras, H.R. Increased SNAIL expression and low syndecan levels are associated with high Gleason grade in prostate cancer. Int. J. Oncol. 2014, 44, 647–654. [Google Scholar] [CrossRef]
- Farfán, N.; Ocarez, N.; Castellón, E.A.; Mejía, N.; de Herreros, A.G.; Contreras, H.R. The transcriptional factor ZEB1 represses Syndecan 1 expression in prostate cancer. Sci. Rep. 2018, 8, 11467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orellana-Serradell, O.; Herrera, D.; Castellon, E.A.; Contreras, H.R. The transcription factor ZEB1 promotes an aggressive phenotype in prostate cancer cell lines. Asian J. Androl. 2018, 20, 294–299. [Google Scholar] [CrossRef] [PubMed]
- Orellana-Serradell, O.; Herrera, D.; Castellón, E.A.; Contreras, H.R. The transcription factor ZEB1 promotes chemoresistance in prostate cancer cell lines. Asian J. Androl. 2019, 21, 460–467. [Google Scholar] [CrossRef] [PubMed]
- Babaei, G.; Aziz, S.G.; Jaghi, N.Z.Z. EMT, cancer stem cells and autophagy; The three main axes of metastasis. Biomed. Pharmacother. 2021, 133, 110909. [Google Scholar] [CrossRef]
- Yin, W.; Wang, J.; Jiang, L.; James Kang, Y. Cancer and stem cells. Exp. Biol. Med. 2021, 246, 1791–1801. [Google Scholar] [CrossRef]
- Ayob, A.Z.; Ramasamy, T.S. Cancer stem cells as key drivers of tumour progression. J. Biomed. Sci. 2018, 25, 20. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.V.; Vanner, R.; Dirks, P.; Eaves, C.J. Cancer stem cells: An evolving concept. Nat. Rev. Cancer 2012, 12, 133–143. [Google Scholar] [CrossRef]
- Fang, D.; Kitamura, H. Cancer stem cells and epithelial-mesenchymal transition in urothelial carcinoma: Possible pathways and potential therapeutic approaches. Int. J. Urol. 2018, 25, 7–17. [Google Scholar] [CrossRef] [Green Version]
- Rycaj, K.; Li, H.; Zhou, J.; Chen, X.; Tang, D.G. Cellular determinants and microenvironmental regulation of prostate cancer metastasis. Semin. Cancer Biol. 2017, 44, 83–97. [Google Scholar] [CrossRef]
- Schilling, D.; Todenhöfer, T.; Hennenlotter, J.; Schwentner, C.; Fehm, T.; Stenzl, A. Isolated, disseminated and circulating tumour cells in prostate cancer. Nat. Rev. Urol. 2012, 9, 448–463. [Google Scholar] [CrossRef]
- Lowes, L.E.; Goodale, D.; Xia, Y.; Postenka, C.; Piaseczny, M.M.; Paczkowski, F.; Allan, A.L. Epithelial-to-mesenchymal transition leads to disease-stage differences in circulating tumor cell detection and metastasis in pre-clinical models of prostate cancer. Oncotarget 2016, 7, 76125–76139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theil, G.; Boehm, C.; Fischer, K.; Bialek, J.; Hoda, R.; Weber, E.; Schönburg, S.; Kawan, F.; Fornara, P. In vivo isolation of circulating tumor cells in patients with different stages of prostate cancer. Oncol. Lett. 2021, 21, 357. [Google Scholar] [CrossRef] [PubMed]
- Ruscetti, M.; Quach, B.; Dadashian, E.L.; Mulholland, D.J.; Wu, H. Tracking and Functional Characterization of Epithelial-Mesenchymal Transition and Mesenchymal Tumor Cells during Prostate Cancer Metastasis. Cancer Res. 2015, 75, 2749–2759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deep, G.; Jain, A.K.; Ramteke, A.; Ting, H.; Vijendra, K.C.; Gangar, S.C.; Agarwal, C.; Agarwal, R. SNAI1 is critical for the aggressiveness of prostate cancer cells with low E-cadherin. Mol. Cancer 2014, 13, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, K.-M.; Su, Z.; Frye, C.; McClellan, S.; Allan, R.W.; Andrejewski, J.T.; Kelley, V.; Jorgensen, M.; Steindler, D.A.; Vieweg, J.; et al. Expression of pluripotent stem cell reprogramming factors by prostate tumor initiating cells. J. Urol. 2010, 183, 2045–2053. [Google Scholar] [CrossRef] [Green Version]
- Bae, K.M.; Parker, N.N.; Dai, Y.; Vieweg, J.; Siemann, D.W. E-cadherin plasticity in prostate cancer stem cell invasion. Am. J. Cancer Res. 2011, 1, 71–84. [Google Scholar]
- Kerr, B.A.; Miocinovic, R.; Smith, A.K.; West, X.Z.; Watts, K.E.; Alzayed, A.W.; Klink, J.C.; Mir, M.C.; Sturey, T.; Hansel, D.E.; et al. CD117⁺ cells in the circulation are predictive of advanced prostate cancer. Oncotarget 2015, 6, 1889–1897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patrawala, L.; Calhoun, T.; Schneider-Broussard, R.; Li, H.; Bhatia, B.; Tang, S.; Reilly, J.G.; Chandra, D.; Zhou, J.; Claypool, K.; et al. Highly purified CD44+ prostate cancer cells from xenograft human tumors are enriched in tumorigenic and metastatic progenitor cells. Oncogene 2006, 25, 1696–1708. [Google Scholar] [CrossRef] [Green Version]
- Klarmann, G.J.; Hurt, E.M.; Mathews, L.A.; Zhang, X.; Duhagon, M.A.; Mistree, T.; Thomas, S.B.; Farrar, W.L. Invasive prostate cancer cells are tumor initiating cells that have a stem cell-like genomic signature. Clin. Exp. Metastasis 2009, 26, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Palapattu, G.S.; Wu, C.; Silvers, C.R.; Martin, H.B.; Williams, K.; Salamone, L.; Bushnell, T.; Huang, L.-S.; Yang, Q.; Huang, J. Selective expression of CD44, a putative prostate cancer stem cell marker, in neuroendocrine tumor cells of human prostate cancer. Prostate 2009, 69, 787–798. [Google Scholar] [CrossRef]
- Chang, Y.-T.; Lin, T.-P.; Campbell, M.; Pan, C.-C.; Lee, S.-H.; Lee, H.-C.; Yang, M.-H.; Kung, H.-J.; Chang, P.-C. REST is a crucial regulator for acquiring EMT-like and stemness phenotypes in hormone-refractory prostate cancer. Sci. Rep. 2017, 7, 42795. [Google Scholar] [CrossRef] [PubMed]
- Davies, A.H.; Beltran, H.; Zoubeidi, A. Cellular plasticity and the neuroendocrine phenotype in prostate cancer. Nat. Rev. Urol. 2018, 15, 271–286. [Google Scholar] [CrossRef]
- Russo, M.V.; Esposito, S.; Tupone, M.G.; Manzoli, L.; Airoldi, I.; Pompa, P.; Cindolo, L.; Schips, L.; Sorrentino, C.; Di Carlo, E. SOX2 boosts major tumor progression genes in prostate cancer and is a functional biomarker of lymph node metastasis. Oncotarget 2016, 7, 12372–12385. [Google Scholar] [CrossRef] [PubMed]
- Esposito, S.; Russo, M.V.; Airoldi, I.; Tupone, M.G.; Sorrentino, C.; Barbarito, G.; Di Meo, S.; Di Carlo, E. SNAI2/Slug gene is silenced in prostate cancer and regulates neuroendocrine differentiation, metastasis-suppressor and pluripotency gene expression. Oncotarget 2015, 6, 17121–17134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soundararajan, R.; Paranjape, A.N.; Maity, S.; Aparicio, A.; Mani, S.A. EMT, stemness and tumor plasticity in aggressive variant neuroendocrine prostate cancers. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Jayachandran, A.; Dhungel, B.; Steel, J.C. Epithelial-to-mesenchymal plasticity of cancer stem cells: Therapeutic targets in hepatocellular carcinoma. J. Hematol. Oncol. 2016, 9, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Sun, B.; Sun, D.; Liu, T.; Che, N.; Gu, Q.; Dong, X.; Li, R.; Liu, Y.; Li, J. Slug promotes hepatocellular cancer cell progression by increasing sox2 and nanog expression. Oncol. Rep. 2015, 33, 149–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mu, P.; Zhang, Z.; Benelli, M.; Karthaus, W.R.; Hoover, E.; Chen, C.-C.; Wongvipat, J.; Ku, S.-Y.; Gao, D.; Cao, Z.; et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 2017, 355, 84–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, T.; Yokoi, A.; Hashimura, M.; Oguri, Y.; Akiya, M.; Saegusa, M. TGF-β-mediated LEFTY/Akt/GSK-3β/Snail axis modulates epithelial-mesenchymal transition and cancer stem cell properties in ovarian clear cell carcinomas. Mol. Carcinog. 2018, 57, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Weinberg, R.A. Epithelial-to-mesenchymal transition in cancer: Complexity and opportunities. Front. Med. 2018, 12, 361–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bocci, F.; Jolly, M.K.; George, J.T.; Levine, H.; Onuchic, J.N. A mechanism-based computational model to capture the interconnections among epithelial-mesenchymal transition, cancer stem cells and Notch-Jagged signaling. Oncotarget 2018, 9, 29906–29920. [Google Scholar] [CrossRef]
- Wang, K.; Ji, W.; Yu, Y.; Li, Z.; Niu, X.; Xia, W.; Lu, S. FGFR1-ERK1/2-SOX2 axis promotes cell proliferation, epithelial-mesenchymal transition, and metastasis in FGFR1-amplified lung cancer. Oncogene 2018, 37, 5340–5354, Erratum in Oncogene 2020, 39, 6619–6620. [Google Scholar] [CrossRef] [PubMed]
- Pradella, D.; Naro, C.; Sette, C.; Ghigna, C. EMT and stemness: Flexible processes tuned by alternative splicing in development and cancer progression. Mol. Cancer 2017, 16, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, T.P.; Gullino, P.M. Quantitation of cell shedding into efferent blood of mammary adenocarcinoma. Cancer Res. 1975, 35, 512–516. [Google Scholar]
- Celià-Terrassa, T.; Kang, Y. Metastatic niche functions and therapeutic opportunities. Nat. Cell Biol. 2018, 20, 868–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganesh, K.; Massagué, J. Targeting metastatic cancer. Nat. Med. 2021, 27, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Cackowski, F.C.; Heath, E.I. Prostate cancer dormancy and recurrence. Cancer Lett. 2022, 524, 103–108. [Google Scholar] [CrossRef]
- Rycaj, K.; Tang, D.G. Metastasis and Metastatic Cells: A Historical Perspective and Current Analysis. In Cancer Stem Cells, 1st ed.; Liu, H., Lathias, D.J., Eds.; Elsevier: Cambridge, MA, USA, 2016; pp. 317–340. [Google Scholar]
- Mei, W.; Lin, X.; Kapoor, A.; Gu, Y.; Zhao, K.; Tang, D. The Contributions of Prostate Cancer Stem Cells in Prostate Cancer Initiation and Metastasis. Cancers 2019, 11, 434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsika, A.; Srinivasan, B.; Day, C.; Mader, S.A.; Kiernan, D.M.; Broomfield, A.; Fu, J.; Hooper, J.D.; Kench, J.G.; Samaratunga, H. Cancer stem cell markers in prostate cancer: An immunohistochemical study of ALDH1, SOX2 and EZH2. Pathology 2015, 47, 622–628. [Google Scholar] [CrossRef] [PubMed]
- Conley-LaComb, M.K.; Huang, W.; Wang, S.; Shi, D.; Jung, Y.S.; Najy, A.; Fridman, R.; Bonfil, R.D.; Cher, M.L.; Chen, Y.Q.; et al. PTEN regulates PDGF ligand switch for β-PDGFR signaling in prostate cancer. Am. J. Pathol. 2012, 180, 1017–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massoner, P.; Thomm, T.; Mack, B.; Untergasser, G.; Martowicz, A.A.; Bobowski, K.; Klocker, H.; Gires, O.O.; Puhr, M. EpCAM is overexpressed in local and metastatic prostate cancer, suppressed by chemotherapy and modulated by MET-associated miRNA-200c/205. Br. J. Cancer 2014, 111, 955–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mainetti, L.E.; Zhe, X.; Diedrich, J.; Saliganan, A.D.; Cho, W.J.; Cher, M.L.; Heath, E.; Fridman, R.; Kim, H.-R.C.; Bonfil, R.D. Bone-induced c-kit expression in prostate cancer: A driver of intraosseous tumor growth. Int. J. Cancer 2015, 136, 11–20. [Google Scholar] [CrossRef]
- Harris, K.S.; Shi, L.; Foster, B.M.; Mobley, M.E.; Elliott, P.L.; Song, C.J.; Watabe, K.; Langefeld, C.D.; Kerr, B.A. CD117/c-kit defines a prostate CSC-like subpopulation driving progression and TKI resistance. Sci. Rep. 2021, 11, 1465. [Google Scholar] [CrossRef] [PubMed]
- Domanska, U.M.; Timmer-Bosscha, H.; Nagengast, W.B.; Munnink, T.H.O.; Kruizinga, R.C.; Ananias, H.J.; Kliphuis, N.M.; Huls, G.; De Vries, E.G.; de Jong, I.J.; et al. CXCR4 inhibition with AMD3100 sensitizes prostate cancer to docetaxel chemotherapy. Neoplasia 2012, 14, 709–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Putzke, A.P.; Ventura, A.P.; Bailey, A.M.; Akture, C.; Opoku-Ansah, J.; Çeliktaş, M.; Hwang, M.S.; Darling, D.S.; Coleman, I.M.; Nelson, P.S.; et al. Metastatic progression of prostate cancer and e-cadherin regulation by zeb1 and SRC family kinases. Am. J. Pathol. 2011, 179, 400–410. [Google Scholar] [CrossRef]
- Celià-Terrassa, T.; Meca-Cortés, O.; Mateo, F.; de Paz, A.M.; Rubio, N.; Arnal-Estapé, A.; Ell, B.J.; Bermudo, R.; Díaz, A.; Guerra-Rebollo, M.; et al. Epithelial-mesenchymal transition can suppress major attributes of human epithelial tumor-initiating cells. J. Clin. Investig. 2012, 122, 1849–1868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakir, B.; Chiarella, A.M.; Pitarresi, J.R.; Rustgi, A.K. EMT, MET, Plasticity, and Tumor Metastasis. Trends Cell Biol. 2020, 30, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Bullock, M.D.; Sayan, A.E.; Packham, G.K.; Mirnezami, A.H. MicroRNAs: Critical regulators of epithelial to mesenchymal (EMT) and mesenchymal to epithelial transition (MET) in cancer progression. Biol. Cell 2012, 104, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Cifuentes, F.F.; Valenzuela, R.H.; Contreras, H.R.; Castellón, E.A. Development of an orthotopic model of human metastatic prostate cancer in the NOD-SCIDγ mouse (Mus musculus) anterior prostate. Oncol. Lett. 2015, 10, 2142–2148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzo, A.; Mollica, V.; Cimadamore, A.; Santoni, M.; Scarpelli, M.; Giunchi, F.; Cheng, L.; Lopez-Beltran, A.; Fiorentino, M.; Montironi, R.; et al. Is There a Role for Immunotherapy in Prostate Cancer? Cells 2020, 9, 2051. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, S.I.M.; Ju, X.; Horvath, L.G.; Clark, G.J. Moving on From Sipuleucel-T: New Dendritic Cell Vaccine Strategies for Prostate Cancer. Front. Immunol. 2021, 12, 641307. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.H.; Rosen, J.M. The mechanisms of therapy resistance in cancer stem cells. In Cancer Stem Cells, 1st ed.; Liu, H., Lathias, D.J., Eds.; Elsevier: Cambridge, MA, USA, 2016; pp. 395–410. [Google Scholar]
- Hoogland, A.M.; Verhoef, E.I.; Roobol, M.J.; Schröder, F.H.; Wildhagen, M.F.; van der Kwast, T.H.; Jenster, G.; van Leenders, G.J. Validation of stem cell markers in clinical prostate cancer: α6-integrin is predictive for non-aggressive disease. Prostate 2014, 74, 488–496. [Google Scholar] [CrossRef]
- Yang, M.; Zhang, X.; Guo, L.; Liu, X.; Wu, J.; Zhu, H. Research Progress for the Clinical Application of Circulating Tumor Cells in Prostate Cancer Diagnosis and Treatment. Biomed. Res. Int. 2021, 2021, 6230826. [Google Scholar] [CrossRef]
- Conteduca, V.; Ku, S.-Y.; Fernandez, L.; Dago-Rodriquez, A.; Lee, J.; Jendrisak, A.; Slade, M.; Gilbertson, C.; Manohar, J.; Sigouros, M.; et al. Circulating tumor cell heterogeneity in neuroendocrine prostate cancer by single cell copy number analysis. NPJ Precis Oncol. 2021, 5, 76, Erratum in NPJ Precis Oncol. 2021, 5, 84. [Google Scholar] [CrossRef] [PubMed]
- Jiao, J.; Hindoyan, A.; Wang, S.; Tran, L.M.; Goldstein, A.S.; Lawson, D.; Chen, D.; Li, Y.; Guo, C.; Zhang, B.; et al. Identification of CD166 as a surface marker for enriching prostate stem/progenitor and cancer initiating cells. PLoS ONE 2012, 7, e42564. [Google Scholar] [CrossRef] [Green Version]
- Park, S.H.; Fong, K.W.; Mong, E.; Martin, M.C.; Schiltz, G.E.; Yu, J. Going beyond Polycomb: EZH2 functions in prostate cancer. Oncogene 2021, 40, 5788–5798. [Google Scholar] [CrossRef] [PubMed]
- Tsao, T.; Beretov, J.; Ni, J.; Bai, X.; Bucci, J.; Graham, P.; Li, Y. Cancer stem cells in prostate cancer radioresistance. Cancer Lett. 2019, 465, 94–104. [Google Scholar] [CrossRef]
- Dubrovska, A.; Elliott, J.; Salamone, R.J.; Telegeev, G.D.; Stakhovsky, A.E.; Schepotin, I.B.; Yan, F.; Wang, Y.; Bouchez, L.C.; Kularatne, S.A.; et al. CXCR4 expression in prostate cancer progenitor cells. PLoS ONE 2012, 7, e31226. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Cozzi, P.; Beretov, J.; Duan, W.; Bucci, J.; Graham, P.; Li, Y. Epithelial cell adhesion molecule (EpCAM) is involved in prostate cancer chemotherapy/radiotherapy response in vivo. BMC Cancer 2018, 18, 1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez, C.; Mendoza, P.; Contreras, H.R.; Vergara, J.; McCubrey, J.A.; Huidobro, C.; Castellón, E.A. Expression of multidrug resistance proteins in prostate cancer is related with cell sensitivity to chemotherapeutic drugs. Prostate 2009, 69, 1448–1459. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, C.; Mercado, A.; Contreras, H.R.; Mendoza, P.; Cabezas, J.; Acevedo, C.; Huidobro, C.; Castellón, E.A. Chemotherapy sensitivity recovery of prostate cancer cells by functional inhibition and knock down of multidrug resistance proteins. Prostate 2011, 71, 1810–1817. [Google Scholar] [CrossRef] [PubMed]
- Patrawala, L.; Calhoun, T.; Schneider-Broussard, R.; Zhou, J.; Claypool, K.; Tang, D.G. Side population is enriched in tumorigenic, stem-like cancer cells, whereas ABCG2+ and ABCG2- cancer cells are similarly tumorigenic. Cancer Res. 2005, 65, 6207–6219. [Google Scholar] [CrossRef] [PubMed]
- Cojoc, M.; Mäbert, K.; Muders, M.H.; Dubrovska, A. A role for cancer stem cells in therapy resistance: Cellular and molecular mechanisms. Semin. Cancer Biol. 2015, 31, 16–27. [Google Scholar] [CrossRef]
- Garcia-Mayea, Y.; Mir, C.; Masson, F.; Paciucci, R.; LLeonart, M.E. Insights into new mechanisms and models of cancer stem cell multidrug resistance. Semin. Cancer Biol. 2020, 60, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Shu, H.; Bin Yuan, B.; Huang, Y.; Wang, L.; He, B.; Sun, Q.; Sun, L. High expression of ABCG2 is associated with chemotherapy resistance of osteosarcoma. J. Orthop. Surg. Res. 2021, 16, 85. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scher, H.I.; Lu, D.; Schreiber, N.A.; Louw, J.; Graf, R.P.; Vargas, H.A.; Johnson, A.; Jendrisak, A.; Bambury, R.; Danila, D.; et al. Association of AR-V7 on Circulating Tumor Cells as a Treatment-Specific Biomarker With Outcomes and Survival in Castration-Resistant Prostate Cancer. JAMA Oncol. 2016, 2, 1441–1449, Erratum in JAMA Oncol. 2016, 2, 1511. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chan, S.C.; Brand, L.J.; Hwang, T.H.; Silverstein, K.A.; Dehm, S.M. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. 2013, 73, 483–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, Q.; Tang, D.G. Androgen receptor and prostate cancer stem cells: Biological mechanisms and clinical implications. Endocr. Relat. Cancer 2015, 22, T209–T220. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.; Gore, C.; Yang, L.; Fazli, L.; Gleave, M.; Pong, R.-C.; Xiao, G.; Zhang, L.; Yun, E.-J.; Tseng, S.-F.; et al. Slug, a unique androgen-regulated transcription factor, coordinates androgen receptor to facilitate castration resistance in prostate cancer. Mol. Endocrinol. 2012, 26, 1496–1507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdolahi, S.; Ghazvinian, Z.; Muhammadnejad, S.; Saleh, M.; Asadzadeh Aghdaei, H.; Baghaei, K. Patient-derived xenograft (PDX) models, applications and challenges in cancer research. J. Transl. Med. 2022, 20, 206. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xue, H.; Cutz, J.-C.; Bayani, J.; Mawji, N.R.; Chen, W.G.; Goetz, L.J.; Hayward, S.W.; Sadar, M.D.; Gilks, C.B.; et al. An orthotopic metastatic prostate cancer model in SCID mice via grafting of a transplantable human prostate tumor line. Lab. Investig. 2005, 85, 1392–1404. [Google Scholar] [CrossRef] [PubMed]
- Tumati, V.; Mathur, S.; Song, K.; Hsieh, J.-T.; Zhao, D.; Takahashi, M.; Dobin, T.; Gandee, L.; Solberg, T.D.; Habib, A.A.; et al. Development of a locally advanced orthotopic prostate tumor model in rats for assessment of combined modality therapy. Int. J. Oncol. 2013, 42, 1613–1619. [Google Scholar] [CrossRef] [Green Version]
- Cifuentes, F.F.; Valenzuela, R.H.; Contreras, H.R.; Castellón, E.A. Surgical cytoreduction of the primary tumor reduces metastatic progression in a mouse model of prostate cancer. Oncol. Rep. 2015, 34, 2837–2844. [Google Scholar] [CrossRef]
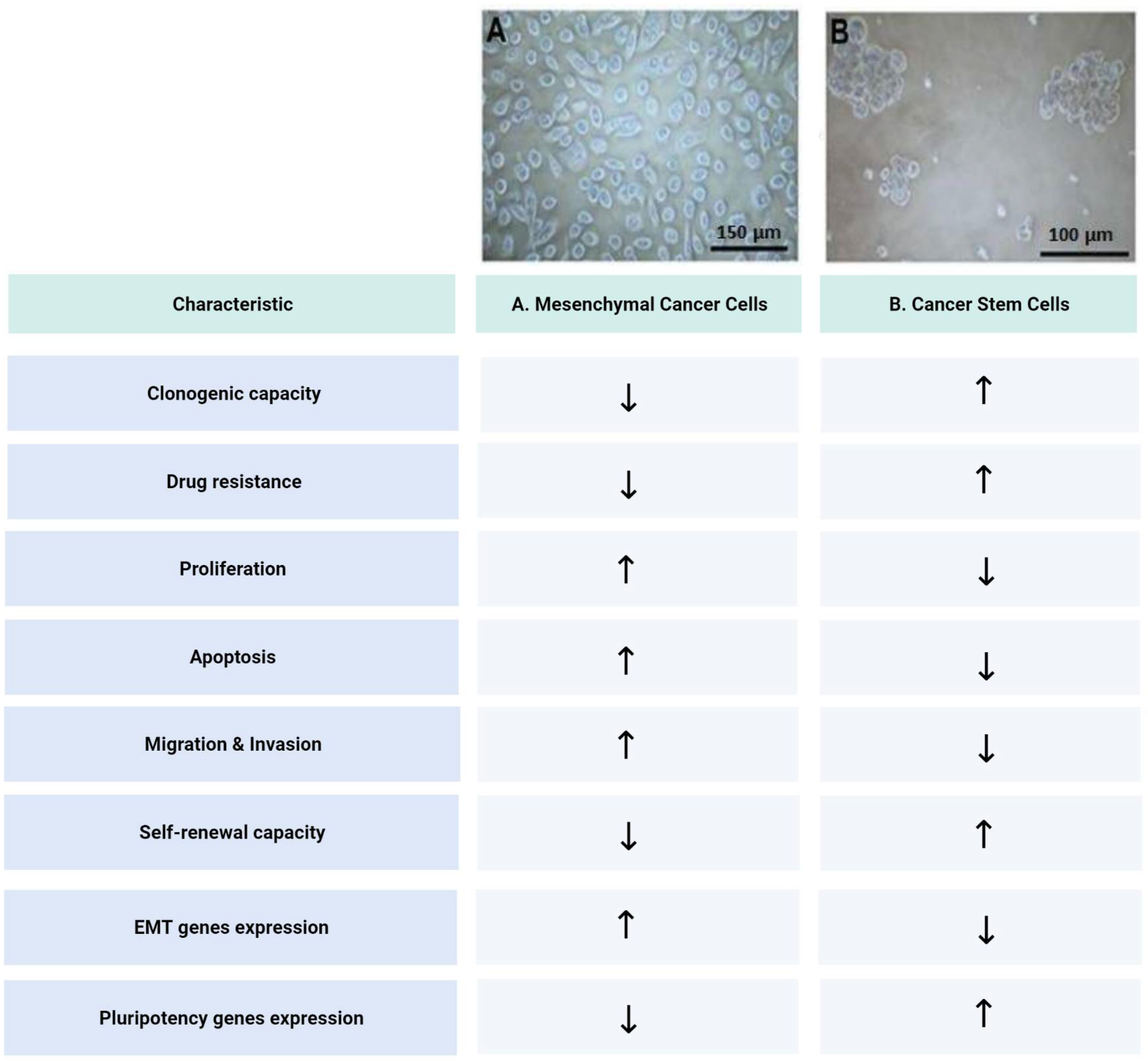
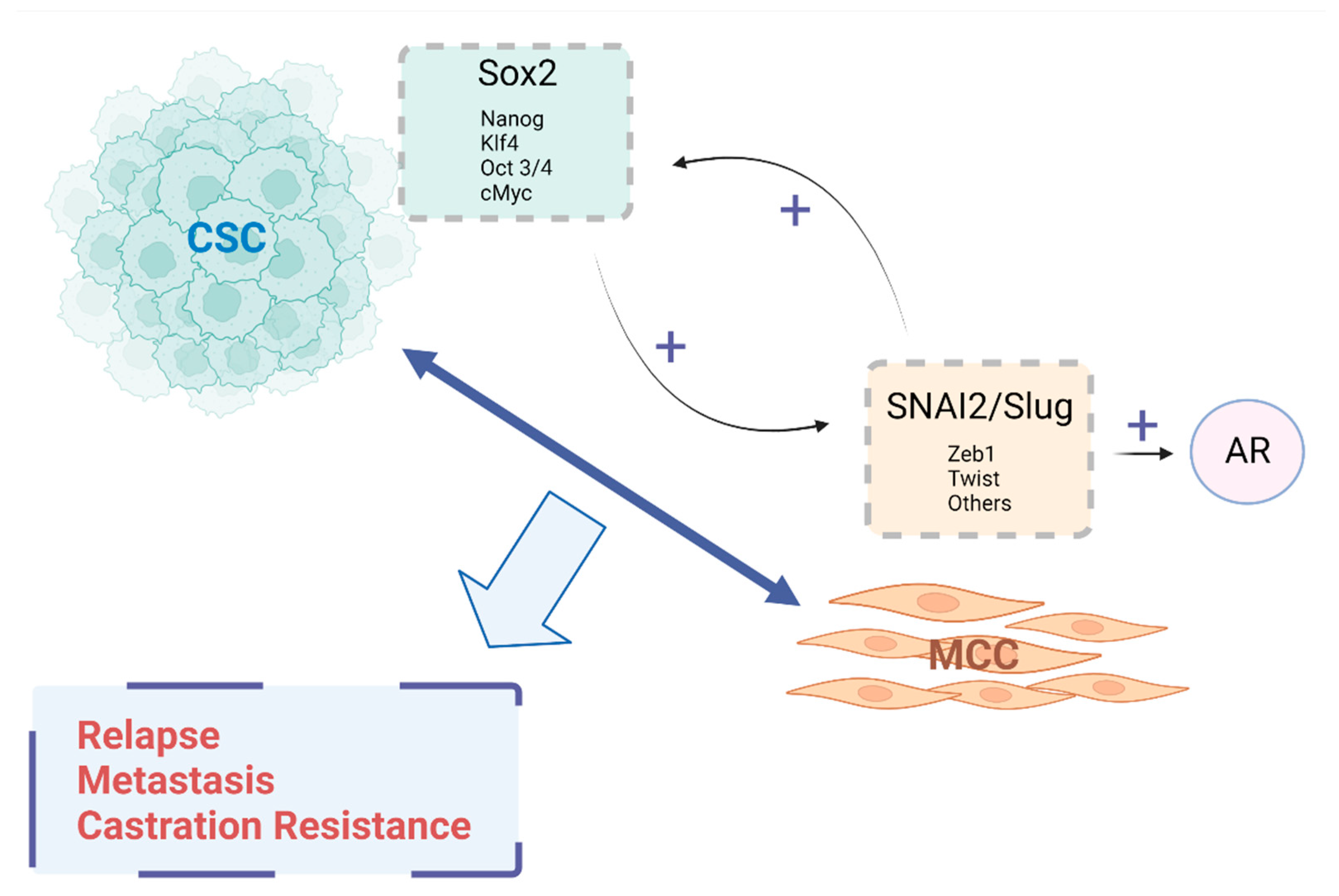
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castellón, E.A.; Indo, S.; Contreras, H.R. Cancer Stemness/Epithelial–Mesenchymal Transition Axis Influences Metastasis and Castration Resistance in Prostate Cancer: Potential Therapeutic Target. Int. J. Mol. Sci. 2022, 23, 14917. https://doi.org/10.3390/ijms232314917
Castellón EA, Indo S, Contreras HR. Cancer Stemness/Epithelial–Mesenchymal Transition Axis Influences Metastasis and Castration Resistance in Prostate Cancer: Potential Therapeutic Target. International Journal of Molecular Sciences. 2022; 23(23):14917. https://doi.org/10.3390/ijms232314917
Chicago/Turabian StyleCastellón, Enrique A., Sebastián Indo, and Héctor R. Contreras. 2022. "Cancer Stemness/Epithelial–Mesenchymal Transition Axis Influences Metastasis and Castration Resistance in Prostate Cancer: Potential Therapeutic Target" International Journal of Molecular Sciences 23, no. 23: 14917. https://doi.org/10.3390/ijms232314917
APA StyleCastellón, E. A., Indo, S., & Contreras, H. R. (2022). Cancer Stemness/Epithelial–Mesenchymal Transition Axis Influences Metastasis and Castration Resistance in Prostate Cancer: Potential Therapeutic Target. International Journal of Molecular Sciences, 23(23), 14917. https://doi.org/10.3390/ijms232314917