
IFNβ-Induced CXCL10 Chemokine Expression Is Regulated by Pellino3 Ligase in Monocytes and Macrophages

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
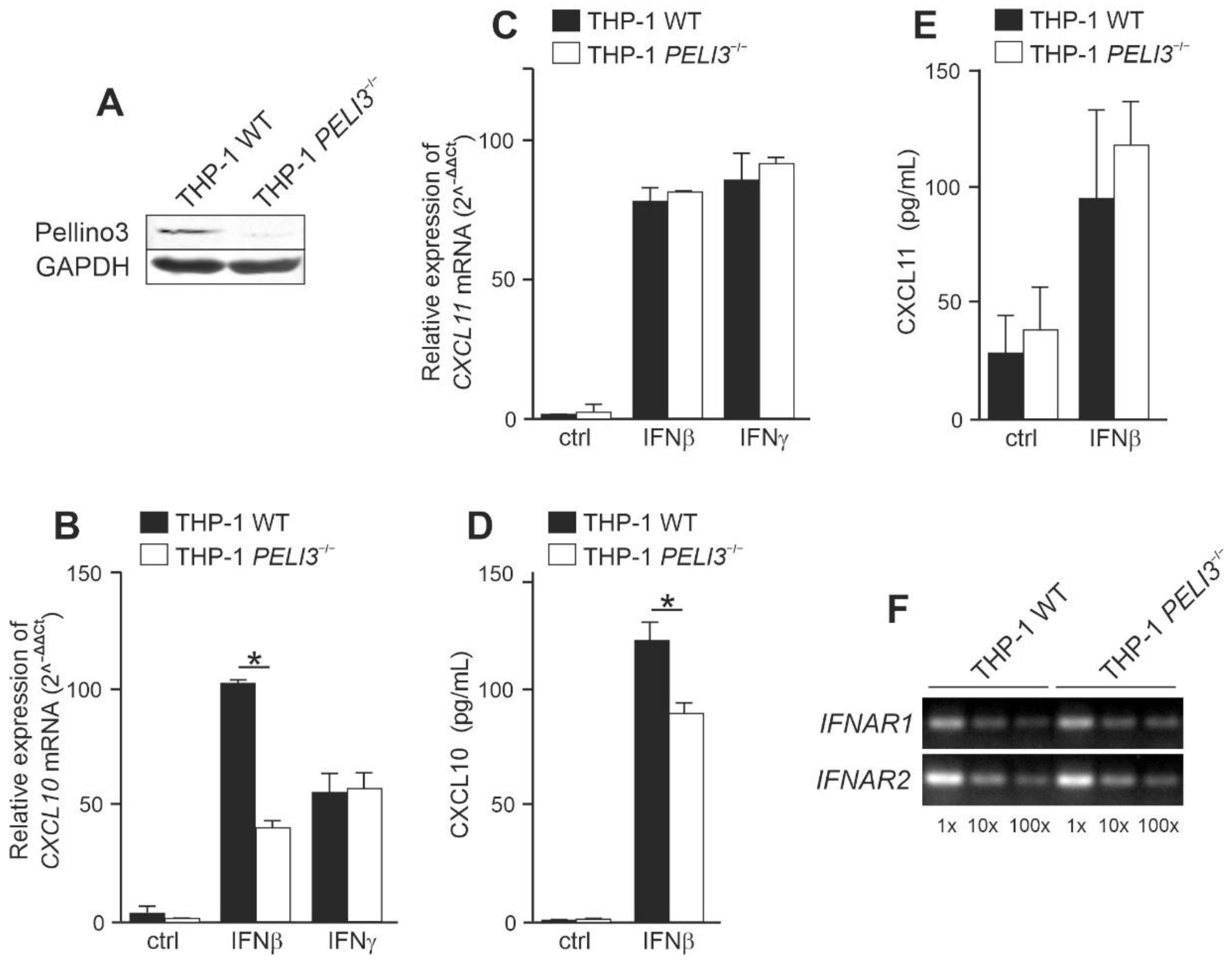
2.1. Pellino3 Positively Regulates CXCL10 Induction via Tyk2 after IFNβ Treatment
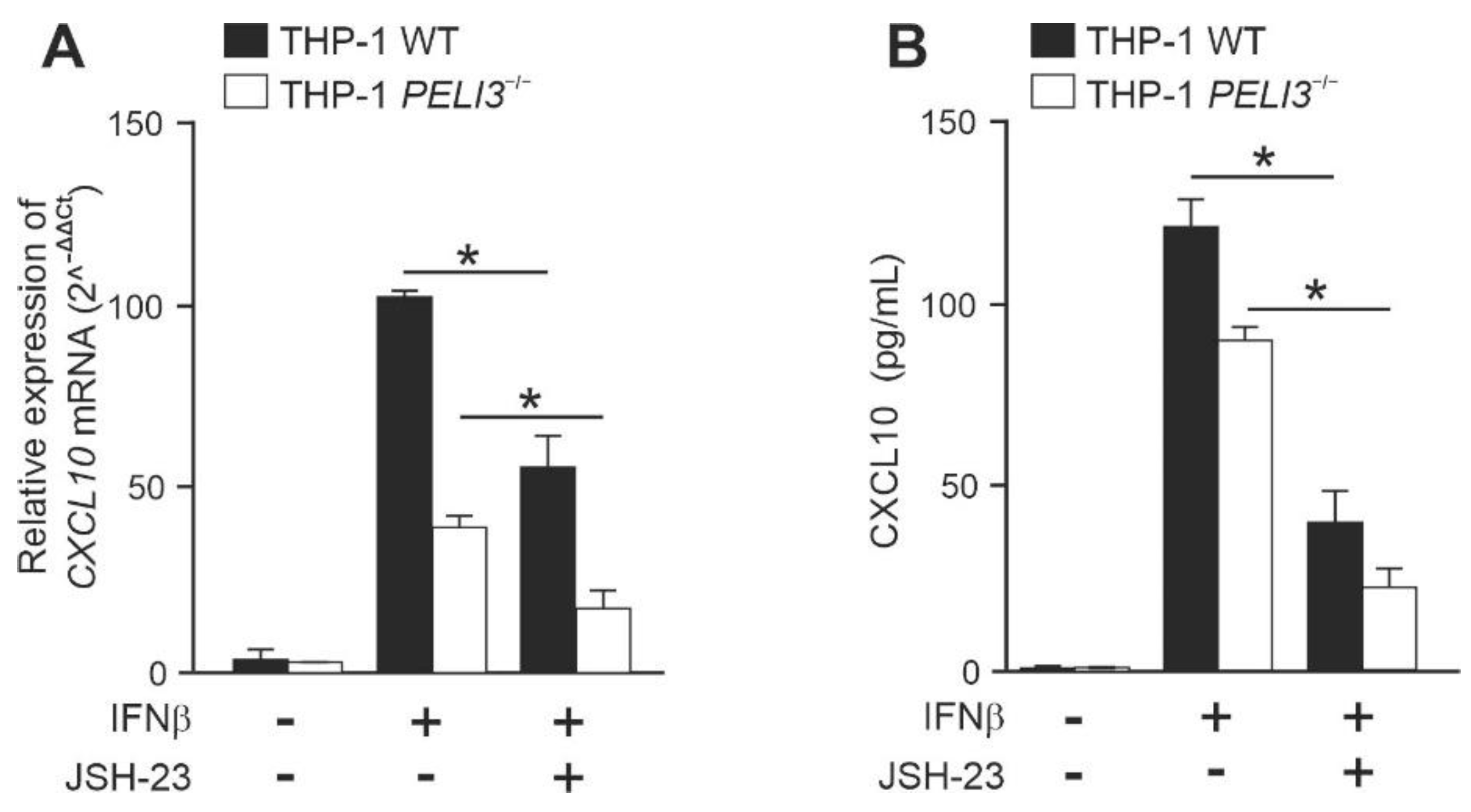
2.2. NF-κB Positively Regulates IFNβ-Induced Expression of CXCL10 in a Pellino3-Independent Way
2.3. IFNβ-Dependent Activation of CXCL10 Requires IRF9 and STAT1/STAT2 Activation
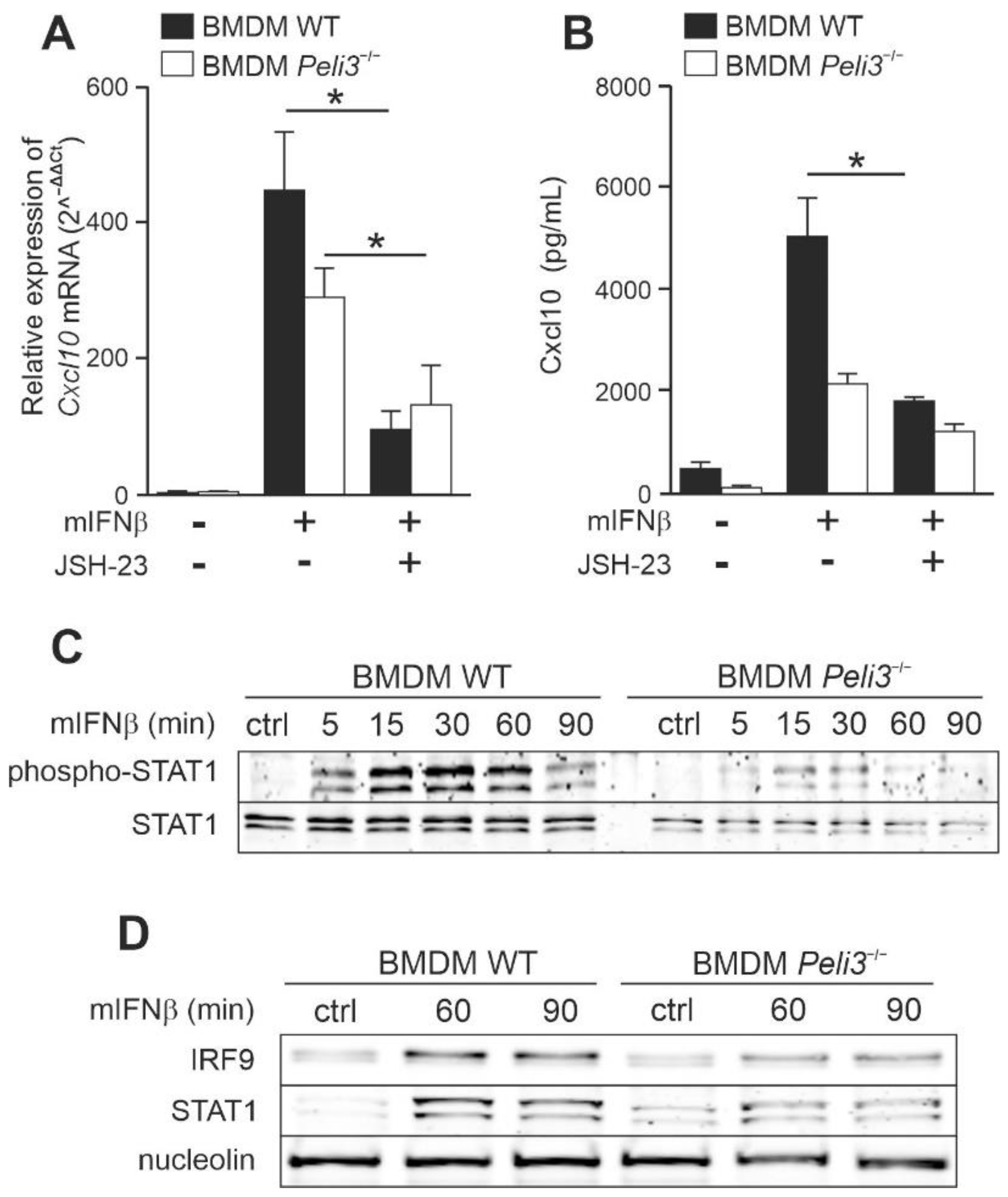
2.4. Knockout of Pellino3 Decreases IFNβ-Induced Expression and Production of CXCL10 in Murine Macrophage Cell Line, BMDM
3. Discussion
4. Materials and Methods
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Mcnab, F.; Mayer-barber, K.; Sher, A.; Wack, A.; Garra, A.O. Type I Interferons in Infectious Disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Zanin, N.; Viaris de Lesegno, C.; Lamaze, C.; Blouin, C.M. Interferon Receptor Trafficking and Signaling: Journey to the Cross Roads. Front. Immunol. 2021, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.H.; Murti, A.; Pfeffer, S.R.; Kim, J.G.; Donner, D.B.; Pfeffer, L.M. Interferon α/β Promotes Cell Survival by Activating Nuclear Factor ΚB through Phosphatidylinositol 3-Kinase and Akt. J. Biol. Chem. 2001, 276, 13756–13761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.H.; Murti, A.; Pfeffer, L.M. Interferon Induces NF-ΚB-Inducing Kinase/Tumor Necrosis Factor Receptor-Associated Factor-Dependent NF-ΚB Activation to Promote Cell Survival. J. Biol. Chem. 2005, 280, 31530–31536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majoros, A.; Platanitis, E.; Kernbauer-Hölzl, E.; Rosebrock, F.; Müller, M.; Decker, T. Canonical and Non-Canonical Aspects of JAK-STAT Signaling: Lessons from Interferons for Cytokine Responses. Front. Immunol. 2017, 8, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.H.; Murti, A.; Valentine, W.J.; Du, Z.; Pfeffer, L.M. Interferonα Activates NF-ΚB in JAK1-Deficient Cells through a TYK2-Dependent Pathway. J. Biol. Chem. 2005, 280, 25849–25853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeffer, L.M. The Role of Nuclear Factor Κb in the Interferon Response. J. Interf. Cytokine Res. 2011, 31, 553–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumaran Satyanarayanan, S.; El Kebir, D.; Soboh, S.; Butenko, S.; Sekheri, M.; Saadi, J.; Peled, N.; Assi, S.; Othman, A.; Schif-Zuck, S.; et al. IFN-β Is a Macrophage-Derived Effector Cytokine Facilitating the Resolution of Bacterial Inflammation. Nat. Commun. 2019, 10, 3471. [Google Scholar] [CrossRef] [Green Version]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of Type I Interferon Responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenton-May, A.E.; Dibben, O.; Emmerich, T.; Ding, H.; Pfafferott, K.; Aasa-Chapman, M.M.; Pellegrino, P.; Williams, I.; Cohen, M.S.; Gao, F.; et al. Relative Resistance of HIV-1 Founder Viruses to Control by Interferon-Alpha. Retrovirology 2013, 10, 146. [Google Scholar] [CrossRef] [PubMed]
- Rudick, R.A.; Fbnsohoff, R.M.; Peppler, R.; Medendorp, S.V.; Lehmann, P.; Alam, J. Interferon Beta Induces Interleukin-10 Expression: Relevance to Multiple Sclerosis. Ann. Neurol. 1996, 40, 618–627. [Google Scholar] [CrossRef] [PubMed]
- Jungo, F.; Dayer, J.M.; Modoux, C.; Hyka, N.; Burger, D. IFN-β Inhibits the Ability of T Lymphocytes to Induce TNF-α and IL-1β Production in Monocytes upon Direct Cell-Cell Contact. Cytokine 2001, 14, 272–282. [Google Scholar] [CrossRef] [PubMed]
- Buttmann, M.; Berberich-Siebelt, F.; Serfling, E.; Rieckmann, P. Interferon-β Is a Potent Inducer of Interferon Regulatory Factor-1/2-Dependent IP-10/CXCL10 Expression in Primary Human Endothelial Cells. J. Vasc. Res. 2007, 44, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Spurrell, J.C.L.; Wiehler, S.; Zaheer, R.S.; Sanders, S.P.; Proud, D. Human Airway Epithelial Cells Produce IP-10 (CXCL10) in Vitro and in Vivo upon Rhinovirus Infection. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 289, 85–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helbig, K.J.; Ruszkiewicz, A.; Semendric, L.; Harley, H.A.J.; McColl, S.R.; Beard, M.R. Expression of the CXCR3 Ligand I-TAC by Hepatocytes in Chronic Hepatitis C and Its Correlation with Hepatic Inflammation. Hepatology 2004, 39, 1220–1229. [Google Scholar] [CrossRef]
- Ahmadi, Z.; Arababadi, M.K.; Hassanshahi, G. CXCL10 Activities, Biological Structure, and Source along with Its Significant Role Played in Pathophysiology of Type i Diabetes Mellitus. Inflammation 2013, 36, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Dufour, J.H.; Dziejman, M.; Liu, M.T.; Leung, J.H.; Lane, T.E.; Luster, A.D. IFN-γ-Inducible Protein 10 (IP-10; CXCL10)-Deficient Mice Reveal a Role for IP-10 in Effector T Cell Generation and Trafficking. J. Immunol. 2002, 168, 3195–3204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohmori, Y.; Hamilton, T.A. Cooperative Interaction between Interferon (IFN) Stimulus Response Element and ΚB Sequence Motifs Controls IFNγ- and Lipopolysaccharide-Stimulated Transcription from the Murine IP-10 Promoter. J. Biol. Chem. 1993, 268, 6677–6688. [Google Scholar] [CrossRef] [PubMed]
- Brownell, J.; Bruckner, J.; Wagoner, J.; Thomas, E.; Loo, Y.-M.; Gale, M.; Liang, T.J.; Polyak, S.J. Direct, Interferon-Independent Activation of the CXCL10 Promoter by NF-ΚB and Interferon Regulatory Factor 3 during Hepatitis C Virus Infection. J. Virol. 2014, 88, 1582–1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siednienko, J.; Jackson, R.; Mellett, M.; Delagic, N.; Yang, S.; Wang, B.; Tang, L.S.; Callanan, J.J.; Mahon, B.P.; Moynagh, P.N. Pellino3 Targets the IRF7 Pathway and Facilitates Autoregulation of TLR3-and Viral-Induced Expression of Type i Interferons. Nat. Immunol. 2012, 13, 1055–1062. [Google Scholar] [CrossRef]
- Tzieply, N.; Kuhn, A.M.; Morbitzer, D.; Namgaladze, D.; Heeg, A.; Schaefer, L.; von Knethen, A.; Jensen, L.E.; Brüne, B. OxLDL Inhibits LPS-Induced IFNβ Expression by Pellino3- and IRAK1/4-Dependent Modification of TANK. Cell. Signal. 2012, 24, 1141–1149. [Google Scholar] [CrossRef]
- Reniewicz, P.; Kula, A.; Makuch, E.; Ochnik, M.; Lipiński, T.; Siednienko, J. Ligase Pellino3 Regulates Macrophage Action and Survival in Response to VSV Infection in RIG-I-Dependent Path. Oxid. Med. Cell. Longev. 2021, 2021, 6668463. [Google Scholar] [CrossRef]
- Butler, M.P.; Hanly, J.A.; Moynagh, P.N. Pellino3 Is a Novel Upstream Regulator of P38 MAPK and Activates CREB in a P38-Dependent Manner. J. Biol. Chem. 2005, 280, 27759–27768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauzzi, M.C.; Velazquez, L.; McKendry, R.; Mogensen, K.E.; Fellous, M.; Pellegrini, S. Interferon-α-Dependent Activation of Tyk2 Requires Phosphorylation of Positive Regulatory Tyrosines by Another Kinase. J. Biol. Chem. 1996, 271, 20494–20500. [Google Scholar] [CrossRef] [Green Version]
- Rani, M.R.S.; Leaman, D.W.; Han, Y.; Leung, S.; Croze, E.; Fish, E.N.; Wolfman, A.; Ransohoff, R.M. Catalytically Active TYK2 Is Essential for Interferon-β-Mediated Phosphorylation of STAT3 and Interferon-α Receptor-1 (IFNAR-1) but Not for Activation of Phosphoinositol 3-Kinase. J. Biol. Chem. 1999, 274, 32507–32511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siednienko, J.; Maratha, A.; Yang, S.; Mitkiewicz, M.; Miggin, S.M.; Moynagh, P.N. Nuclear Factor ΚB Subunits RelB and CRel Negatively Regulate Toll-like Receptor 3-Mediated β-Interferon Production via Induction of Transcriptional Repressor Protein YY1. J. Biol. Chem. 2011, 286, 44750–44763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leszczyńska, E.; Makuch, E.; Mitkiewicz, M.; Jasyk, I.; Narita, M.; Górska, S.; Lipiński, T.; Siednienko, J. Absence of Mal/Tirap Results in Abrogated Imidazoquinolinones-Dependent Activation of Irf7 and Suppressed Ifnβ and Ifn-i Activated Gene Production. Int. J. Mol. Sci. 2020, 21, 8925. [Google Scholar] [CrossRef]
- Siednienko, J.; Halle, A.; Nagpal, K.; Golenbock, D.T.; Miggin, S.M. TLR3-Mediated IFN-β Gene Induction Is Negatively Regulated by the TLR Adaptor MyD88 Adaptor-Like. Eur. J. Immunol. 2010, 40, 3150–3160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.H.; Murti, A.; Pfeffer, S.R.; Basu, L.; Kim, J.G.; Pfeffer, L.M. IFNα/β Promotes Cell Survival by Activating NF-ΚB. Proc. Natl. Acad. Sci. USA 2000, 97, 13631–13636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andzinski, L.; Wu, C.F.; Lienenklaus, S.; Kröger, A.; Weiss, S.; Jablonska, J. Delayed Apoptosis of Tumor Associated Neutrophils in the Absence of Endogenous IFN-β. Int. J. Cancer 2015, 136, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.Y.; Li, Y.; Kumagai, Y.; Xu, Y.; Weinstein, J.S.; Kellner, E.S.; Nacionales, D.C.; Butfiloski, E.J.; Van Rooijen, N.; Akira, S.; et al. Type I Interferon Modulates Monocyte Recruitment and Maturation in Chronic Inflammation. Am. J. Pathol. 2009, 175, 2023–2033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, H.; Okabe, Y.; Kawane, K.; Fukuyama, H.; Nagata, S. Lethal Anemia Caused by Interferon-β Produced in Mouse Embryos Carrying Undigested DNA. Nat. Immunol. 2005, 6, 49–56. [Google Scholar] [CrossRef]
- Thompson, M.R.; Kaminski, J.J.; Kurt-Jones, E.A.; Fitzgerald, K.A. Pattern Recognition Receptors and the Innate Immune Response to Viral Infection. Viruses 2011, 3, 920–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, W.J.; Han, S.H.; Kim, D.H.; Song, Y.J.; Lee, J.B.; Park, S.Y.; Song, C.S.; Lee, S.W.; Choi, I.S. Induction of IFN-β through TLR-3- A Nd RIG-I-Mediated Signaling Pathways in Canine Respiratory Epithelial Cells Infected with H3N2 Canine Influenza Virus. J. Microbiol. Biotechnol. 2021, 31, 942–948. [Google Scholar] [CrossRef] [PubMed]
- Utay, N.S.; Douek, D.C. Interferons and Hiv Infection: The Good, the Bad, and the Ugly. Pathog. Immun. 2016, 1, 107–116. [Google Scholar] [CrossRef] [Green Version]
- Filipi, M.; Jack, S. Interferons in the Treatment of Multiple Sclerosis: A Clinical Efficacy, Safety, and Tolerability Update. Int. J. MS Care 2020, 22, 165–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platanitis, E.; Demiroz, D.; Schneller, A.; Fischer, K.; Capelle, C.; Hartl, M.; Gossenreiter, T.; Müller, M.; Novatchkova, M.; Decker, T. A Molecular Switch from STAT2-IRF9 to ISGF3 Underlies Interferon-Induced Gene Transcription. Nat. Commun. 2019, 10, 2921. [Google Scholar] [CrossRef] [Green Version]
- Cheon, H.; Holvey-Bates, E.G.; Schoggins, J.W.; Forster, S.; Hertzog, P.; Imanaka, N.; Rice, C.M.; Jackson, M.W.; Junk, D.J.; Stark, G.R. IFNβ-Dependent Increases in STAT1, STAT2, and IRF9 Mediate Resistance to Viruses and DNA Damage. EMBO J. 2013, 32, 2751–2763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.; Ohmori, Y.; Bandyopadhyay, S.; Sen, G.; Hamilton, T. Interferon-Stimulated Response Element and NFϰB Sites Cooperate to Regulate Double-Stranded RNA-Induced Transcription of the IP-10 Gene. J. Interferon Res. 1994, 14, 357–363. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Makuch, E.; Jasyk, I.; Kula, A.; Lipiński, T.; Siednienko, J. IFNβ-Induced CXCL10 Chemokine Expression Is Regulated by Pellino3 Ligase in Monocytes and Macrophages. Int. J. Mol. Sci. 2022, 23, 14915. https://doi.org/10.3390/ijms232314915
Makuch E, Jasyk I, Kula A, Lipiński T, Siednienko J. IFNβ-Induced CXCL10 Chemokine Expression Is Regulated by Pellino3 Ligase in Monocytes and Macrophages. International Journal of Molecular Sciences. 2022; 23(23):14915. https://doi.org/10.3390/ijms232314915
Chicago/Turabian StyleMakuch, Edyta, Izabella Jasyk, Anna Kula, Tomasz Lipiński, and Jakub Siednienko. 2022. "IFNβ-Induced CXCL10 Chemokine Expression Is Regulated by Pellino3 Ligase in Monocytes and Macrophages" International Journal of Molecular Sciences 23, no. 23: 14915. https://doi.org/10.3390/ijms232314915
APA StyleMakuch, E., Jasyk, I., Kula, A., Lipiński, T., & Siednienko, J. (2022). IFNβ-Induced CXCL10 Chemokine Expression Is Regulated by Pellino3 Ligase in Monocytes and Macrophages. International Journal of Molecular Sciences, 23(23), 14915. https://doi.org/10.3390/ijms232314915