
The Role of the Coagulation System in Peripheral Arterial Disease: Interactions with the Arterial Wall and Its Vascular Microenvironment and Implications for Rational Therapies



{kind=link}
Abstract
1. Introduction
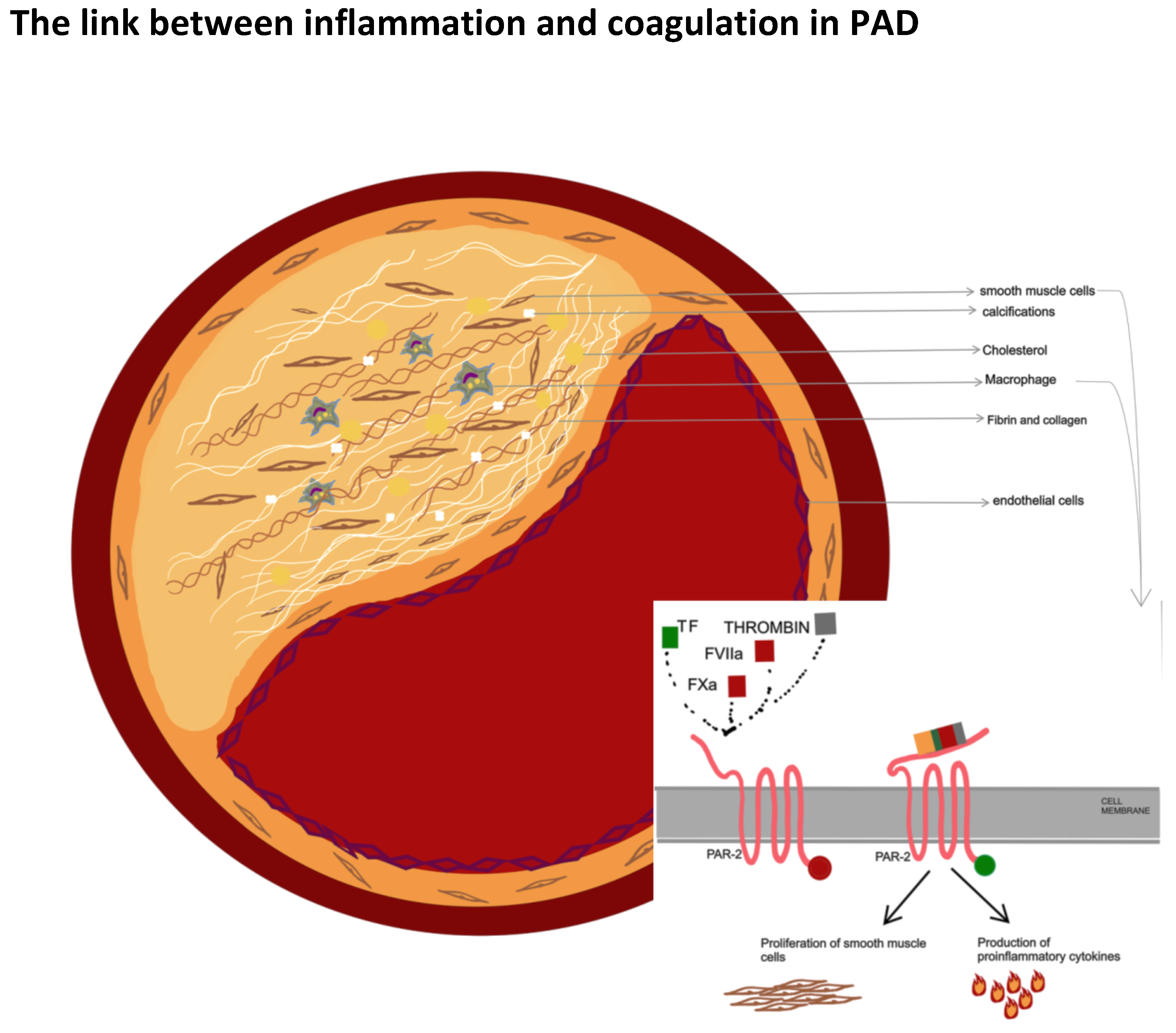
2. Role of Coagulation in the Formation and Progression of Atherosclerosis
3. Plaque Composition and Vulnerability: Differences in Coronary and Peripheral Atherosclerotic Disease
4. Endothelial Disfunction in Chronic Kidney Disease and the Risk of Lower Extremity Atherosclerosis
5. Pad: An Inflammatory or a Procoagulant State?
6. COVID-19: A Potential Model of the Hyperinflammation and Dysfunction of Coagulation
7. The Role of Inflammation and the Immune System in Atherosclerosis
Involvement of the Immune System in Atherosclerosis
8. Inflammatory Cytokines in the Atherosclerotic Disease
9. Coagulation Factors as Markers of Risk in Peripheral Artery Disease
9.1. Tissue Factor and Tissue Factor Pathway Inhibitor
9.2. Fibrinogen
9.3. D-dimer
9.4. Von Villebrand Factor and ADAMTS13
9.5. Tissue Plasminogen Activator
10. The Role of Genetics in Peripheral Artery Disease
10.1. Prothrombin G20210A Mutation
10.2. Factor V Leiden
10.3. Methylenetetrahydrofolate Reductase (MTHFR) C677T Mutation
11. Rational Therapy
12. Anti-Inflammatory and Immunomodulatory Drugs as a New Potential Therapeutic Strategy: A Hope for The Future?
12.1. ANTI-IL1 Agents
12.2. Colchicine
12.3. ANTI-IL6 Agents
12.4. ANTI-TNF Agents
12.5. Immunomodulatory Drugs
13. Future Perspectives
14. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Fowkes, F.G.; Rudan, D.; Rudan, I.; Aboyans, V.; Denenberg, J.O.; McDermott, M.M.; Norman, P.E.; Sampson, U.K.; Williams, L.J.; Mensah, G.A.; et al. Comparison of global estimates of prevalence and risk factors for peripheral artery disease in 2000 and 2010: A systematic review and analysis. Lancet 2013, 382, 1329–1340. [Google Scholar] [CrossRef] [PubMed]
- Tran, B. Assessment and management of peripheral arterial disease: What every cardiologist should know. Heart 2021, 107, 1835–1843. [Google Scholar] [CrossRef] [PubMed]
- Sampson, U.K.; Fowkes, F.G.; McDermott, M.M.; Criqui, M.H.; Aboyans, V.; Norman, P.E.; Forouzanfar, M.H.; Naghavi, M.; Song, Y.; Harrell, F.E., Jr.; et al. Global and regional burden of death and disability from peripheral artery disease: 21 world regions, 1990 to 2010. Glob. Heart 2014, 9, 145–158. [Google Scholar] [CrossRef]
- Novo, S. Classification, epidemiology, risk factors, and natural history of peripheral arterial disease. Diabetes Obes. Metab. 2002, 4 (Suppl. S2), S1–S6. [Google Scholar] [CrossRef] [PubMed]
- Rooke, T.W.; Hirsch, A.T.; Misra, S.; Sidawy, A.N.; Beckman, J.A.; Findeiss, L.K.; Golzarian, J.; Gornik, H.L.; Halperin, J.L.; Jaff, M.R.; et al. 2011 ACCF/AHA focused update of the guideline for the management of patients with peripheral artery disease (updating the 2005 guideline): A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: Developed in collaboration with the Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society for Vascular Medicine, and Society for Vascular Surgery. Catheter. Cardiovasc. Interv. 2012, 79, 501–531. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.L.; Halperin, J.L.; Albert, N.M.; Bozkurt, B.; Brindis, R.G.; Curtis, L.H.; DeMets, D.; Guyton, R.A.; Hochman, J.S.; Kovacs, R.J.; et al. Management of patients with peripheral artery disease (compilation of 2005 and 2011 ACCF/AHA guideline recommendations): A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2013, 127, 1425–1443. [Google Scholar] [CrossRef] [PubMed]
- Steg, P.G.; Bhatt, D.L.; Wilson, P.W.; D’Agostino, R., Sr.; Ohman, E.M.; Röther, J.; Liau, C.S.; Hirsch, A.T.; Mas, J.L.; Ikeda, Y.; et al. REACH Registry Investigators. One-year cardiovascular event rates in outpatients with atherothrombosis. JAMA 2007, 297, 1197–2206. [Google Scholar] [CrossRef] [PubMed]
- Andersson, C.; Nayor, M.; Tsao, C.W.; Levy, D.; Vasan, R.S. Framingham Heart Study, JACC Focus Seminar, 1/8. J. Am. Coll. Cardiol. 2021, 77, 2680–2692. [Google Scholar] [CrossRef]
- Stamler, J.; Naton, J.D. The Multiple Risk Factor Intervention Trial (MRFIT)—Importance then and now. JAMA 2008, 300, 1343–1345. [Google Scholar] [CrossRef]
- Makin, A.J.; Chung, N.A.; Silverman, S.H.; Lip, G.Y. Vascular endothelial growth factor and tissue factor in patients with established peripheral artery disease: A link between angiogenesis and thrombogenesis? Clin. Sci. 2003, 104, 397–404. [Google Scholar] [CrossRef]
- Lassila, R. Role and Management of Coagulation Disorders in Peripheral Arterial Disease. Scand. J. Surg. 2012, 101, 94–99. [Google Scholar] [CrossRef]
- Olie, R.H.; van der Meijden, P.E.J.; Ten Cate, H. The coagulation system in atherothrombosis: Implications for new therapeutic strategies. Res. Pract. Thromb. Haemost. 2018, 2, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, J.N.; Smith, K.M.; Schwartz, S.M.; Gordon, D. Localization of tissue factor in the normal vessel wall and the atherosclerotic plaque. Proc. Natl. Acad. Sci. USA 1989, 86, 2839–2843. [Google Scholar] [CrossRef] [PubMed]
- Borissoff, J.I.; Heeneman, S.; Kilinç, E.; Kaššák, P.; Van Oerle, R.; Winckers, K.; Govers-Riemslag, J.W.; Hamulyák, K.; Hackeng, T.M.; Daemen, M.J.; et al. Early Atherosclerosis Exhibits an Enhanced Procoagulant State. Circulation 2010, 122, 821–830. [Google Scholar] [CrossRef] [PubMed]
- Gertow, K.; Amato, M.; Werba, J.P.; Bianchi, E.; Parolari, A.; Colnago, D.; Brambilla, M.; Ravani, A.; Veglia, F.; Baldassarre, D.; et al. Tissue factor gene promoter haplotype associates with carotid intima-media thickness in subjects in cardiovascular risk prevention. Atherosclerosis 2009, 207, 168–173. [Google Scholar] [CrossRef]
- Westmuckett, A.D.; Lupu, C.; Goulding, D.A.; Das, S.; Kakkar, V.V.; Lupu, F. In situ analysis of tissue factor-dependent thrombin generation in human atherosclerotic vessels. Thromb. Haemost. 2000, 84, 904–911. [Google Scholar] [CrossRef] [PubMed]
- Narula, N.; Dannenberg, A.J.; Olin, J.W.; Bhatt, D.L.; Johnson, K.W.; Nadkarni, G.; Min, J.; Torii, S.; Poojary, P.; Anand, S.S.; et al. Pathology of Peripheral Artery Disease in Patients with Critical Limb Ischemia. J. Am. Coll. Cardiol. 2018, 72, 18. [Google Scholar] [CrossRef] [PubMed]
- Kockx, M.M.; De Meyer, G.R.; Muhring, J.; Bult, H.; Bultinck, J.; Herman, A.G. Distribution of cell replication and apoptosis in atherosclerotic plaques of cholesterol-fed rabbits. Atherosclerosis 1996, 120, 115–124. [Google Scholar] [CrossRef]
- Shah, P.K.; Falk, E.; Badimon, J.J.; Fernandez-Ortiz, A.; Mailhac, A.; Villareal-Levy, G.; Fallon, J.T.; Regnstrom, J.; Fuster, V. Human monocyte derived macrophages induce collagen breakdown in fibrous caps of atherosclerotic plaques: Potential role of matrix-degrading metalloproteinases and implications for plaque rupture. Circulation 1995, 92, 1565–1569. [Google Scholar] [PubMed]
- Hetterich, H.; Webber, N.; Willner, M.; Herzen, J.; Birnbacher, L.; Hipp, A.; Marschner, M.; Auweter, S.D.; Habbel, C.; Schüller, U.; et al. AHA classification of coronary and carotid atherosclerotic plaques by grating-based phase-contrast computed tomography. Eur. Radiol. 2016, 26, 3223–3233. [Google Scholar] [CrossRef]
- Derksen, W.J.; de Vries, J.P.; Vink, A.; Velema, E.; Vos, J.A.; de Kleijn, D.; Moll, F.L.; Pasterkamp, G. Histologic atherosclerotic plaque characteristics are associated with restenosis rates after endarterectomy of the common and superficial femoral arteries. J. Vasc. Surg. 2010, 52, 592–599. [Google Scholar] [CrossRef]
- Ross, R.; Wight, T.N.; Strandness, E.; Thiele, B. Human atherosclerosis. I. Cell constitution and characteristics of advanced lesions of the superficial femoral artery. Am. J. Pathol. 1984, 114, 79–93. [Google Scholar]
- McDermott, M.M.; Kramer, C.M.; Tian, L.; Carr, J.; Guralnik, J.M.; Polonsky, T.; Carroll, T.; Kibbe, M.; Criqui, M.H.; Ferrucci, L.; et al. Plaque Composition in the Proximal Superficial Femoral Artery and Peripheral Artery Disease Events. J. Am. Coll. Cardiol. Cardiovasc. Imaging 2017, 10, 1003–1012. [Google Scholar] [CrossRef] [PubMed]
- Abbas, A.E.; Zacharias, S.K.; Goldstein, J.A.; Hanson, I.D.; Safian, R.D. Invasive characterization of atherosclerotic plaque in patients with peripheral arterial disease using near-infrared spectroscopy intravascular ultrasound. Catheter. Cardiovasc. Interv. 2017, 90, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Polonsky, T.S.; Liu, K.; Tian, L.; Carr, J.; Carroll, T.J.; Berry, J.; Criqui, M.H.; Ferrucci, L.; Guralnik, J.M.; Kibbe, M.R.; et al. High-Risk Plaque in the Superficial Femoral Artery of People with Peripheral Artery Disease: Prevalence and Associated Clinical Characteristics. Atherosclerosis 2014, 237, 169–176. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jezovnik, M.K.; Zidar, N.; Lezaic, L.; Gersak, B.; Poredos, P. Identification of inflamed atherosclerotic lesions in vivo using PET-CT. Inflammation 2014, 37, 426–434. [Google Scholar] [CrossRef] [PubMed]
- Rudd, J.H.; Myers, K.S.; Bansilal, S.; Machac, J.; Woodward, M.; Fuster, V.; Farkouh, M.E.; Fayad, Z.A. Relationships among regional arterial inflammation, calcification, risk factors, and biomarkers: A prospective fluorodeoxyglucose positron-emission tomography/computed tomography imaging study. Circ. Cardiovasc. Imaging 2009, 2, 107–115. [Google Scholar] [CrossRef]
- Fischer, A.; Gutstein, D.E.; Fuster, V. Thrombosis and coagulation abnormalities in the acute coronary syndromes. Cardiol. Clin. 1999, 17, 283–294. [Google Scholar] [CrossRef]
- Marynenko, T.; Halenova, T.; Raksha, N.; Vovk, T.; Tyravska, Y.; Savchuk, O.; Ostapchenko, L. Coagulation markers in patients with coronary artery disease. J. Biol. Res. 2022, 95, 10259. [Google Scholar] [CrossRef]
- Sarnak, M.J.; Levey, A.S.; Schoolwerth, A.C.; Coresh, J.; Culleton, B.; Hamm, L.L.; McCullough, P.A.; Kasiske, B.L.; Kelepouris, E.; Klag, M.J.; et al. Kidney disease as a risk factor for development of cardiovascular disease: A statement from the American Heart Association Councils on Kidney in Cardiovascular Disease, High Blood Pressure Research, Clinical Cardiology, and Epidemiology and Prevention. Circulation 2003, 108, 2154–2169. [Google Scholar] [CrossRef]
- O’Hare, A.; Johansen, K. Lower-extremity peripheral arterial disease among patients with end-stage renal disease. J. Am. Soc. Nephrol. 2001, 12, 2838–2847. [Google Scholar] [CrossRef]
- Roumeliotis, S.; Mallamaci, F.; Zoccal, C. Endothelial Dysfunction in Chronic Kidney Disease, from Biology to Clinical Outcomes: A 2020 Update. J. Clin. Med. 2020, 9, 2359. [Google Scholar] [CrossRef]
- Wattanakit, K.; Folsom, A.R.; Selvin, E.; Coresh, J.; Hirsch, A.T.; Weatherley, B.D. Kidney function and risk of peripheral arterial disease: Results from the Atherosclerosis Risk in Communities (ARIC) Study. J. Am. Soc. Nephrol. 2007, 18, 629–636. [Google Scholar] [CrossRef] [PubMed]
- O’Hare, A.M.; Hsu, C.Y.; Bacchetti, P.; Johansen, K.L. Peripheral vascular disease risk factors among patients undergoing hemodialysis. J. Am. Soc. Nephrol. 2002, 13, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Jaar, B.G.; Plantinga, L.C.; Astor, B.C.; Fink, N.E.; Longenecker, C.; Tracy, R.P.; Marcovina, S.M.; Powe, N.R.; Coresh, J. Novel and traditional cardiovascular risk factors for peripheral arterial disease in incident-dialysis patients. Adv. Chronic. Kidney. Dis. 2007, 14, 304–313. [Google Scholar] [CrossRef]
- Gimbrone, M.A.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [PubMed]
- Düsing, P.; Zietzer, A.; Goody, P.R.; Hosen, M.R.; Kurts, C.; Nickenig, G.; Jansen, F. Vascular pathologies in chronic kidney disease: Pathophysiological mechanisms and novel therapeutic approaches. J. Mol. Med. 2021, 99, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Stenvinkel, A.; Alvestrand, A. Inflammation in end-stage renal disease: Sources, consequences and therapy. Semin. Dial 2022, 15, 330–338. [Google Scholar]
- Stenvinkel, P. Endothelial dysfunction and inflammation—is there a link? Nephrol. Dial Transpl. 2001, 16, 1968–1971. [Google Scholar] [CrossRef] [PubMed]
- Igari, K.; Kudo, T.; Toyofuku, T.; Inoue, Y. The relationship between endothelial dysfunction and endothelial cell markers in peripheral arterial disease. PLoS ONE 2016, 11, e0166840. [Google Scholar] [CrossRef]
- Cuenca, M.V.; Van Bezu, J.; Beelen, R.H.J.; Vervloet, M.G.; Hordijk, P.L. Stabilization of cell-Cell junctions by active Vitamin D ameliorates uraemia-Induced loss of human endothelial barrier function. Nephrol. Dial. Transplant. 2019, 34, 252–264. [Google Scholar] [CrossRef]
- Himmelfarb, J.; Stenvinkel, P.; Ikizler, T.A.; Hakim, R.M. The elephant of uremia: Oxidative stress as a unifying concept of cardiovascular disease in uremia. Kidney Int. 2022, 62, 1524–1538. [Google Scholar] [CrossRef] [PubMed]
- Annuk, M.; Zilmer, M.; Lind, L.; Linde, T.; Fellström, B. Oxidative stress and endothelial function in chronic renal failure. J. Am. Soc. Nephrol. 2001, 12, 2747–2752. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.I.M.I.; Saglam, M.; Caglar, K.; Cakir, E.; Sonmez, A.; Ozgurtas, T.; Aydin, A.; Eyileten, T.; Ozcan, O.; Acikel, C.; et al. The determinants of endothelial dysfunction in CKD: Oxidative stress and asymmetric dimethylarginine. Am. J. Kidney Dis. 2006, 47, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.I.; Sonmez, A.; Karaman, M.; Ay, S.A.; Saglam, M.; Yaman, H.; Kilic, S.; Eyileten, T.; Caglar, K.; Oguz, Y.; et al. Low triiodothyronine alters flow-Mediated vasodilatation in advanced nondiabetic kidney disease. Am. J. Nephrol. 2011, 33, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Shuto, E.; Taketani, Y.; Tanaka, R.; Harada, N.; Isshiki, M.; Sato, M.; Nashiki, K.; Amo, K.; Yamamoto, H.; Higashi, Y.; et al. Dietary phosphorus acutely impairs endothelial function. J. Am. Soc. Nephrol. 2009, 20, 1504–1512. [Google Scholar] [CrossRef]
- Vervloet, M. Renal and extrarenal effects of fibroblast growth factor 23. Nat. Rev. Nephrol. 2019, 15, 109–120. [Google Scholar] [CrossRef]
- Silswal, N.; Touchberry, C.D.; Daniel, D.R.; McCarthy, D.L.; Zhang, S.; Andresen, J.; Stubbs, J.R.; Wacker, M.J. FGF23 directly impairs endothelium-Dependent vasorelaxation by increasing superoxide levels and reducing nitric oxide bioavailability. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E426–E436. [Google Scholar] [CrossRef]
- Baylis, C. Arginine, arginine analogs and nitric oxide production in chronic kidney disease. Nat. Clin. Pract. Nephrol. 2006, 2, 209–220. [Google Scholar] [CrossRef]
- Batkoa, K.; Krzanowskia, M.; Gajdab, M. Endothelial injury is closely related to osteopontin and TNF receptormediated inflammation in end-stage renal disease. Cytokine 2019, 121, 54729. [Google Scholar]
- Yoshii, Y.; Okada, Y.; Sasaki, S.; Mori, H.; Oida, K.; Ishii, H. Expression of thrombomodulin in human aortic smooth muscle cells with special reference to atherosclerotic lesion types and age differences. Med. Electron. Microsc. 2003, 36, 165–172. [Google Scholar] [CrossRef]
- Ishii, H.; Uchiyama, H.; Kazama, M. Soluble thrombomodulin antigen in conditioned medium is increased by damage of endothelial cells. Thromb. Haemost. 1991, 65, 618–623. [Google Scholar] [CrossRef]
- Pircher, J.; Merkle, M.; Wörnle, M.; Ribeiro, A.; Czermak, T.; Stampnik, Y.; Mannell, H.; Niemeyer, M.; Vielhauer, V.; Krötz, F. Prothrombotic effects of tumor necrosis factor alpha in vivo are amplified by the absence of TNF-alpha receptor subtype 1 and require TNF-alpha receptor subtype 2. Arthritis Res. Ther. 2012, 14, R225. [Google Scholar] [CrossRef]
- El-Abbadi, M.M.; Pai, A.S.; Leaf, E.M.; Yang, H.Y.; Bartley, B.A.; Quan, K.K.; Ingalls, C.M.; Liao, H.W.; Giachelli, C.M. Phosphate feeding induces arterial medial calcification in uremic mice: Role of serum phosphorus, fibroblast growth factor-23, and osteopontin. Kidney Int. 2009, 75, 1297–1307. [Google Scholar] [CrossRef]
- Pedersen, T.X.; Madsen, M.; Junker, N.; Christoffersen, C.; Vikeså, J.; Bro, S.; Hultgårdh-Nilsson, A.; Nielsen, L.B. Osteopontin deficiency dampens the pro-atherogenic effect of uraemia. Cardiovasc. Res. 2013, 98, 352–359. [Google Scholar] [CrossRef][Green Version]
- Barreto, D.V.; Lenglet, A.; Liabeuf, S.; Kretschmer, A.; Barreto, F.C.; Nollet, A.; Slama, M.; Choukroun, G.; Brazier, M.; Massy, Z. Prognostic implication of plasma osteopontin levels in patients with chronic kidney disease. Nephron. Clin. Pract. 2011, 117, c363–c372. [Google Scholar] [CrossRef]
- van der Vorm, L.N.; Visser, R.; Huskens, D.; Veninga, A.; Adams, D.L.; Remijn, J.A.; Hemker, H.C.; Rensma, P.L.; van Horssen, R.; de Laat, B. Circulating active von Willebrand factor levels are increased in chronic kidney disease and end-Stage renal disease. Clin. Kidney J. 2020, 13, 72–74. [Google Scholar] [CrossRef]
- Péquériaux, N.C.; Fijnheer, R.; Gemen, E.F.; Barendrecht, A.D.; Dekker, F.W.; Krediet, R.T.; Beutler, J.J.; Boeschoten, E.W.; Roest, M. Plasma concentration of von Willebrand factor predicts mortality in patients on chronic renal replacement therapy. Nephrol. Dial. Transplant. 2012, 27, 2452–2457. [Google Scholar] [CrossRef]
- Nascimento, M.-M.; Pecoits-Filho, R.; Lindholm, B. Inflammation, Malnutrition and Atherosclerosis in End-Stage Renal Disease: A Global Perspective. Blood Purif. 2002, 20, 454–458. [Google Scholar] [CrossRef]
- Hirsch, A.T.; Hartman, L.; Town, R.J.; Virnig, B.A. National health care costs of peripheral arterial disease in the Medicare population. Vasc. Med. 2008, 13, 209–215. [Google Scholar] [CrossRef]
- Matsushita, K.; Ballew, S.H.; Coresh, J.; Arima, H.; Ärnlöv, J.; Cirillo, M.; Ebert, N.; Hiramoto, J.S.; Kimm, H.; Shlipak, M.G.; et al. Article measures of chronic kidney disease and risk of incident peripheral artery disease: A collaborative meta-analysis of individual partecipant data. Lancet Diabetes Endocrinol. 2017, 5, 718–728. [Google Scholar] [CrossRef] [PubMed]
- Bourrier, M.; Ferguson, T.W.; Embil, J.M.; Rigatto, C.; Komenda, P.; Tangri, N. Peripheral artery disease: Its adverse consequences with and without CKD. Am. J. Kidney Dis. 2020, 75, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Gilhotra, R.A.; Rodrigues, B.T.; Vangaveti, V.N.; Kan, G.; Porter, D.; Sangla, K.S.; Malabu, U.H. Non-traumatic lower limb amputation in patients with end-stage renal failure on dialysis: An Australian perspective. Ren. Fail. 2016, 38, 1036–1043. [Google Scholar] [CrossRef] [PubMed]
- Cooper, B.A.; Penne, E.L.; Bartlett, L.H.; Pollock, C.A. Protein malnutrition and hypoalbuminemia as predictors of vascular events and mortality in ESRD. Am. J. Kidney Diseases. 2004, 43, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Anantha-Narayanan, M.; Sheikh, A.B.; Nagpal, S.; Jelani, Q.-u.-A.; Smolderen, K.G.; Regan, C.; Ionescu, C.; Ochoa Chaar, C.I.; Schneider, M.; Llanos-Chea, F.; et al. Systematic Review and Meta analysis of Outcomes of Lower Extremity Peripheral Arterial Interventions in Patients with and without Chronic Kidney Disease or End Stage Renal Disease. J. Vasc. Surg. 2021, 73, 331–340. [Google Scholar] [CrossRef]
- Ardissino, D.; Merlini, P.A.; Bauer, K.A.; Bramucci, E.; Ferrario, M.; Coppola, R.; Fetiveau, R.; Lucreziotti, S.; Rosenberg, R.D.; Mannucci, P.M. Thrombogenic potential of human coronary atherosclerotic plaques. Blood 2001, 98, 2726–2729. [Google Scholar] [CrossRef]
- Hollenberg, M.D.; Saifeddine, M.; Al-Ani, B.; Gui, Y. Proteinase-activated receptor 4 (PAR4): Action of PAR4-activating peptidesin vascular and gastric tissue and lack of cross-reactivity withPAR1 and PAR2. Can. J. Physiol. Pharmacol. 1999, 77, 458–464. [Google Scholar] [CrossRef]
- Emilsson, K.; Wahlestedt, C.; Sun, M.K.; Nystedt, S.; Owman, C.; Sun-delin, J. Vascular effects of proteinase-activated receptor 2 ago-nist peptide. J. Vasc. Res. 1997, 34, 267–272. [Google Scholar] [CrossRef]
- Indrakusuma, I.; Romacho, T.; Eckel, J. Protease-activated recep-tor 2 promotes pro-atherogenic effects through transactivation of the VEGF receptor 2 in human vascular smooth muscle cells. Front. Pharmacol. 2016, 7, 497. [Google Scholar]
- Lorence, J.M.; Krupa, A.; Booshehri, L.M.; Allen, T.C.; Kurdowska, A.K. Metalloproteinase-9 contributes to endothelial dysfunctionin atherosclerosis via protease activated receptor-1. PLoS ONE 2017, 12, e0171427. [Google Scholar]
- Zuo, P.; Zuo, Z.; Zheng, Y.; Wang, X.; Zhou, Q.; Chen, L.; Ma, G. Protease-activated receptor-2 deficiency attenuates atherosclerotic lesion progression and instability in apolipoprotein E-deficien mice. Front. Pharmacol. 2017, 8, 647. [Google Scholar] [CrossRef] [PubMed]
- Riewald, M.; Petrovan, R.J.; Donner, A.; Mueller, B.M.; Ruf, W. Activation of endothelial cell protease activated receptor 1 by the protein C pathway. Science 2002, 296, 1880–1882. [Google Scholar] [CrossRef] [PubMed]
- Ravid, J.D.; Leiva, O.; Chitalia, V.C. Janus Kinase Signaling Pathway and Its Role in COVID-19 Inflammatory, Vascular, and Thrombotic Manifestations. Cells 2022, 11, 306. [Google Scholar] [CrossRef] [PubMed]
- Cevik, M.; Tate, M.; Lloyd, O.; Maraolo, A.E.; Schafers, J.; Ho, A. SARS-CoV-2, SARS-CoV, and MERS-CoV viral load dynamics, duration of viral shedding, and infectiousness: A systematic review and meta-analysis. Lancet Microbe 2021, 2, e13–e22. [Google Scholar] [CrossRef]
- Giamarellos-Bourboulis, E.J.; Netea, M.G.; Rovina, N.; Akinosoglou, K.; Antoniadou, A.; Antonakos, N.; Damoraki, G.; Gkavogianni, T.; Adami, M.E.; Katsaounou, P.; et al. Complex Immune Dysregulation in COVID-19 Patients with Severe Respiratory Failure. Cell Host Microbe 2020, 27, 992–1000. [Google Scholar] [CrossRef] [PubMed]
- Sims, J.T.; Krishnan, V.; Chang, C.Y.; Engle, S.M.; Casalini, G.; Rodgers, G.H.; Bivi, N.; Nickoloff, B.J.; Konrad, R.J.; de Bono, S.; et al. Characterization of the cytokine storm reflects hyperinflammatory endothelial dysfunction in COVID-19. J. Allergy Clin. Immunol. 2021, 147, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Tay, M.Z.; Poh, C.M.; Renia, L.; MacAry, P.A.; Ng, L.F.P. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef]
- Group, R.C.; Horby, P.; Lim, W.S.; Emberson, J.R.; Mafham, M.; Bell, J.L.; Linsell, L.; Staplin, N.; Brightling, C.; Ustianowski, A.; et al. Dexamethasone in Hospitalized Patients with COVID-19. N. Engl. J. Med. 2021, 384, 693–704. [Google Scholar]
- Rosas, I.O.; Brau, N.; Waters, M.; Go, R.C.; Hunter, B.D.; Bhagani, S.; Skiest, D.; Aziz, M.S.; Cooper, N.; Douglas, I.S.; et al. Tocilizumab in Hospitalized Patients with Severe COVID-19 Pneumonia. N. Engl. J. Med. 2021, 384, 1503–1516. [Google Scholar] [CrossRef]
- Reman-Cap Investigators; Gordon, A.C.; Mouncey, P.R.; Al-Beidh, F.; Rowan, K.M.; Nichol, A.D.; Arabi, Y.M.; Annane, D.; Beane, A.; van Bentum-Puijk, W.; et al. Interleukin-6 Receptor Antagonists in Critically Ill Patients with COVID-19. N. Engl. J. Med. 2021, 384, 1491–1502. [Google Scholar]
- Kalil, A.C.; Patterson, T.F.; Mehta, A.K.; Tomashek, K.M.; Wolfe, C.R.; Ghazaryan, V.; Marconi, V.C.; Ruiz-Palacios, G.M.; Hsieh, L.; Kline, S.; et al. Baricitinib plus Remdesivir for Hospitalized Adults with COVID-19. N. Engl. J. Med. 2021, 384, 795–807. [Google Scholar] [CrossRef] [PubMed]
- Mondal, R.; Lahiri, D.; Deb, S.; Bandyopadhyay, D.; Shome, G.; Sarkar, S.; Paria, S.R.; Thakurta, T.G.; Singla, P.; Biswas, S.C. COVID-19: Are we dealing with a multisystem vasculopathy in disguise of a viral infection? J. Thromb. Thrombolysis 2020, 50, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.M.; Xu, G.; Wang, B.; Liu, B.C. Cytokine storm syndrome in coronavirus disease 2019: A narrative review. J. Intern. Med. 2021, 289, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Zanoli, L.; Gaudio, A.; Mikhailidis, D.P.; Katsiki, N.; Castellino, N.; Lo Cicero, L.; Geraci, G.; Sessa, C.; Fiorito, L.; Marino, F.; et al. Vascular Dysfunction of COVID-19 Is Partially Reverted in the Long-Term. Circ. Res. 2022, 130, 1276–1285. [Google Scholar] [CrossRef] [PubMed]
- Nägele, M.P.; Haubner, B.; Tanner, F.C.; Ruschitzka, F.; Flammer, A.J. Endothelial dysfunction in COVID-19: Current findings and therapeutic implications. Atherosclerosis 2020, 314, 58–62. [Google Scholar] [CrossRef]
- Kaur, S.; Tripathi, D.M.; Yadav, A. The Enigma of Endothelium in COVID-19. Front Physiol. 2020, 11, 89. [Google Scholar] [CrossRef]
- Pearce, L.; Davidson, S.M.; Yellon, D.M. The cytokine storm of COVID19: A spotlight on prevention and protection. Expert Opin. Ther. Targets 2020, 24, 723–730. [Google Scholar] [CrossRef]
- Sardu, C.; Gambardella, J.; Morelli, M.B.; Wang, X.; Marfella, R.; Santulli, G. Hypertension, thrombosis, kidney failure, and diabetes: Is COVID-19 an endothelial disease? A comprehensive evaluation of clinical and basic evidence. J. Clin. Med. 2020, 9, E1417. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Zanoli, L.; Briet, M.; Empana, J.P.; Cunha, P.G.; Mäki-Petäjä, K.M.; Protogerou, A.D.; Tedgui, A.; Touyz, R.M.; Schiffrin, E.L.; Spronck, B.; et al. Association for Research into Arterial Structure, Physiology (ARTERY) Society, the European Society of Hypertension (ESH) Working Group on Vascular Structure and Function, and the European Network for Noninvasive Investigation of Large Arteries. Vascular consequences of inflammation: A position statement from the ESH Working Group on Vascular Structure and Function and the ARTERY Society. J. Hypertens. 2020, 38, 1682–1698. [Google Scholar] [CrossRef] [PubMed]
- Potteaux, S.; Gautier, E.L.; Hutchison, S.B.; van Rooijen, N.; Rader, D.J.; Thomas, M.J. Suppressed monocyte recruitment drives macrophage removal from atherosclerotic plaques of ApoE/mice during disease regression. J. Clin. Investig. 2011, 121, 2025–2036. [Google Scholar] [CrossRef]
- Zernecke, A.; Winkels, H.; Cochain, C.; Williams, J.W.; Wolf, D.; Soehnlein, O.; Robbins, C.S.; Monaco, C.; Park, I.; McNamara, C.A.; et al. Meta-analysis of leukocyte diversity in atherosclerotic mouse aortas. Circ. Res. 2020, 127, 402–426. [Google Scholar] [CrossRef] [PubMed]
- Depuydt, M.A.C.; Prange, K.H.M.; Slenders, L.; Örd, T.; Elbersen, D.; Boltjes, A.; de Jager, S.C.A.; Asselbergs, F.W.; de Borst, G.J.; Aavik, E.; et al. Microanatomy of the human atherosclerotic plaque by single-cell transcriptomics. Circ. Res. 2020, 127, 1437–1455. [Google Scholar] [CrossRef] [PubMed]
- Merad, M.; Sathe, P.; Helft, J.; Miller, J.; Mortha, A. The dendritic cell lineage: Ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu. Rev. Immunol. 2013, 31, 563–604. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Sobenin, I.A.; Orekhov, A.N.; Bobryshev, Y.V. Dendritic cells in atherosclerotic inflammation: The complexity of functions and the peculiarities 114 Ashok Munjal and Rekha Khandia of pathophysiological effects. Front. Physiol. 2014, 5, 196. [Google Scholar] [CrossRef] [PubMed]
- Vainio, S.; Ikonen, E. Macrophage cholesterol transport: A critical player in foam cell formation. Ann. Med. 2003, 35, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Birck, M.M.; Saraste, A.; Hyttel, P.; Odermarsky, M.; Liuba, P.; Saukko, P.; Hansen, A.K.; Pesonen, E. Endothelial cell death and intimal foam cell accumulation in the coronary artery of infected hypercholesterolemic minipigs. J. Cardiovasc. Transl. Res. 2013, 6, 579–587. [Google Scholar] [CrossRef]
- Wang, L.; Ai, Z.; Khoyratty, T.; Zec, K.; Eames, H.L.; van Grinsven, E.; Hudak, A.; Morris, S.; Ahern, D.; Monaco, C.; et al. ROS-producing immature neutrophils in giant cell arteritis are linked to vascular pathologies. JCI Insight 2020, 5, e139163. [Google Scholar] [CrossRef] [PubMed]
- Warnatsch, A.; Ioannou, M.; Wang, Q.; Papayannopoulos, V. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science 2015, 349, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Ketelhuth, D.F.J.; Hansson, G.K. Adaptive response of T and B cells in atherosclerosis. Circ. Res. 2016, 118, 668–678. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, M.; Tabas, I. Dendritic cells in atherosclerosis. In Seminars in Immunopathology; Springer: Berlin/Heidelberg, Germany, 2014; Volume 36, pp. 93–102. [Google Scholar]
- Saigusa, R.; Winkels, H.; Ley, K. T cell subsets and functions in atherosclerosis. Nat. Rev. Cardiol. 2020, 17, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Nus, M.; Sage, A.P.; Lu, Y.; Masters, L.; Lam, B.Y.H.; Newland, S.; Weller, S.; Tsiantoulas, D.; Raffort, J.; Marcus, D.; et al. Marginal zone B cells control the response of follicular helper T cells to a high-cholesterol diet. Nat. Med. 2017, 23, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Castell, J.V.; Gómez-Lechón, M.J.; David, M.; Fabra, R.; Trullenque, R.; Heinrich, P.C. Acute-phase response of human hepatocytes: Regulation of acute-phase protein synthesis by interleukin-6. Hepatology 1990, 12, 1179–1186. [Google Scholar] [CrossRef] [PubMed]
- Kamari, Y.; Werman-Venkert, R.; Shaish, A.; Werman, A.; Harari, A.; Gonen, A.; Voronov, E.; Grosskopf, I.; Sharabi, Y.; Grossman, E.; et al. Differential role and tissue specificity of interleukin-1alpha gene expression in atherogenesis and lipid metabolism. Atherosclerosis 2007, 195, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Vromman, A.; Ruvkun, V.; Shvartz, E.; Wojtkiewicz, G.; Santos Masson, G.; Tesmenitsky, Y.; Folco, E.; Gram, H.; Nahrendorf, M.; Swirski, F.K.; et al. Stage-dependent differential effects of interleukin-1 isoforms on experimental atherosclerosis. Eur. Heart J. 2019, 40, 2482–2491. [Google Scholar] [CrossRef] [PubMed]
- Mallat, Z.; Corbaz, A.; Scoazec, A.; Graber, P.; Alouani, S.; Esposito, B.; Humbert, Y.; Chvatchko, Y.; Tedgui, A. Interleukin-18/interleukin-18 binding protein signaling modulates atherosclerotic lesion development and stability. Circ. Res. 2001, 89, E41–E45. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Alcaide, P.; Liu, L.; Sun, J.; He, A.; Luscinskas, F.W.; Shi, G.P. Regulation of endothelial cell adhesion molecule expression by mast cells, macrophages, and neutrophils. PLoS ONE 2011, 6, e14525. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, J.; Liu, X.; Awar, L.; McMickle, A.; Bai, F.; Yu, S. IL-17 induces expression of vascular cell adhesion molecule through signalling pathway of NF-kB, but not Akt1 and TAK1 in vascular smooth muscle cells. Scand. J. Immunol. 2013, 77, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Tousoulis, D.; Oikonomou, E.; Economou, E.K.; Crea, F.; Kaski, J.C. Inflammatory cytokines in atherosclerosis: Current therapeutic approaches. Eur. Heart J. 2016, 37, 1723–1732. [Google Scholar] [CrossRef]
- Barath, P.; Fishbein, M.C.; Cao, J.; Berenson, J.; Helfant, R.H.; Forrester, J.S. Detection and localisation of tumor necrosis factor in human atheroma. Am. J. Cardiol. 1990, 65, 297–302. [Google Scholar] [CrossRef]
- Brånén, L.; Hovgaard, L.; Nitulescu, M.; Bengtsson, E.; Nilsson, J.; Jovinge, S. Inhibition of tumor necrosis factor-α reduces atherosclerosis in apolipoprotein E knockout mice. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2137–2142. [Google Scholar] [CrossRef]
- Ridker, P.M.; Rifai, N.; Pfeffer, M.; Sacks, F.; Lepage, S.; Braunwald, E. Elevation of tumor necrosis factor-α and increased risk of recurrent coronary events after myocardial infarction. Circulation 2000, 101, 2149–2153. [Google Scholar] [CrossRef]
- Strom, A.C.; Cross, A.J.; Cole, J.E.; Blair, P.A.; Leib, C.; Goddard, M.E.; Rosser, E.C.; Park, I.; Nilsson, A.H.; Nilsson, J.; et al. B regulatory cells are increased in hypercholesterolaemic mice and protect from lesion development via IL-10. Thromb. Haemost. 2015, 114, 835–847. [Google Scholar] [PubMed]
- Smith, F.B.; Lee, A.J.; Hau, C.M.; Rumley, A.; Lowe, G.D.; Fowkes, F.G. Plasma fibrinogen, haemostatic factors and prediction of peripheral arterial disease in the Edinburgh Artery Study. Blood Coagul. Fibrinolysis 2000, 11, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Undas, A.; Nowakowski, T.; Cieśla-Dul, M.; Sadowski, J. Abnormal plasma fibrin clot characteristics are associated with worse clinical outcome in patients with peripheral arterial disease and thromboangiitis obliterans. Atherosclerosis 2011, 215, 481–486. [Google Scholar] [CrossRef] [PubMed]
- McDermott, M.M.; Green, D.; Greenland, P.; Liu, K.; Criqui, M.H.; Chan, C.; Guralnik, J.M.; Pearce, W.H.; Ridker, P.M.; Taylor, L.; et al. Relation of levels of hemostatic factors and inflammatory markers to the ankle brachial index. Am. J. Cardiol. 2003, 92, 194–199. [Google Scholar] [CrossRef]
- Lee, A.J.; MacGregor, A.S.; Hau, C.M.; Price, J.F.; Rumley, A.; Lowe, G.D.; Fowkes, F.G. The role of haematological factors in diabetic peripheral arterial disease: The Edinburgh artery study. Br. J. Haematol. 1999, 105, 648–654. [Google Scholar] [CrossRef] [PubMed]
- Blann, A.D.; Amiral, J.; McCollum, C.N.; Lip, G.Y. Differences in free and total tissue factor pathway inhibitor, and tissue factor in peripheral artery disease compared to healthy controls. Atherosclerosis 2000, 152, 29–34. [Google Scholar] [CrossRef]
- Wieczór, R.; Kulwas, A.; Rość, D. Implications of Hemostasis Disorders in Patients with Critical Limb Ischemia-An In-Depth Comparison of Selected Factors. J. Clin. Med. 2020, 9, 659. [Google Scholar] [CrossRef] [PubMed]
- Caplice, N.M.; Mueske, C.S.; Kleppe, L.S.; Simari, R.D. Presence of tissue factor pathway inhibitor in human atherosclerotic plaques is associated with reduced tissue factor activity. Circulation 1998, 98, 1051–1057. [Google Scholar] [CrossRef]
- Falciani, M.; Gori, A.M.; Fedi, S.; Chiarugi, L.; Simonetti, I.; Dabizzi, R.P.; Prisco, D.; Pepe, G.; Abbate, R.; Gensini, G.F.; et al. Elevated tissue factor and tissue factor pathway inhibitor circulating levels in ischaemic heart disease patients. Thromb Haemost. 1998, 79, 495–499. [Google Scholar]
- Lane, J.S.; Vittinghoff, E.; Lane, K.T.; Hiramoto, J.S.; Messina, L.M. Risk factors for premature peripheral vascular disease: Results for the National Health and Nutritional Survey, 1999–2002. J. Vasc. Surg. 2006, 44, 319–324. [Google Scholar] [CrossRef]
- Unlu, Y.; Karapolat, S.; Karaca, Y.; Kiziltunc, A. Comparison of levels of inflammatory markers and hemostatic factors in the patients with and without peripheral arterial disease. Thromb. Res. 2006, 117, 357–364. [Google Scholar] [CrossRef]
- Makin, A.J.; Chung, N.A.; Silverman, S.H.; Lip, G.Y. Thrombogenesis and endothelial damage/dysfunction in peripheral artery disease. Relationship to ethnicity and disease severity. Thromb. Res. 2003, 111, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Altes, P.; Perez, P.; Esteban, C. Raised Fibrinogen Levels and Outcome in Outpatients with Peripheral Artery Disease. Angiology 2018, 69, 9720. [Google Scholar] [CrossRef] [PubMed]
- Arfan, S.; Zamzam, A.; Syed, M.H.; Jain, S.; Jahanpour, N.; Abdin, R.; Qadura, M. The Clinical Utility of D-Dimer and Prothrombin Fragment (F1+2) for Peripheral Artery Disease: A Prospective Study. Biomedicines 2022, 10, 878. [Google Scholar] [CrossRef]
- McDermott, M.M.; Liu, K.; Green, D.; Greenland, P.; Tian, L.; Kibbe, M.; Tracy, R.; Shah, S.; Wilkins, J.T.; Huffman, M.; et al. Changes in D-dimer and inflammatory biomarkers before ischemic events in patients with peripheral artery disease: The BRAVO Study. Vasc. Med. 2016, 21, 12–20. [Google Scholar] [CrossRef]
- Soares, R.P.; Bydlowski, S.P.; Nascimento, N.M.; Thomaz, A.M.; Bastos EN, M.; Lopes, A.A. Plasmatic ADAMTS-13 metalloprotease and von Willebrand factor in children with cyanotic congenital heart disease. Braz. J. Med. Biol. Res. 2013, 46, 375–381. [Google Scholar] [CrossRef]
- Crawley, J.T.; Lane, D.A.; Woodward, M.; Rumley, A.; Lowe, G.D.O. Evidence that high von Willebrand factor and low ADAMTS-13 levels independently increase the risk of a nonfatal heart attack. J. Thromb. Haemost. 2008, 6, 583–588. [Google Scholar] [CrossRef]
- Andersson, H.M.; Siegerink, B.; Luken, B.M.; Crawley, J.T.; Algra, A.; Lane, D.A.; Rosendaal, F.R. High VWF, low ADAMTS13, and oral contraceptives increase the risk of ischemic stroke and myocardial infarction in young women. Blood 2012, 119, 1555–1560. [Google Scholar] [CrossRef]
- Bongers, T.N.; de Bruijne, E.L.; Dippel, D.W.; De Jong, A.J.; Deckers, J.W.; Poldermans, D.; de Maat, M.P.M.; Leebeek FW, G. Lower levels of ADAMTS13 are associated with cardiovascular disease in young patients. Atherosclerosis 2009, 207, 250–254. [Google Scholar] [CrossRef]
- Chion, C.K.; Doggen, C.J.; Crawley, J.T.; Lane, D.A.; Rosendaal, F.R. ADAMTS13 and von Willebrand factor and the risk of myocardial infarction in men. Blood 2007, 109, 1998–2000. [Google Scholar] [CrossRef] [PubMed]
- Green, D.; Tian, L.; Greenland, P.; Liu, K.; Kibbe, M.; Tracy, R.; Shah, S.; Wilkins, J.T.; Huffman, M.D.; Liao, Y.; et al. Association of the von Willebrand Factor–ADAMTS13 Ratio with Incident Cardiovascular Events in Patients with Peripheral Arterial Disease. Clin. Appl. Thromb. Hemost. 2017, 23, 807–813. [Google Scholar] [CrossRef]
- Polok, K.; Gorka, J.; Fronczek, J.; Iwaniec, T.; Górka, K.; Szczeklik, W. Perioperative cardiovascular complications rate and activity of coagulation and fibrinolysis among patients undergoing vascular surgery for peripheral artery disease and abdominal aortic aneurysm. Vascular 2021, 29, 134–142. [Google Scholar] [CrossRef]
- Atiq, F.; van de Wouw, J.; Sorop, O.; Heinonen, I.; De Maat, M.P.; Merkus, D.; Duncker, D.J.; Leebeek, F.W. Endothelial dysfunction, atherosclerosis, and increase of von Willebrand factor and Factor VIII: A randomized controlled trial inswine. Thromb Haemost. 2021, 121, 676–686. [Google Scholar] [CrossRef] [PubMed]
- Mehta, J.; Mehta, P.; Lawson, D.; Saldeen, T. Plasma tissue plasminogen activator inhibitor levels in coronary artery disease: Correlation with age and serum tryceride concentrations. J. Am. Coll. Cardiol. 1987, 9, 263–268. [Google Scholar] [CrossRef]
- Verheugt, F.W.A.; Cate, J.W.; Sturk, A.; Imandt, L.; Verhorst, P.M.; Van Eenige, M.J.; Verwey, W.; Roos, J.P. Tissue plasminogen activator activity and inhibition in acute myocardial infarction and angiographically normal coronary arteries. Am. J. Cardiol. 1987, 59, 1075–1079. [Google Scholar] [CrossRef] [PubMed]
- Huber, K.; Rose, D.; Schuster, E.; Glogar, D.H.; Kaindl, F.; Binder, B.R. Orcadian fluctuations of plasminogen activator inhibitor and tissue plasminogen activator levels in plasma of patients with unstable coronary artery disease and acute myocardial infarction. Thromb Haemost. 1988, 60, 372–376. [Google Scholar]
- Phillips, G.B.; Pinkernell, B.H.; Jing, T.Y. The association of hypotestosteronemia with coronary artery disease in men. Arterioscler. Thromb. 1994, 14, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Vanderkerckhove, Y.; Baele, G.; De Puydt, H.; Weyne, A.; Clement, D. Plasma tissue plasminogen activator levels in patients with coronary artery disease. Thromb. Res. 1988, 50, 449–453. [Google Scholar] [CrossRef]
- Wieczór, R.; Wieczór, A.M.; Rość, D. Tissue-type plasminogen activator and plasminogen activator inhibitor type 1 in patients with symptomatic lower extremity artery disease. Minerva Cardiol. Angiol. 2021, 69, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Sacco, R.L.; Kasner, S.E.; Broderick, J.P.; Caplan, L.R.; Connors, J.J.; Culebras, A.; Elkind, M.S.; George, M.G.; Hamdan, A.D.; Higashida, R.T.; et al. An updated definition of stroke for the 21st century: A statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2013, 44, 2064–2089. [Google Scholar] [CrossRef]
- McGurgan, I.J.; Ziai, W.C.; Werring, D.J.; Al-Shahi Salman, R.; Parry-Jones, A.R. Acute Intracerebral Haemorrhage: Diagnosis and Management. Pract. Neurol. 2021, 21, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Hemphill, J.C.; Bonovich, D.C.; Besmertis, L.; Manley, G.T.; Johnston, S.C. The ICH Score: A Simple, Reliable Grading Scale for Intracerebral Hemorrhage. Stroke 2001, 32, 891–897. [Google Scholar] [CrossRef] [PubMed]
- van Asch, C.J.; Luitse, M.J.; Rinkel, G.J.; van der Tweel, I.; Algra, A.; Klijn, C.J. Incidence, Case Fatality, and Functional Outcome of Intracerebral Haemorrhage over Time, According to Age, Sex, and Ethnic Origin: A Systematic Review and Meta-Analysis. Lancet Neurol. 2010, 9, 167–176. [Google Scholar] [CrossRef]
- Woodruff, T.M.; Thundyil, J.; Tang, S.C.; Sobey, C.G.; Taylor, S.M.; Arumugam, T.V. Pathophysiology, treatment and animal and cellular models of human ischemic stroke. Mol. Neurodegener. 2011, 6, 11. [Google Scholar] [CrossRef]
- Jeyaseelan, K.; Lim, K.Y.; Armugam, A. MicroRNA expression in the blood and brain of rats subjected to transient focal ischemia by middle cerebral artery occlusion. Stroke 2008, 39, 959–966. [Google Scholar] [CrossRef]
- Dharap, A.; Bowen, K.; Place, R.; Li, L.C.; Vemuganti, R. Transient focal ischemia induces extensive temporal changes in rat cerebral microRNAome. J. Cereb. Blood Flow Metab. 2009, 29, 675–687. [Google Scholar] [CrossRef]
- Sorensen, S.S.; Nygaard, A.B.; Nielsen, M.Y.; Jensen, K.; Christensen, T. miRNA expression profiles in cerebrospinal fluid and blood of patients with acute ischemic stroke. Transl. Stroke Res. 2014, 5, 711–718. [Google Scholar] [CrossRef]
- Li, S.H.; Su, S.Y.; Liu, J.L. Differential regulation of microRNAs in patients with ischemic stroke. Curr. Neurovasc. Res. 2015, 12, 214–221. [Google Scholar] [CrossRef]
- Aumiller, V.; Forstemann, K. Roles of microRNAs beyond development metabolism and neural plasticity. Biochim. Biophys. Acta 2008, 1779, 692–696. [Google Scholar] [CrossRef] [PubMed]
- Merkerova, M.; Belickova, M.; Bruchova, H. Differential expression of microRNAs in hematopoietic cell lineages. Eur. J. Haematol. 2008, 81, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Giordano, M.; Trotta, M.C.; Ciarambino, T.; D’Amico, M.; Schettini, F.; Sisto, A.D.; D’Auria, V.; Voza, A.; Malatino, L.S.; Biolo, G.; et al. Circulating miRNA-195-5p and -451a in Patients with Acute Hemorrhagic Stroke in Emergency Department. Life 2022, 12, 763. [Google Scholar] [CrossRef] [PubMed]
- Giordano, M.; Ciarambino, T.; D’Amico, M.; Trotta, M.C.; Di Sette, A.M.; Marfella, R.; Malatino, L.; Paolisso, G.; Adinolfi, L.E. Circulating MiRNA-195-5p and -451a in Transient and Acute Ischemic Stroke Patients in an Emergency Department. J. Clin. Med. 2019, 8, 130. [Google Scholar] [CrossRef] [PubMed]
- Camaioni, C.; Gustapane, M.; Cialdella, P.; Della Bona, R.; Biasucci, R.M. Microparticles and microRNAs: New players in the complex field of coagulation. Intern. Emerg. Med. 2013, 8, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, A.; Xiang, J.; Lv, Y.; Zhang, X. MiR-451 Acts as a Suppressor of Angiogenesis in Hepatocellular Carcinoma by Targeting the IL-6R-STAT3 Pathway. Oncol. Rep. 2016, 36, 1385–1392. [Google Scholar] [CrossRef]
- Zhao, W.-J.; Zhang, H.-F.; Su, J.-Y. Downregulation of MicroRNA-195 Promotes Angiogenesis Induced by Cerebral Infarction via Targeting VEGFA. Mol. Med. Rep. 2017, 16, 5434–5440. [Google Scholar]
- Xiong, Y.; Mahmood, A.; Chopp, M. Angiogenesis, Neurogenesis and Brain Recovery of Function Following Injury. Curr. Opin. Investig. Drugs 2010, 11, 298–308. [Google Scholar]
- Ergul, A.; Alhusban, A.; Fagan, S.C. Angiogenesis: A Harmonized Target for Recovery after Stroke. Stroke 2012, 43, 2270–2274. [Google Scholar] [CrossRef]
- Greenberg, D.A.; Jin, K. Vascular Endothelial Growth Factors (VEGFs) and Stroke. Cell. Mol. Life Sci. 2013, 70, 1753–1761. [Google Scholar] [CrossRef]
- Giordano, M.; Trotta, M.C.; Ciarambino, T.; D’Amico, M.; Galdiero, M.; Schettini, F.; Paternosto, D.; Salzillo, M.; Alfano, R.; Andreone, V.; et al. Circulating miRNA-195-5p and -451a in Diabetic Patients with Transient and Acute Ischemic Stroke in the Emergency Department. Int. J. Mol. Sci. 2020, 21, 7615. [Google Scholar] [CrossRef]
- Tuttolomondo, A.; Maida, C.; Pinto, A. Diabetic foot syndrome: Immune-inflammatory features as possible cardiovascular markers in diabetes. World J. Orthop. 2015, 6, 62–76. [Google Scholar] [CrossRef] [PubMed]
- Di Raimondo, D.; Tuttolomondo, A.; Buttà, C.; Casuccio, A.; Giarrusso, L.; Miceli, G.; Licata, G.; Pinto, A. Metabolic and anti-inflammatory effects of a home-based programme of aerobic physical exercise. Int. J. Clin. Pract. 2013, 67, 1247–1253. [Google Scholar] [CrossRef] [PubMed]
- Mueller, T.; Marschon, R.; Dieplinger, B.; Haidinger, D.; Gegenhuber, A.; Poelz, W.; Webersinke, G.; Haltmayer, M. Factor V Leiden, prothrombin G20210A, and methylenetetrahydrofolate reductase C677T mutations are not associated with chronic limb ischemia: The Linz Peripheral Arterial Disease (LIPAD) Study. J. Vasc. Surg. 2005, 41, 808–815. [Google Scholar] [CrossRef][Green Version]
- Vazquez, F.; Rodger, M.; Carrier, M.; Le Gal, G.; Reny, J.L.; Sofi, F.; Mueller, T.; Nagpal, S.; Jetty, P.; Gandara, E. Prothrombin G20210A Mutation and Lower Extremity Peripheral Arterial Disease: A Systematic Review and Meta-analysis. Eur. J. Vasc. Endovasc. Surg. 2015, 50, 232–240. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Klarin, D.; Lynch, G.; Aragam, K. Genome-wide Association Study of Peripheral Artery Disease in the Million Veteran Program. Nat. Med. 2019, 25, 1274–1279. [Google Scholar] [CrossRef] [PubMed]
- Khandanpour, N.; Willis, G.; Meyer, F.J.; Armon, M.P.; Loke, Y.K.; Wright, A.J.; Finglas, P.M.; Jennings, B.A. Peripheral arterial disease and methylenetetrahydrofolate reductase (MTHFR) C677T mutations: A case-control study and meta-analysis. J. Vasc. Surg. 2009, 49, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Memtsas, V.; Arachchillage, D.; Gorog, D. Role, Laboratory Assessment and Clinical Relevance of Fibrin, Factor XIII and Endogenous Fibrinolysis in Arterial and Venous Thrombosis. Int. J. Mol. Sci. 2021, 22, 1472. [Google Scholar] [CrossRef] [PubMed]
- Signorelli, S.S.; Anzaldi, M.; Fiore, V. Inflammation in peripheral arterial disease (PAD). Curr. Pharm. Des. 2012, 18, 4350–4357. [Google Scholar] [CrossRef] [PubMed]
- Cassar, K.; Bachoo, P.; Ford, I.; Greaves, M.; Brittenden, J. Markers of Coagulation Activation, Endothelial Stimulation and Inflammation in Patients with Peripheral Arterial Disease. Eur. J. Vasc. Endovasc. Surg. 2005, 29, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Caravaca, J.M.; Camelo-Castillo, A.; Ramírez-Macías, I.; Gil-Pérez, P.; López-García, C.; Esteve-Pastor, M.A.; Orenes-Piñero, E.; Tello-Montoliu, A.; Marín, F. Antithrombotic Therapy in Patients with Peripheral Artery Disease: A Focused Review on Oral Anticoagulation. Int. J. Mol. Sci. 2021, 22, 7113. [Google Scholar] [CrossRef] [PubMed]
- Aboyans, V.; Ricco, J.B.; Bartelink, M.E.L.; Björck, M.; Brodmann, M.; Cohnert, T.; Collet, J.P.; Czerny, M.; De Carlo, M.; Debus, S.; et al. 2017 ESC Guidelines on the Diagnosis and Treatment of Peripheral Arterial Diseases, in collaboration with the European Society for Vascular Surgery (ESVS). Eur. Heart J. 2017, 39, 763–816. [Google Scholar] [CrossRef] [PubMed]
- Gerhard-Herman, M.D.; Gornik, H.L.; Barrett, C.; Barshes, N.R.; Corriere, M.A.; Drachman, D.E.; Fleisher, L.A.; Fowkes, F.G.; Hamburg, N.M.; Kinlay, S.; et al. 2016 AHA/ACC Guideline on the Management of Patients with Lower Extremity Peripheral Artery Disease: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2017, 135, e686–e725. [Google Scholar] [CrossRef] [PubMed]
- Meadows, T.A.; Bhatt, D.L. Clinical aspects of platelet inhibitors and thrombus formation. Circ. Res. 2007, 100, 1261–1275. [Google Scholar] [CrossRef] [PubMed]
- Patti, G.; Micieli, G.; Cimminiello, C.; Bolognese, L. The Role of Clopidogrel in 2020: A Reappraisal. Cardiovasc. Ther. 2020, 2020, 8703627. [Google Scholar] [CrossRef]
- Dorsam, R.T.; Kunapuli, S.P. Central role of the P2Y12 receptor in platelet activation. J. Clin. Investig. 2004, 113, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Damman, P.; Woudstra, P.; Kuijt, W.J.; de Winter, R.J.; James, S.K. P2Y12 platelet inhibition in clinical practice. J. Thromb. Thrombolysis. 2012, 33, 143–153. [Google Scholar] [CrossRef]
- Wallentin, L. P2Y(12) inhibitors: Differences in properties and mechanisms of action and potential consequences for clinical use. Eur. Heart J. 2009, 30, 1964–1977. [Google Scholar] [CrossRef] [PubMed]
- Bonaca, M.P.; Scirica, B.M.; Creager, M.A.; Olin, J.; Bounameaux, H.; Dellborg, M.; Lamp, J.M.; Murphy, S.A.; Braunwald, E.; Morrow, D.A. Vorapaxar in patients with peripheral artery disease: Results from TRA2P-TIMI 50. Circulation 2013, 127, 1522–1529. [Google Scholar] [CrossRef] [PubMed]
- Bonaca, M.P.; Gutierrez, J.A.; Creager, M.A.; Scirica, B.M.; Olin, J.; Murphy, S.A.; Braunwald, E.; Morrow, D.A. Acute limb ischemia and outcomes with vorapaxar in patients with peripheral artery disease: Results from the Trial to Assess the Effects of Vorapaxar in Preventing Heart Attack and Stroke in Patients with Atherosclerosis-Thrombolysis in Myocardial Infarction 50 (TRAP-TIMI 50). Circulation 2016, 133, 997–1005. [Google Scholar] [PubMed]
- Mackman, N.; Bergmeier, W.; Stouffer, G.A.; Weitz, J.I. Therapeutic strategies for thrombosis: New targets and approaches. Nat. Rev. Drug Discov. 2020, 19, 333–352. [Google Scholar] [CrossRef] [PubMed]
- Posma, J.J.; Posthuma, J.J.; Spronk, H.M. Coagulation and non-coagulation effects of thrombin. J. Thromb. Haemost. 2016, 14, 1908–1916. [Google Scholar] [CrossRef] [PubMed]
- Koo, B.H.; Kim, D.S. Factor Xa induces mitogenesis of vascular smooth muscle cells via autocrine production of epiregulin. J. Biol. Chem. 2003, 278, 52578–52586. [Google Scholar] [CrossRef] [PubMed]
- Capell, W.H.; Bonaca, M.P.; Nehler, M.R.; Chen, E.; Kittelson, J.M.; Anand, S.S.; Berkowitz, S.D.; Debus, E.S.; Fanelli, F.; Haskell, L.; et al. Rationale and design for the Vascular Outcomes study of ASA along with rivaroxaban in endovascular or surgical limb revascularization for peripheral artery disease (VOYAGER PAD). Am. Heart J. 2018, 199, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Weitz, J.I. Insights into the role of thrombin in the pathogenesis of recurrent ischaemia after acute coronary syndrome. Thromb. Haemost. 2014, 112, 924–931. [Google Scholar] [CrossRef] [PubMed]
- Falk, E.; Nakano, M.; Bentzon, J.F.; Finn, A.V.; Virmani, R. Update on acute coronary syndromes: The pathologists’ view. Eur. Heart J. 2013, 34, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Hindricks, G.; Potpara, T.; Dagres, N.; Arbelo, E.; Bax, J.J.; Blomström-Lundqvist, C.; Boriani, G.; Castella, M.; Dan, G.-A.; Dilaveris, P.E.; et al. 2020 ESC Guidelines for the diagnosis and management of atrial fibrillation developed in collaboration with the European Association for Cardio-Thoracic Surgery (EACTS): The Task Force for the diagnosis and management of atrial fibrillation of the European Society of Cardiology (ESC) Developed with the special contribution of the European Heart Rhythm Association (EHRA) of the ESC. Eur. Heart J. 2021, 42, 373–498. [Google Scholar] [CrossRef] [PubMed]
- Weitz, J.I.; Angiolillo, D.J.; Geisler, T.; Heitmeier, S. Dual Pathway Inhibition for Vascular Protection in Patients with Atherosclerotic Disease: Rationale and Review of the Evidence. Thromb. Haemost. 2020, 120, 1147–1158. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.K.; Stafford, D.W. Structural and functional insights into enzymes of the vitamin K cycle. J. Thromb. Haemost. 2016, 14, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Warfarin Antiplatelet Vascular Evaluation Trial Investigators; Anand, S.; Yusuf, S.; Xie, C.; Pogue, J.; Eikelboom, J.; Budaj, A.; Sussex, B.; Liu, L.; Guzman, R.; et al. Oral anticoagulant and antiplatelet therapy and peripheral arterial disease. N. Engl. J. Med. 2007, 357, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Moll, F.; Baumgartner, I.; Jaff, M.; Nwachuku, C.; Tangelder, M.; Ansel, G.; Adams, G.; Zeller, T.; Rundback, J.; Grosso, M.; et al. Edoxaban Plus Aspirin vs Dual Antiplatelet Therapy in Endovascular Treatment of Patients with Peripheral Artery Disease: Results of the ePAD Trial. J. Endovasc. Ther. 2018, 25, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Biagioni, R.B.; Lopes, R.D.; Agati, L.B.; Sacilotto, R.; Wolosker, N.; Sobreira, M.L.; de Freitas Soares, B.L.; Joviliano, E.E.; Bernardi, W.H.; Junior, V.C.; et al. Rationale and design for the study Apixaban versus ClopidoGRel on a background of aspirin in patient undergoing InfraPoPliteal angioplasty for critical limb ischemia: AGRIPPA trial. Am. Heart J. 2020, 227, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Korjian, S.; Braunwald, E.; Daaboul, Y.; Verheugt, F.; Bode, C.; Tendera, M.; Jain, P.; Plotnikov, A.; Burton, P.; Gibson, C.M. Safety and efficacy of rivaroxaban for the secondary prevention following acute coronary syndromes among biomarker-positive patients: Insights from the ATLAS ACS 2-TIMI 51 trial. Eur. Heart J. Acute Cardiovasc. Care. 2019, 8, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Steffel, J.; Eikelboom, J.W.; Anand, S.S.; Shestakovska, O.; Yusuf, S.; Fox, K.A.A. The COMPASS Trial: Net Clinical Benefit of Low-Dose Rivaroxaban Plus Aspirin as Compared with Aspirin in Patients with Chronic Vascular Disease. Circulation 2020, 142, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Anti-inflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Morton, A.C.; Rothman, A.M.; Greenwood, J.P.; Gunn, J.; Chase, A.; Clarke, B.; Hall, A.S.; Fox, K.; Foley, C.; Banya, W. The effect of interleukin-1 receptor antagonist therapy on markers of inflammation in non-ST elevation acute coronary syndromes: The MRC-ILA heart study. Eur. Heart J. 2015, 36, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Abbate, A.; Trankle, C.R.; Buckley, L.F.; Lipinski, M.J.; Appleton, D.; Kadariya, D.; Canada, J.M.; Carbone, S.; Roberts, C.S.; Abouzaki, N.; et al. Interleukin-1 blockade inhibits the acute inflammatory response in patients with ST-segment-elevation myocardial infarction. J. Am. Heart Assoc. 2020, 9, e014941. [Google Scholar] [CrossRef] [PubMed]
- El Sayed, H.; Kerensky, R.; Stecher, M.; Mohanty, P.; Davies, M. A randomised phase II study of Xilonix, a targeted therapy against interleukin 1α, for the prevention of superficial femoral artery restenosis after percutaneous revascularisation. J. Vasc. Surg. 2016, 63, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Tardif, J.-C.; Kouz, S.; Waters, D.D.; Bertrand, O.F.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Ibrahim, R.; Gamra, H.; Kiwan, G.S.; et al. Efficacy and safety of low-dose colchicine after myocardial infarction. N. Engl. J. Med. 2019, 381, 2497–2505. [Google Scholar] [CrossRef] [PubMed]
- Nidorf, S.M.; Eikelboom, J.W.; Budgeon, C.A.; Thompson, P.L. Low-dose colchicine for secondary prevention of cardiovascular disease. J. Am. Coll. Cardiol. 2013, 61, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Interleukin-6 Receptor Mendelian Randomisation Analysis (IL6R MR) Consortium; Swerdlow, D.I.; Holmes, M.V.; Kuchenbaecker, K.B.; Engmann, J.E.; Shah, T.; Sofat, R.; Guo, Y.; Chung, C.; Peasey, A.; et al. The interleukin-6 receptor as a target for prevention of coronary heart disease: A mendelian randomisation analysis. Lancet 2012, 379, 1214–1224. [Google Scholar] [PubMed]
- Broch, K.; Anstensrud, A.K.; Woxholt, S.; Sharma, K.; Tøllefsen, I.M.; Bendz, B.; Aakhus, S.; Ueland, T.; Amundsen, B.H.; Damås, J.K.; et al. Randomised trial of interleukin-6 receptor inhibition in patients with acute ST-segment elevation myocardial infarction. J. Am. Coll. Cardiol. 2021, 77, 1845–1855. [Google Scholar] [CrossRef]
- Kleveland, O.; Kunszt, G.; Bratlie, M.; Ueland, T.; Broch, K.; Holte, E.; Michelsen, A.E.; Bendz, B.; Amundsen, B.H.; Espevik, T.; et al. Effect of a single dose of the interleukin-6 receptor antagonist tocilizumab on inflammation and troponin T release in patients with non-ST-elevation myocardial infarction: A doubleblind, randomised, placebo-controlled phase 2 trial. Eur. Heart J. 2016, 37, 2406–2413. [Google Scholar] [CrossRef]
- Giles, J.T.; Sattar, N.; Gabriel, S.; Ridker, P.M.; Gay, S.; Warne, C.; Musselman, D.; Brockwell, L.; Shittu, E.; Klearman, M.; et al. Cardiovascular Safety of Tocilizumab Versus Etanercept in Rheumatoid Arthritis: A Randomized Controlled Trial. Arthritis Rheumatol. 2019, 72, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Chung, E.S.; Packer, M.; Lo, K.H.; Fasanmade, A.A.; Willerson, J.T. Randomized, double-blind, placebocontrolled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-α, in patients with moderate-to-severe heart failure: Results of the Anti-TNF Therapy Against Congestive Heart Failure (ATTACH) trial. Circulation 2003, 107, 3133–3140. [Google Scholar]
- Mann, D.L.; McMurray, J.J.; Packer, M.; Swedberg, K.; Borer, J.S.; Colucci, W.S.; Djian, J.; Drexler, H.; Feldman, A.; Kober, L.; et al. Targeted anticytokine therapy in patients with chronic heart failure: Results of the randomised etanercept worldwide evaluation (RENEWAL). Circulation 2004, 109, 1594–1602. [Google Scholar] [CrossRef]
- Ma, X.; Xu, S. TNF inhibitor therapy for rheumatoid arthritis. Biomed. Rep. 2012, 1, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Schrezenmeier, E.; Dörner, T. Mechanisms of action of hydroxychloroquine and chloroquine: Implications for rheumatology. Nat. Rev. Rheumatol. 2020, 16, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Sharma, T.S.; Wasko, M.C.M.; Tang, X.; Vedamurthy, D.; Yan, X.; Cote, J.; Bili, A. Hydroxychloroquine use is associated with decreased incident cardiovascular events in rheumatoid arthritis patients. J. Am. Heart Assoc. 2016, 5, e002867. [Google Scholar] [CrossRef]
- US National Library of Medicine. ClinicalTrials.gov 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT02874287 (accessed on 10 October 2022).
- Hilgendorf, I.; Swirski, F.K.; Robbins, C.S. Monocyte fate in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Soehnlein, O.; Drechsler, M.; Döring, Y.; Lievens, D.; Hartwig, H.; Kemmerich, K.; Ortega-Gómez, A.; Mandl, M.; Vijayan, S.; Projahn, D.; et al. Distinct functions of chemokine receptor axes in the atherogenic mobilisation and recruitment of classical monocytes. EMBO Mol. Med. 2013, 5, 471–481. [Google Scholar] [CrossRef]
- Lawrence, T.; Natoli, G. Transcriptional regulation of macrophage polarisation: Enabling diversity with identity. Nat. Rev. Immunol. 2011, 11, 750–761. [Google Scholar] [CrossRef]
- Fidler, T.P.; Xue, C.; Yalcinkaya, M.; Hardaway, B.; Abramowicz, S.; Xiao, T.; Liu, W.; Thomas, D.G.; Hajebrahimi, M.A.; Pircher, J.; et al. The AIM2 inflammasome exacerbates atherosclerosis in clonal haematopoiesis. Nature 2021, 592, 296–301. [Google Scholar] [CrossRef]
- Monaco, C.; Gregan, S.M.; Navin, T.J.; Foxwell, B.M.; Davies, A.H.; Feldmann, M. Toll-like receptor-2 mediates inflammation and matrix degradation in human atherosclerosis. Circulation 2009, 120, 2462–2469. [Google Scholar] [CrossRef]
- Chyu, K.Y.; Zhao, X.; Dimayuga, P.C.; Zhou, J.; Li, X.; Yano, J.; Lio, W.M.; Chan, L.F.; Kirzner, J.; Trinidad, P.; et al. CD8+ T cells mediate the atheroprotective effect of immunisation with an ApoB-100 peptide. PLoS ONE 2012, 7, e30780. [Google Scholar] [CrossRef]
- Dunér, P.; Mattisson, I.Y.; Fogelstrand, P.; Glise, L.; Ruiz, S.; Farina, C.; Borén, J.; Nilsson, J.; Bengtsson, E. Antibodies against apoB100 peptide 210 inhibit atherosclerosis in apoE−/−mice. Sci. Rep. 2021, 11, 9022. [Google Scholar] [CrossRef]
- Herbin, O.; Ait-Oufella, H.; Yu, W.; Fredrikson, G.N.; Aubier, B.; Perez, N.; Barateau, V.; Nilsson, J.; Tedgui, A.; Mallat, Z. Regulatory T-cell response to apolipoprotein B100-derived peptides reduces the development and progression of atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 605–612. [Google Scholar] [CrossRef]
- Bauersachs, R.M.; Szarek, M.; Brodmann, M.; Gudz, I.; Debus, E.S.; Nehler, M.R.; Anand, S.S.; Patel, M.R.; Hess, C.N.; Capell, W.H.; et al. Total Ischemic Event Reduction with Rivaroxaban After Peripheral Arterial Revascularization in the VOYAGER PAD Trial. J. Am. Coll. Cardiol. 2021, 78, 317–326. [Google Scholar] [CrossRef]
- Gurbel, P.A.; Bliden, K.P.; Turner, S.E.; Tantry, U.S.; Gesheff, M.G.; Barr, T.P.; Covic, L.; Kuliopulos, A. Cell-Penetrating Pepducin Therapy Targeting PAR1 in Subjects with Coronary Artery Disease. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 189–197. [Google Scholar] [CrossRef]
- Nieman, M.T. Protease-activated receptors in hemostasis. Blood 2016, 128, 169–177. [Google Scholar] [CrossRef]
- Wong, P.C.; Seiffert, D.; Bird, J.E.; Watson, C.A.; Bostwick, J.S.; Giancarli, M.; Allegretto, N.; Hua, J.; Harden, D.; Guay, J.; et al. Blockade of protease-activated receptor-4 (PAR4) provides robust antithrombotic activity with low bleeding. Sci. Transl. Med. 2017, 9, eaaf5294. [Google Scholar] [CrossRef]
- Martins Lima, A.; Martins Cavaco, A.C.; Fraga-Silva, R.A.; Eble, J.A.; Stergiopulos, N. From Patients to Platelets and Back Again: Pharmacological Approaches to Glycoprotein, V.I.; a Thrilling Antithrombotic Target with Minor Bleeding Risks. Thromb. Haemost. 2019, 119, 1720–1739. [Google Scholar] [CrossRef]
- Rayes, J.; Watson, S.P.; Nieswandt, B. Functional significance of the platelet immune receptors GPVI and CLEC-2. J. Clin. Investig. 2019, 129, 12–23. [Google Scholar] [CrossRef]
- Nurden, A.T. Clinical significance of altered collagen-receptor functioning in platelets with emphasis on glycoprotein VI. Blood Rev. 2019, 38, 100592. [Google Scholar] [CrossRef]
- Ungerer, M.; Rosport, K.; Bültmann, A.; Piechatzek, R.; Uhland, K.; Schlieper, P.; Gawaz, M.; Münch, G. Novel antiplatelet drug revacept (Dimeric Glycoprotein VI-Fc) specifically and efficiently inhibited collagen-induced platelet aggregation without affecting general hemostasis in humans. Circulation 2011, 123, 1891–1899. [Google Scholar] [CrossRef]
- Voors-Pette, C.; Lebozec, K.; Dogterom, P.; Jullien, L.; Billiald, P.; Ferlan, P.; Renaud, L.; Favre-Bulle, O.; Avenard, G.; Machacek, M.; et al. Safety and Tolerability, Pharmacokinetics, and Pharmacodynamics of ACT017, an Antiplatelet GPVI (Glycoprotein VI) Fab. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 956–964. [Google Scholar] [CrossRef]
- Bergmeier, W.; Chauhan, A.K.; Wagner, D.D. Glycoprotein Ibalpha and von Willebrand factor in primary platelet adhesion and thrombus formation: Lessons from mutant mice. Thromb. Haemost. 2008, 99, 264–270. [Google Scholar] [CrossRef]
- Denorme, F.; De Meyer, S.F. The VWF-GPIb axis in ischaemic stroke: Lessons from animal models. Thromb. Haemost. 2016, 116, 597–604. [Google Scholar] [CrossRef]
- Quach, M.E.; Chen, W.; Li, R. Mechanisms of platelet clearance and translation to improve platelet storage. Blood 2018, 131, 1512–1521. [Google Scholar] [CrossRef]
- Gratacap, M.P.; Guillermet-Guibert, J.; Martin, V.; Chicanne, G.; Tronchère, H.; Gaits-Iacovoni, F.; Payrastre, B. Regulation and roles of PI3Kβ, a major actor in platelet signaling and functions. Adv. Enzyme Regul. 2011, 51, 106–116. [Google Scholar] [CrossRef]
- Zheng, Z.; Pinson, J.A.; Mountford, S.J.; Orive, S.; Schoenwaelder, S.M.; Shackleford, D.; Powell, A.; Nelson, E.M.; Hamilton, J.R.; Jackson, S.P.; et al. Discovery and antiplatelet activity of a selective PI3Kβ inhibitor (MIPS-9922). Eur. J. Med. Chem. 2016, 122, 339–351. [Google Scholar] [CrossRef]
- Nylander, S.; Wågberg, F.; Andersson, M.; Skärby, T.; Gustafsson, D. Exploration of efficacy and bleeding with combined phosphoinositide 3-kinase β inhibition and aspirin in man. J. Thromb. Haemost. 2015, 13, 1494–1502. [Google Scholar] [CrossRef]
- Wheeler, A.P.; Gailani, D. The intrinsic pathway of coagulation as a target for antithrombotic therapy. Hematol. Oncol. Clin. N. Am. 2016, 30, 1099–1114. [Google Scholar] [CrossRef]
- Nickel, K.F.; Long, A.T.; Fuchs, T.A.; Butler, L.M.; Renne, T. Factor XII as a therapeutic target in thromboembolic and inflammatory diseases. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 13–20. [Google Scholar] [CrossRef]
- Weitz, J.I.; Fredenburgh, J.C. Platelet polyphosphate: The long and the short of it. Blood 2017, 129, 1574–1575. [Google Scholar] [CrossRef]
- Kenne, E.; Renne, T. Factor XII: A drug target for safe interference with thrombosis and inflammation. Drug. Discov. Today 2014, 19, 1459–1464. [Google Scholar] [CrossRef]
- Toomey, J.R.; Blackburn, M.N.; Storer, B.L.; Valocik, R.E.; Koster, P.F.; Feuerstein, G.Z. Comparing the antithrombotic efficacy of a humanized anti-factor IX(a) monoclonal antibody (SB 249417) to the low molecular weight heparin enoxaparin in a rat model of arterial thrombosis. Thromb. Res. 2000, 100, 73–79. [Google Scholar] [CrossRef]
- Toomey, J.R.; Valocik, R.E.; Koster, P.F.; Gabriel, M.A.; McVey, M.; Hart, T.K.; Ohlstein, E.H.; Parsons, A.A.; Barone, F.C. Inhibition of factor IX(a) is protective in a rat model of thromboembolic stroke. Stroke 2002, 33, 578–585. [Google Scholar] [CrossRef]
- Weitz, J.I.; Harenberg, J. New developments in anticoagulants: Past, present and future. Thromb. Haemost. 2017, 117, 1283–1288. [Google Scholar] [CrossRef]
- Weitz, J.I.; Fredenburgh, J.C. 2017 scientific sessions sol sherry distinguished lecture in thrombosis: Factor xi as a target for new anticoagulants. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 304–310. [Google Scholar] [CrossRef]
- Pireaux, V.; Tassignon, J.; Demoulin, S.; Derochette, S.; Borenstein, N.; Ente, A.; Fiette, L.; Douxfils, J.; Lancellotti, P.; Guyaux, M.; et al. Anticoagulation with an Inhibitor of Factors XIa and XIIa During Cardiopulmonary Bypass. J. Am. Coll. Cardiol. 2019, 74, 2178–2189. [Google Scholar] [CrossRef] [PubMed]
- Bristol-Myers-Squibb. A Study on BMS-986177 for the Prevention of a Stroke in Patients Receiving Aspirin and Clopidogrel (AXIOMATIC-SSP) (NCT03766581). Available online: https://clinicaltrials.gov/ct2/show/study/NCT03766581 (accessed on 10 October 2022).
- Bayer. Study to Gather Information about the Proper Dosing and Safety of the Oral FXIa Inhibitor BAY 2433334 in Patients Following an Acute Heart Attack (PACIFIC-AMI) (NCT04304534). Available online: https://clinicaltrials.gov/ct2/show/NCT04304534 (accessed on 1 March 2021).
- Jain, S.; Pitoc, G.A.; Holl, E.K.; Zhang, Y.; Borst, L.; Leong, K.W.; Lee, J.; Sullenger, B.A. Nucleic acid scavengers inhibit thrombosis without increasing bleeding. Proc. Natl. Acad. Sci. USA 2012, 109, 12938–12943. [Google Scholar] [CrossRef]
- Travers, R.J.; Shenoi, R.A.; Kalathottukaren, M.T.; Kizhakkedathu, J.N.; Morrissey, J.H. Nontoxic polyphosphate inhibitors reduce thrombosis while sparing hemostasis. Blood 2014, 124, 3183–3190. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miceli, G.; Basso, M.G.; Rizzo, G.; Pintus, C.; Tuttolomondo, A. The Role of the Coagulation System in Peripheral Arterial Disease: Interactions with the Arterial Wall and Its Vascular Microenvironment and Implications for Rational Therapies. Int. J. Mol. Sci. 2022, 23, 14914. https://doi.org/10.3390/ijms232314914
Miceli G, Basso MG, Rizzo G, Pintus C, Tuttolomondo A. The Role of the Coagulation System in Peripheral Arterial Disease: Interactions with the Arterial Wall and Its Vascular Microenvironment and Implications for Rational Therapies. International Journal of Molecular Sciences. 2022; 23(23):14914. https://doi.org/10.3390/ijms232314914
Chicago/Turabian StyleMiceli, Giuseppe, Maria Grazia Basso, Giuliana Rizzo, Chiara Pintus, and Antonino Tuttolomondo. 2022. "The Role of the Coagulation System in Peripheral Arterial Disease: Interactions with the Arterial Wall and Its Vascular Microenvironment and Implications for Rational Therapies" International Journal of Molecular Sciences 23, no. 23: 14914. https://doi.org/10.3390/ijms232314914
APA StyleMiceli, G., Basso, M. G., Rizzo, G., Pintus, C., & Tuttolomondo, A. (2022). The Role of the Coagulation System in Peripheral Arterial Disease: Interactions with the Arterial Wall and Its Vascular Microenvironment and Implications for Rational Therapies. International Journal of Molecular Sciences, 23(23), 14914. https://doi.org/10.3390/ijms232314914