
OSCA Genes in Bread Wheat: Molecular Characterization, Expression Profiling, and Interaction Analyses Indicated Their Diverse Roles during Development and Stress Response

 , , ,
, , , 
 and
and 
Abstract
:1. Introduction
2. Results
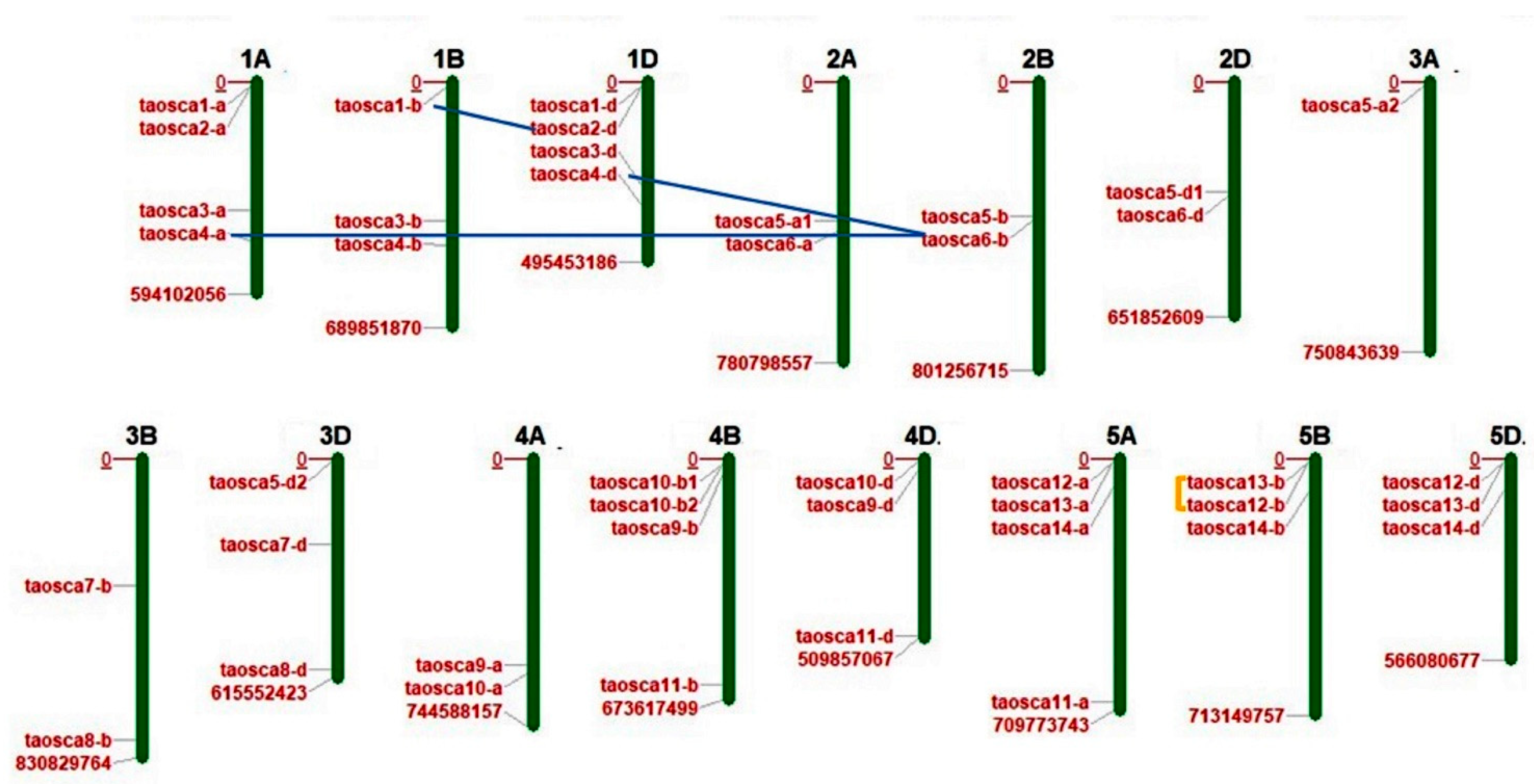
2.1. In Silico Identification, Chromosomal Distribution, and Protein Characterization
2.2. Prediction of Duplication Events and Ka/Ks Analysis
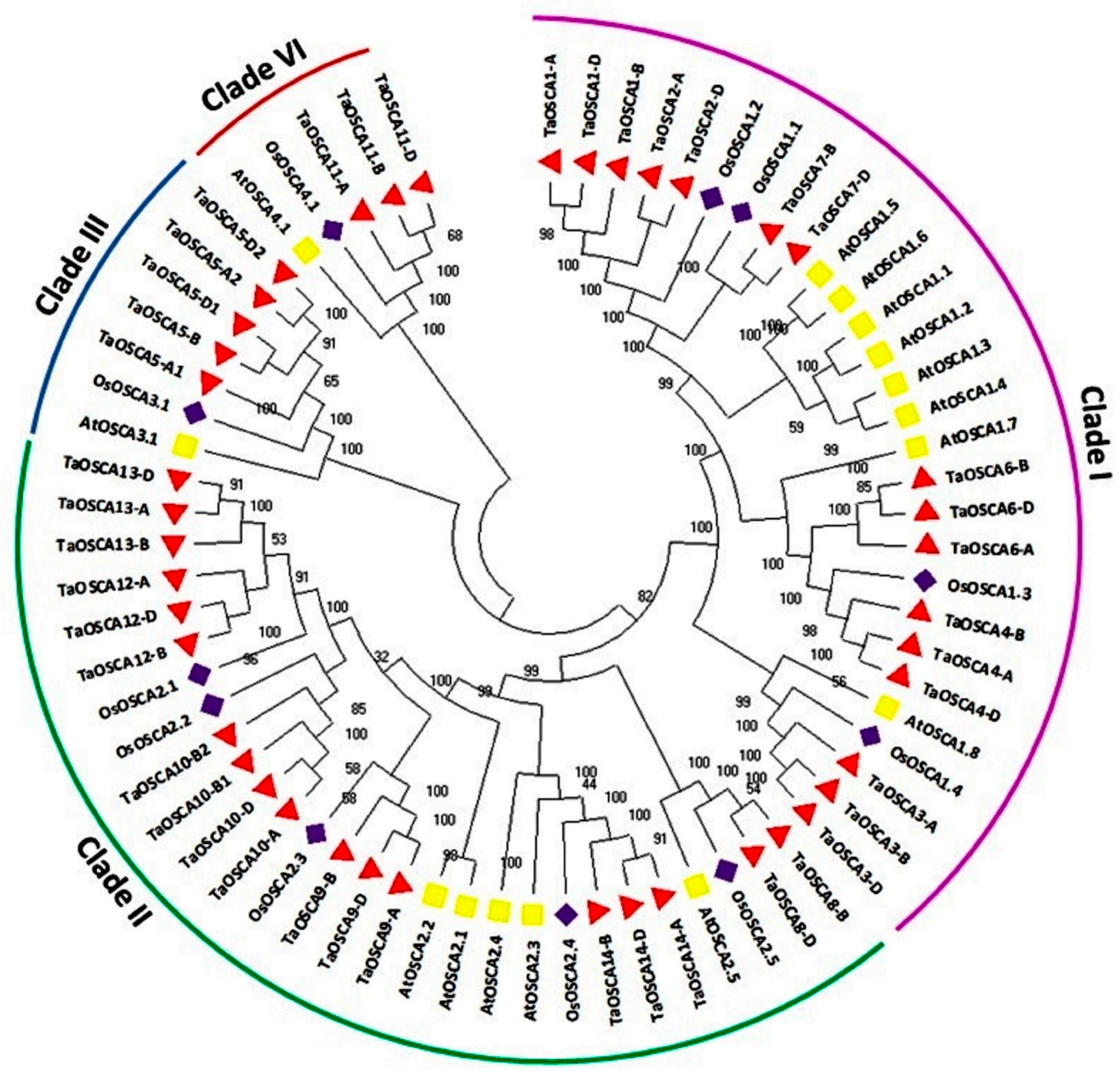
2.3. Phylogenetic Analysis
2.4. Expression Profiling of TaOSCA Genes in Tissue Developmental Stages
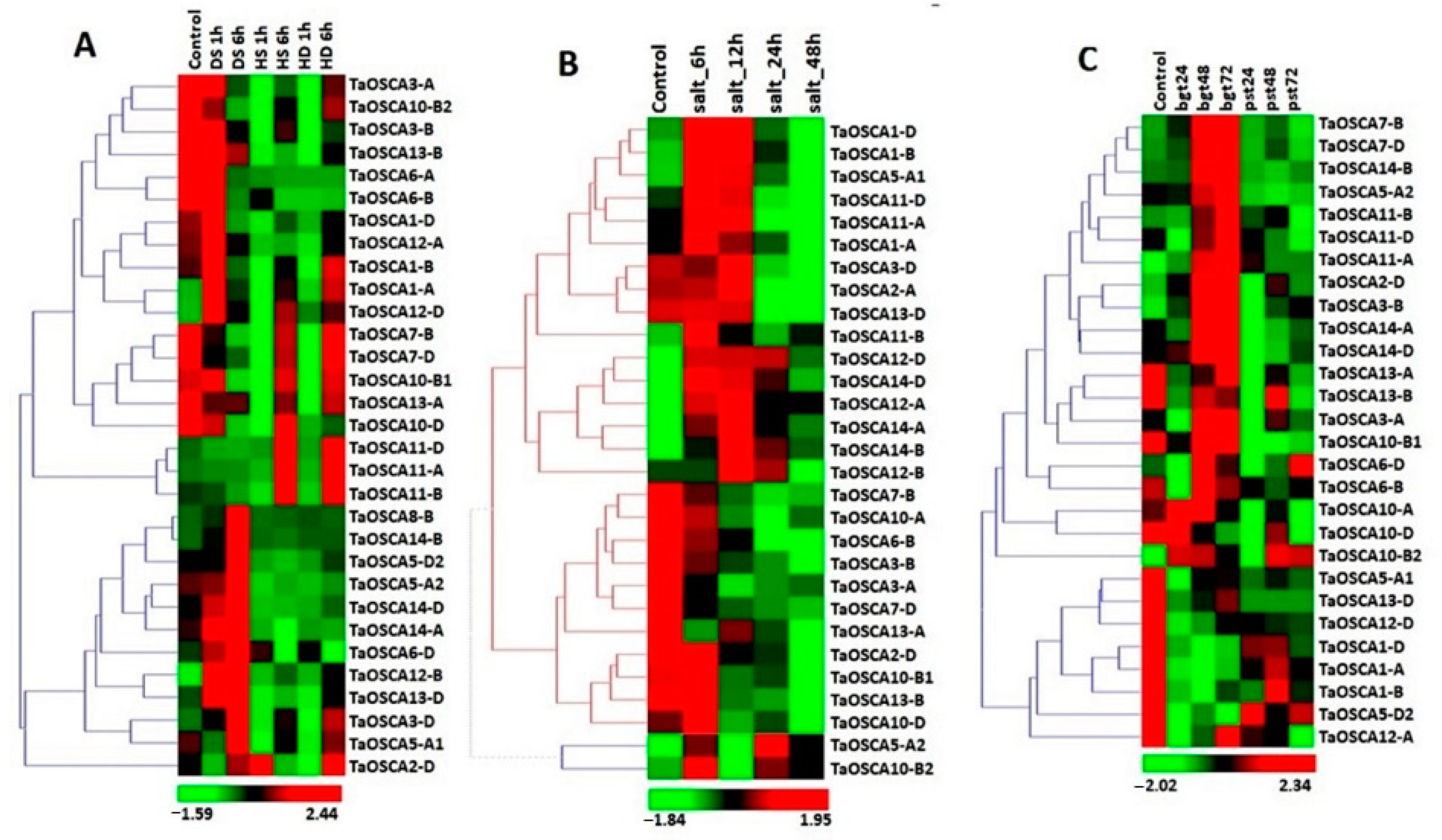
2.5. Expression Profiling of TaOSCA Genes in Abiotic Stresses
2.6. Expression Profiling of TaOSCA Genes in Biotic Stresses
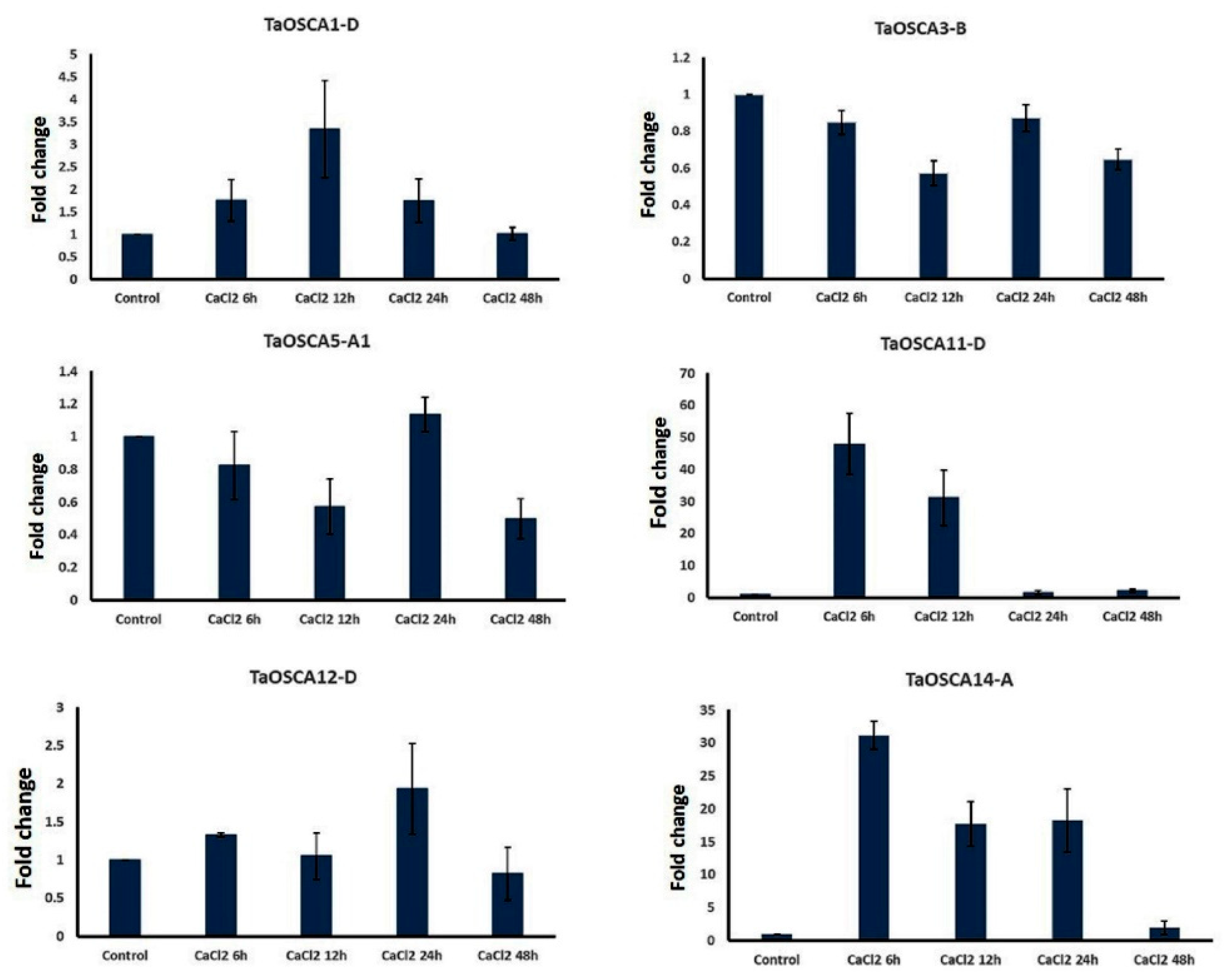
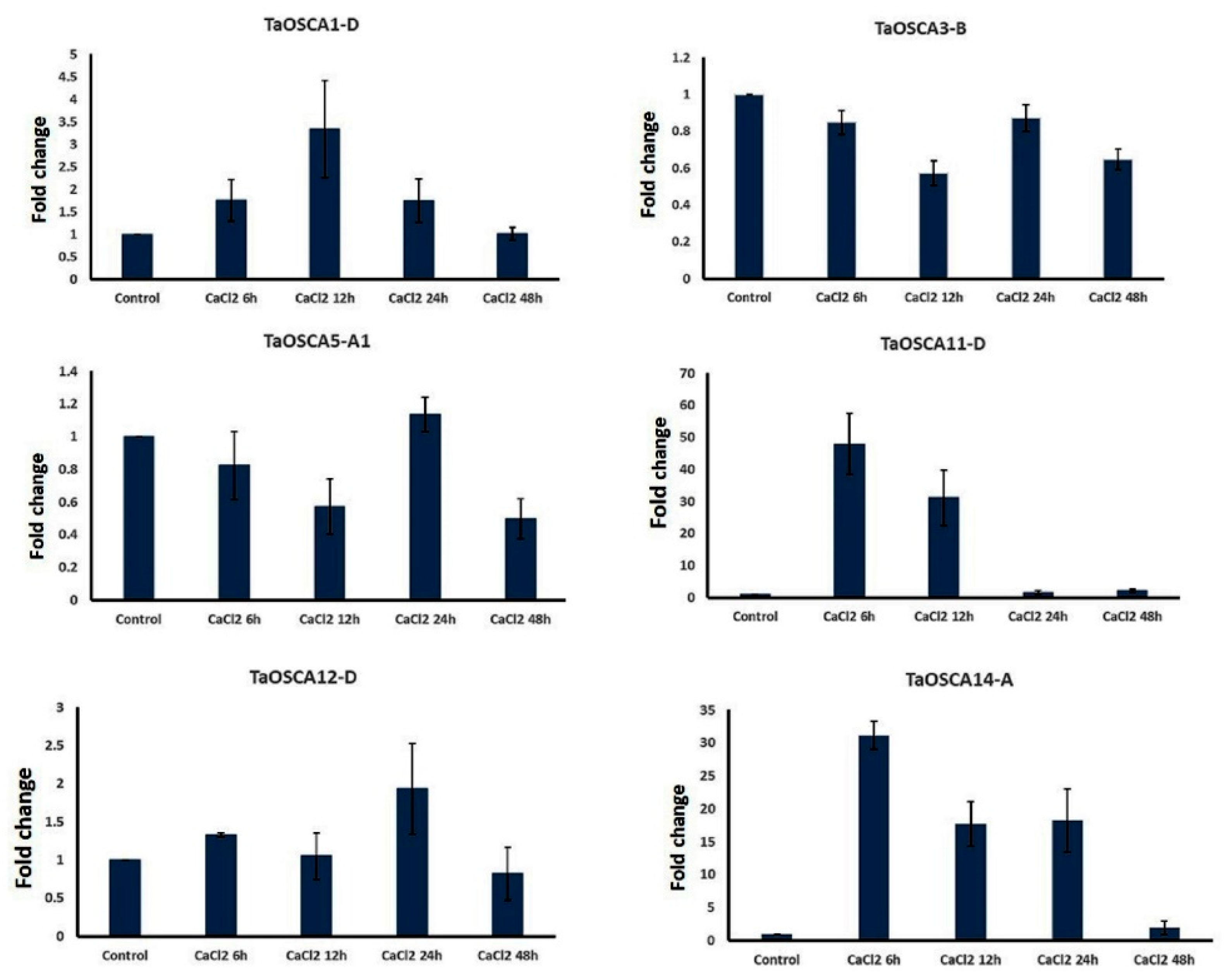
2.7. qRT-PCR Analysis under CaCl2 Treatment
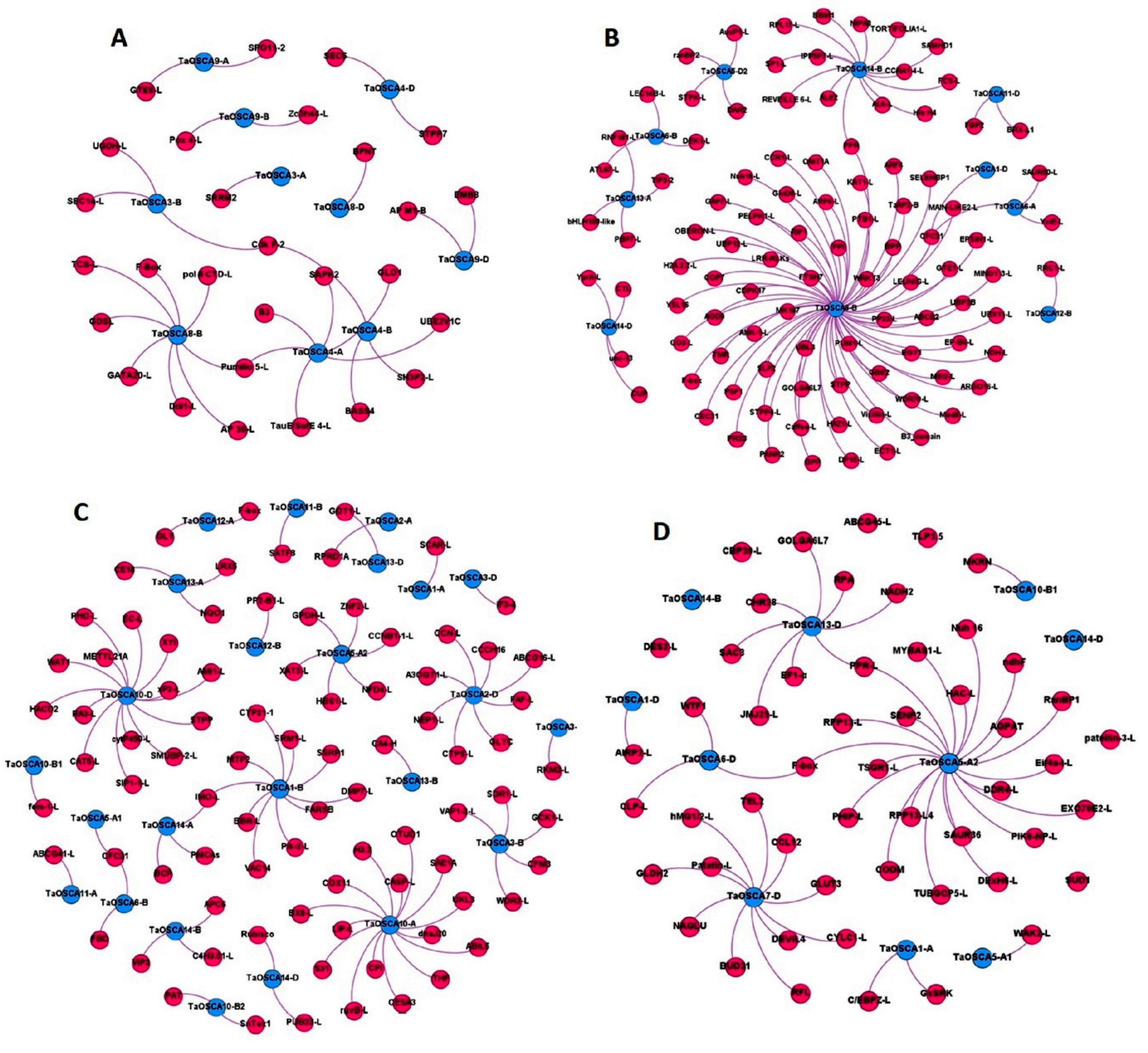
2.8. Co-Expression Analysis and Gene Ontology Mapping
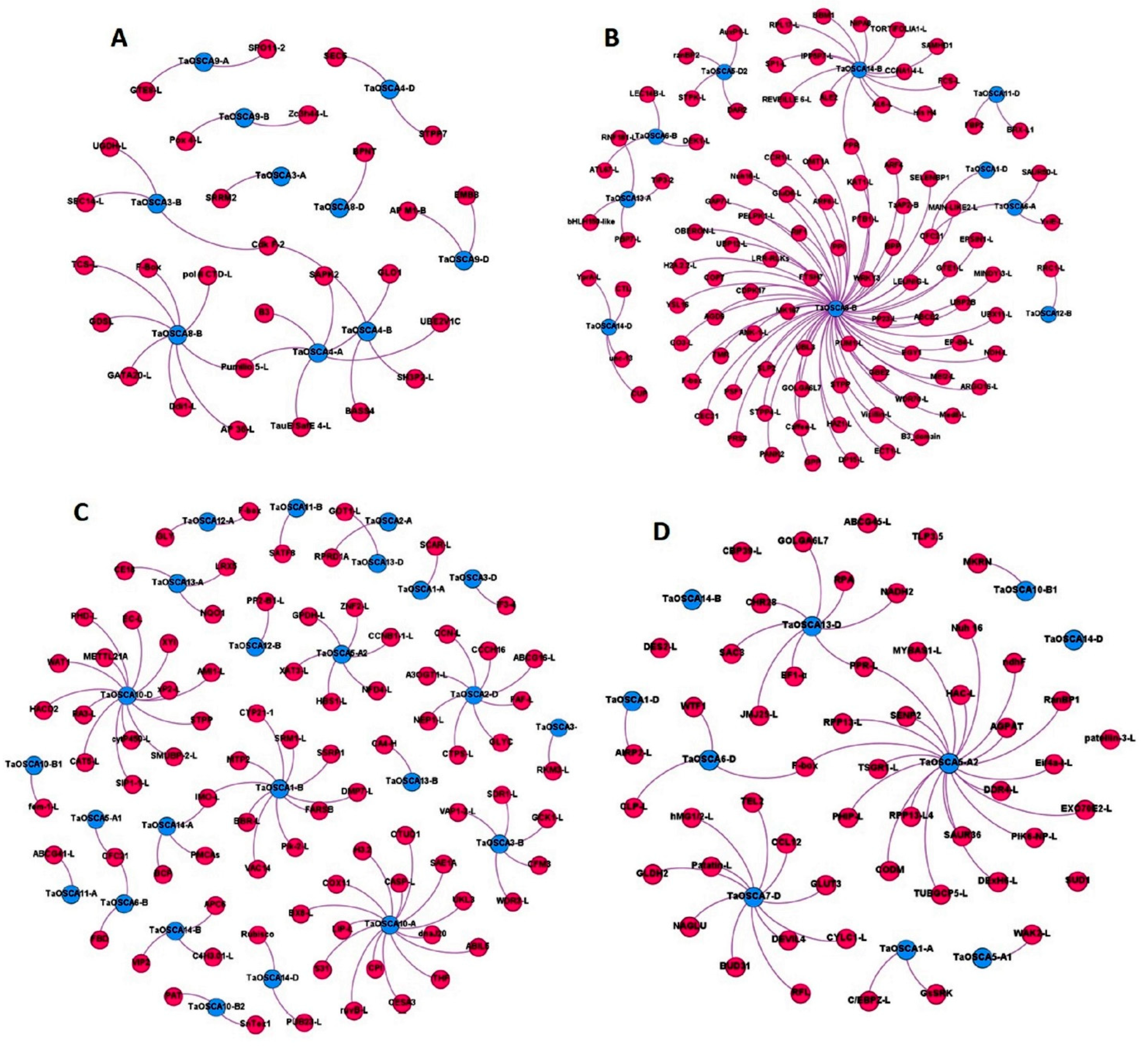
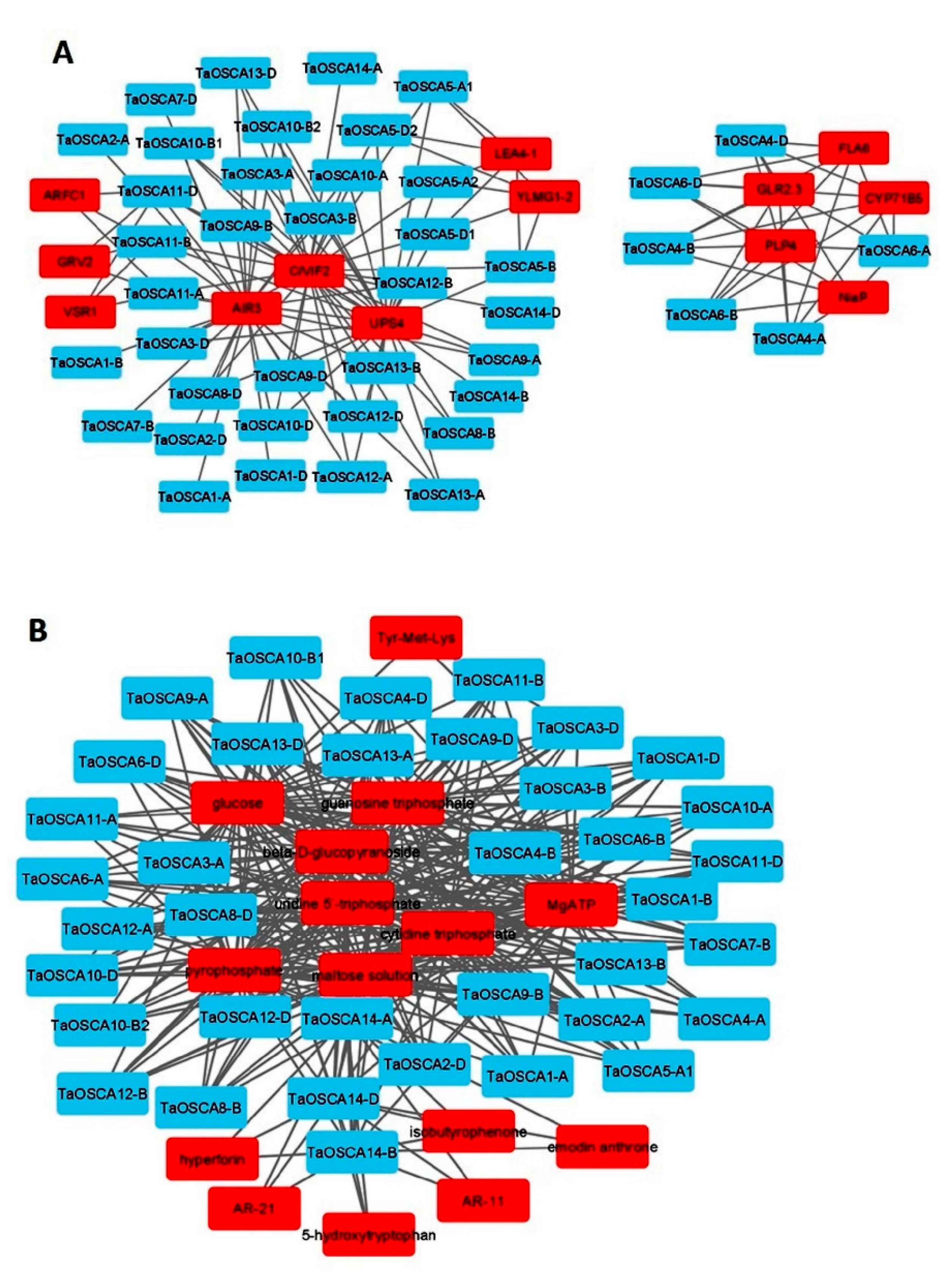
2.9. Protein–Protein Interaction Analysis
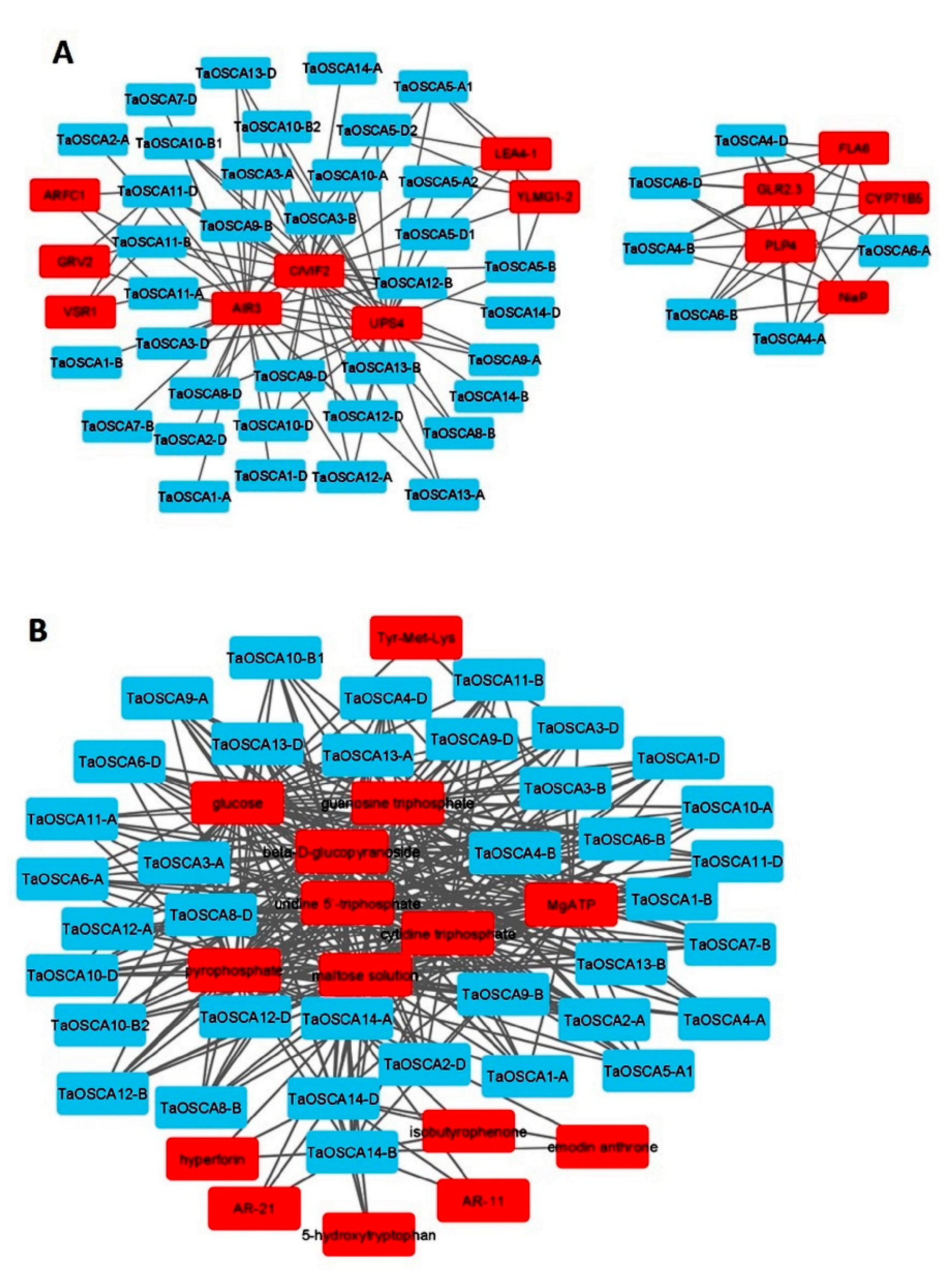
2.10. Protein–Chemical Interaction Analysis
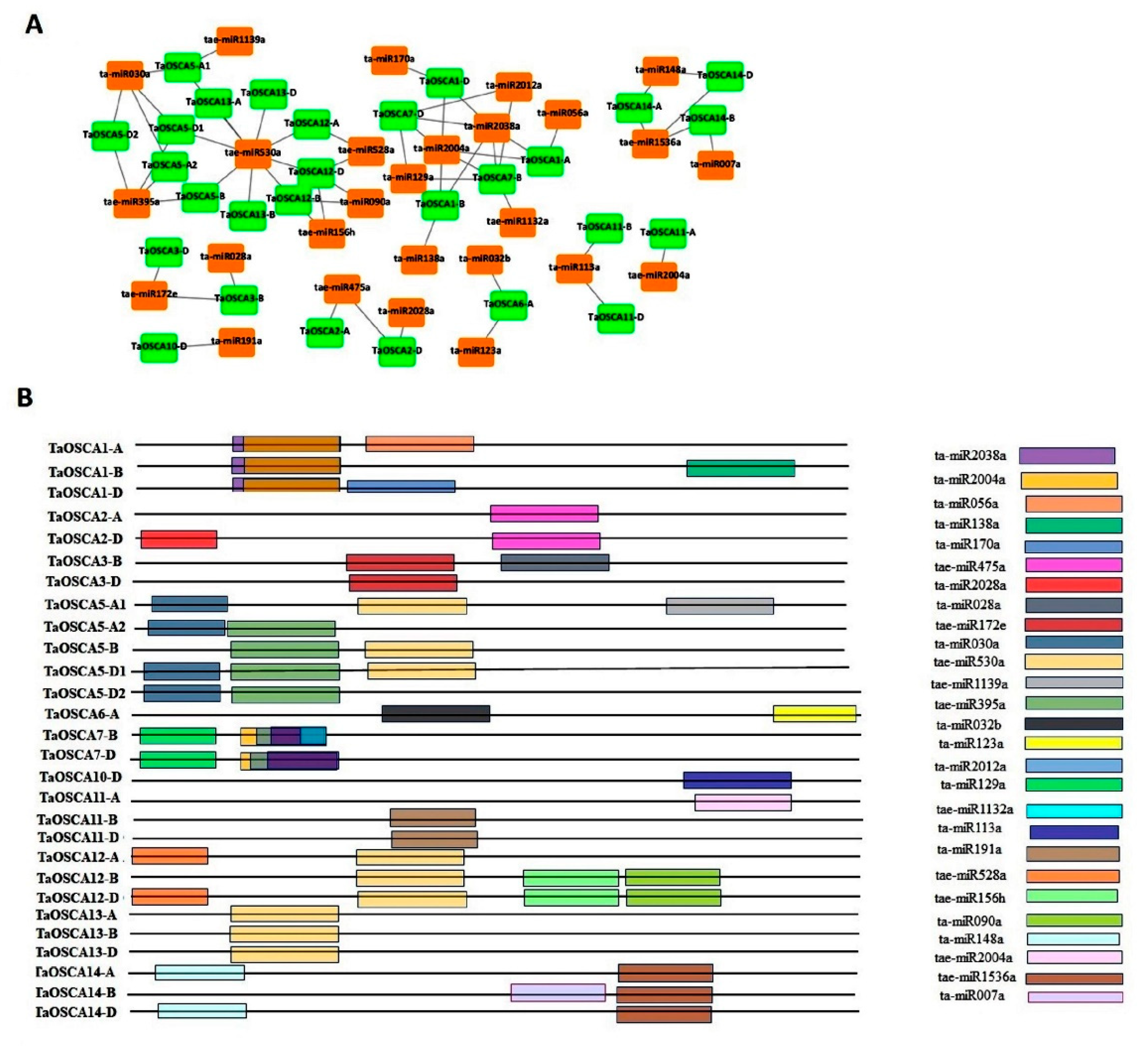
2.11. miRNAs Interaction Analysis
3. Discussion
4. Materials and Methods
4.1. Identification and Chromosomal Distribution
4.2. In Silico Protein Characterization
4.3. Prediction of Duplication Events and Ka/Ks Analysis
4.4. Phylogenetic Analysis
4.5. Expression Profiling of TaOSCA Genes
4.6. qRT-PCR Analysis
4.7. Co-Expression Analysis and Gene Ontology Mapping
4.8. Protein–Protein, Protein–Chemical, and miRNA Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bartels, D.; Sunkar, R. Drought and salt tolerance in plants. Crit. Rev. Plant Sci. 2005, 24, 23–58. [Google Scholar] [CrossRef]
- McAinsh, M.R.; Pittman, J.K. Shaping the calcium signature. New Phytol. 2009, 181, 275–294. [Google Scholar] [CrossRef]
- White, P.J. Calcium channels in higher plants. BBA-Biomembr. 2000, 1465, 171–189. [Google Scholar] [CrossRef] [Green Version]
- Taneja, M.; Upadhyay, S.K. An introduction to the calcium transport elements in plants. In Calcium Transport Elements in Plants; Academic Press: Cambridge, MA, USA, 2021; pp. 1–18. [Google Scholar]
- Smyth, J.T.; DeHaven, W.I.; Jones, B.F.; Mercer, J.C.; Trebak, M.; Vazquez, G.; Putney, J.W., Jr. Emerging perspectives in store-operated Ca2+ entry: Roles of Orai, Stim and TRP. BBA-Mol. Cell Res. 2006, 1763, 1147–1160. [Google Scholar]
- Upadhyay, S.K. Calcium Transport Elements in Plants; Academic Press: Cambridge, MA, USA, 2021; pp. 1–464. [Google Scholar]
- Zhang, M.; Wang, D.; Kang, Y.; Wu, J.X.; Yao, F.; Pan, C.; Yan, Z.; Song, C.; Chen, L. Structure of the mechanosensitive OSCA channels. Nat. Struct Mol. Biol 2018, 25, 850–858. [Google Scholar] [CrossRef]
- Batistic, O.; Kudla, J. Analysis of calcium signaling pathways in plants. Biochim. Biophys. Acta 2012, 1820, 1283–1293. [Google Scholar] [CrossRef] [PubMed]
- Trewavas, A.; Knight, M. Mechanical signalling, calcium and plant form. Plant Mol. Biol. 1994, 26, 1329–1341. [Google Scholar] [CrossRef]
- Booth, I.R.; Edwards, M.D.; Black, S.; Schumann, U.; Miller, S. Mechanosensitive channels in bacteria: Signs of closure? Nat. Rev. Microbiol. 2007, 5, 431–440. [Google Scholar] [CrossRef]
- Árnadóttir, J.; Chalfie, M. Eukaryotic mechanosensitive channels. Annu Rev. Biophys 2010, 39, 111–137. [Google Scholar] [CrossRef] [PubMed]
- Hou, C.; Tian, W.; Kleist, T.; He, K.; Garcia, V.; Bai, F.; Hao, Y.; Luan, S.; Li, L. DUF221 proteins are a family of osmosensitive calcium-permeable cation channels conserved across eukaryotes. Cell Res. 2014, 24, 632–635. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Wang, J.; Sun, L. Structure of the hyperosmolality-gated calcium permeable channel OSCA1. 2. Nat. Commun. 2018, 9, 1–9. [Google Scholar] [CrossRef]
- Yuan, F.; Yang, H.; Xue, Y.; Kong, D.; Ye, R.; Li, C.; Zhang, J.; Theprungsirikul, L.; Shrift, T.; Krichilsky, B.; et al. OSCA1 mediates osmotic-stress-evoked Ca2+ increases vital for osmosensing in Arabidopsis. Nature 2014, 514, 367–371. [Google Scholar] [CrossRef]
- Li, Y.; Yuan, F.; Wen, Z.; Li, Y.; Wang, F.; Zhu, T.; Zhuo, W.; Jin, X.; Wang, Y.; Zhao, H.; et al. Genome-wide survey and expression analysis of the OSCA gene family in rice. BMC Plant Biol. 2015, 15, 261. [Google Scholar] [CrossRef] [Green Version]
- Gu, X.; Wang, P.; Liu, Z.; Wang, L.; Huang, Z.; Zhang, S.; Wu, J. Genome-wide identification and expression analysis of the OSCA gene family in Pyrus bretschneideri. Can. J. Plant Sci. 2018, 98, 918–929. [Google Scholar] [CrossRef] [Green Version]
- Yin, L.; Zhang, M.; Wu, R.; Chen, X.; Liu, F.; Xing, B. Genome-wide analysis of OSCA gene family members in Vigna radiata and their involvement in the osmotic response. BMC Plant Biol 2021, 21, 408. [Google Scholar] [CrossRef]
- Ding, S.; Feng, X.; Du, H.; Wang, H. Genome-wide analysis of maize OSCA family members and their involvement in drought stress. PeerJ 2019, 7, e6765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Xu, Y.; Yang, F.; Magwanga, R.O.; Cai, X.; Wang, X.; Wang, Y.; Hou, Y.; Wang, K.; Liu, F.; et al. Genome-wide identification of OSCA gene family and their potential function in the regulation of dehydration and salt stress in Gossypium hirsutum. J. Cotton Res. 2019, 2, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Zeng, H.; Zhao, B.; Wu, H.; Zhu, Y.; Chen, H. Comprehensive in silico characterization and expression profiling of nine gene families associated with calcium transport in soybean. Agronomy 2020, 10, 1539. [Google Scholar] [CrossRef]
- Cao, L.; Zhang, P.; Lu, X.; Wang, G.; Wang, Z.; Zhang, Q.; Zhang, X.; Wei, X.; Mei, F.; Wei, L.; et al. Systematic analysis of the maize OSCA genes revealing ZmOSCA family members involved in osmotic stress and ZmOSCA2.4 confers enhanced drought tolerance in transgenic arabidopsis. Int. J. Mol. Sci. 2020, 21, 351. [Google Scholar] [CrossRef] [Green Version]
- Zhai, Y.; Wen, Z.; Han, Y.; Zhuo, W.; Wang, F.; Xi, C.; Liu, J.; Gao, P.; Zhao, H.; Wang, Y.; et al. Heterogeneous expression of plasma-membrane-localised OsOSCA1.4 complements osmotic sensing based on hyperosmolality and salt stress in arabidopsis osca1 mutant. Cell Calcium 2020, 91, 102261. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; Wen, Z.; Fang, W.; Wang, Y.; Xi, C.; Liu, J.; Zhao, H.; Wang, Y.; Han, S. Functional analysis of rice OSCA genes overexpressed in the arabidopsis osca1 mutant due to drought and salt stresses. Transgenic Res. 2021, 30, 811–820. [Google Scholar] [CrossRef]
- Thor, K.; Jiang, S.; Michard, E.; George, J.; Scherzer, S.; Huang, S.; Dindas, J.; Derbyshire, P.; Leitão, N.; DeFalco, T.A.; et al. The calcium-permeable channel OSCA1. 3 regulates plant stomatal immunity. Nature 2020, 585, 569–573. [Google Scholar] [CrossRef]
- Yoshioka, K.; Moeder, W. Calcium channel in plants helps shut the door on intruders. Nature 2020, 585, 507–508. [Google Scholar] [CrossRef] [PubMed]
- Tajima, F. Simple methods for testing the molecular evolutionary clock hypothesis. Genetics 1993, 135, 599–607. [Google Scholar] [CrossRef]
- Choulet, F.; Alberti, A.; Theil, S.; Glover, N.; Barbe, V.; Daron, J.; Pingault, L.; Sourdille, P.; Couloux, A.; Paux, E.; et al. Structural and functional partitioning of bread wheat chromosome 3B. Science 2014, 345, 1249721. [Google Scholar] [CrossRef]
- Pingault, L.; Choulet, F.; Alberti, A.; Glover, N.; Wincker, P.; Feuillet, C.; Paux, E. Deep transcriptome sequencing provides new insights into the structural and functional organization of the wheat genome. Genome Biol. 2015, 16, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Xin, M.; Qin, J.; Peng, H.; Ni, Z.; Yao, Y.; Sun, Q. Temporal transcriptome profiling reveals expression partitioning of homeologous genes contributing to heat and drought acclimation in wheat (Triticum aestivum L.). BMC Plant Biol. 2015, 15, 152. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Liu, Z.; Khan, A.A.; Lin, Q.; Han, Y.; Mu, P.; Liu, Y.; Zhang, H.; Li, L.; Meng, X.; et al. Expression partitioning of homeologs and tandem duplications contribute to salt tolerance in wheat (Triticum aestivum L.). Sci. Rep. 2016, 6, 21476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Yang, Y.; Wang, C.; Liu, M.; Li, H.; Fu, Y.; Wang, Y.; Nie, Y.; Liu, X.; Ji, W. Large-scale transcriptome comparison reveals distinct gene activations in wheat responding to stripe rust and powdery mildew. BMC Genom. 2014, 15, 898. [Google Scholar] [CrossRef] [Green Version]
- Tong, K.; Wu, X.; He, L.; Qiu, S.; Liu, S.; Cai, L.; Rao, S.; Chen, J. Genome-Wide Identification and Expression Profile of OSCA Gene Family Members in Triticum aestivum L. Int. J. Mol. Sci. 2021, 23, 469. [Google Scholar] [CrossRef]
- Kaur, A.; Taneja, M.; Tyagi, S.; Sharma, A.; Singh, K.; Upadhyay, S.K. Genome-wide characterization and expression analysis suggested diverse functions of the mechanosensitive channel of small conductance-like (MSL) genes in cereal crops. Sci. Rep. 2020, 10, 16583. [Google Scholar] [CrossRef] [PubMed]
- Madhu; Kaur, A.; Tyagi, S.; Singh, K.; Upadhyay, S.K. Exploration of glutathione reductase for abiotic stress response in bread wheat (Triticum aestivum L.). Plant Cell Rep. 2022, 41, 639–654. [Google Scholar] [CrossRef] [PubMed]
- Appels, R.; Eversole, K.; Feuillet, C.; Keller, B.; Rogers, J.; Stein, N.; Pozniak, C.J.; Choulet, F.; Distelfeld, A.; Poland, J. Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 2018, 361, eaar7191. [Google Scholar]
- Ellegren, H. Comparative genomics and the study of evolution by natural selection. Mol. Ecol. 2008, 17, 4586–4596. [Google Scholar] [CrossRef]
- Marcussen, T.; Sandve, S.R.; Heier, L.; Spannagl, M.; Pfeifer, M.; International Wheat Genome Sequencing Consortium; Jakobsen, K.S.; Wulff, B.B.; Steuernagel, B.; Mayer, K.F.; et al. Ancient hybridizations among the ancestral genomes of bread wheat. Science 2014, 345, 1250092. [Google Scholar] [CrossRef] [PubMed]
- Kärkönen, A.; Murigneux, A.; Martinant, J.P.; Pepey, E.; Tatout, C.; Dudley, B.J.; Fry, S.C. UDP-glucose dehydrogenases of maize: A role in cell wall pentose biosynthesis. Biochem. J. 2005, 391, 409–415. [Google Scholar] [CrossRef]
- Chen, Y.; Li, C.; Zhang, B.; Yi, J.; Yang, Y.; Kong, C.; Lei, C.; Gong, M. The role of the late embryogenesis-abundant (LEA) protein family in development and the abiotic stress response: A comprehensive expression analysis of potato (Solanum tuberosum). Genes 2019, 10, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, F.; Hu, Y.; Vannozzi, A.; Wu, K.; Cai, H.; Qin, Y.; Mullis, A.; Lin, Z.; Zhang, L. The WRKY transcription factor family in model plants and crops. Crit. Rev. Plant Sci. 2017, 36, 311–335. [Google Scholar] [CrossRef]
- Romeis, T. Protein kinases in the plant defence response. Curr. Opin. Plant Biol. 2001, 4, 407–414. [Google Scholar] [CrossRef]
- Sharma, A.; Sharma, H.; Rajput, R.; Pandey, A.; Upadhyay, S.K. Molecular Characterization Revealed the Role of Thaumatin-Like Proteins of Bread Wheat in Stress Response. Front. Plant Sci. 2022, 12, 807448. [Google Scholar] [CrossRef]
- Cho, D.; Kim, S.A.; Murata, Y.; Lee, S.; Jae, S.K.; Nam, H.G.; Kwak, J.M. De-regulated expression of the plant glutamate receptor homolog AtGLR3. 1 impairs long-term Ca2+-programmed stomatal closure. Plant J. 2009, 58, 437–449. [Google Scholar] [CrossRef]
- Mowla, S.B.; Cuypers, A.; Driscoll, S.P.; Kiddle, G.; Thomson, J.; Foyer, C.H.; Theodoulou, F.L. Yeast complementation reveals a role for an Arabidopsis thaliana late embryogenesis abundant (LEA)-like protein in oxidative stress tolerance. Plant J. 2006, 48, 743–756. [Google Scholar] [CrossRef]
- Olvera-Carrillo, Y.; Campos, F.; Reyes, J.L.; Garciarrubio, A.; Covarrubias, A.A. Functional analysis of the group 4 late embryogenesis abundant proteins reveals their relevance in the adaptive response during water deficit in Arabidopsis. Plant Physiol. 2010, 154, 373–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Chen, S.; Cheng, Y.; Zhang, N.; Ma, Y.; Wang, W.; Tian, H.; Li, Y.; Hussain, S.; Wang, S. Cell wall/vacuolar inhibitor of fructosidase 1 regulates ABA response and salt tolerance in Arabidopsis. Plant Signal. Behav. 2020, 15, 1744293. [Google Scholar] [CrossRef] [PubMed]
- Rietz, S.; Dermendjiev, G.; Oppermann, E.; Tafesse, F.G.; Effendi, Y.; Holk, A.; Parker, J.E.; Teige, M.; Scherer, G.F. Roles of Arabidopsis patatin-related phospholipases a in root development are related to auxin responses and phosphate deficiency. Mol. Plant 2010, 3, 524–538. [Google Scholar] [CrossRef] [PubMed]
- Neuteboom, L.W.; Veth-Tello, L.M.; Clijdesdale, O.R.; Hooykaas, P.J.; van der Zaal, B.J. A novel subtilisin-like protease gene from Arabidopsis thaliana is expressed at sites of lateral root emergence. DNA Res. 1999, 6, 13–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemtiri-Chlieh, F.; Arold, S.T.; Gehring, C. Mg2+ is a missing link in plant cell Ca2+ signalling and homeostasis—A study on Vicia faba guard cells. Int. J. Mol. Sci. 2020, 21, 3771. [Google Scholar] [CrossRef]
- Briskin, D.P.; Poole, R.J. Role of magnesium in the plasma membrane ATPase of red beet. Plant Physiol. 1983, 71, 969–971. [Google Scholar] [CrossRef]
- Cordewener, J.; Krüse-Wolters, M.; Wassink, H.; Haaker, H.; Veeger, C. The role of MgATP hydrolysis in nitrogenase catalysis. Eur. J. Biochem. 1988, 172, 739–745. [Google Scholar] [CrossRef]
- Nuoffer, C.; Balch, W.E. GTPases: Multifunctional molecular switches regulating vesicular traffic. Annu. Rev. Biochem 1994, 63, 949–990. [Google Scholar] [CrossRef]
- Carthew, R.W.; Sontheimer, E.J. Origins and mechanisms of miRNAs and siRNAs. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contreras-Cubas, C.; Palomar, M.; Arteaga-Vázquez, M.; Reyes, J.L.; Covarrubias, A.A. Non-coding RNAs in the plant response to abiotic stress. Planta 2012, 236, 943–958. [Google Scholar] [CrossRef] [PubMed]
- Bai, Q.; Wang, X.; Chen, X.; Shi, G.; Liu, Z.; Guo, C.; Xiao, K. Wheat miRNA TaemiR408 acts as an essential mediator in plant tolerance to Pi deprivation and salt stress via modulating stress-associated physiological processes. Front. Plant Sci. 2018, 9, 499. [Google Scholar] [CrossRef]
- Lin, Y.; Lai, Z.; Tian, Q.; Lin, L.; Lai, R.; Yang, M.; Zhang, D.; Chen, Y.; Zhang, Z. Endogenous target mimics down-regulate miR160 mediation of ARF10,-16, and-17 cleavage during somatic embryogenesis in Dimocarpus longan Lour. Front. Plant Sci. 2015, 6, 956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, F.; Wei, H.; Zhao, S.; Wang, L.; Zheng, H.; Lu, M. Identification of microRNAs involved in regeneration of the secondary vascular system in Populus tomentosa Carr. Front. Plant Sci. 2016, 7, 724. [Google Scholar] [CrossRef] [Green Version]
- Meng, F.; Liu, H.; Wang, K.; Liu, L.; Wang, S.; Zhao, Y.; Yin, J.; Li, Y. Development-associated microRNAs in grains of wheat (Triticum aestivum L.). BMC Plant Biol. 2013, 13, 140. [Google Scholar] [CrossRef] [Green Version]
- Sun, F.; Guo, G.; Du, J.; Guo, W.; Peng, H.; Ni, Z.; Sun, Q.; Yao, Y. Whole-genome discovery of miRNAs and their targets in wheat (Triticum aestivum L. ). BMC Plant Biol 2014, 14, 142. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Q.H.; Helliwell, C.A. Regulation of flowering time and floral patterning by miR172. J. Exp. Bot. 2011, 62, 487–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budak, H.; Akpinar, B.A. Plant miRNAs: Biogenesis, organization and origins. Funct. Integr. Genom. 2015, 15, 523–531. [Google Scholar] [CrossRef]
- Akdogan, G.; Tufekci, E.D.; Uranbey, S.; Unver, T. miRNA-based drought regulation in wheat. Funct. Integr. Genom. 2016, 16, 221–233. [Google Scholar] [CrossRef]
- Bakhshi, B.; Fard, E.M.; Gharechahi, J.; Safarzadeh, M.; Nikpay, N.; Fotovat, R.; Azimi, M.R.; Salekdeh, G.H. The contrasting microRNA content of a drought tolerant and a drought susceptible wheat cultivar. J. Plant Physiol. 2017, 216, 35–43. [Google Scholar] [CrossRef] [PubMed]
- She, K.; Pan, W.; Yan, Y.; Shi, T.; Chu, Y.; Cheng, Y.; Ma, B.; Song, W. Genome-Wide Identification, Evolution and Expressional Analysis of OSCA Gene Family in Barley (Hordeum vulgare L.). Int. J. Mol. Sci. 2022, 23, 13027. [Google Scholar] [CrossRef]
- Letunic, I.; Doerks, T.; Bork, P. SMART: Recent updates, new developments and status in 2015. Nucleic Acids Res. 2015, 43, D257–D260. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Bo, Y.; Han, L.; He, J.; Lanczycki, C.J.; Lu, S.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; et al. CDD/SPARCLE: Functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2017, 45, D200–D203. [Google Scholar] [CrossRef] [Green Version]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein identification and analysis tools on the ExPASy server. In The Proteomics Protocols Handbook; Humana Press: New York, NY, USA, 2005; pp. 571–607. [Google Scholar]
- Yu, C.S.; Chen, Y.C.; Lu, C.H.; Hwang, J.K. Prediction of protein subcellular localization. Proteins: Struct. Funct. Bioinform. 2006, 64, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Suyama, M.; Torrents, D.; Bork, P. PAL2NAL: Robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006, 34 (Suppl. 2), W609–W612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaut, B.S.; Morton, B.R.; McCaig, B.C.; Clegg, M.T. Substitution rate comparisons between grasses and palms: Synonymous rate differences at the nuclear gene Adh parallel rate differences at the plastid gene rbcL. Proc Natl Acad Sci. USA 1996, 93, 10274–10279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547. [Google Scholar] [CrossRef]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Papatheodorou, I.; Fonseca, N.A.; Keays, M.; Tang, Y.A.; Barrera, E.; Bazant, W.; Burke, M.; Füllgrabe, A.; Fuentes, A.M.P.; George, N.; et al. Expression Atlas: Gene and protein expression across multiple studies and organisms. Nucleic Acids Res. 2018, 46, D246–D251. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.; Gordish-Dressman, H.; Hoffman, E.P. An interactive power analysis tool for microarray hypothesis testing and generation. Bioinformatics 2006, 22, 808–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shumayla; Mendu, V.; Singh, K.; Upadhyay, S.K. Insight into the Roles of Proline-Rich Extensin-like Receptor Protein Kinases of Bread Wheat (Triticum aestivum L.). Life 2022, 12, 941. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Nazarov, P.; Muller, A.; Khutko, V.; Vallar, L. Co-Expression Analysis of Large Microarray Data Sets Using CoExpress Software Tool. In Proceedings of the Conference: WCSB 2010, Luxembourg, 16–18 June 2010. [Google Scholar]
- Conesa, A.; Götz, S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genom. 2008, 2008, 619832. [Google Scholar] [CrossRef] [Green Version]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An open source software for exploring and manipulating networks visualization and exploration of large graphs. In Proceedings of the Third International AAAI Conference on Weblogs and Social Media, San Jose, CA, USA, 17–20 May 2009. [Google Scholar]
- Franceschini, A.; Szklarczyk, D.; Frankild, S.; Kuhn, M.; Simonovic, M.; Roth, A.; Lin, J.; Minguez, P.; Bork, P.; Von Mering, C.; et al. STRING v9. 1: Protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2012, 41, D808–D815. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, M.; Szklarczyk, D.; Franceschini, A.; Von Mering, C.; Jensen, L.J.; Bork, P. STITCH 3: Zooming in on protein–chemical interactions. Nucleic Acids Res. 2012, 40, D876–D880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, X.; Zhuang, Z.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server (2017 release). Nucleic Acids Res. 2018, 46, W49–W54. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene A | Gene B | Ka | Ks | Ka/Ks | T = Ks/2r | Selection Pressure |
|---|---|---|---|---|---|---|
| TaOSCA4-A | TaOSCA6-B | 0.0885 | 0.5033 | 0.1758 | 13.5 MYA | Purifying |
| TaOSCA1-B | TaOSCA2-D | 0.0697 | 0.2413 | 0.2886 | 22.2 MYA | Purifying |
| TaOSCA12-B | TaOSCA13-B | 0.1297 | 0.3029 | 0.4282 | 32.9 MYA | Purifying |
| Group A | Group B | Outgroup | Nt | Na | Nb | χ2 | p |
|---|---|---|---|---|---|---|---|
| TaOSCA4-A | TaOSCA6-B | TaOSCA11-B | 93 | 10 | 6 | 1.00 | 0.31731 |
| TaOSCA1-B | TaOSCA2-D | TaOSCA5-B | 201 | 13 | 15 | 0.14 | 0.70546 |
| TaOSCA12-B | TaOSCA13-B | TaOSCA11-D | 112 | 13 | 11 | 0.17 | 0.68309 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaur, A.; Sharma, A.; Madhu; Dixit, S.; Singh, K.; Upadhyay, S.K. OSCA Genes in Bread Wheat: Molecular Characterization, Expression Profiling, and Interaction Analyses Indicated Their Diverse Roles during Development and Stress Response. Int. J. Mol. Sci. 2022, 23, 14867. https://doi.org/10.3390/ijms232314867
Kaur A, Sharma A, Madhu, Dixit S, Singh K, Upadhyay SK. OSCA Genes in Bread Wheat: Molecular Characterization, Expression Profiling, and Interaction Analyses Indicated Their Diverse Roles during Development and Stress Response. International Journal of Molecular Sciences. 2022; 23(23):14867. https://doi.org/10.3390/ijms232314867
Chicago/Turabian StyleKaur, Amandeep, Alok Sharma, Madhu, Sameer Dixit, Kashmir Singh, and Santosh Kumar Upadhyay. 2022. "OSCA Genes in Bread Wheat: Molecular Characterization, Expression Profiling, and Interaction Analyses Indicated Their Diverse Roles during Development and Stress Response" International Journal of Molecular Sciences 23, no. 23: 14867. https://doi.org/10.3390/ijms232314867
APA StyleKaur, A., Sharma, A., Madhu, Dixit, S., Singh, K., & Upadhyay, S. K. (2022). OSCA Genes in Bread Wheat: Molecular Characterization, Expression Profiling, and Interaction Analyses Indicated Their Diverse Roles during Development and Stress Response. International Journal of Molecular Sciences, 23(23), 14867. https://doi.org/10.3390/ijms232314867