
A Novel Aryl Hydrocarbon Receptor Antagonist HBU651 Ameliorates Peripheral and Hypothalamic Inflammation in High-Fat Diet-Induced Obese Mice
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. In Vitro Study
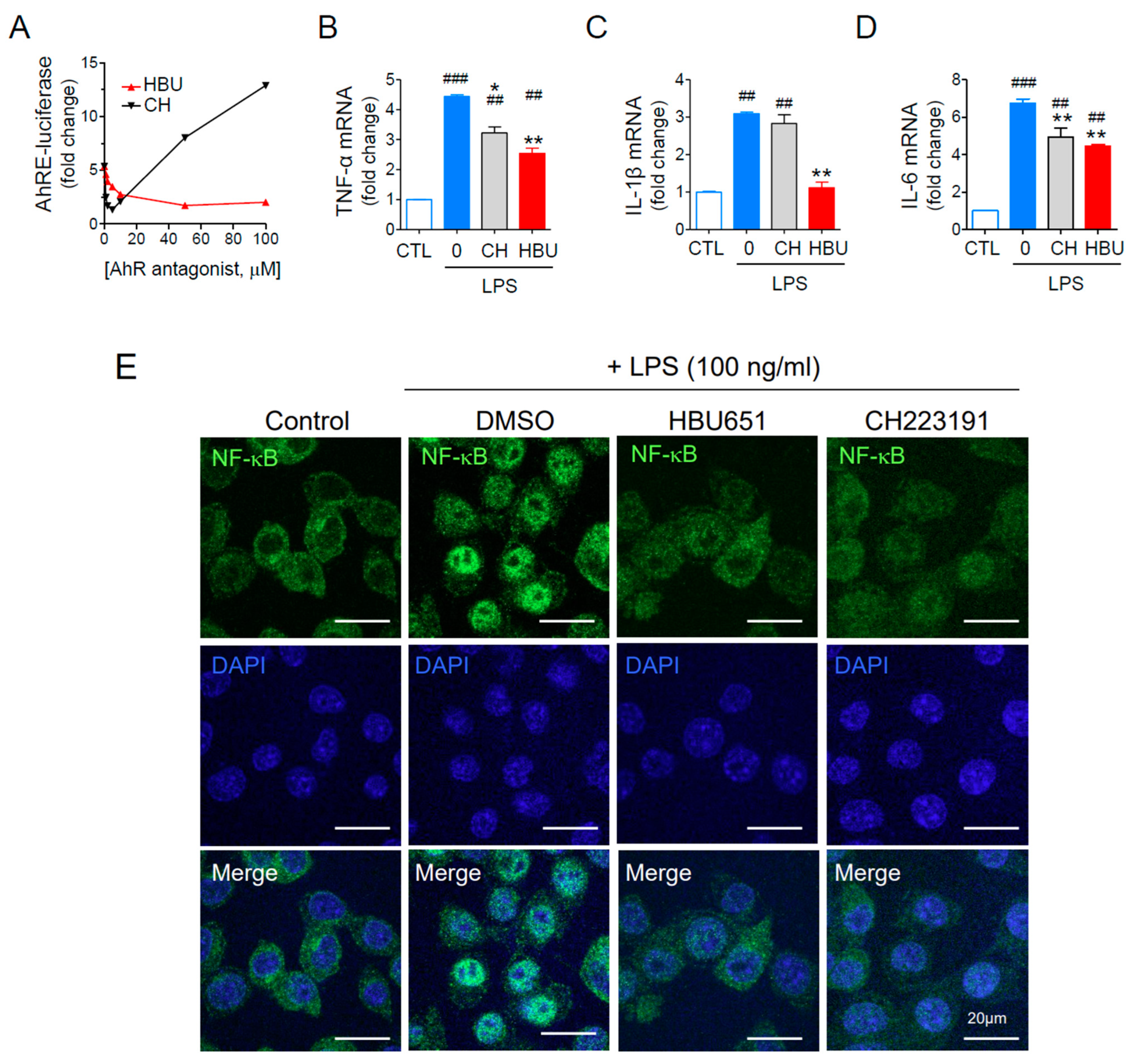
2.1.1. HBU651 Is an AhR Antagonist
2.1.2. Anti-Neuroinflammatory Effects of HBU651 on LPS-Stimulated BV2 Cells
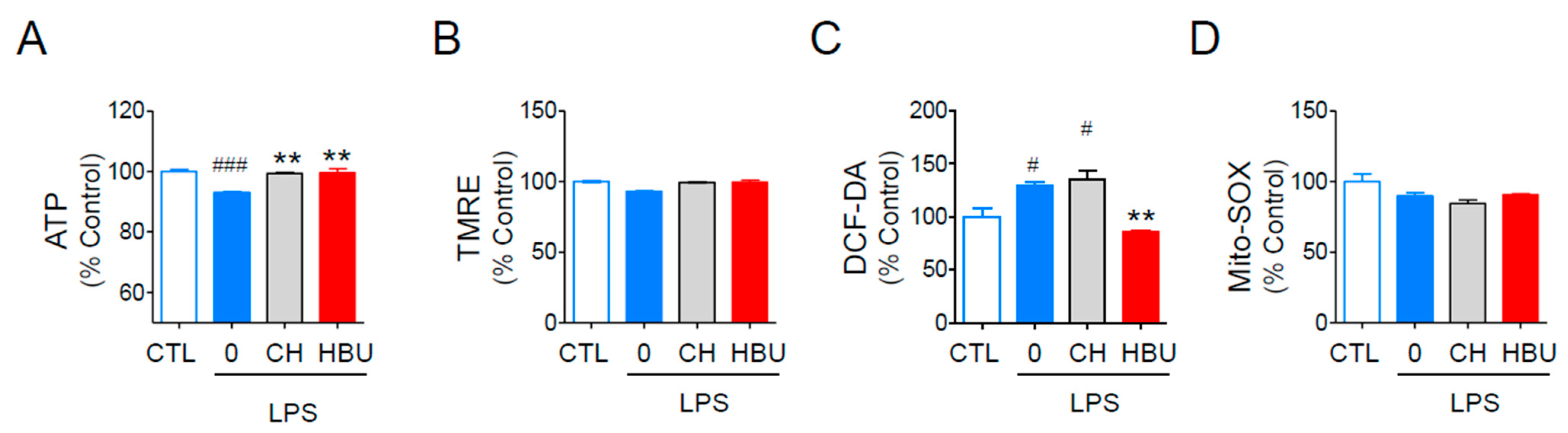
2.1.3. The Effects of HBU651 on Mitochondrial Function in BV2 Cells
2.2. In Vivo Study
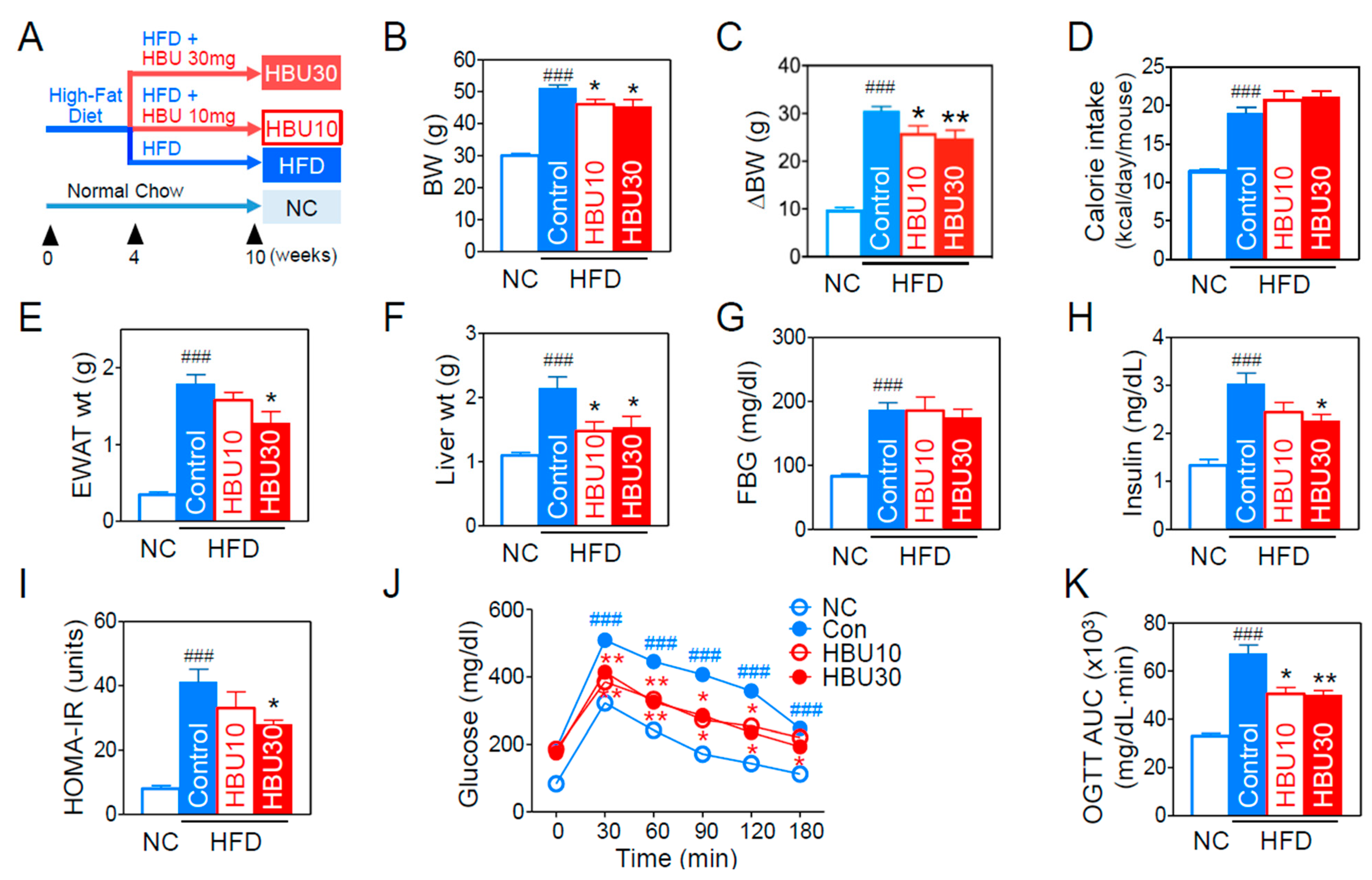
2.2.1. Metabolic Phenotypes
2.2.2. Glucose Metabolism and Insulin Resistance
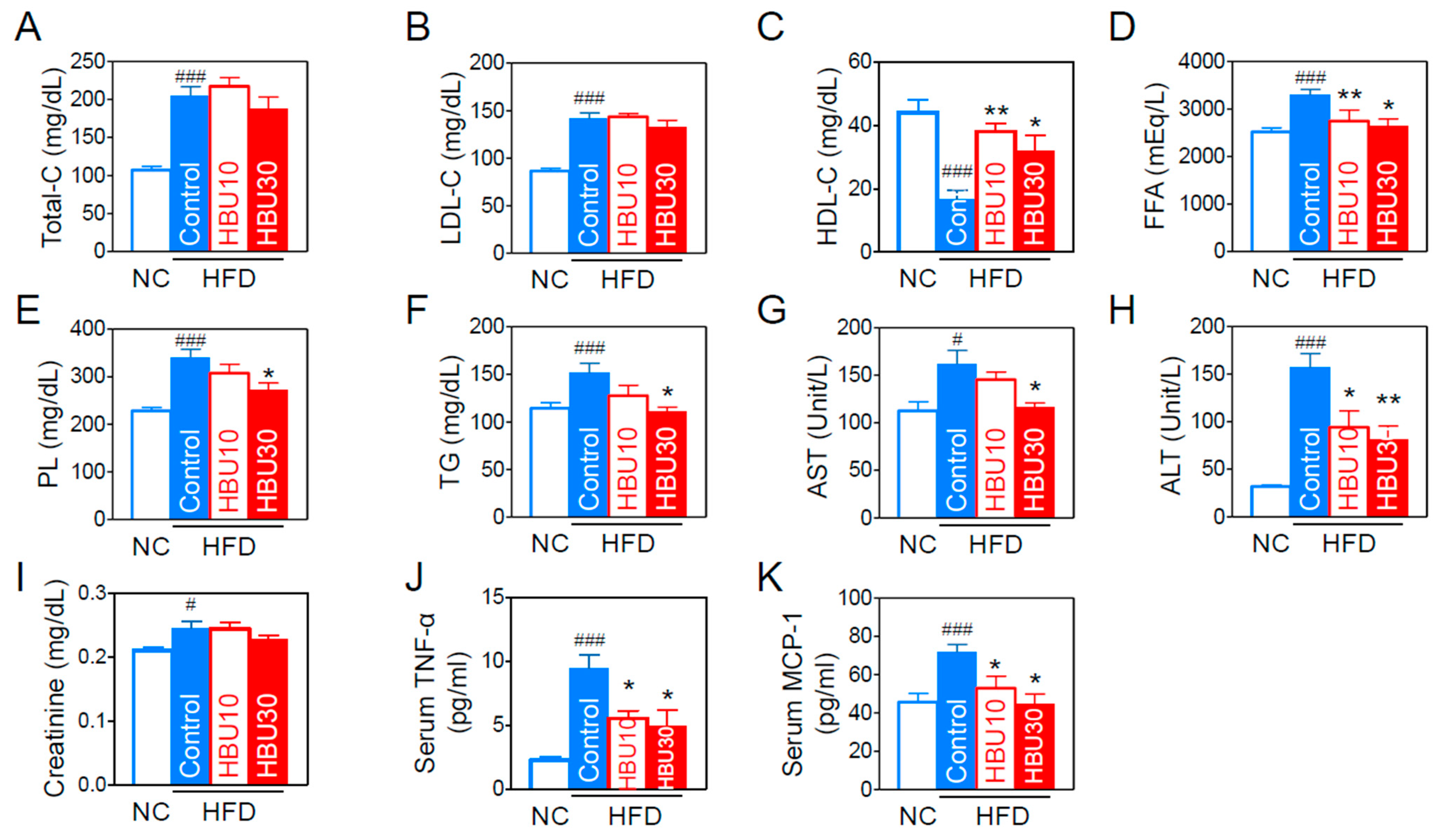
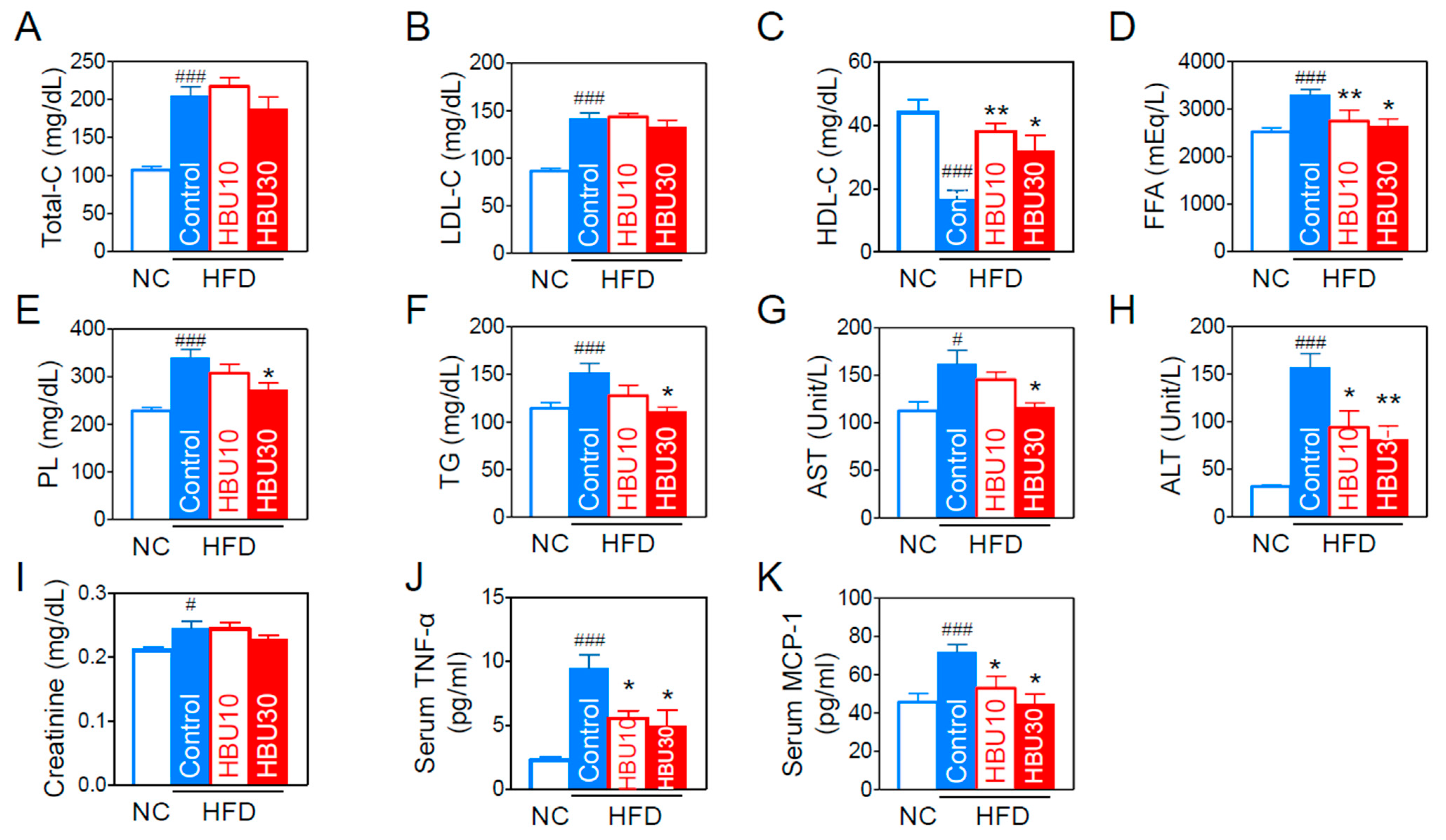
2.2.3. Lipid and Biochemical Parameters
2.2.4. Serum Inflammatory Cytokines and Chemokines
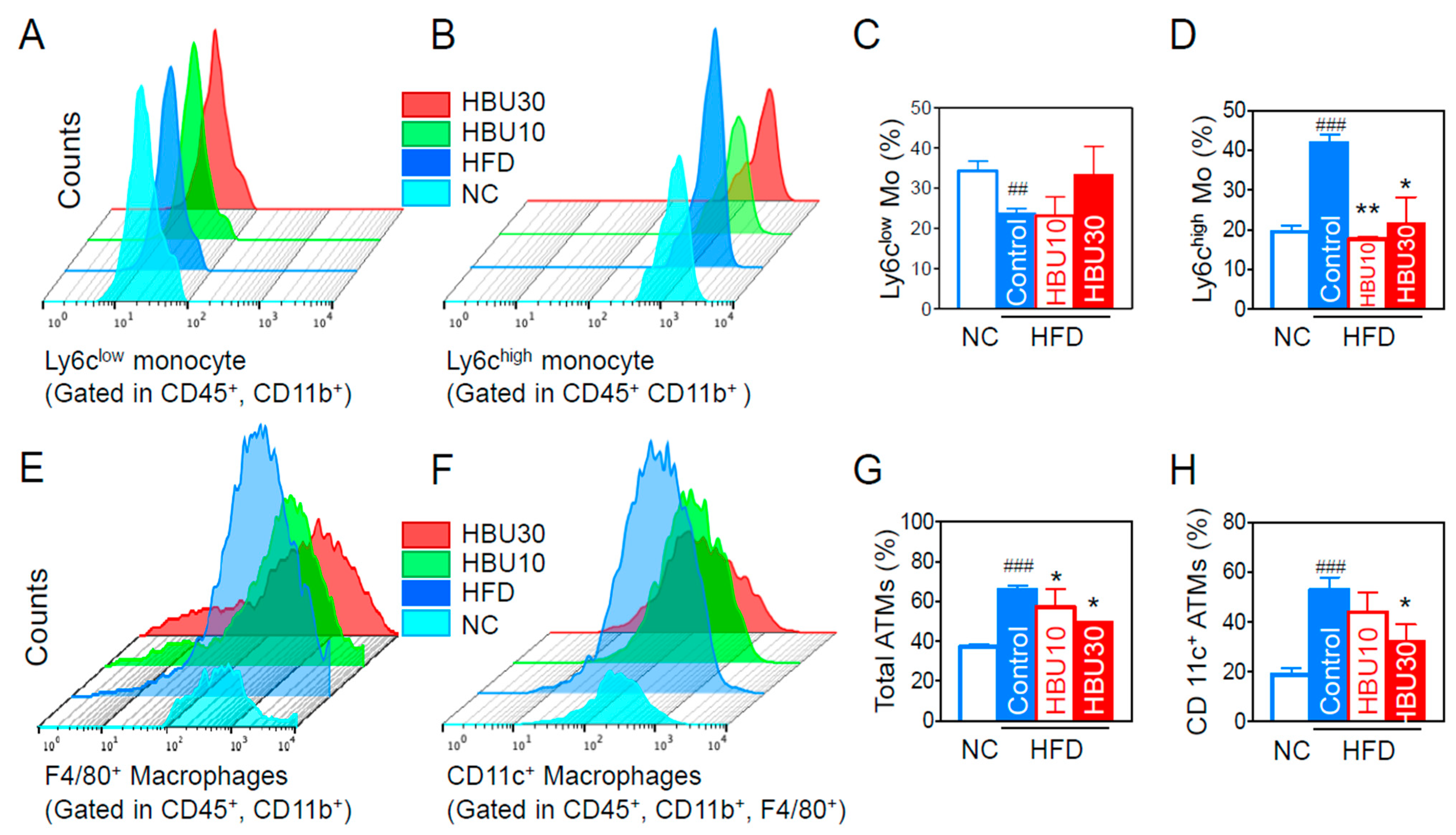
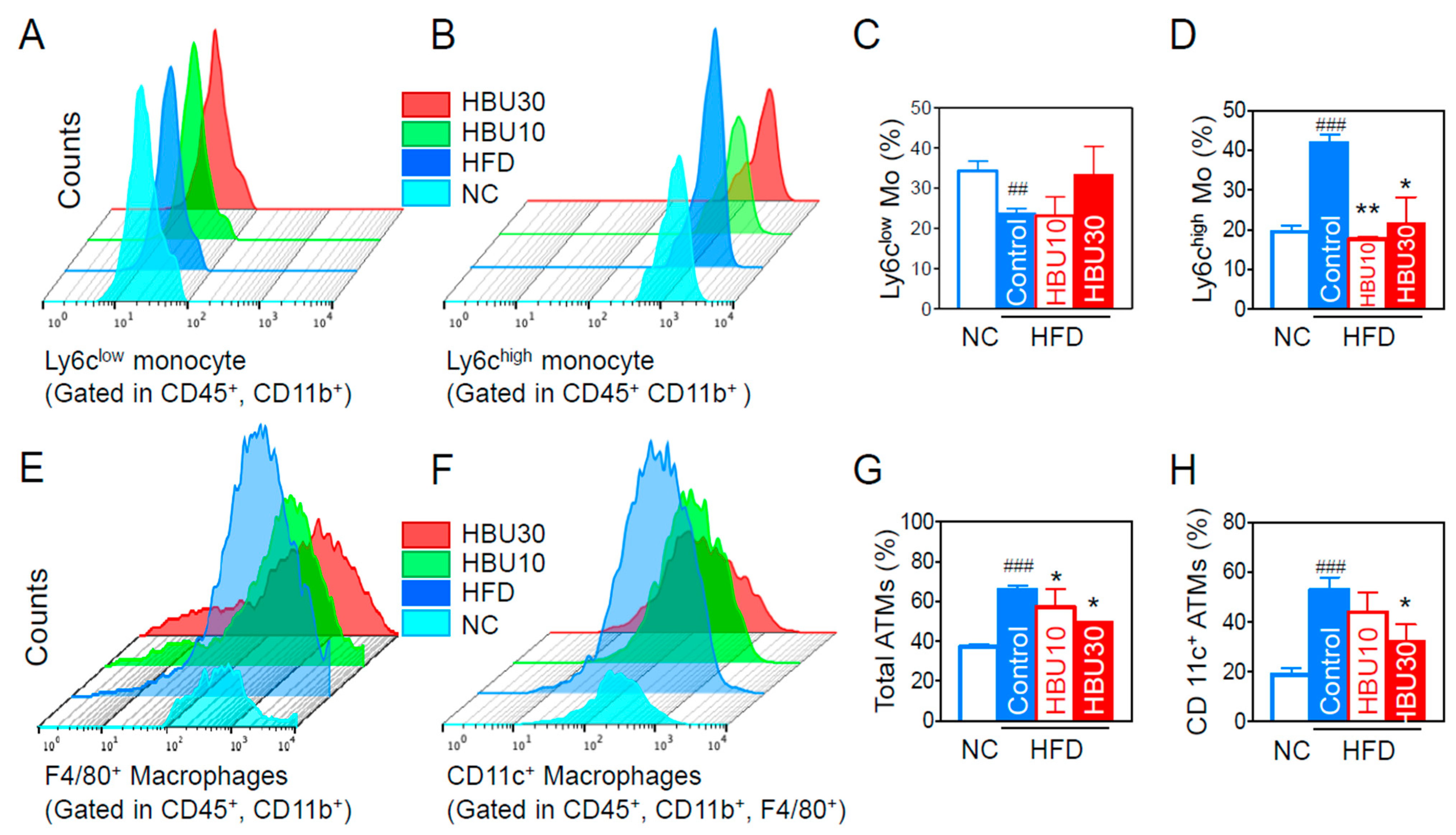
2.2.5. Inflammatory Monocytes and Adipose Tissue Macrophages (ATMs)
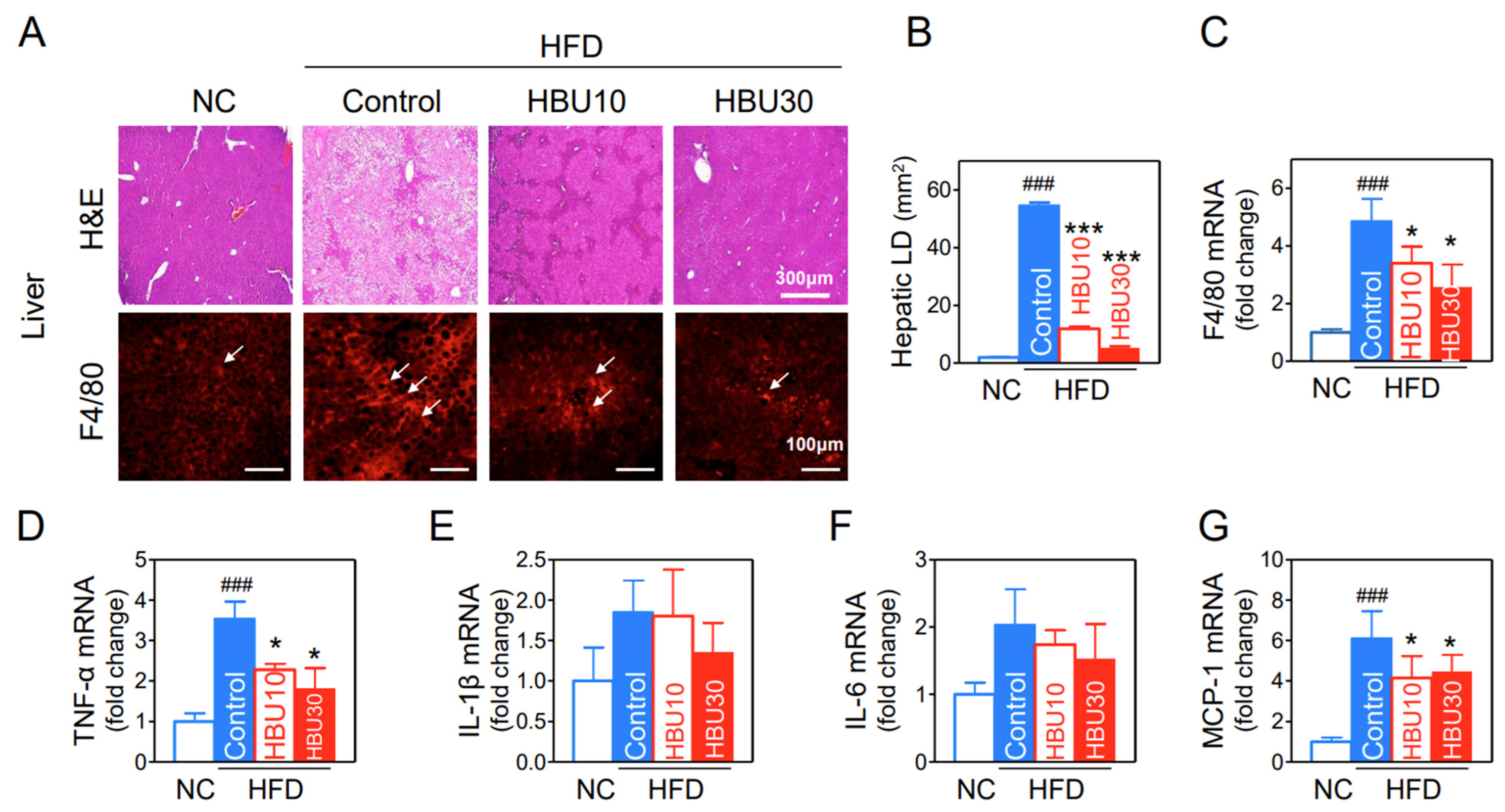
2.2.6. Hepatic Steatosis and Cytokine mRNAs
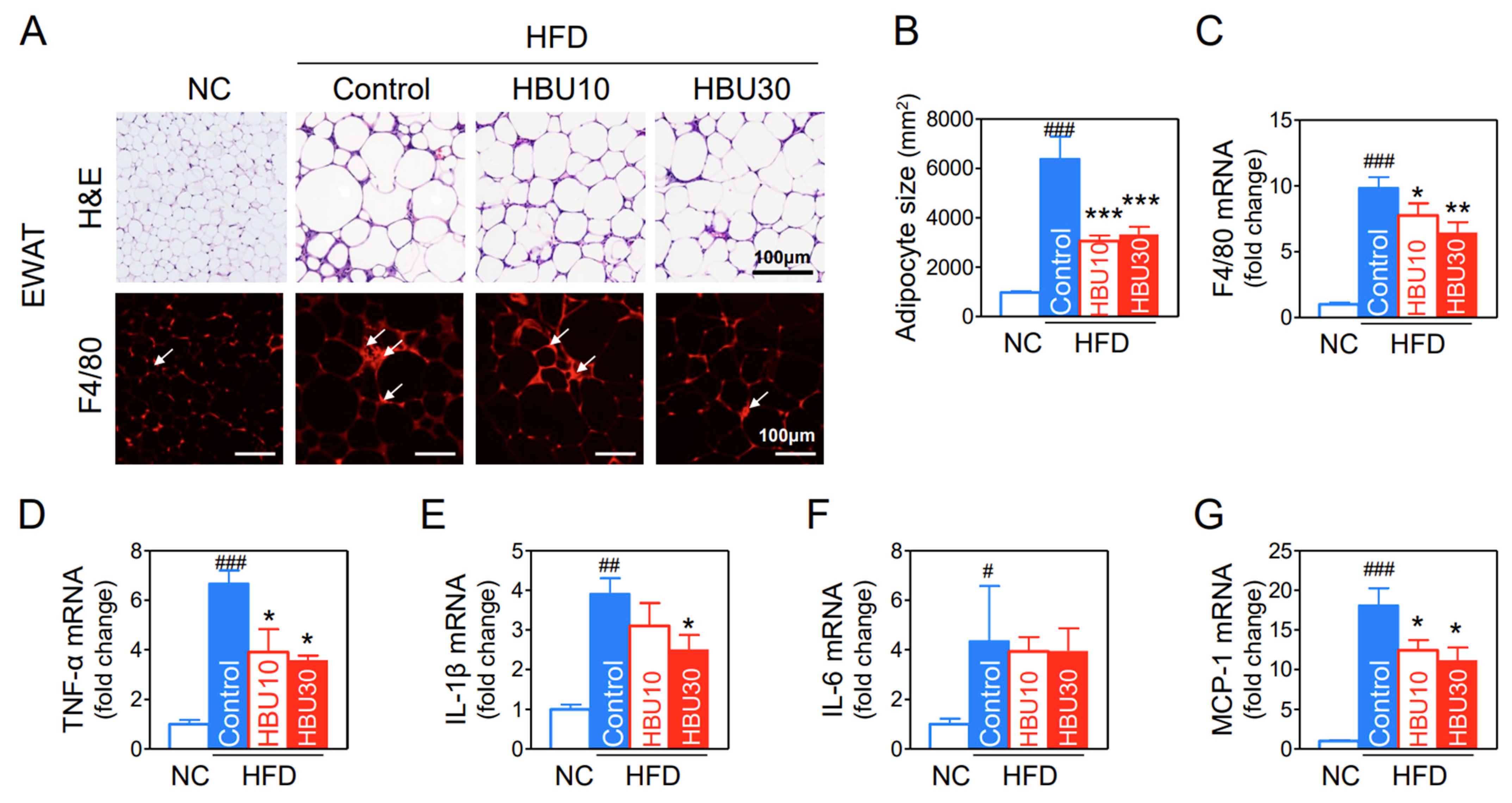
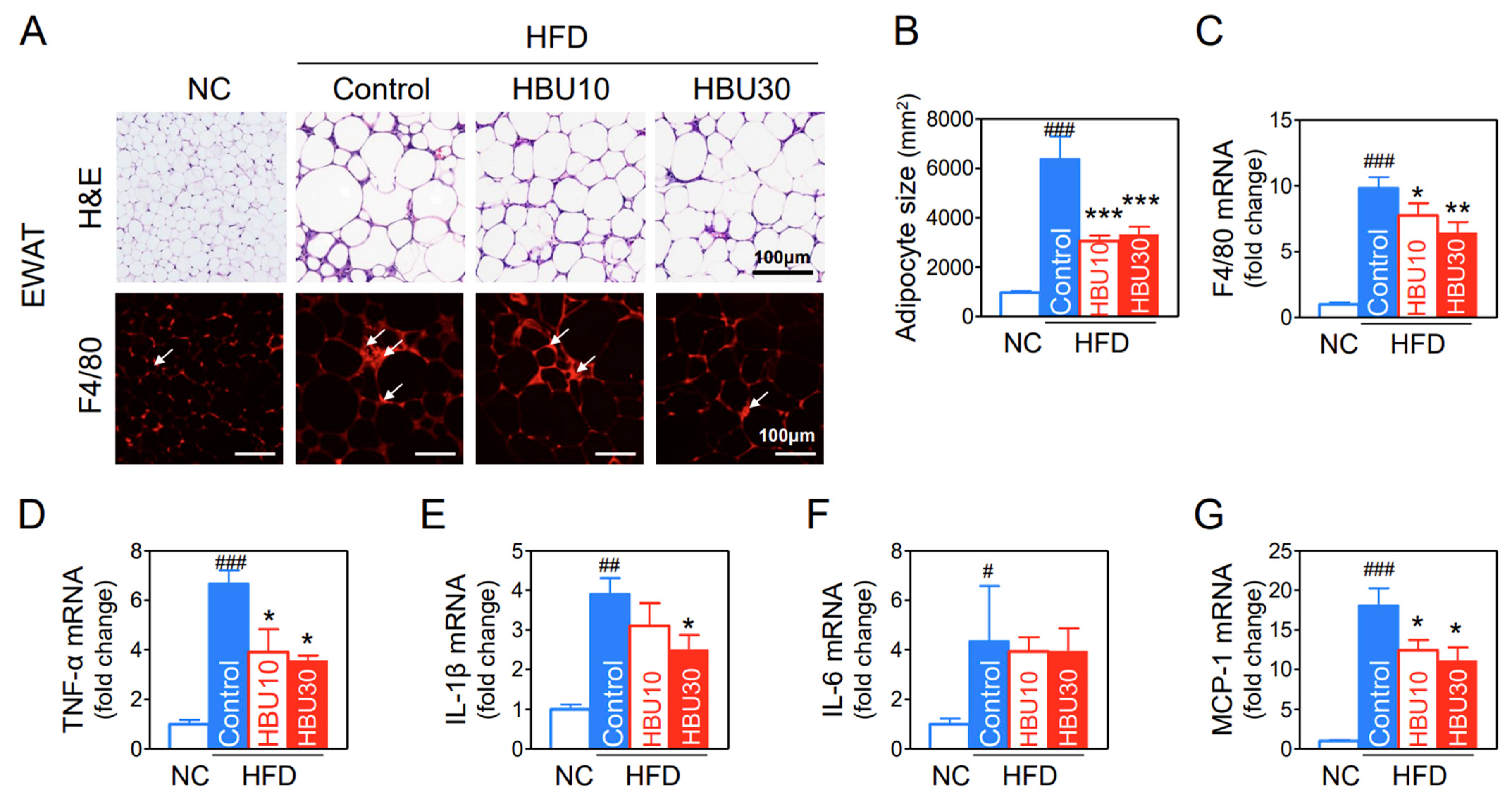
2.2.7. Fat Accumulation and Macrophage Infiltration in Adipose Tissue
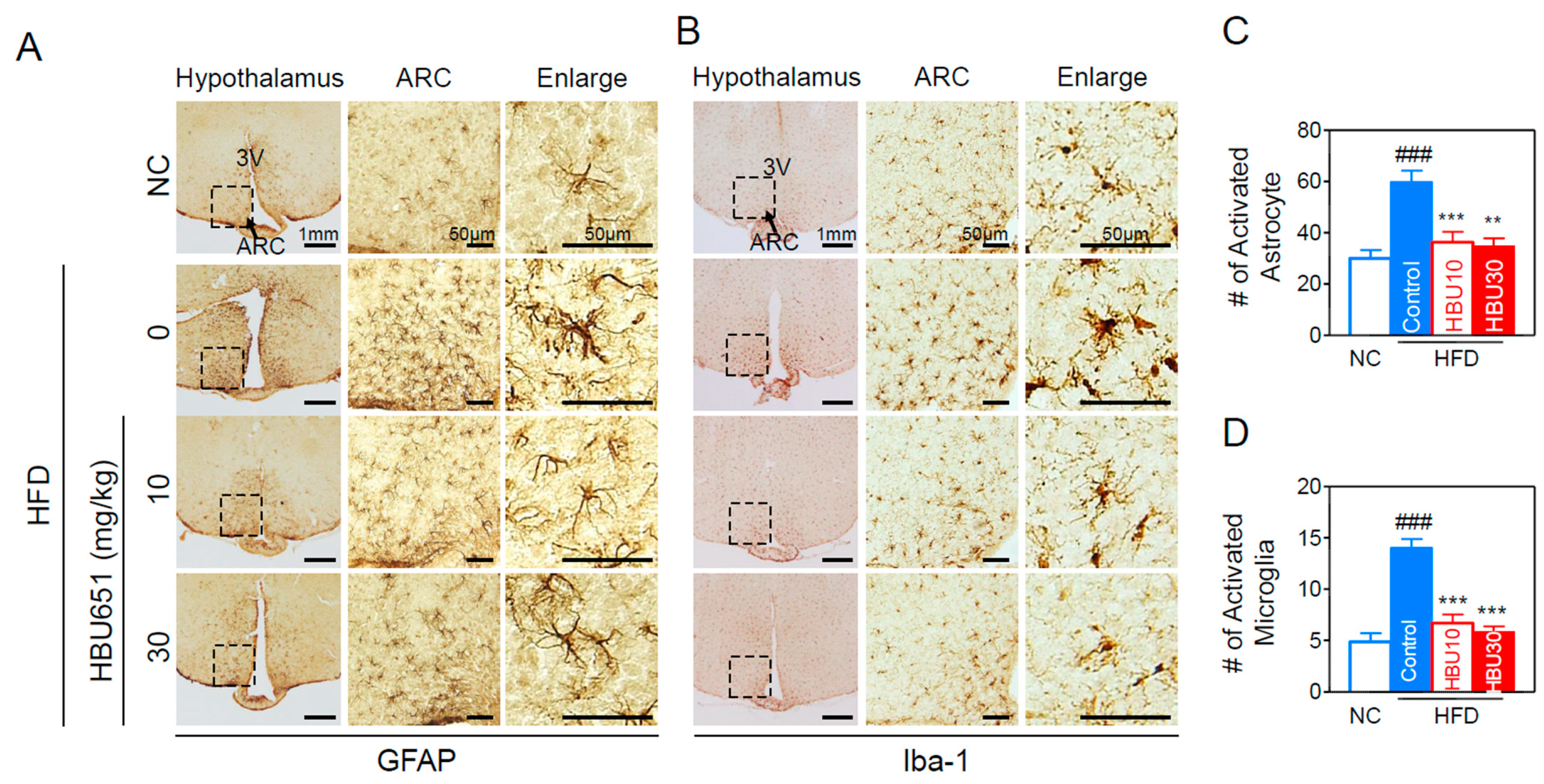
2.2.8. Neuroinflammation in the Hypothalamus
3. Discussion
4. Materials and Methods
4.1. Preparation of HBU651
4.2. AhR-Dependent Luciferase Reporter Assay
4.3. Cell Culture and Treatment
4.4. Assays for Mitochondrial Function
4.5. Assays for NF-κB Activation
4.6. Real-Time Quantitative Reverse Transcription-PCR (qRT-PCR)
4.7. Experimental Scheme of High Fat Diet (HFD)-Fed Mice
4.8. Metabolic Phenotype Measurements
4.9. Biochemical Analysis
4.10. Serum TNF-α and MCP-1 Protein Levels
4.11. Preparation of Stromal Vascular Cells (SVCs)
4.12. Flow Cytometric Analysis of Blood Immune Cells and Adipose Tissue Macrophages (ATMs)
4.13. Histological Analysis of the Liver and Epididymal Fat Pad
4.14. Brain Tissue Preparation and Immunohistochemical Staining
4.15. Statistical Analysis
5. Conclusions
6. Patents
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Gao, H.M.; Hong, J.S. Why neurodegenerative diseases are progressive: Uncontrolled inflammation drives disease progression. Trends Immunol. 2008, 29, 357–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baquero, M.; Martín, N. Depressive symptoms in neurodegenerative diseases. World J. Clin. Cases 2015, 3, 682–693. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Guzman-Martinez, L.; Maccioni, R.B.; Andrade, V.; Navarrete, L.P.; Pastor, M.G.; Ramos-Escobar, N. Neuroinflammation as a Common Feature of Neurodegenerative Disorders. Front. Pharmacol. 2019, 10, 1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kölliker-Frers, R.; Udovin, L.; Otero-Losada, M.; Kobiec, T.; Herrera, M.I.; Palacios, J.; Razzitte, G.; Capani, F. Neuroinflammation: An Integrating Overview of Reactive-Neuroimmune Cell Interactions in Health and Disease. Mediat. Inflamm. 2021, 2021, 9999146. [Google Scholar] [CrossRef]
- Terrone, G.; Balosso, S.; Pauletti, A.; Ravizza, T.; Vezzani, A. Inflammation and reactive oxygen species as disease modifiers in epilepsy. Neuropharmacology 2020, 167, 107742. [Google Scholar] [CrossRef]
- Shih, R.H.; Wang, C.Y.; Yang, C.M. NF-kappaB Signaling Pathways in Neurological Inflammation: A Mini Review. Front. Mol. Neurosci. 2015, 8, 77. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Hussain, B.; Chang, J. Peripheral inflammation and blood-brain barrier disruption: Effects and mechanisms. CNS Neurosci. Ther. 2021, 27, 36–47. [Google Scholar] [CrossRef]
- Rhea, E.M.; Salameh, T.S.; Logsdon, A.F.; Hanson, A.J.; Erickson, M.A.; Banks, W.A. Blood-Brain Barriers in Obesity. AAPS J. 2017, 19, 921–930. [Google Scholar] [CrossRef]
- Saltiel, A.R.; Olefsky, J.M. Inflammatory mechanisms linking obesity and metabolic disease. J. Clin. Investig. 2017, 127, 1–4. [Google Scholar] [CrossRef]
- Guillemot-Legris, O.; Muccioli, G.G. Obesity-Induced Neuroinflammation: Beyond the Hypothalamus. Trends Neurosci. 2017, 40, 237–253. [Google Scholar] [CrossRef] [PubMed]
- Ye, J. Emerging role of adipose tissue hypoxia in obesity and insulin resistance. Int. J. Obes. 2009, 33, 54–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castanon, N.; Luheshi, G.; Layé, S. Role of neuroinflammation in the emotional and cognitive alterations displayed by animal models of obesity. Front. Neurosci. 2015, 9, 229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tucsek, Z.; Toth, P.; Tarantini, S.; Sosnowska, D.; Gautam, T.; Warrington, J.P.; Giles, C.B.; Wren, J.D.; Koller, A.; Ballabh, P.; et al. Aging exacerbates obesity-induced cerebromicrovascular rarefaction, neurovascular uncoupling, and cognitive decline in mice. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2014, 69, 1339–1352. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.F.A.; Van Winkle, L.S.; Esser, C.; Haarmann-Stemmann, T. The aryl hydrocarbon receptor as a target of environmental stressors-Implications for pollution mediated stress and inflammatory responses. Redox Biol. 2020, 34, 101530. [Google Scholar] [CrossRef]
- Favennec, M.; Hennart, B.; Caiazzo, R.; Leloire, A.; Yengo, L.; Verbanck, M.; Arredouani, A.; Marre, M.; Pigeyre, M.; Bessede, A.; et al. The kynurenine pathway is activated in human obesity and shifted toward kynurenine monooxygenase activation. Obesity 2015, 23, 2066–2074. [Google Scholar] [CrossRef]
- Wincent, E.; Bengtsson, J.; Bardbori, A.M.; Alsberg, T.; Luecke, S.; Rannug, U.; Rannug, A. Inhibition of cytochrome P4501-dependent clearance of the endogenous agonist FICZ as a mechanism for activation of the aryl hydrocarbon receptor. Proc. Natl. Acad. Sci. USA 2012, 109, 4479–4484. [Google Scholar] [CrossRef] [Green Version]
- McMillan, B.J.; Bradfield, C.A. The aryl hydrocarbon receptor is activated by modified low-density lipoprotein. Proc. Natl. Acad. Sci. USA 2007, 104, 1412–1417. [Google Scholar] [CrossRef] [Green Version]
- Kerley-Hamilton, J.S.; Trask, H.W.; Ridley, C.J.; Dufour, E.; Ringelberg, C.S.; Nurinova, N.; Wong, D.; Moodie, K.L.; Shipman, S.L.; Moore, J.H.; et al. Obesity is mediated by differential aryl hydrocarbon receptor signaling in mice fed a Western diet. Environ. Health Perspect. 2012, 120, 1252–1259. [Google Scholar] [CrossRef] [Green Version]
- Joisten, N.; Ruas, J.L.; Braidy, N.; Guillemin, G.J.; Zimmer, P. The kynurenine pathway in chronic diseases: A compensatory mechanism or a driving force? Trends Mol. Med. 2021, 27, 946–954. [Google Scholar] [CrossRef]
- Tanaka, M.; Fujikawa, M.; Oguro, A.; Itoh, K.; Vogel, C.F.A.; Ishihara, Y. Involvement of the Microglial Aryl Hydrocarbon Receptor in Neuroinflammation and Vasogenic Edema after Ischemic Stroke. Cells 2021, 10, 718. [Google Scholar] [CrossRef] [PubMed]
- Neavin, D.R.; Liu, D.; Ray, B.; Weinshilboum, R.M. The Role of the Aryl Hydrocarbon Receptor (AHR) in Immune and Inflammatory Diseases. Int. J. Mol. Sci. 2018, 19, 3851. [Google Scholar] [CrossRef] [Green Version]
- Rothhammer, V.; Quintana, F.J. The aryl hydrocarbon receptor: An environmental sensor integrating immune responses in health and disease. Nat. Rev. Immunol. 2019, 19, 184–197. [Google Scholar] [CrossRef] [PubMed]
- Andreeva-Gateva, P.; Bakalov, D.; Sabit, Z.; Tafradjiiska-Hadjiolova, R. Aryl hydrocarbon receptors as potential therapeutic targets. Pharmacia 2020, 67, 311–315. [Google Scholar] [CrossRef]
- Lee, H.K.; Park, W.H.; Kang, Y.C.; Kang, S.; Im, S.; Park, S.; Kim, J.T.; Lee, M.; Seok, J.; Oh, M.S.; et al. Serum biomarkers from cell-based assays for AhRL and MIS strongly predicted the future development of diabetes in a large community-based prospective study in Korea. Sci. Rep. 2020, 10, 6339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pak, Y.K.; Choi, H.S.; Park, W.H.; Im, S.; Lind, P.M.; Lind, L.; Lee, H.K. High Serum-Induced AhRL Is Associated with Prevalent Metabolic Syndrome and Future Impairment of Glucose Tolerance in the Elderly. Endocrinol. Metab. 2021, 36, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Degroot, D.E.; Hayashi, A.; He, G.; Denison, M.S. CH223191 is a ligand-selective antagonist of the Ah (Dioxin) receptor. Toxicol. Sci. Off. J. Soc. Toxicol. 2010, 117, 393–403. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Ishihara, Y.; Kado, S.Y.; Hoeper, C.; Harel, S.; Vogel, C.F.A. Role of NF-kB RelB in Aryl Hydrocarbon Receptor-Mediated Ligand Specific Effects. Int. J. Mol. Sci. 2019, 20, 2652. [Google Scholar] [CrossRef] [Green Version]
- Vogel, C.F.; Matsumura, F. A new cross-talk between the aryl hydrocarbon receptor and RelB, a member of the NF-kappaB family. Biochem. Pharmacol. 2009, 77, 734–745. [Google Scholar] [CrossRef]
- Kang, S.; Piao, Y.; Kang, Y.C.; Lim, S.; Pak, Y.K. DA-9805 protects dopaminergic neurons from endoplasmic reticulum stress and inflammation. Biomed. Pharmacother. 2022, 145, 112389. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Xu, X.; Zheng, L.; Mo, J.; Jin, X.; Bao, Y. Nilotinib inhibits microglia-mediated neuroinflammation to protect against dopaminergic neuronal death in Parkinson’s disease models. Int. Immunopharmacol. 2021, 99, 108025. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Piao, Y.; Kang, Y.C.; Lim, S.; Pak, Y.K. Qi-activating quercetin alleviates mitochondrial dysfunction and neuroinflammation in vivo and in vitro. Arch. Pharmacal Res. 2020, 43, 553–566. [Google Scholar] [CrossRef] [PubMed]
- Park, W.H.; Kang, S.; Piao, Y.; Pak, C.J.; Oh, M.S.; Kim, J.; Kang, M.S.; Pak, Y.K. Ethanol. extract of Bupleurum falcatum and saikosaponins inhibit neuroinflammation via inhibition of NF-kappaB. J. Ethnopharmacol. 2015, 174, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.G.; Kang, S.; Im, S.; Pak, Y.K. Cinnamic Acid Attenuates Peripheral and Hypothalamic Inflammation in High-Fat Diet-Induced Obese Mice. Pharmaceutics 2022, 14, 1675. [Google Scholar] [CrossRef]
- Surmi, B.K.; Hasty, A.H. Macrophage infiltration into adipose tissue: Initiation, propagation and remodeling. Future Lipidol. 2008, 3, 545–556. [Google Scholar] [CrossRef] [Green Version]
- Cuartero, M.I.; Ballesteros, I.; de la Parra, J.; Harkin, A.L.; Abautret-Daly, A.; Sherwin, E.; Fernandez-Salguero, P.; Corbi, A.L.; Lizasoain, I.; Moro, M.A. L-kynurenine/aryl hydrocarbon receptor pathway mediates brain damage after experimental stroke. Circulation 2014, 130, 2040–2051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaiser, H.; Parker, E.; Hamrick, M.W. Kynurenine signaling through the aryl hydrocarbon receptor: Implications for aging and healthspan. Exp. Gerontol. 2020, 130, 110797. [Google Scholar] [CrossRef]
- Schwarcz, R.; Bruno, J.P.; Muchowski, P.J.; Wu, H.Q. Kynurenines in the mammalian brain: When physiology meets pathology. Nat. Rev. Neurosci. 2012, 13, 465–477. [Google Scholar] [CrossRef]
- Kawano, Y.; Nishiumi, S.; Tanaka, S.; Nobutani, K.; Miki, A.; Yano, Y.; Seo, Y.; Kutsumi, H.; Ashida, H.; Azuma, T.; et al. Activation of the aryl hydrocarbon receptor induces hepatic steatosis via the upregulation of fatty acid transport. Arch. Biochem. Biophys. 2010, 504, 221–227. [Google Scholar] [CrossRef]
- Roh, E.; Kwak, S.H.; Jung, H.S.; Cho, Y.M.; Pak, Y.K.; Park, K.S.; Kim, S.Y.; Lee, H.K. Serum aryl hydrocarbon receptor ligand activity is associated with insulin resistance and resulting type 2 diabetes. Acta Diabetol. 2015, 52, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Chon, S.; Park, S.Y.; Yun, S.; Baik, S.H.; Woo, J.T.; Rhee, S.Y.; Pak, Y.K.; Kim, S.H. Association of aryl hydrocarbon receptor transactivating activity, a potential biomarker for persistent organic pollutants, with the risk of gestational diabetes mellitus. Sci. Rep. 2021, 11, 3185. [Google Scholar] [CrossRef] [PubMed]
- Park, W.H.; Kang, S.; Lee, H.K.; Salihovic, S.; Bavel, B.V.; Lind, P.M.; Pak, Y.K.; Lind, L. Relationships between serum-induced AhR bioactivity or mitochondrial inhibition and circulating polychlorinated biphenyls (PCBs). Sci. Rep. 2017, 7, 9383. [Google Scholar] [CrossRef] [Green Version]
- Opitz, C.A.; Litzenburger, U.M.; Sahm, F.; Ott, M.; Tritschler, I.; Trump, S.; Schumacher, T.; Jestaedt, L.; Schrenk, D.; Weller, M.; et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 2011, 478, 197–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L. AHR Function in Lymphocytes: Emerging Concepts. Trends Immunol. 2016, 37, 17–31. [Google Scholar] [CrossRef] [Green Version]
- Gutiérrez-Vázquez, C.; Quintana, F.J. Regulation of the Immune Response by the Aryl Hydrocarbon Receptor. Immunity 2018, 48, 19–33. [Google Scholar] [CrossRef] [Green Version]
- Ishihara, Y.; Kado, S.Y.; Bein, K.J.; He, Y.; Pouraryan, A.A.; Urban, A.; Haarmann-Stemmann, T.; Sweeney, C.; Vogel, C.F.A. Aryl Hydrocarbon Receptor Signaling Synergizes with TLR/NF-κB-Signaling for Induction of IL-22 Through Canonical and Non-Canonical AhR Pathways. Front. Toxicol. 2022, 3, 787360. [Google Scholar] [CrossRef]
- La Merrill, M.; Kuruvilla, B.S.; Pomp, D.; Birnbaum, L.S.; Threadgill, D.W. Dietary fat alters body composition, mammary development, and cytochrome p450 induction after maternal TCDD exposure in DBA/2J mice with low-responsive aryl hydrocarbon receptors. Environ. Health Perspect. 2009, 117, 1414–1419. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Ballantyne, C.M. Metabolic Inflammation and Insulin Resistance in Obesity. Circ. Res. 2020, 126, 1549–1564. [Google Scholar] [CrossRef]
- Lee, B.C.; Lee, J. Cellular and molecular players in adipose tissue inflammation in the development of obesity-induced insulin resistance. Biochim. Et Biophys. Acta 2014, 1842, 446–462. [Google Scholar] [CrossRef]
- Freeman, A.M.; Pennings, N. Insulin Resistance. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Samuel, V.T.; Shulman, G.I. The pathogenesis of insulin resistance: Integrating signaling pathways and substrate flux. J. Clin. Investig. 2016, 126, 12–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honka, M.J.; Latva-Rasku, A.; Bucci, M.; Virtanen, K.A.; Hannukainen, J.C.; Kalliokoski, K.K.; Nuutila, P. Insulin-stimulated glucose uptake in skeletal muscle, adipose tissue and liver: A positron emission tomography study. Eur. J. Endocrinol. 2018, 178, 523–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trouwborst, I.; Bowser, S.M.; Goossens, G.H.; Blaak, E.E. Ectopic Fat Accumulation in Distinct Insulin Resistant Phenotypes; Targets for Personalized Nutritional Interventions. Front. Nutr. 2018, 5, 77. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.X.; Wang, C.; Zhang, Z.M.; Jaeger, C.D.; Krager, S.L.; Bottum, K.M.; Liu, J.; Liao, D.F.; Tischkau, S.A. Aryl hydrocarbon receptor deficiency protects mice from diet-induced adiposity and metabolic disorders through increased energy expenditure. Int. J. Obes. 2015, 39, 1300–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Sundaram, K.; Mu, J.; Dryden, G.W.; Sriwastva, M.K.; Lei, C.; Zhang, L.; Qiu, X.; Xu, F.; Yan, J.; et al. High-fat diet-induced upregulation of exosomal phosphatidylcholine contributes to insulin resistance. Nat. Commun. 2021, 12, 213. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Tripathy, D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 2009, 32 (Suppl. 2), S157–S163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swirski, F.K.; Libby, P.; Aikawa, E.; Alcaide, P.; Luscinskas, F.W.; Weissleder, R.; Pittet, M.J. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J. Clin. Investig. 2007, 117, 195–205. [Google Scholar] [CrossRef] [Green Version]
- Mills, C.D. M1 and M2 Macrophages: Oracles of Health and Disease. Crit. Rev. Immunol. 2012, 32, 463–488. [Google Scholar] [CrossRef] [Green Version]
- Covarrubias, A.; Byles, V.; Horng, T. ROS sets the stage for macrophage differentiation. Cell Res. 2013, 23, 984–985. [Google Scholar] [CrossRef] [Green Version]
- Taki, Y.; Kinomura, S.; Sato, K.; Inoue, K.; Goto, R.; Okada, K.; Uchida, S.; Kawashima, R.; Fukuda, H. Relationship between body mass index and gray matter volume in 1428 healthy individuals. Obes. Silver Spring Md. 2008, 16, 119–124. [Google Scholar] [CrossRef]
- Cai, D.; Liu, T. Inflammatory cause of metabolic syndrome via brain stress and NF-κB. Aging 2012, 4, 98–115. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef] [PubMed]
- Moyse, E.; Krantic, S.; Djellouli, N.; Roger, S.; Angoulvant, D.; Debacq, C.; Leroy, V.; Fougere, B.; Aidoud, A. Neuroinflammation: A Possible Link Between Chronic Vascular Disorders and Neurodegenerative Diseases. Front. Aging Neurosci. 2022, 14, 827263. [Google Scholar] [CrossRef] [PubMed]
- Jais, A.; Brüning, J.C. Hypothalamic inflammation in obesity and metabolic disease. J. Clin. Investig. 2017, 127, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Dionysopoulou, S.; Charmandari, E.; Bargiota, A.; Vlahos, N.; Mastorakos, G.; Valsamakis, G. The Role of Hypothalamic Inflammation in Diet-Induced Obesity and Its Association with Cognitive and Mood Disorders. Nutrients 2021, 13, 498. [Google Scholar] [CrossRef]
- Jeong, J.S.; Piao, Y.; Kang, S.; Son, M.; Kang, Y.C.; Du, X.F.; Ryu, J.; Cho, Y.W.; Jiang, H.H.; Oh, M.S.; et al. Triple herbal extract DA-9805 exerts a neuroprotective effect via amelioration of mitochondrial damage in experimental models of Parkinson’s disease. Sci. Rep. 2018, 8, 15953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noh, J.W.; Kwon, O.J.; Lee, B.C. The Immunomodulating Effect of Baicalin on Inflammation and Insulin Resistance in High-Fat-Diet-Induced Obese Mice. Evid. Based Complement. Altern. Med. 2021, 2021, 5531367. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, S.; Lee, A.G.; Im, S.; Oh, S.J.; Yoon, H.J.; Park, J.H.; Pak, Y.K. A Novel Aryl Hydrocarbon Receptor Antagonist HBU651 Ameliorates Peripheral and Hypothalamic Inflammation in High-Fat Diet-Induced Obese Mice. Int. J. Mol. Sci. 2022, 23, 14871. https://doi.org/10.3390/ijms232314871
Kang S, Lee AG, Im S, Oh SJ, Yoon HJ, Park JH, Pak YK. A Novel Aryl Hydrocarbon Receptor Antagonist HBU651 Ameliorates Peripheral and Hypothalamic Inflammation in High-Fat Diet-Induced Obese Mice. International Journal of Molecular Sciences. 2022; 23(23):14871. https://doi.org/10.3390/ijms232314871
Chicago/Turabian StyleKang, Sora, Aden Geonhee Lee, Suyeol Im, Seung Jun Oh, Hye Ji Yoon, Jeong Ho Park, and Youngmi Kim Pak. 2022. "A Novel Aryl Hydrocarbon Receptor Antagonist HBU651 Ameliorates Peripheral and Hypothalamic Inflammation in High-Fat Diet-Induced Obese Mice" International Journal of Molecular Sciences 23, no. 23: 14871. https://doi.org/10.3390/ijms232314871
APA StyleKang, S., Lee, A. G., Im, S., Oh, S. J., Yoon, H. J., Park, J. H., & Pak, Y. K. (2022). A Novel Aryl Hydrocarbon Receptor Antagonist HBU651 Ameliorates Peripheral and Hypothalamic Inflammation in High-Fat Diet-Induced Obese Mice. International Journal of Molecular Sciences, 23(23), 14871. https://doi.org/10.3390/ijms232314871