
Cigarette Smoke Extract Induces p38 MAPK-Initiated, Fas-Mediated Eryptosis

 , , , , and
, , , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
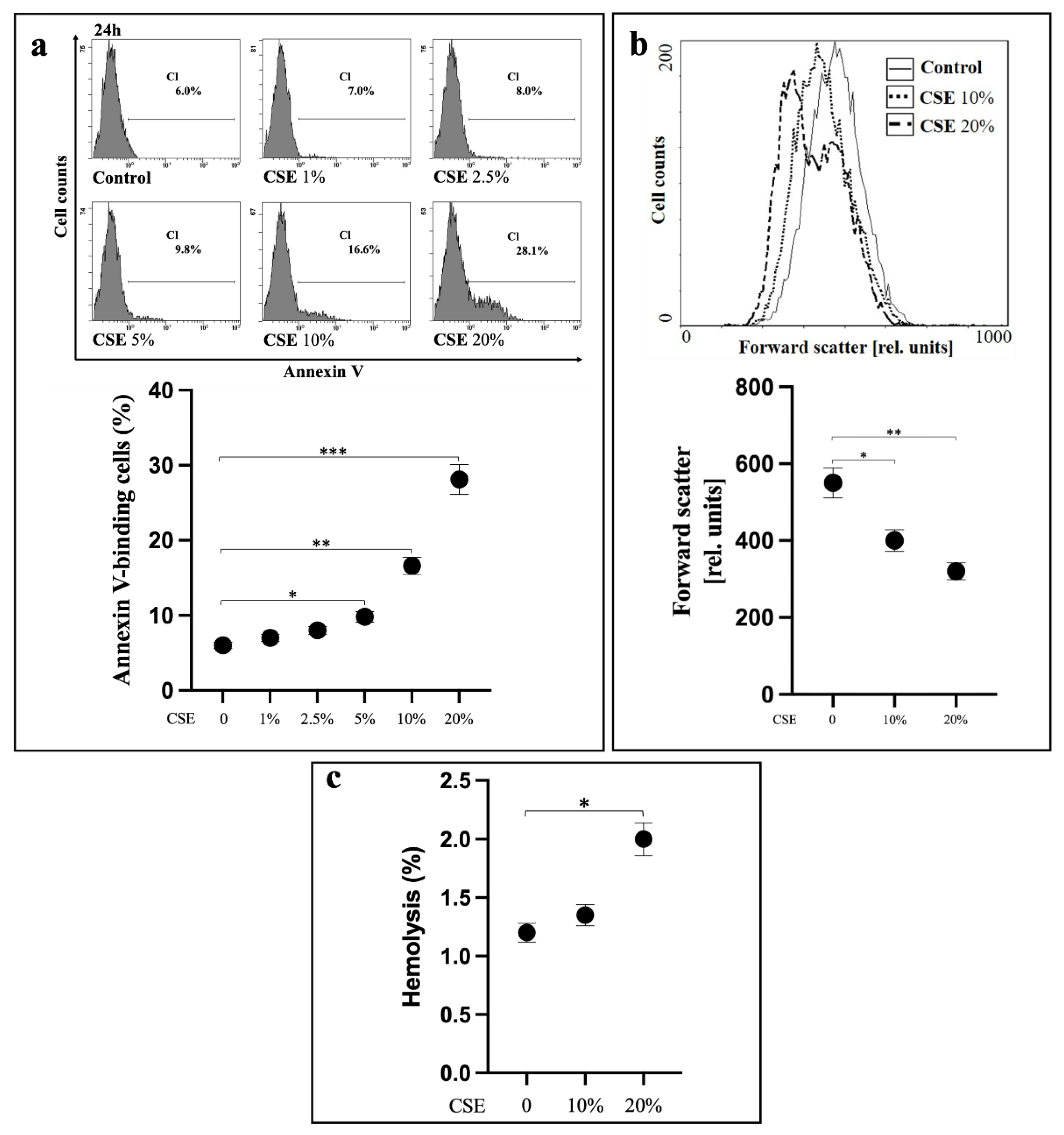
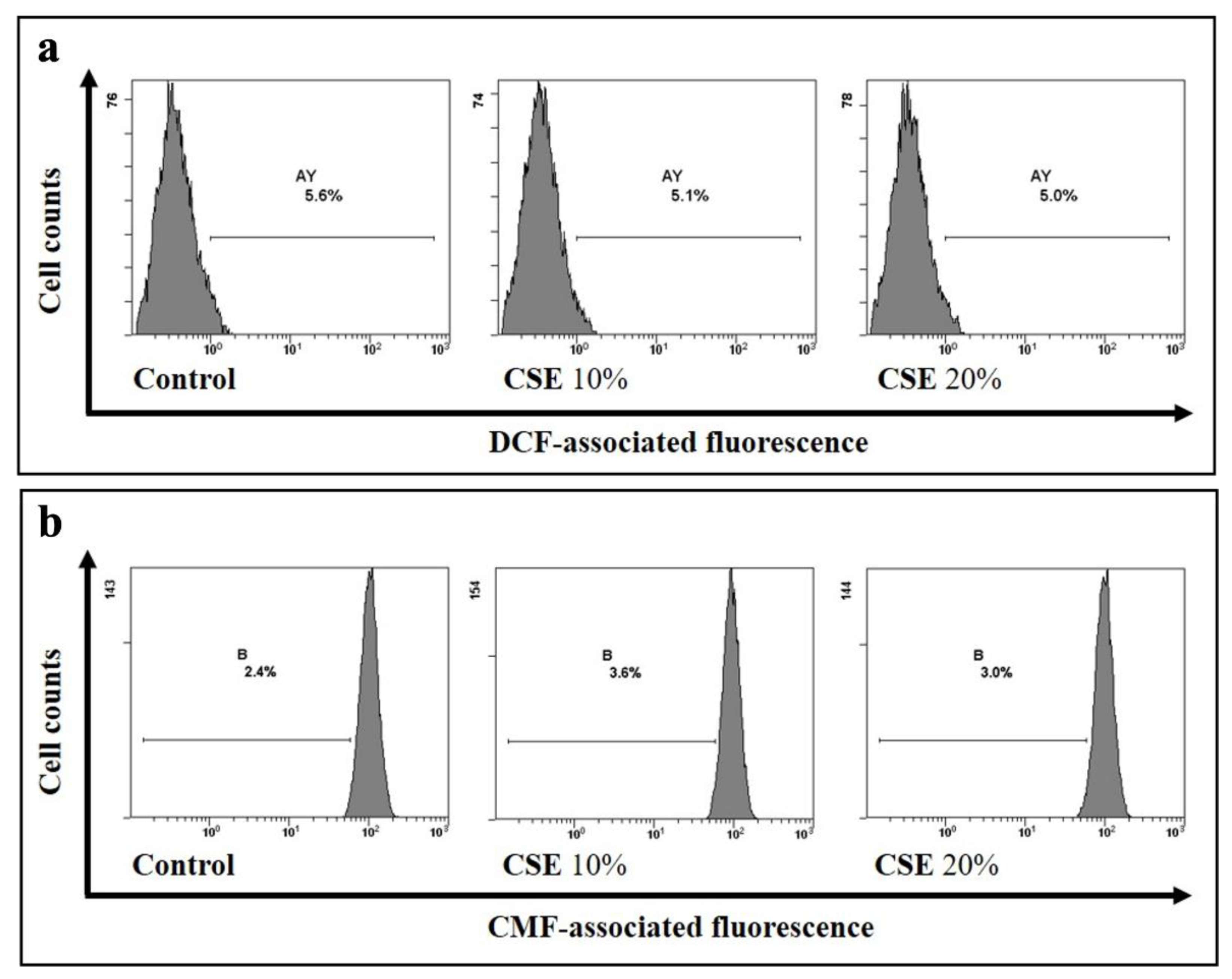
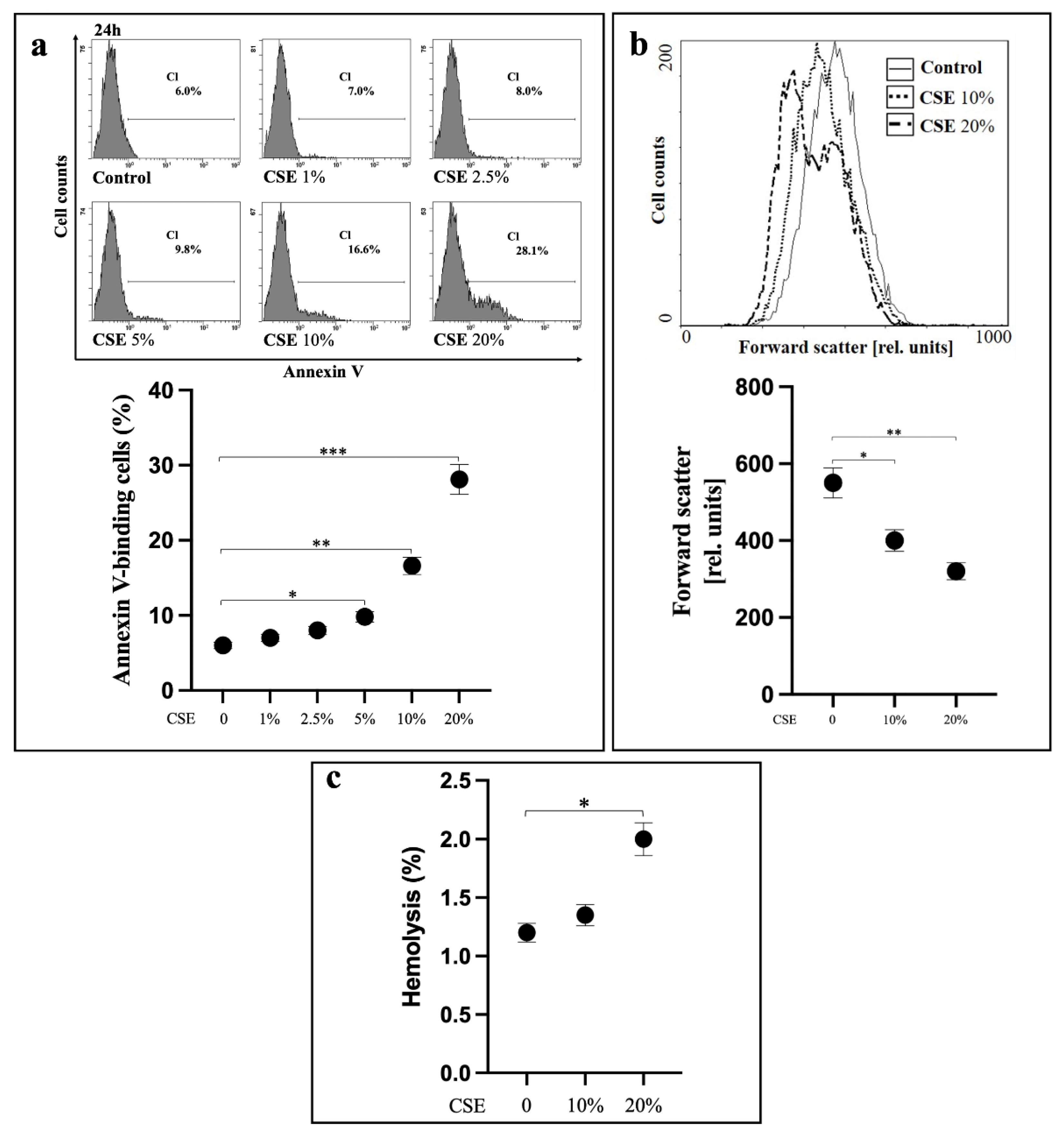
2.1. CSE Induces Oxidative Stress-Independent Eryptosis
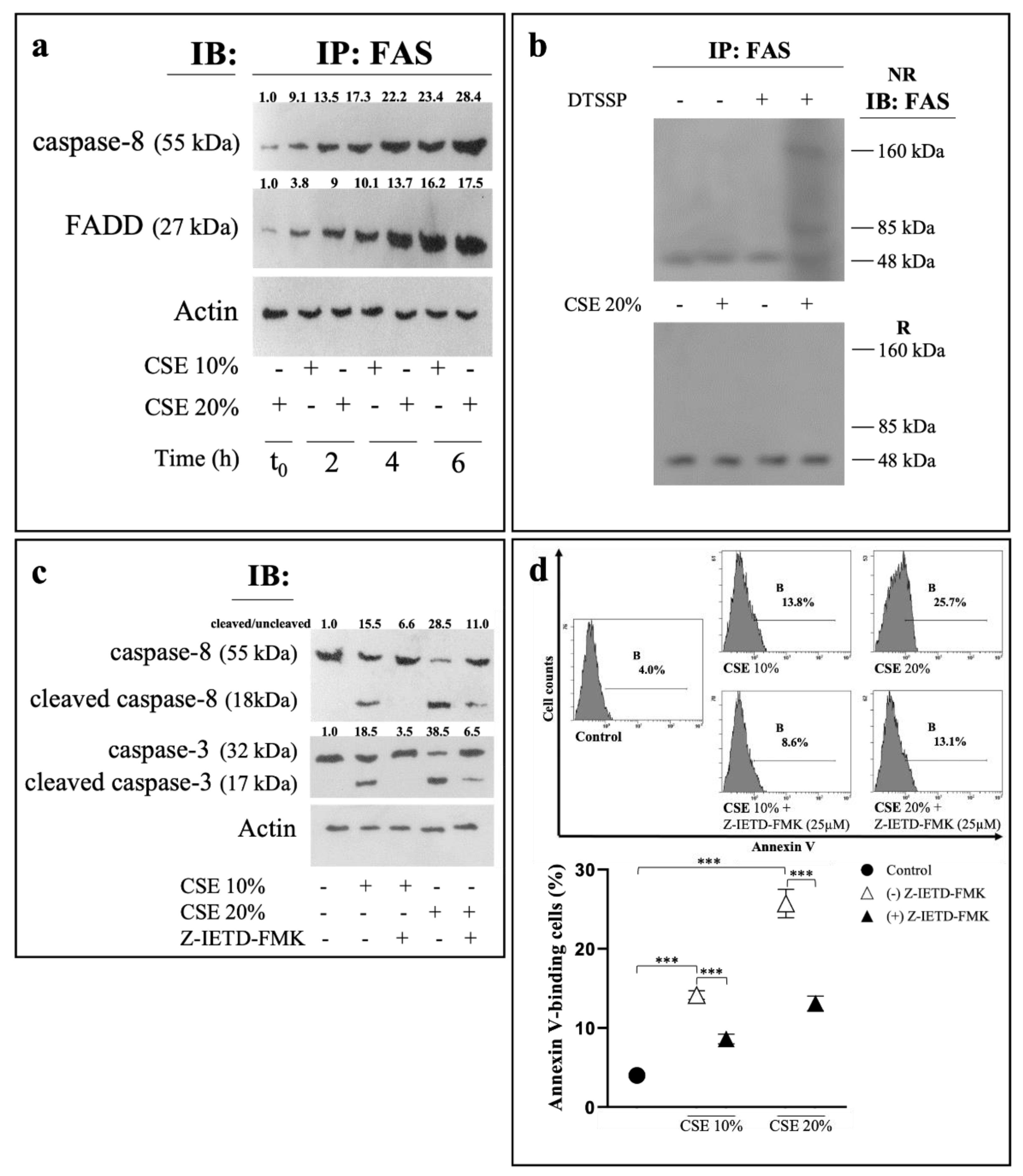
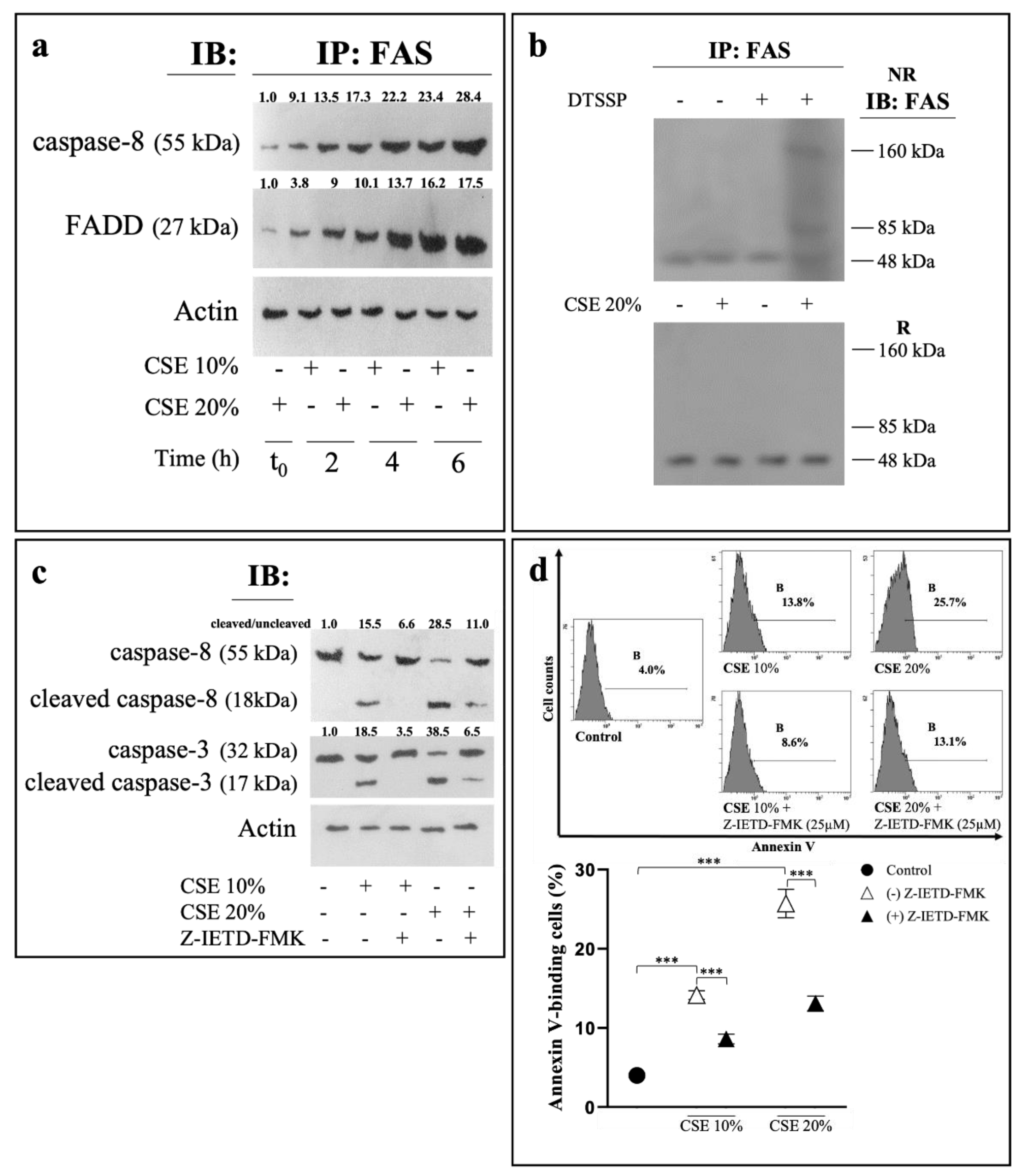
2.2. CSE Induces Eryptosis through Extrinsic Apoptotic Pathway
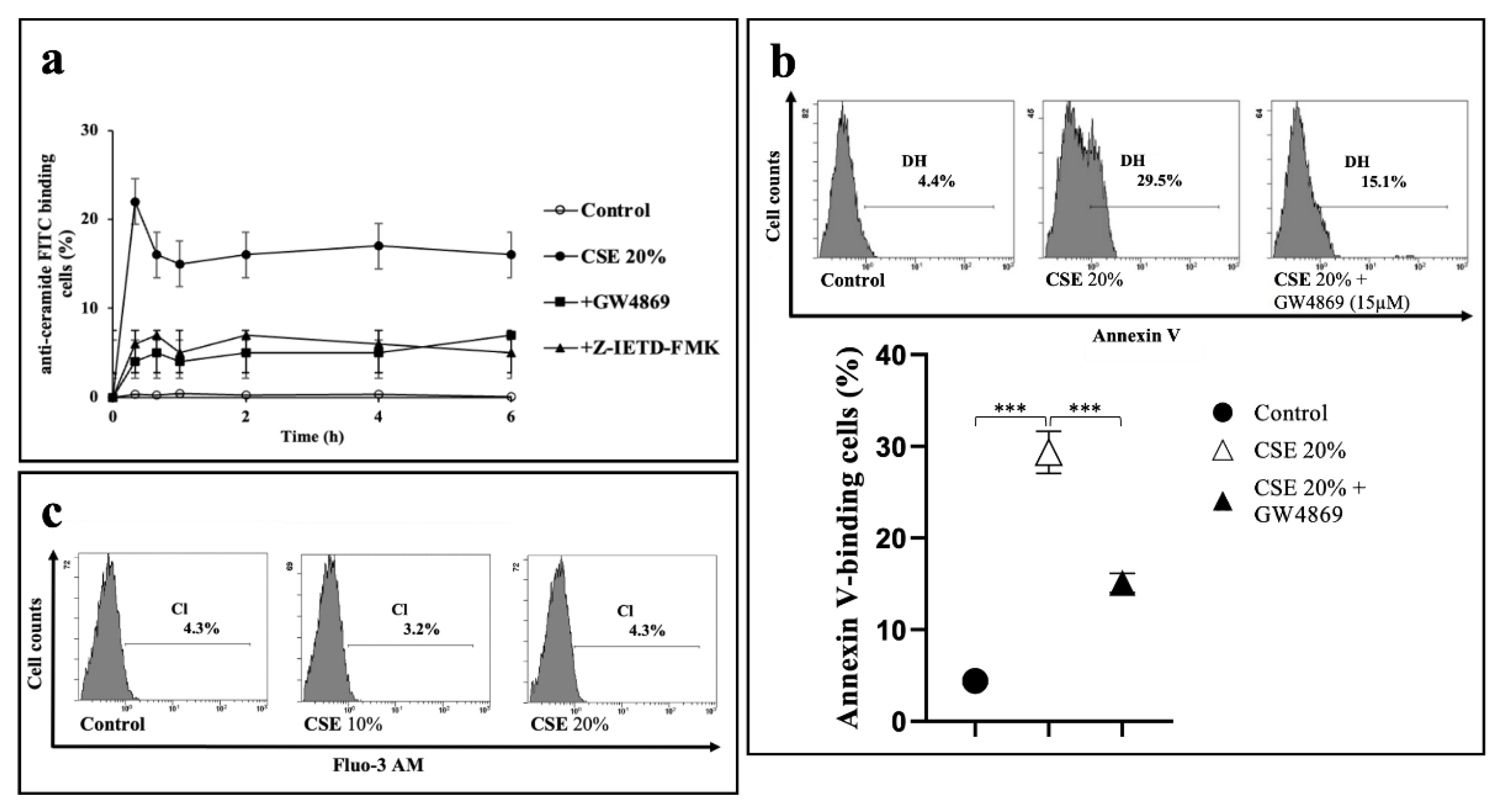
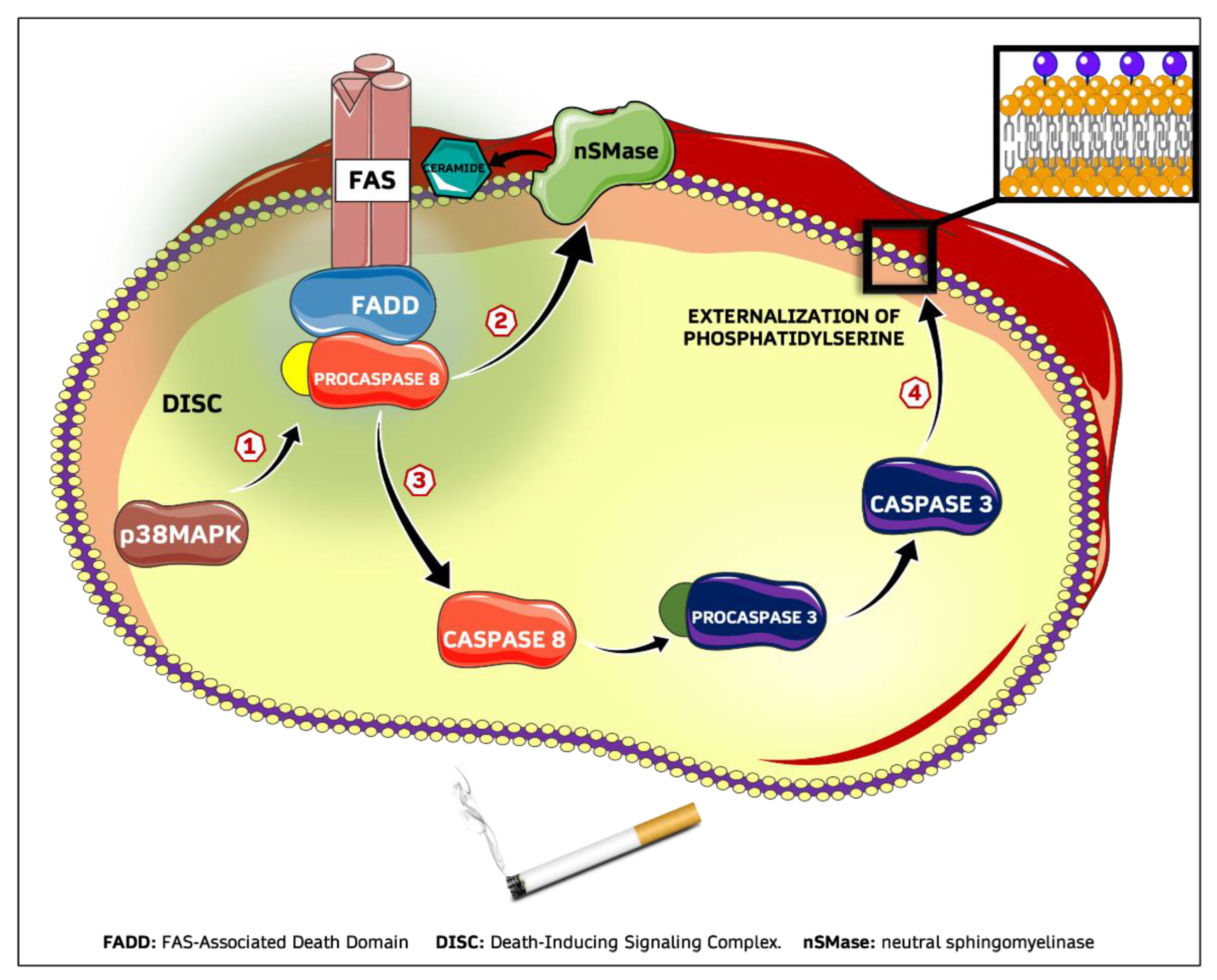
2.3. CSE-Induced Eryptosis Requires Caspase-8-Mediated Ceramide Formation
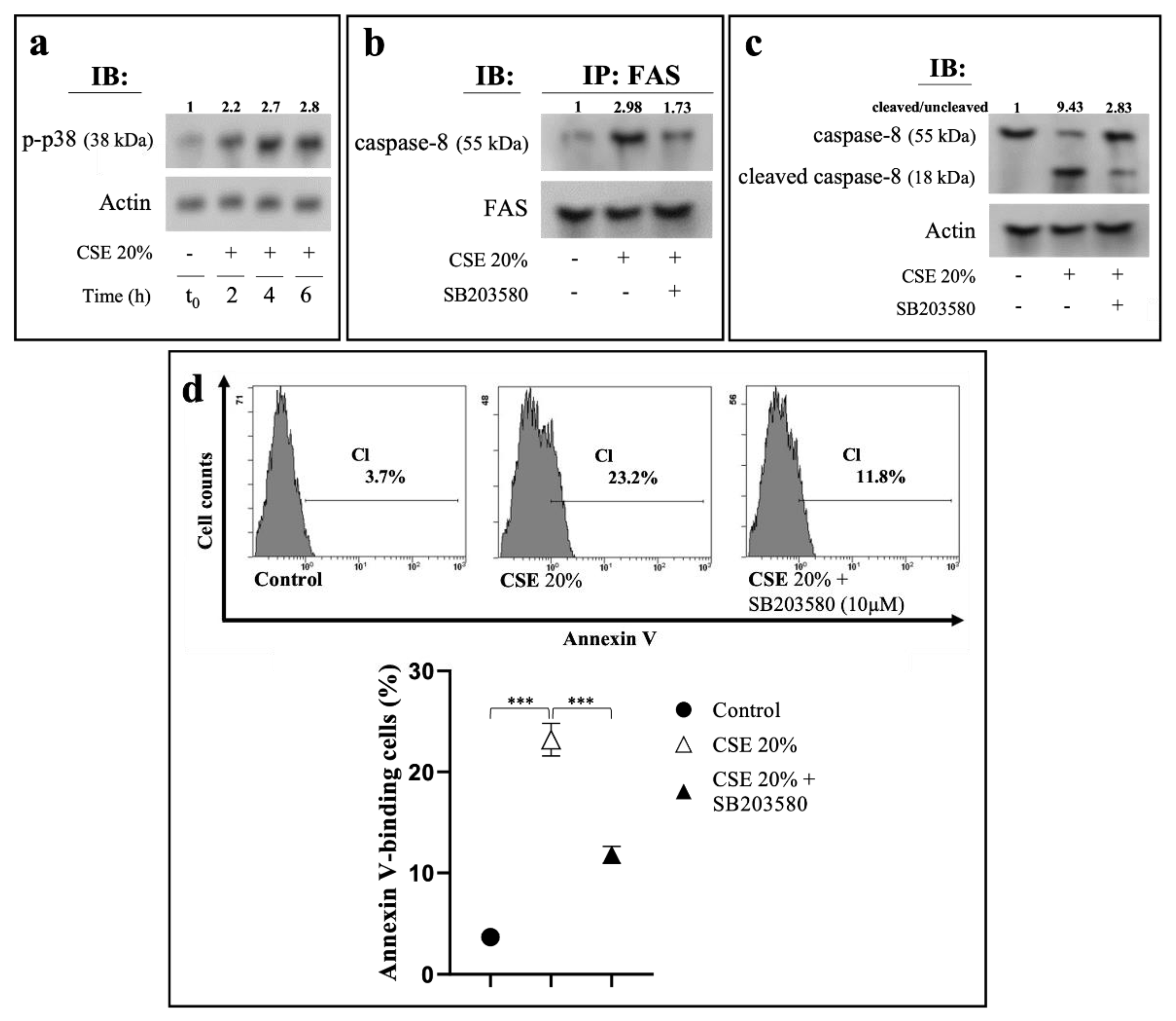
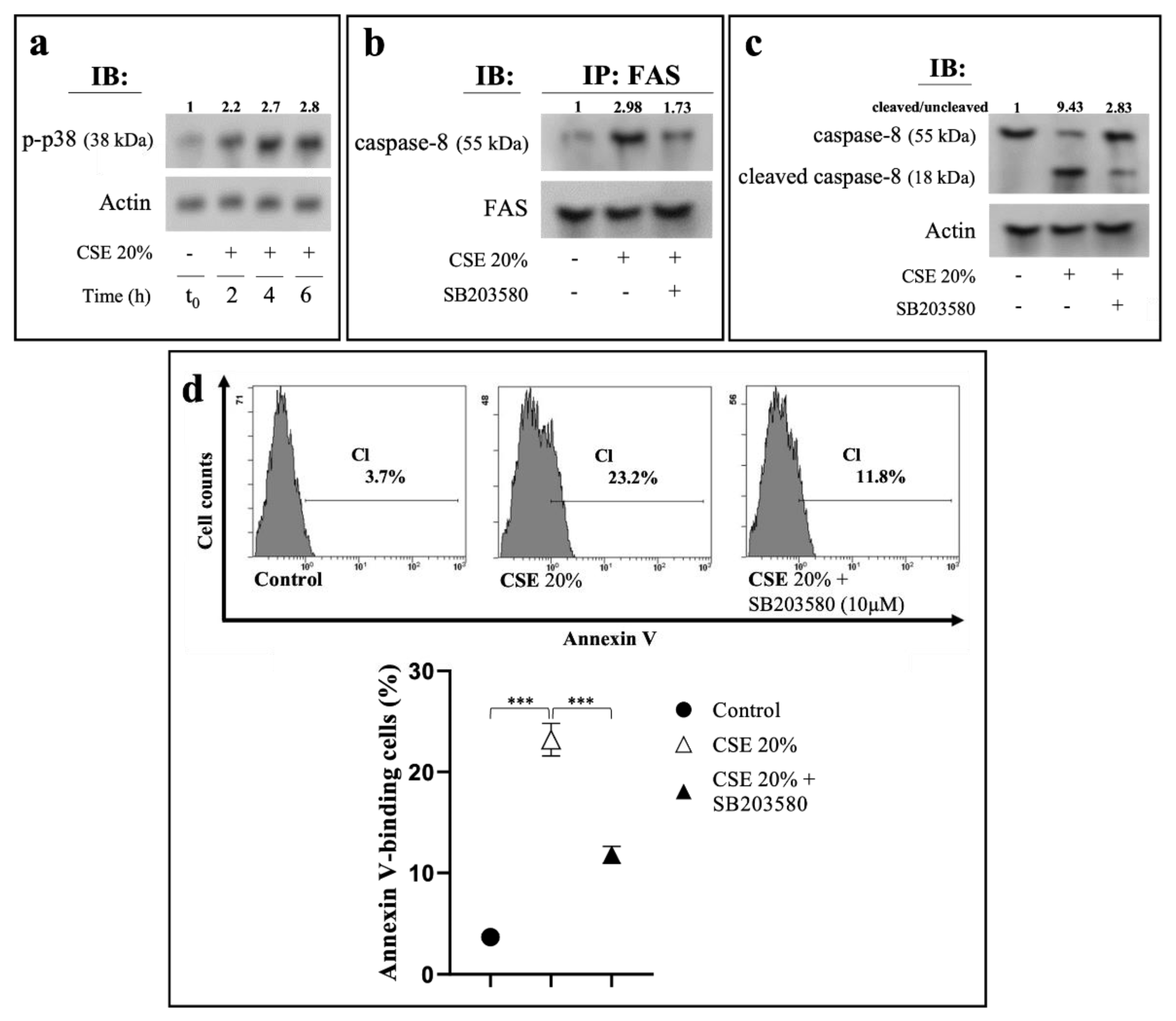
2.4. CSE-Induced Extrinsic Eryptosis Is Initiated via p38 MAPK
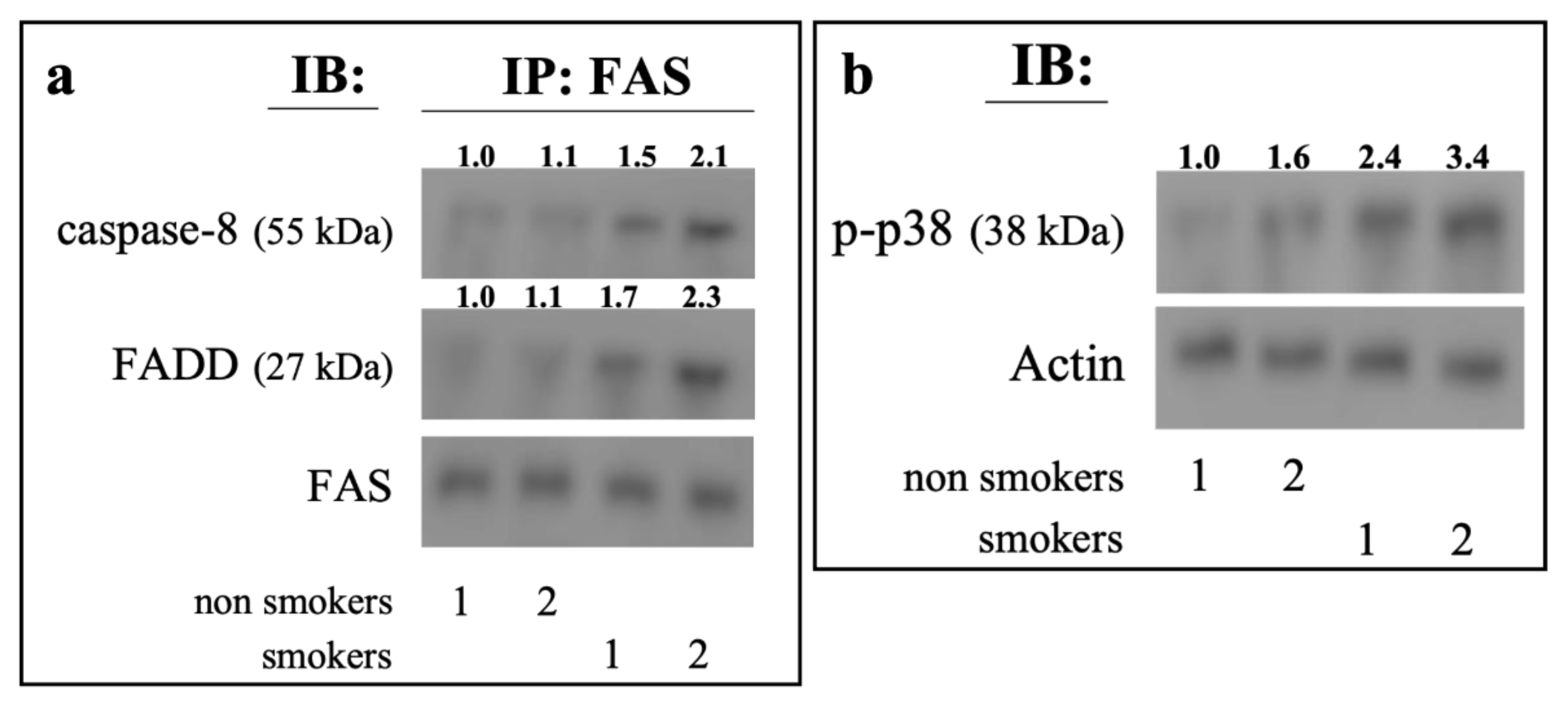
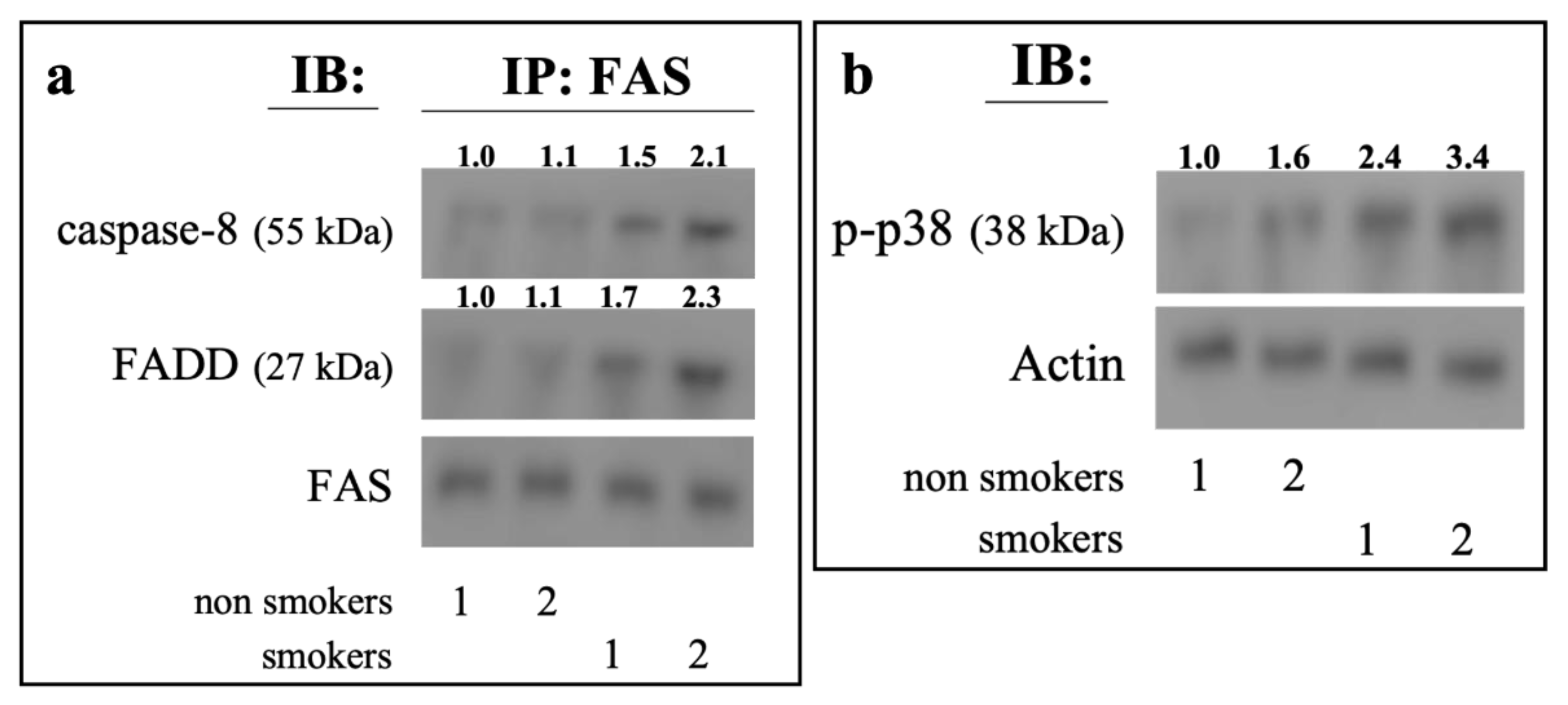
2.5. Ex Vivo Analysis of Erythrocytes from Smokers
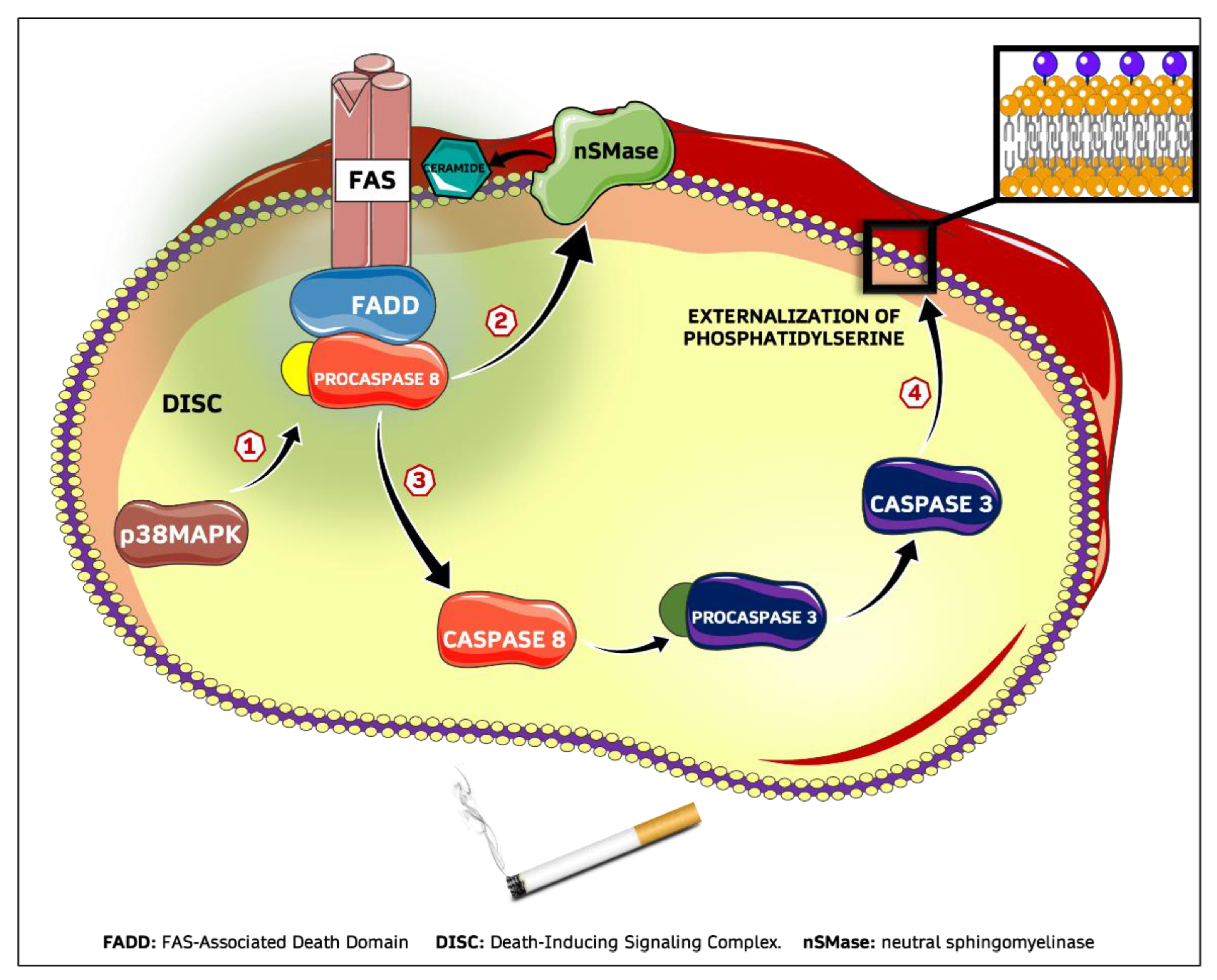
3. Discussion
Study Limitations
4. Materials and Methods
4.1. Preparation of CSE
4.2. RBCs and Treatment
4.3. Haemolysis
4.4. Flow Cytometry
4.4.1. Measurement of PS Externalization and FSC
4.4.2. Measurement of Intracellular ROS
4.4.3. Measurement of Intracellular GSH
4.4.4. Measurement of Intracellular Calcium
4.4.5. Measurement of Ceramide
4.5. Immunoprecipitation
4.6. Western Blotting
4.7. Cross-Linking Reaction by (3,3′-Dithiobis (Sulfosuccinimidyl Propionate)) DTSSP
4.8. Ex Vivo Analysis
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Restivo, I.; Attanzio, A.; Tesoriere, L.; Allegra, M.; Garcia-Llatas, G.; Cilla, A. Anti-Eryptotic Activity of Food-Derived Phytochemicals and Natural Compounds. Int. J. Mol. Sci. 2022, 23, 3019. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.; Qadri, S.M. Mechanisms and Significance of Eryptosis, the Suicidal Death of Erythrocytes. Blood Purif. 2012, 33, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Qadri, S.M.; Bissinger, R.; Solh, Z.; Oldenborg, P.-A. Eryptosis in health and disease: A paradigm shift towards understanding the (patho)physiological implications of programmed cell death of erythrocytes. Blood Rev. 2017, 31, 349–361. [Google Scholar] [CrossRef]
- Lang, F.; Bissinger, R.; Abed, M.; Artunc, F. Eryptosis–The Neglected Cause of Anemia in End Stage Renal Disease. Kidney Blood Press. Res. 2017, 42, 749–760. [Google Scholar] [CrossRef]
- Dias, G.F.; Grobe, N.; Rogg, S.; Jörg, D.J.; Pecoits-Filho, R.; Moreno-Amaral, A.N.; Kotanko, P. The Role of Eryptosis in the Pathogenesis of Renal Anemia: Insights from Basic Research and Mathematical Modeling. Front. Cell Dev. Biol. 2020, 8, 598148. [Google Scholar] [CrossRef] [PubMed]
- Restivo, I.; Attanzio, A.; Tesoriere, L.; Allegra, M. Suicidal Erythrocyte Death in Metabolic Syndrome. Antioxidants 2021, 10, 154. [Google Scholar] [CrossRef] [PubMed]
- Borst, O.; Abed, M.; Alesutan, I.; Towhid, S.T.; Qadri, S.M.; Föller, M.; Gawaz, M.; Lang, F. Dynamic adhesion of eryptotic erythrocytes to endothelial cells via CXCL16/SR-PSOX. Am. J. Physiol.-Cell Physiol. 2012, 302, C644–C651. [Google Scholar] [CrossRef] [Green Version]
- Abed, M.; Towhid, S.T.; Pakladok, T.; Alesutan, I.; Götz, F.; Gulbins, E.; Lang, F. Effect of bacterial peptidoglycan on erythrocyte death and adhesion to endothelial cells. Int. J. Med. Microbiol. 2013, 303, 182–189. [Google Scholar] [CrossRef]
- Walker, B.; Towhid, S.T.; Schmid, E.; Hoffmann, S.M.; Abed, M.; Münzer, P.; Vogel, S.; Neis, F.; Brucker, S.; Gawaz, M.; et al. Dynamic adhesion of eryptotic erythrocytes to immobilized platelets via platelet phosphatidylserine receptors. Am. J. Physiol.-Cell Physiol. 2014, 306, C291–C297. [Google Scholar] [CrossRef]
- Lang, E.; Qadri, S.M.; Lang, F. Killing Me Softly–Suicidal Erythrocyte Death. Int. J. Biochem. Cell Biol. 2012, 44, 1236–1243. [Google Scholar] [CrossRef]
- Lang, E.; Lang, F. Mechanisms and pathophysiological significance of eryptosis, the suicidal erythrocyte death. Semin. Cell Dev. Biol. 2015, 39, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Tesoriere, L.; Attanzio, A.; Allegra, M.; Cilla, A.; Gentile, C.; Livrea, M.A. Oxysterol Mixture in Hypercholesterolemia-Relevant Proportion Causes Oxidative Stress-Dependent Eryptosis. Cell. Physiol. Biochem. 2014, 34, 1075–1089. [Google Scholar] [CrossRef] [Green Version]
- Berg, C.P.; Engels, I.H.; Rothbart, A.; Lauber, K.; Renz, A.; Schlosser, S.F.; Schulze-Osthoff, K.; Wesselborg, S. Human mature red blood cells express caspase-3 and caspase-8, but are devoid of mitochondrial regulators of apoptosis. Cell Death Differ. 2001, 8, 1197–1206. [Google Scholar] [CrossRef]
- Mandal, D.; Mazumder, A.; Das, P.; Kundu, M.; Basu, J. Fas-, Caspase 8-, and Caspase 3-dependent Signaling Regulates the Activity of the Aminophospholipid Translocase and Phosphatidylserine Externalization in Human Erythrocytes. J. Biol. Chem. 2005, 280, 39460–39467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sagan, D.; Jermnim, N.; Tangvarasittichai, O. CD95 is not functional in human erythrocytes. Int. J. Lab. Hematol. 2010, 32, e244–e247. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.T.; Creager, M.A. The relationship of cigarette smoking to peripheral arterial disease. Rev. Cardiovasc. Med. 2004, 5, 189–193. [Google Scholar]
- Law, M.R.; Morris, J.; Wald, N.J. Environmental tobacco smoke exposure and ischaemic heart disease: An evaluation of the evidence. BMJ 1997, 315, 973–980. [Google Scholar] [CrossRef] [Green Version]
- Messner, B.; Bernhard, D. Smoking and Cardiovascular Disease: Mechanisms of Endothelial Dysfunction and Early Atherogenesis. Arter. Thromb. Vasc. Biol. 2014, 34, 509–515. [Google Scholar] [CrossRef] [Green Version]
- Shah, R.S.; Cole, J.W. Smoking and stroke: The more you smoke the more you stroke. Expert Rev. Cardiovasc. Ther. 2010, 8, 917–932. [Google Scholar] [CrossRef]
- Nowak, J.; Murray, J.J.; Oates, J.A.; FitzGerald, G.A. Biochemical evidence of a chronic abnormality in platelet and vascular function in healthy individuals who smoke cigarettes. Circulation 1987, 76, 6–14. [Google Scholar] [CrossRef] [Green Version]
- Blann, A.D.; McCollum, C.N. Adverse Influence of Cigarette Smoking on the Endothelium. Thromb. Haemost. 1993, 70, 707–711. [Google Scholar] [CrossRef] [PubMed]
- Kiowski, W.; Linder, L.; Stoschitzky, K.; Pfisterer, M.; Burckhardt, D.; Burkart, F.; Bühler, F.R. Diminished vascular response to inhibition of endothelium-derived nitric oxide and enhanced vasoconstriction to exogenously administered endothelin-1 in clinically healthy smokers. Circulation 1994, 90, 27–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Mahdy, M.A.; Abdelghany, T.M.; Hemann, C.; Ewees, M.G.; Mahgoup, E.M.; Eid, M.S.; Shalaan, M.T.; Alzarie, Y.A.; Zweier, J.L. Chronic cigarette smoke exposure triggers a vicious cycle of leukocyte and endothelial-mediated oxidant stress that results in vascular dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2020, 319, H51–H65. [Google Scholar] [CrossRef]
- Attanzio, A.; Frazzitta, A.; Vasto, S.; Tesoriere, L.; Pintaudi, A.M.; Livrea, M.A.; Cilla, A.; Allegra, M. Increased eryptosis in smokers is associated with the antioxidant status and C-reactive protein levels. Toxicology 2018, 411, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Pryor, W.A.; Stone, K. Oxidants in Cigarette Smoke Radicals, Hydrogen Peroxide, Peroxynitrate, and Peroxynitrite. Ann. N. Y. Acad. Sci. 1993, 686, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Stennicke, H.R.; Salvesen, G.S. Caspases–Controlling Intracellular Signals by Protease Zymogen Activation. Biochim. Et Biophys. Acta–Protein Struct. Mol. Enzymol. 2000, 1477, 299–306. [Google Scholar] [CrossRef]
- LaRocca, T.; Stivison, E.A.; Hod, E.A.; Spitalnik, S.; Cowan, P.; Randis, T.; Ratner, A.J. Human-Specific Bacterial Pore-Forming Toxins Induce Programmed Necrosis in Erythrocytes. mBio 2014, 5, e01251-14. [Google Scholar] [CrossRef] [Green Version]
- Lang, E.; Bissinger, R.; Gulbins, E.; Lang, F. Ceramide in the regulation of eryptosis, the suicidal erythrocyte death. Apoptosis 2015, 20, 758–767. [Google Scholar] [CrossRef]
- López, D.J.; Egido-Gabas, M.; López-Montero, I.; Busto, J.V.; Casas, J.; Garnier, M.; Monroy, F.; Larijani, B.; Goñi, F.M.; Alonso, A. Accumulated Bending Energy Elicits Neutral Sphingomyelinase Activity in Human Red Blood Cells. Biophys. J. 2012, 102, 2077–2085. [Google Scholar] [CrossRef] [Green Version]
- Gatidis, S.; Zelenak, C.; Fajol, A.; Lang, E.; Jilani, K.; Michael, D.; Qadri, S.M.; Lang, F. p38 MAPK Activation and Function following Osmotic Shock of Erythrocytes. Cell. Physiol. Biochem. 2011, 28, 1279–1286. [Google Scholar] [CrossRef]
- Vona, R.; Gambardella, L.; Ortona, E.; Santulli, M.; Malorni, W.; Carè, A.; Pietraforte, D.; Straface, E. Functional Estrogen Receptors of Red Blood Cells. Do They Influence Intracellular Signaling? Cell. Physiol. Biochem. 2019, 53, 186–199. [Google Scholar] [CrossRef]
- Aoshiba, K.; Tamaoki, J.; Nagai, A. Acute cigarette smoke exposure induces apoptosis of alveolar macrophages. Am. J. Physiol. Cell. Mol. Physiol. Lung 2001, 281, L1392–L1401. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, Y.; Mio, T.; Nagai, S.; Miki, H.; Ito, I.; Izumi, T. Cytotoxic effects of cigarette smoke extract on an alveolar type II cell-derived cell line. Am. J. Physiol. Cell. Mol. Physiol. 2001, 281, L509–L516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carnevali, S.; Petruzzelli, S.; Longoni, B.; Vanacore, R.; Barale, R.; Cipollini, M.; Scatena, F.; Paggiaro, P.; Celi, A.; Giuntini, C. Cigarette smoke extract induces oxidative stress and apoptosis in human lung fibroblasts. Am. J. Physiol. Cell. Mol. Physiol. Lung 2003, 284, L955–L963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Y.; Han, W.; Giraldo, C.; De Li, Y.; Block, E.R. Effect of Cigarette Smoke Extract on Nitric Oxide Synthase in Pulmonary Artery Endothelial Cells. Am. J. Respir. Cell Mol. Biol. 1998, 19, 819–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messner, B.; Frotschnig, S.; Steinacher-Nigisch, A.; Winter, B.; Eichmair, E.; Gebetsberger, J.; Schwaiger, S.; Ploner, C.; Laufer, G.; Bernhard, D. Apoptosis and necrosis: Two different outcomes of cigarette smoke condensate-induced endothelial cell death. Cell Death Dis. 2012, 3, e424. [Google Scholar] [CrossRef] [Green Version]
- Attanzio, A.; Frazzitta, A.; Cilla, A.; Livrea, M.A.; Tesoriere, L.; Allegra, M. 7-Keto-Cholesterol and Cholestan-3beta, 5alpha, 6beta-Triol Induce Eryptosis through Distinct Pathways Leading to NADPH Oxidase and Nitric Oxide Synthase Activation. Cell. Physiol. Biochem. 2019, 53, 933–947. [Google Scholar] [CrossRef] [Green Version]
- Park, J.-W.; Kim, H.P.; Lee, S.-J.; Wang, X.; Wang, Y.; Ifedigbo, E.; Watkins, S.C.; Ohba, M.; Ryter, S.W.; Vyas, Y.M.; et al. Protein Kinase Cα and ζ Differentially Regulate Death-Inducing Signaling Complex Formation in Cigarette Smoke Extract-Induced Apoptosis. J. Immunol. 2008, 180, 4668–4678. [Google Scholar] [CrossRef] [Green Version]
- De Maria, R.; Zeuner, A.; Eramo, A.; Domenichelli, C.; Bonci, D.; Grignani, F.; Srinivasula, S.M.; Alnemri, E.S.; Testa, U.; Peschle, C. Negative regulation of erythropoiesis by caspase-mediated cleavage of GATA-1. Nature 1999, 401, 489–493. [Google Scholar] [CrossRef]
- De Maria, R.; Testa, U.; Luchetti, L.; Zeuner, A.; Stassi, G.; Pelosi, E.; Riccioni, R.; Felli, N.; Samoggia, P.; Peschle, C. Apoptotic Role of Fas/Fas Ligand System in the Regulation of Erythropoiesis. Blood 1999, 93, 796–803. [Google Scholar] [CrossRef]
- Biswas, D.; Sen, G.; Sarkar, A.; Biswas, T. Atorvastatin acts synergistically with N-acetyl cysteine to provide therapeutic advantage against Fas-activated erythrocyte apoptosis during chronic arsenic exposure in rats. Toxicol. Appl. Pharmacol. 2011, 250, 39–53. [Google Scholar] [CrossRef]
- Mandal, S.; Mukherjee, S.; Chowdhury, K.D.; Sarkar, A.; Basu, K.; Paul, S.; Karmakar, D.; Chatterjee, M.; Biswas, T.; Sadhukhan, G.C.; et al. S-allyl cysteine in combination with clotrimazole downregulates Fas induced apoptotic events in erythrocytes of mice exposed to lead. Biochim. Biophys. Acta (BBA)–Gen. Subj. 2012, 1820, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Khan, E.; Careaga, M.; Goldkorn, T. Neutral sphingomyelinase 2 is activated by cigarette smoke to augment ceramide-induced apoptosis in lung cell death. Am. J. Physiol. Cell. Mol. Physiol. 2009, 297, L125–L133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schweitzer, K.S.; Hatoum, H.; Brown, M.B.; Gupta, M.; Justice, M.J.; Beteck, B.; Van Demark, M.; Gu, Y.; Presson, R.G., Jr.; Hubbard, W.C.; et al. Mechanisms of lung endothelial barrier disruption induced by cigarette smoke: Role of oxidative stress and ceramides. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 301, L836–L846. [Google Scholar] [CrossRef]
- Filosto, S.; Castillo, S.; Danielson, A.; Franzi, L.; Khan, E.; Kenyon, N.; Last, J.; Pinkerton, K.; Tuder, R.; Goldkorn, T. Neutral Sphingomyelinase 2: A Novel Target in Cigarette Smoke-Induced Apoptosis and Lung Injury. Am. J. Respir. Cell Mol. Biol. 2011, 44, 350–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahyayauch, H.; García-Arribas, A.B.; Sot, J.; González-Ramírez, E.J.; Busto, J.V.; Monasterio, B.G.; Jiménez-Rojo, N.; Contreras, F.X.; Rendón-Ramírez, A.; Martin, C.; et al. Pb(II) Induces Scramblase Activation and Ceramide-Domain Generation in Red Blood Cells. Sci. Rep. 2018, 8, 7456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grassmé, H.; Riethmüller, J.; Gulbins, E. Biological aspects of ceramide-enriched membrane domains. Prog. Lipid Res. 2007, 46, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Grullich, C.; Sullards, M.; Fuks, Z.; Merrill, A.H.; Kolesnick, R. CD95(Fas/APO-1) Signals Ceramide Generation Independent of the Effector Stage of Apoptosis. J. Biol. Chem. 2000, 275, 8650–8656. [Google Scholar] [CrossRef] [Green Version]
- Cremesti, A.; Paris, F.; Grassmé, H.; Holler, N.; Tschopp, J.; Fuks, Z.; Gulbins, E.; Kolesnick, R. Ceramide Enables Fas to Cap and Kill. J. Biol. Chem. 2001, 276, 23954–23961. [Google Scholar] [CrossRef] [Green Version]
- Chaigne-Delalande, B.; Moreau, J.F.; Legembre, P. Rewinding the DISC. Arch. Immunol. Ther. Exp. (Warsz.) 2008, 56. [Google Scholar] [CrossRef] [PubMed]
- Yue, J.; López, J.M. Understanding MAPK Signaling Pathways in Apoptosis. Int. J. Mol. Sci. 2020, 21, 2346. [Google Scholar] [CrossRef] [PubMed]
- Kuo, W.-H.; Chen, J.-H.; Lin, H.-H.; Chen, B.-C.; Hsu, J.-D.; Wang, C.-J. Induction of apoptosis in the lung tissue from rats exposed to cigarette smoke involves p38/JNK MAPK pathway. Chem. Biol. Interact. 2005, 155, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Rubiolo, C.; Piazzolla, D.; Meissl, K.; Beug, H.; Huber, J.C.; Kolbus, A.; Baccarini, M. A balance between Raf-1 and Fas expression sets the pace of erythroid differentiation. Blood 2006, 108, 152–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attanzio, A.; D’Anneo, A.; Pappalardo, F.; Bonina, F.P.; Livrea, M.A.; Allegra, M.; Tesoriere, L. Phenolic Composition of Hydrophilic Extract of Manna from Sicilian Fraxinus angustifolia Vahl and its Reducing, Antioxidant and Anti-Inflammatory Activity in Vitro. Antioxidants 2019, 8, 494. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Restivo, I.; Attanzio, A.; Giardina, I.C.; Di Gaudio, F.; Tesoriere, L.; Allegra, M. Cigarette Smoke Extract Induces p38 MAPK-Initiated, Fas-Mediated Eryptosis. Int. J. Mol. Sci. 2022, 23, 14730. https://doi.org/10.3390/ijms232314730
Restivo I, Attanzio A, Giardina IC, Di Gaudio F, Tesoriere L, Allegra M. Cigarette Smoke Extract Induces p38 MAPK-Initiated, Fas-Mediated Eryptosis. International Journal of Molecular Sciences. 2022; 23(23):14730. https://doi.org/10.3390/ijms232314730
Chicago/Turabian StyleRestivo, Ignazio, Alessandro Attanzio, Ilenia Concetta Giardina, Francesca Di Gaudio, Luisa Tesoriere, and Mario Allegra. 2022. "Cigarette Smoke Extract Induces p38 MAPK-Initiated, Fas-Mediated Eryptosis" International Journal of Molecular Sciences 23, no. 23: 14730. https://doi.org/10.3390/ijms232314730