
sFlt-1 in Chronic Kidney Disease: Friend or Foe?
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. PlGF/Flt-1 Pathway
2.1. PlGF
2.2. Flt-1 and the Soluble Isoform of Flt-1 (sFlt-1)
3. PlGF/sFlt-1 Imbalance in CKD
3.1. Circulating PlGF in CKD
3.2. PlGF Production in CKD
3.3. Circulating sFlt-1 in CKD
3.4. Heparin Loading Test
3.5. Production of sFlt-1 in CKD
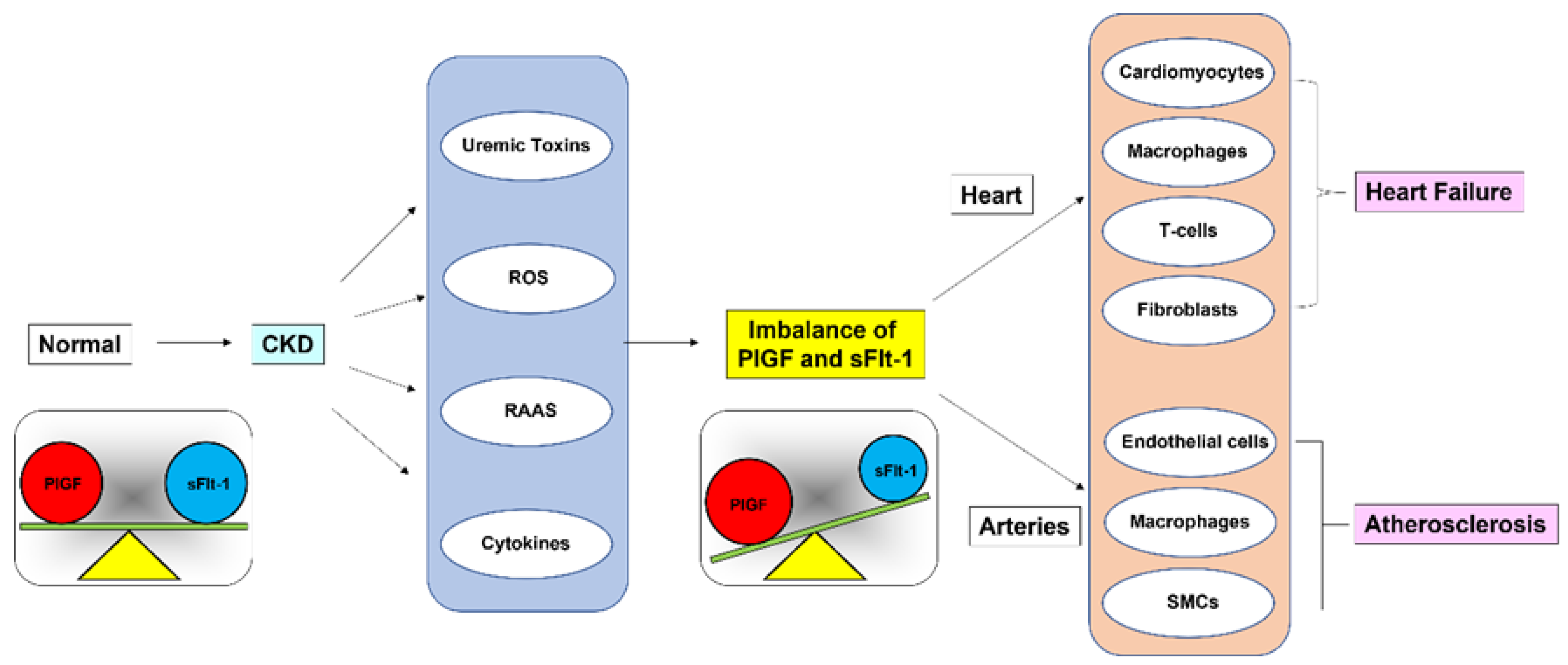
4. Role of PlGF/sFlt-1 Imbalance
4.1. Atherosclerosis in CKD Model Mice
4.2. Generation of sFlt-1 KO Mice
4.3. Increased sFlt-1 and HF
4.4. Decreased sFlt-1 and HF
5. Distribution of sFlt-1
6. Imbalance between PlGF and sFlt-1
7. Perspectives and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bikbov, B.; Perico, N.; Remuzzi, G.; on behalf of the GBDGDEG. Disparities in Chronic Kidney Disease Prevalence among Males and Females in 195 Countries: Analysis of the Global Burden of Disease 2016 Study. Nephron 2018, 139, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Adler, A.I.; Stevens, R.J.; Manley, S.E.; Bilous, R.W.; Cull, C.A.; Holman, R.R.; UKPDS Group. Development and progression of nephropathy in type 2 diabetes: The United Kingdom Prospective Diabetes Study (UKPDS 64). Kidney Int. 2003, 63, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Ninomiya, T.; Kiyohara, Y.; Kubo, M.; Tanizaki, Y.; Doi, Y.; Okubo, K.; Wakugawa, Y.; Hata, J.; Oishi, Y.; Shikata, K.; et al. Chronic kidney disease and cardiovascular disease in a general Japanese population: The Hisayama Study. Kidney Int. 2005, 68, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Ronco, C.; Haapio, M.; House, A.A.; Anavekar, N.; Bellomo, R. Cardiorenal syndrome. J. Am. Coll. Cardiol. 2008, 52, 1527–1539. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, R.; Ali, Y.; Hashizume, R.; Suzuki, N.; Ito, M. BNP as a Major Player in the Heart-Kidney Connection. Int. J. Mol. Sci. 2019, 20, 3581. [Google Scholar] [CrossRef]
- Ivey-Miranda, J.B.; Stewart, B.; Cox, Z.L.; McCallum, W.; Maulion, C.; Gleason, O.; Meegan, G.; Amatruda, J.G.; Moreno-Villagomez, J.; Mahoney, D.; et al. FGF-23 (Fibroblast Growth Factor-23) and Cardiorenal Interactions. Circ. Heart Fail. 2021, 14, e008385.7. [Google Scholar] [CrossRef] [PubMed]
- Clementi, A.; Virzi, G.M.; Battaglia, G.G.; Ronco, C. Neurohormonal, Endocrine, and Immune Dysregulation and Inflammation in Cardiorenal Syndrome. Cardiorenal Med. 2019, 9, 265–273. [Google Scholar] [CrossRef]
- Silva-Hucha, S.; Pastor, A.M.; Morcuende, S. Neuroprotective Effect of Vascular Endothelial Growth Factor on Motoneurons of the Oculomotor System. Int. J. Mol. Sci. 2021, 22, 814. [Google Scholar] [CrossRef]
- Alitalo, K.; Tammela, T.; Petrova, T.V. Lymphangiogenesis in development and human disease. Nature 2005, 438, 946–953. [Google Scholar] [CrossRef]
- Autiero, M.; Waltenberger, J.; Communi, D.; Kranz, A.; Moons, L.; Lambrechts, D.; Kroll, J.; Plaisance, S.; De Mol, M.; Bono, F.; et al. Role of PlGF in the intra- and intermolecular cross talk between the VEGF receptors Flt1 and Flk1. Nat. Med. 2003, 9, 936–943. [Google Scholar] [CrossRef]
- Shibuya, M. Vascular endothelial growth factor and its receptor system: Physiological functions in angiogenesis and pathological roles in various diseases. J. Biochem. 2013, 153, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Dabravolski, S.A.; Khotina, V.A.; Omelchenko, A.V.; Kalmykov, V.A.; Orekhov, A.N. The Role of the VEGF Family in Atherosclerosis Development and Its Potential as Treatment Targets. Int. J. Mol. Sci. 2022, 23, 931. [Google Scholar] [CrossRef] [PubMed]
- Maglione, D.; Guerriero, V.; Viglietto, G.; Delli-Bovi, P.; Persico, M.G. Isolation of a human placenta cDNA coding for a protein related to the vascular permeability factor. Proc. Natl. Acad. Sci. USA 1991, 88, 9267–9271. [Google Scholar] [CrossRef]
- Yonekura, H.; Sakurai, S.; Liu, X.; Migita, H.; Wang, H.; Yamagishi, S.; Nomura, M.; Abedin, M.J.; Unoki, H.; Yamamoto, Y.; et al. Placenta growth factor and vascular endothelial growth factor B and C expression in microvascular endothelial cells and pericytes. Implication in autocrine and paracrine regulation of angiogenesis. J. Biol. Chem. 1999, 274, 35172–35178. [Google Scholar] [CrossRef]
- Persico, M.G.; Vincenti, V.; Di Palma, T. Structure, expression and receptor-binding properties of placenta growth factor (PlGF). Curr. Top. Microbiol. Immunol. 1999, 237, 31–40. [Google Scholar]
- Iwama, H.; Uemura, S.; Naya, N.; Imagawa, K.; Takemoto, Y.; Asai, O.; Onoue, K.; Okayama, S.; Somekawa, S.; Kida, Y.; et al. Cardiac expression of placental growth factor predicts the improvement of chronic phase left ventricular function in patients with acute myocardial infarction. J. Am. Coll. Cardiol. 2006, 47, 1559–1567. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Moons, L.; Luttun, A.; Vincenti, V.; Compernolle, V.; De Mol, M.; Wu, Y.; Bono, F.; Devy, L.; Beck, H.; et al. Synergism between vascular endothelial growth factor and placental growth factor contributes to angiogenesis and plasma extravasation in pathological conditions. Nat. Med. 2001, 7, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Khurana, R.; Moons, L.; Shafi, S.; Luttun, A.; Collen, D.; Martin, J.F.; Carmeliet, P.; Zachary, I.C. Placental growth factor promotes atherosclerotic intimal thickening and macrophage accumulation. Circulation 2005, 111, 2828–2836. [Google Scholar] [CrossRef]
- Roncal, C.; Buysschaert, I.; Gerdes, N.; Georgiadou, M.; Ovchinnikova, O.; Fischer, C.; Stassen, J.M.; Moons, L.; Collen, D.; De Bock, K.; et al. Short-term delivery of anti-PlGF antibody delays progression of atherosclerotic plaques to vulnerable lesions. Cardiovasc. Res. 2010, 86, 29–36. [Google Scholar] [CrossRef]
- Shibuya, M.; Yamaguchi, S.; Yamane, A.; Ikeda, T.; Tojo, A.; Matsushime, H.; Sato, M. Nucleotide sequence and expression of a novel human receptor-type tyrosine kinase gene (flt) closely related to the fms family. Oncogene 1990, 5, 519–524. [Google Scholar]
- Sawano, A.; Takahashi, T.; Yamaguchi, S.; Aonuma, M.; Shibuya, M. Flt-1 but not KDR/Flk-1 tyrosine kinase is a receptor for placenta growth factor, which is related to vascular endothelial growth factor. Cell Growth Differ. 1996, 7, 213–221. [Google Scholar] [PubMed]
- Kendall, R.L.; Thomas, K.A. Inhibition of vascular endothelial cell growth factor activity by an endogenously encoded soluble receptor. Proc. Natl. Acad. Sci. USA 1993, 90, 10705–10709. [Google Scholar] [CrossRef] [PubMed]
- Maharaj, A.S.; Saint-Geniez, M.; Maldonado, A.E.; D’Amore, P.A. Vascular endothelial growth factor localization in the adult. Am. J. Pathol. 2006, 168, 639–648. [Google Scholar] [CrossRef]
- Maharaj, A.S.; D’Amore, P.A. Roles for VEGF in the adult. Microvasc. Res. 2007, 74, 100–113. [Google Scholar] [CrossRef] [PubMed]
- D’Amore, P.A. Vascular endothelial cell growth factor-a: Not just for endothelial cells anymore. Am. J. Pathol. 2007, 171, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Oura, H.; Bertoncini, J.; Velasco, P.; Brown, L.F.; Carmeliet, P.; Detmar, M. A critical role of placental growth factor in the induction of inflammation and edema formation. Blood 2003, 101, 560–567. [Google Scholar] [CrossRef]
- Sela, S.; Itin, A.; Natanson-Yaron, S.; Greenfield, C.; Goldman-Wohl, D.; Yagel, S.; Keshet, E. A novel human-specific soluble vascular endothelial growth factor receptor 1: Cell-type-specific splicing and implications to vascular endothelial growth factor homeostasis and preeclampsia. Circ. Res. 2008, 102, 1566–1574. [Google Scholar] [CrossRef] [PubMed]
- Jebbink, J.; Keijser, R.; Veenboer, G.; van der Post, J.; Ris-Stalpers, C.; Afink, G. Expression of placental FLT1 transcript variants relates to both gestational hypertensive disease and fetal growth. Hypertension 2011, 58, 70–76. [Google Scholar] [CrossRef]
- Rosenberg, V.A.; Buhimschi, I.A.; Lockwood, C.J.; Paidas, M.J.; Dulay, A.T.; Ramma, W.; Abdel-Razeq, S.S.; Zhao, G.; Ahmad, S.; Ahmed, A.; et al. Heparin elevates circulating soluble fms-like tyrosine kinase-1 immunoreactivity in pregnant women receiving anticoagulation therapy. Circulation 2011, 124, 2543–2553. [Google Scholar] [CrossRef]
- Sela, S.; Natanson-Yaron, S.; Zcharia, E.; Vlodavsky, I.; Yagel, S.; Keshet, E. Local retention versus systemic release of soluble VEGF receptor-1 are mediated by heparin-binding and regulated by heparanase. Circ. Res. 2011, 108, 1063–1070. [Google Scholar] [CrossRef]
- Pilarczyk, K.; Sattler, K.J.E.; Galili, O.; Versari, D.; Olson, M.L.; Meyer, F.B.; Zhu, X.Y.; Lerman, L.O.; Lerman, A. Placenta growth factor expression in human atherosclerotic carotid plaques is related to plaque destabilization. Atherosclerosis 2008, 196, 333–340. [Google Scholar] [CrossRef] [PubMed]
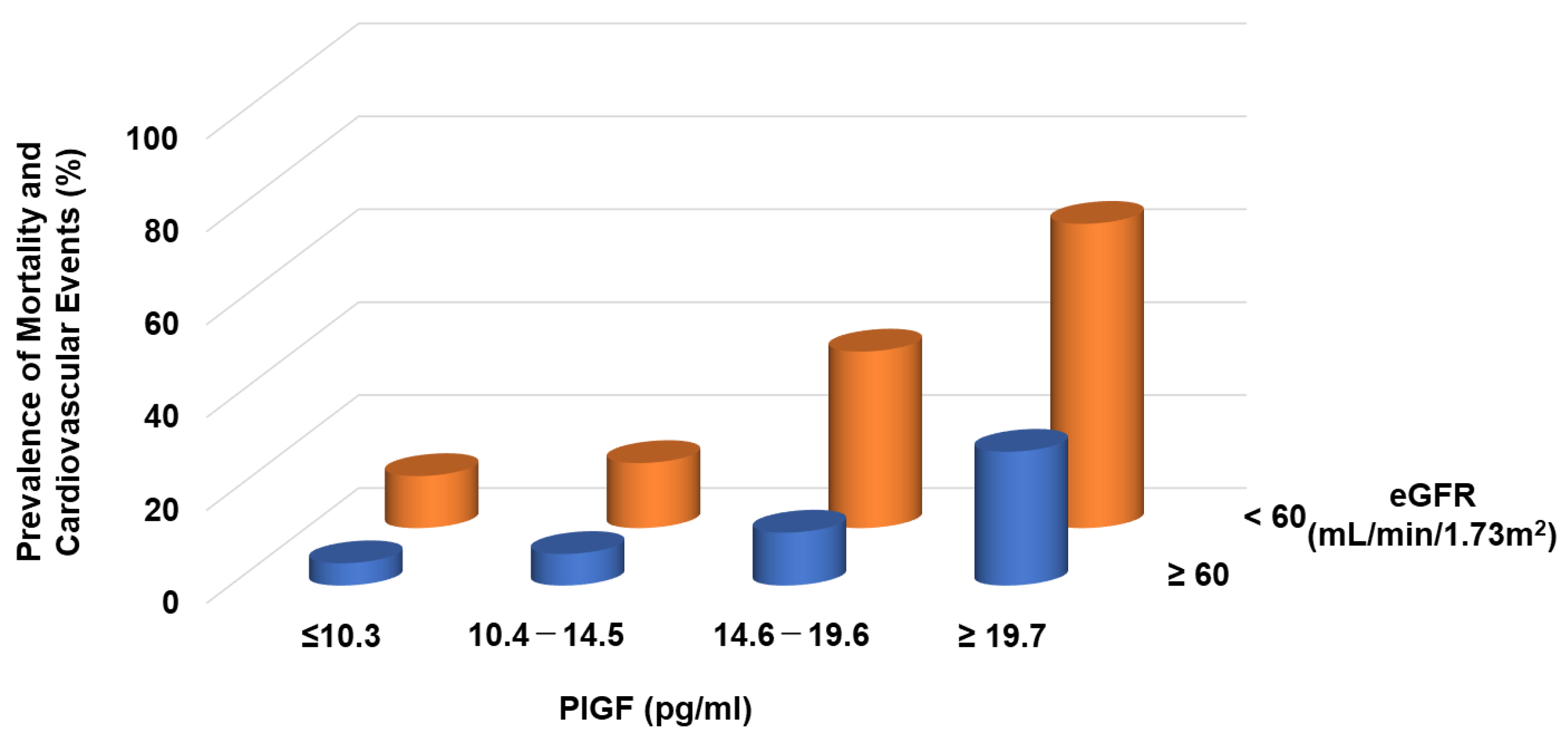
- Matsui, M.; Takeda, Y.; Uemura, S.; Matsumoto, T.; Seno, A.; Onoue, K.; Tsushima, H.; Morimoto, K.; Soeda, T.; Okayama, S.; et al. Placental Growth Factor as a Predictor of Cardiovascular Events in Patients with CKD from the NARA-CKD Study. J. Am. Soc. Nephrol. 2015, 26, 2871–2881. [Google Scholar] [CrossRef] [PubMed]
- Peiskerova, M.; Kalousova, M.; Danzig, V.; Mikova, B.; Hodkova, M.; Nemecek, E.; Bani-Hani, A.; Ambroz, D.; Benakova, H.; Linhart, A.; et al. Placental growth factor may predict increased left ventricular mass index in patients with mild to moderate chronic kidney disease--a prospective observational study. BMC Nephrol. 2013, 14, 142. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Du, Y.; Yang, N.; Liang, Y.; Li, Y. Microvasculature change and placenta growth factor expression in the early stage of a rat remnant kidney model. Am. J. Nephrol. 2006, 26, 97–104. [Google Scholar] [CrossRef]
- Matsui, M.; Takeda, Y.; Uemura, S.; Matsumoto, T.; Seno, A.; Onoue, K.; Tsushima, H.; Morimoto, K.; Soeda, T.; Okayama, S.; et al. Suppressed soluble Fms-like tyrosine kinase-1 production aggravates atherosclerosis in chronic kidney disease. Kidney Int. 2014, 85, 393–403. [Google Scholar] [CrossRef]
- Onoue, K.; Uemura, S.; Takeda, Y.; Somekawa, S.; Iwama, H.; Imagawa, K.; Nishida, T.; Morikawa, Y.; Takemoto, Y.; Asai, O.; et al. Reduction of circulating soluble fms-like tyrosine kinase-1 plays a significant role in renal dysfunction-associated aggravation of atherosclerosis. Circulation 2009, 120, 2470–2477. [Google Scholar] [CrossRef]
- McGraw, A.P.; Bagley, J.; Chen, W.S.; Galayda, C.; Nickerson, H.; Armani, A.; Caprio, M.; Carmeliet, P.; Jaffe, I.Z. Aldosterone increases early atherosclerosis and promotes plaque inflammation through a placental growth factor-dependent mechanism. J. Am. Heart Assoc. 2013, 2, e000018. [Google Scholar] [CrossRef]
- Bakris, G.L.; Agarwal, R.; Anker, S.D.; Pitt, B.; Ruilope, L.M.; Rossing, P.; Kolkhof, P.; Nowack, C.; Schloemer, P.; Joseph, A.; et al. Effect of Finerenone on Chronic Kidney Disease Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2020, 383, 2219–2229. [Google Scholar] [CrossRef]
- Pitt, B.; Filippatos, G.; Agarwal, R.; Anker, S.D.; Bakris, G.L.; Rossing, P.; Joseph, A.; Kolkhof, P.; Nowack, C.; Schloemer, P.; et al. Cardiovascular Events with Finerenone in Kidney Disease and Type 2 Diabetes. N. Engl. J. Med. 2021, 385, 2252–2263. [Google Scholar] [CrossRef]
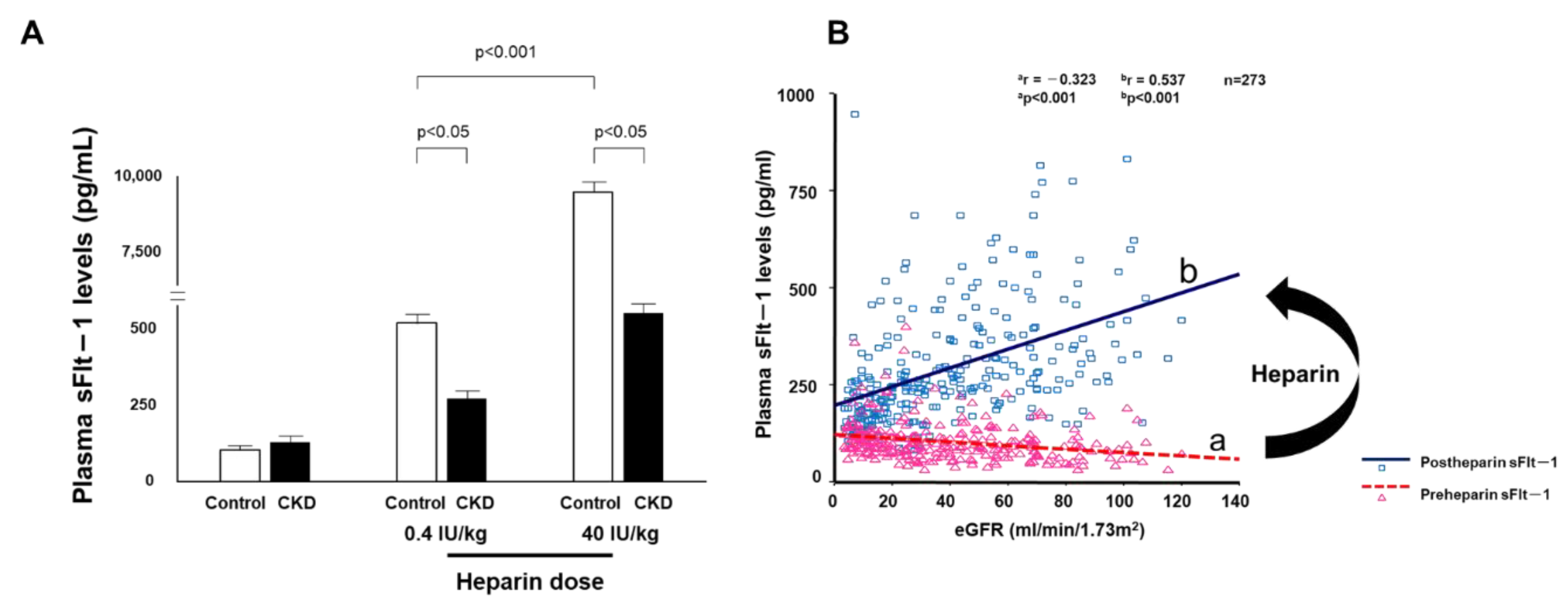
- Di Marco, G.S.; Reuter, S.; Hillebrand, U.; Amler, S.; Konig, M.; Larger, E.; Oberleithner, H.; Brand, E.; Pavenstadt, H.; Brand, M. The soluble VEGF receptor sFlt1 contributes to endothelial dysfunction in CKD. J. Am. Soc. Nephrol. 2009, 20, 2235–2245. [Google Scholar] [CrossRef]
- Ky, B.; French, B.; Ruparel, K.; Sweitzer, N.K.; Fang, J.C.; Levy, W.C.; Sawyer, D.B.; Cappola, T.P. The vascular marker soluble fms-like tyrosine kinase 1 is associated with disease severity and adverse outcomes in chronic heart failure. J. Am. Coll. Cardiol. 2011, 58, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Wada, H.; Shinozaki, T.; Suzuki, M.; Sakagami, S.; Ajiro, Y.; Funada, J.; Matsuda, M.; Shimizu, M.; Takenaka, T.; Morita, Y.; et al. Impact of Chronic Kidney Disease on the Associations of Cardiovascular Biomarkers With Adverse Outcomes in Patients with Suspected or Known Coronary Artery Disease: The EXCEED-J Study. J. Am. Heart Assoc. 2022, 11, e023464. [Google Scholar] [CrossRef] [PubMed]
- Rambod, M.; Heine, G.H.; Seiler, S.; Dominic, E.A.; Rogacev, K.S.; Dwivedi, R.; Ramezani, A.; Wing, M.R.; Amdur, R.L.; Fliser, D.; et al. Association of vascular endothelial factors with cardiovascular outcome and mortality in chronic kidney disease patients: A 4-year cohort study. Atherosclerosis 2014, 236, 360–365. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Moore, K.H.; Chapman, H.; George, E.M. Unfractionated heparin displaces sFlt-1 from the placental extracellular matrix. Biol. Sex Differ. 2020, 11, 34. [Google Scholar] [CrossRef]
- Nakada, Y.; Onoue, K.; Nakano, T.; Ishihara, S.; Kumazawa, T.; Nakagawa, H.; Ueda, T.; Nishida, T.; Soeda, T.; Okayama, S.; et al. AST-120, an Oral Carbon Absorbent, Protects against the Progression of Atherosclerosis in a Mouse Chronic Renal Failure Model by Preserving sFlt-1 Expression Levels. Sci. Rep. 2019, 9, 15571. [Google Scholar] [CrossRef]
- House, A.A.; Wanner, C.; Sarnak, M.J.; Pina, I.L.; McIntyre, C.W.; Komenda, P.; Kasiske, B.L.; Deswal, A.; deFilippi, C.R.; Cleland, J.G.F.; et al. Heart failure in chronic kidney disease: Conclusions from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2019, 95, 1304–1317. [Google Scholar] [CrossRef]
- Draker, N.; Torry, D.S.; Torry, R.J. Placenta growth factor and sFlt-1 as biomarkers in ischemic heart disease and heart failure: A review. Biomark. Med. 2019, 13, 785–799. [Google Scholar] [CrossRef]
- Hammadah, M.; Georgiopoulou, V.V.; Kalogeropoulos, A.P.; Weber, M.; Wang, X.; Samara, M.A.; Wu, Y.; Butler, J.; Tang, W.H. Elevated Soluble Fms-Like Tyrosine Kinase-1 and Placental-Like Growth Factor Levels Are Associated With Development and Mortality Risk in Heart Failure. Circ. Heart Fail. 2016, 9, e002115. [Google Scholar] [CrossRef][Green Version]
- Di Marco, G.S.; Kentrup, D.; Reuter, S.; Mayer, A.B.; Golle, L.; Tiemann, K.; Fobker, M.; Engelbertz, C.; Breithardt, G.; Brand, E.; et al. Soluble Flt-1 links microvascular disease with heart failure in CKD. Basic Res. Cardiol. 2015, 110, 30. [Google Scholar] [CrossRef]
- Seno, A.; Takeda, Y.; Matsui, M.; Okuda, A.; Nakano, T.; Nakada, Y.; Kumazawa, T.; Nakagawa, H.; Nishida, T.; Onoue, K.; et al. Suppressed Production of Soluble Fms-Like Tyrosine Kinase-1 Contributes to Myocardial Remodeling and Heart Failure. Hypertension 2016, 68, 678–687. [Google Scholar] [CrossRef]
- Maynard, S.E.; Min, J.Y.; Merchan, J.; Lim, K.H.; Li, J.; Mondal, S.; Libermann, T.A.; Morgan, J.P.; Sellke, F.W.; Stillman, I.E.; et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J. Clin. Investig. 2003, 111, 649–658. [Google Scholar] [CrossRef]
- Nakano, T.; Onoue, K.; Seno, A.; Ishihara, S.; Nakada, Y.; Nakagawa, H.; Ueda, T.; Nishida, T.; Soeda, T.; Watanabe, M.; et al. Involvement of chronic inflammation via monocyte chemoattractant protein-1 in uraemic cardiomyopathy: A human biopsy study. ESC Heart Fail. 2021, 8, 3156–3167. [Google Scholar] [CrossRef] [PubMed]
- Savira, F.; Magaye, R.; Liew, D.; Reid, C.; Kelly, D.J.; Kompa, A.R.; Sangaralingham, S.J.; Burnett, J.C.; Kaye, D., Jr.; Wang, B.H. Cardiorenal syndrome: Multi-organ dysfunction involving the heart, kidney and vasculature. Br. J. Pharmacol. 2020, 177, 2906–2922. [Google Scholar] [CrossRef] [PubMed]
- Eddy, A.C.; Chapman, H.; George, E.M. Heparanase regulation of sFLT-1 release in trophoblasts in vitro. Placenta 2019, 85, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Wewers, T.M.; Schulz, A.; Nolte, I.; Pavenstädt, H.; Brand, M.; Di Marco, G.S. Circulating Soluble Fms-like Tyrosine Kinase in Renal Diseases Other than Preeclampsia. J. Am. Soc. Nephrol. 2021, 32, 1853–1863. [Google Scholar] [CrossRef]
- Wewers, T.M.; Mayer, A.B.; Pfleiderer, A.; Beul, K.; Schmidt, R.; Heitplatz, B.; Van Marck, V.; Nolte, I.; Pavenstädt, H.; Reuter, S.; et al. Increased soluble fms-like tyrosine kinase 1 after ischemia reperfusion contributes to adverse clinical outcomes following kidney transplantation. Kidney Int. 2019, 95, 1091–1102. [Google Scholar] [CrossRef]
- Levine, R.J.; Lam, C.; Qian, C.; Yu, K.F.; Maynard, S.E.; Sachs, B.P.; Sibai, B.M.; Epstein, F.H.; Romero, R.; Thadhani, R.; et al. Soluble endoglin and other circulating antiangiogenic factors in preeclampsia. N. Engl. J. Med. 2006, 355, 992–1005. [Google Scholar] [CrossRef]
- Karumanchi, S.A.; Epstein, F.H. Placental ischemia and soluble fms-like tyrosine kinase 1: Cause or consequence of preeclampsia? Kidney Int. 2007, 71, 959–961. [Google Scholar] [CrossRef]
- Zeisler, H.; Llurba, E.; Chantraine, F.; Vatish, M.; Staff, A.C.; Sennstrom, M.; Olovsson, M.; Brennecke, S.P.; Stepan, H.; Allegranza, D.; et al. Predictive Value of the sFlt-1: PlGF Ratio in Women with Suspected Preeclampsia. N. Engl. J. Med. 2016, 374, 13–22. [Google Scholar] [CrossRef]
- Yoo, S.A.; Kim, M.; Kang, M.C.; Kong, J.S.; Kim, K.M.; Lee, S.; Hong, B.K.; Jeong, G.H.; Lee, J.; Shin, M.G.; et al. Placental growth factor regulates the generation of TH17 cells to link angiogenesis with autoimmunity. Nat. Immunol. 2019, 20, 1348–1359. [Google Scholar] [CrossRef]
- Lekawanvijit, S. Cardiotoxicity of Uremic Toxins: A Driver of Cardiorenal Syndrome. Toxins 2018, 10, 352. [Google Scholar] [CrossRef] [PubMed]
- Winterberg, P.D.; Robertson, J.M.; Kelleman, M.S.; George, R.P.; Ford, M.L. T Cells Play a Causal Role in Diastolic Dysfunction during Uremic Cardiomyopathy. J. Am. Soc. Nephrol. 2019, 30, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Thadhani, R.; Hagmann, H.; Schaarschmidt, W.; Roth, B.; Cingoez, T.; Karumanchi, S.A.; Wenger, J.; Lucchesi, K.J.; Tamez, H.; Lindner, T.; et al. Removal of soluble fms-like tyrosine kinase-1 by dextran sulfate apheresis in preeclampsia. J. Am. Soc. Nephrol. 2016, 27, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Kosugi, T.; Nakayama, T.; Li, Q.; Chiodo, V.A.; Zhang, L.; Campbell-Thompson, M.; Grant, M.; Croker, B.P.; Nakagawa, T. Soluble Flt-1 gene therapy ameliorates albuminuria but accelerates tubulointerstitial injury in diabetic mice. Am. J. Physiol. Renal Physiol. 2010, 298, F609–F616. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsui, M.; Onoue, K.; Saito, Y. sFlt-1 in Chronic Kidney Disease: Friend or Foe? Int. J. Mol. Sci. 2022, 23, 14187. https://doi.org/10.3390/ijms232214187
Matsui M, Onoue K, Saito Y. sFlt-1 in Chronic Kidney Disease: Friend or Foe? International Journal of Molecular Sciences. 2022; 23(22):14187. https://doi.org/10.3390/ijms232214187
Chicago/Turabian StyleMatsui, Masaru, Kenji Onoue, and Yoshihiko Saito. 2022. "sFlt-1 in Chronic Kidney Disease: Friend or Foe?" International Journal of Molecular Sciences 23, no. 22: 14187. https://doi.org/10.3390/ijms232214187
APA StyleMatsui, M., Onoue, K., & Saito, Y. (2022). sFlt-1 in Chronic Kidney Disease: Friend or Foe? International Journal of Molecular Sciences, 23(22), 14187. https://doi.org/10.3390/ijms232214187