
Neurodegenerative Diseases: From Dysproteostasis, Altered Calcium Signalosome to Selective Neuronal Vulnerability to AAV-Mediated Gene Therapy



 , ,
, ,
Abstract
1. Introduction
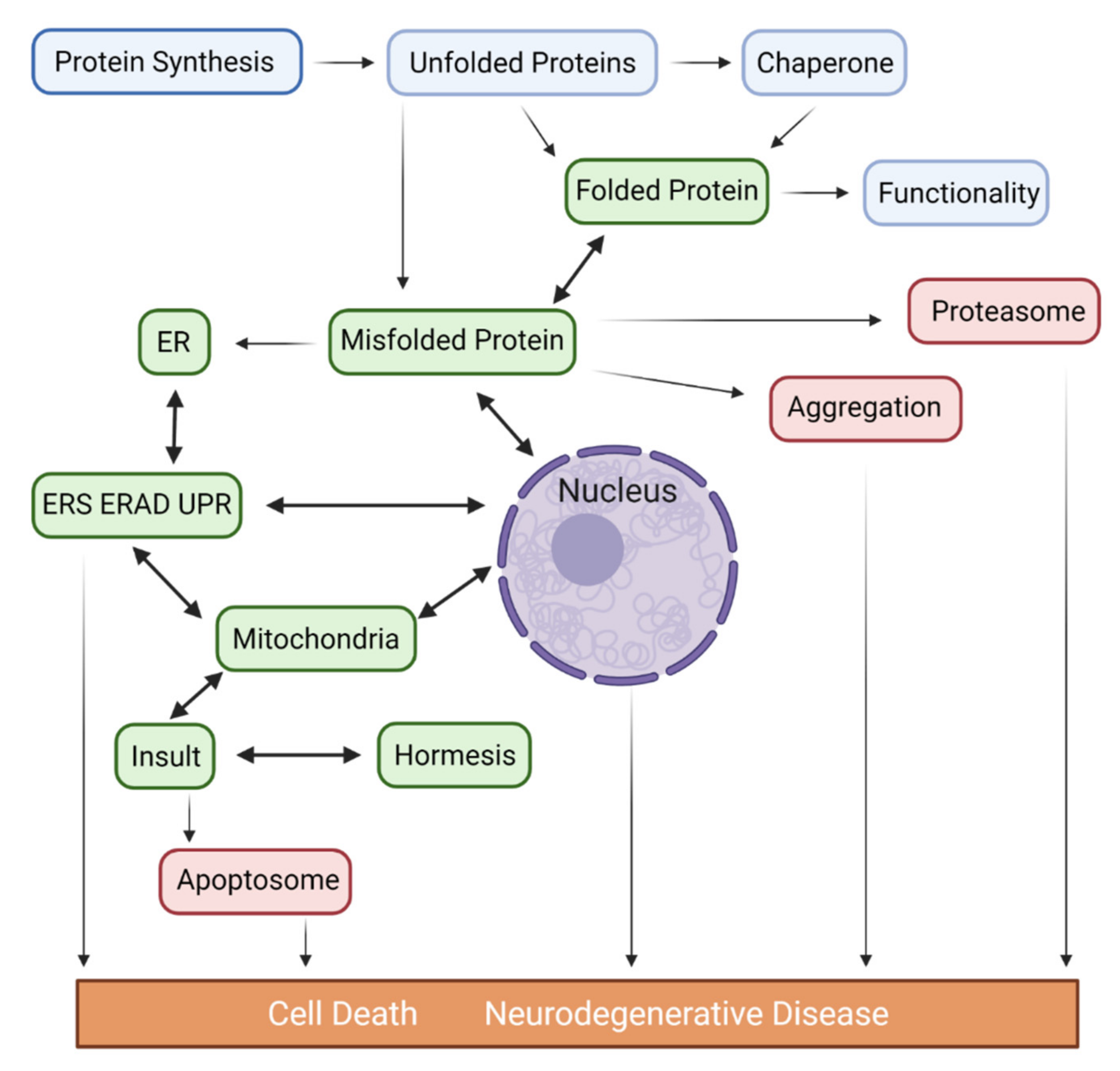
2. Endoplasmic Reticulum Stress (ERS) and Dysproteostasis in Major Neurodegenerative Diseases
2.1. Proteostasis Imbalance in AD
2.2. Proteostasis Imbalance in PD
2.3. Proteostasis Imbalance in HD
2.4. Proteostasis Imbalance in ALS
2.5. Proteostasis Imbalance in CJD
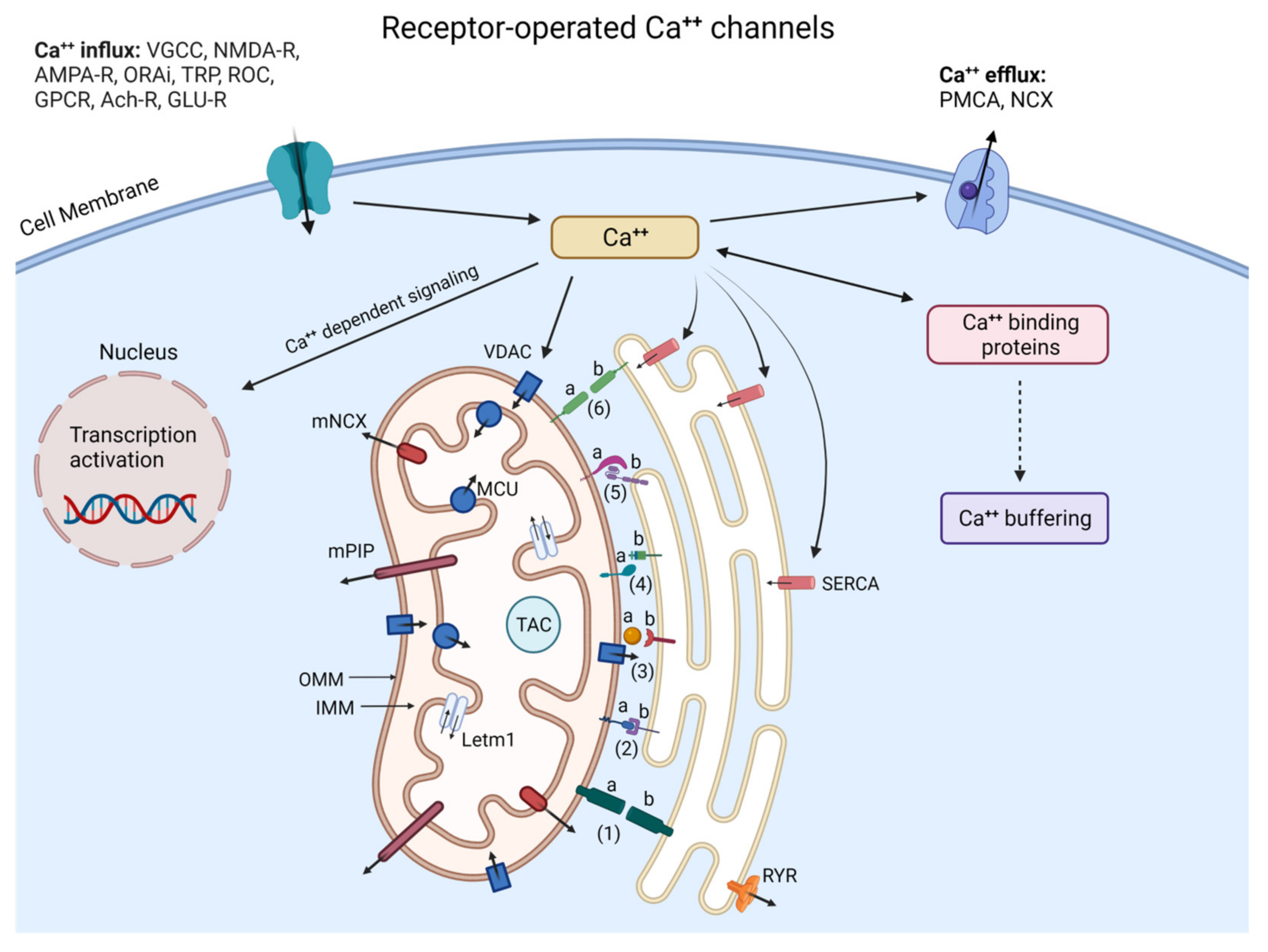
3. Pathology of Calcium Signaling in Major Neurodegenerative Diseases
3.1. Dysfunction of Calcium Signaling in AD
3.2. Dysfunction of Calcium Signaling in PD
3.3. Dysfunction of Calcium Signaling in HD
3.4. Dysfunction of Calcium Signaling in ALS
3.5. Dysfunction of Calcium Signaling in CJD
4. Selective Neuronal Vulnerability in Major Neurodegenerative Diseases
4.1. Dendritic Morphology Underlying Neuronal Vulnerability
4.2. Intracellular Signaling Mechanisms Underlying Neuronal Vulnerability
5. CRMP3/DPYSL4 as a Potential Neuroprotective Target for ND
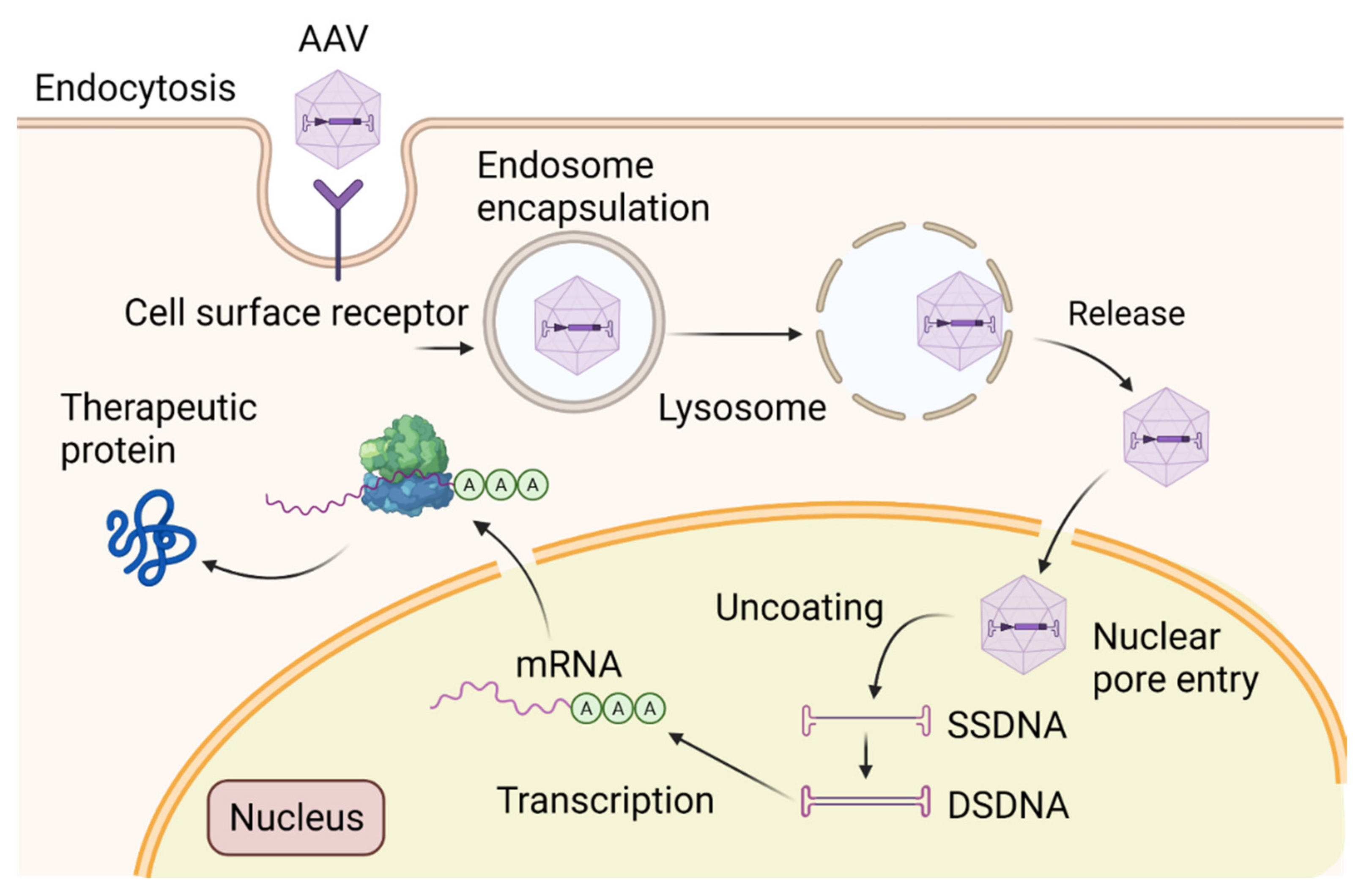
6. Adeno-Associated Virus (AAV)-Mediated Gene Therapy: From Pre-Clinical Studies to Clinical Trials
7. Closing Thoughts: Accomplishments and Expectations
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Voet, S.; Srinivasan, S.; Lamkanfi, M.; van Loo, G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol. Med. 2019, 11, e10248. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef] [PubMed]
- Schrank, S.; Barrington, N.; Stutzmann, G.E. Calcium-Handling Defects and Neurodegenerative Disease. Cold Spring Harb. Perspect. Biol. 2020, 12, a035212. [Google Scholar] [CrossRef] [PubMed]
- Rossmann, M.P.; Dubois, S.M.; Agarwal, S.; Zon, L.I. Mitochondrial function in development and disease. Dis. Models Mech. 2021, 14, dmm048912. [Google Scholar] [CrossRef] [PubMed]
- Oakes, S.A.; Papa, F.R. The role of endoplasmic reticulum stress in human pathology. Annu. Rev. Pathol. 2015, 10, 173–194. [Google Scholar] [CrossRef]
- Özdinler, P.H. Expanded access: Opening doors to personalized medicine for rare disease patients and patients with neuro-degenerative diseases. FEBS J. 2021, 288, 1457–1461. [Google Scholar] [CrossRef]
- Abulimiti, A.; Lai, M.S.; Chang, R.C. Applications of adeno-associated virus vector-mediated gene delivery for neurodegenerative diseases and psychiatric diseases: Progress, advances, and challenges. Mech. Ageing Dev. 2021, 199, 111549. [Google Scholar] [CrossRef]
- Wanker, E.E.; Ast, A.; Schindler, F.; Trepte, P.; Schnoegl, S. The pathobiology of perturbed mutant huntingtin protein-protein inter-actions in Huntington’s disease. J. Neurochem. 2019, 151, 507–519. [Google Scholar] [CrossRef]
- Shacham, T.; Sharma, N.; Lederkremer, G.Z. Protein Misfolding and ER Stress in Huntington’s Disease. Front. Mol. Biosci. 2019, 6, 20. [Google Scholar] [CrossRef]
- Lin, C.H.; Tallaksen-Greene, S.; Chien, W.M.; Cearley, J.A.; Jackson, W.S.; Crouse, A.B.; Ren, S.; Li, X.J.; Albin, R.L.; Detloff, P.J. Neurological abnormalities in a knock-in mouse model of Huntington’s disease. Hum. Mol. Genet. 2001, 10, 137–144. [Google Scholar] [CrossRef]
- Świtońska, K.; Szlachcic, W.J.; Handschuh, L.; Wojciechowski, P.; Stelmaszczuk, Ł.M.M.; Figlerowicz, M.; Figiel, M. Identification of Altered Developmental Pathways in Human Juvenile HD iPSC with 71Q and 109Q Using Transcriptome Profiling. Front. Cell. Neurosci. 2019, 12, 528. [Google Scholar] [CrossRef]
- Keo, A.; Aziz, N.A.; Dzyubachyk, O.; Van der Grond, J.; van Roon-Mom, W.M.; Lelieveldt, B.P.; Reinders, M.J.; Mahfouz, A. Co-expression Patterns between ATN1 and ATXN2 Coincide with Brain Regions Affected in Huntington’s Disease. Front. Mol. Neurosci. 2017, 10, 399. [Google Scholar] [CrossRef]
- Bhattacharyya, N.P.; Banerjee, M.; Majumder, P. Huntington’s disease: Roles of huntingtin-interacting protein 1 (HIP-1) and its molecular partner HIPPI in the regulation of apoptosis and transcription. FEBS J. 2008, 275, 4271–4279. [Google Scholar] [CrossRef]
- Buendía, G.A.R.; Leleu, M.; Marzetta, F.; Vanzan, L.; Tan, J.Y.; Ythier, V.; Randall, E.L.; Marques, A.C.; Baubec, T.; Murr, R.; et al. Three-dimensional chromatin interactions remain stable upon CAG/CTG repeat expansion. Sci. Adv. 2020, 6, eaaz4012. [Google Scholar] [CrossRef]
- Oswald, F.; Klöble, P.; Ruland, A.; Rosenkranz, D.; Hinz, B.; Butter, F.; Ramljak, S.; Zechner, U.; Herlyn, H. The FOXP2-Driven Network in Developmental Disorders and Neurodegeneration. Front. Cell. Neurosci. 2017, 11, 212. [Google Scholar] [CrossRef]
- Costa, M.D.C.; Teixeira-Castro, A.; Constante, M.; Magalhães, M.; Magalhães, P.; Cerqueira, J.; Vale, J.; Passão, V.; Barbosa, C.; Robalo, C.; et al. Exclusion of mutations in the PRNP, JPH3, TBP, ATN1, CREBBP, POU3F2 and FTL genes as a cause of disease in Portuguese patients with a Huntington-like phenotype. J. Hum. Genet. 2006, 51, 645–651. [Google Scholar] [CrossRef]
- Iqbal, K.; Alonso, A.C.; Gong, C.X.; Khatoon, S.; Pei, J.J.; Wang, J.Z.; Grundke-Iqbal, I. Mechanisms of neurofibrillary degeneration and the formation of neurofibrillary tangles. J. Neural. Transm. Suppl. 1998, 53, 169–180. [Google Scholar] [CrossRef]
- Kjældgaard, A.L.; Pilely, K.; Olsen, K.S.; Lauritsen, A.Ø.; Pedersen, S.W.; Svenstrup, K.; Karlsborg, M.; Thagesen, H.; Blaabjerg, M.; Theodorsdottir, A.; et al. Complement Profiles in Patients with Amyotrophic Lateral Sclerosis: A Prospective Observational Cohort Study. J. Inflamm. Res. 2021, 14, 1043–1053. [Google Scholar] [CrossRef]
- Fairless, R.; Williams, S.K.; Diem, R. Calcium-Binding Proteins as Determinants of Central Nervous System Neuronal Vulnerability to Disease. Int. J. Mol. Sci. 2019, 20, 2146. [Google Scholar] [CrossRef]
- Yuan, H.H.; Chen, R.J.; Zhu, Y.H.; Peng, C.L.; Zhu, X.R. The neuroprotective effect of overexpression of calbindin-D(28k) in an animal model of Parkinson’s disease. Mol. Neurobiol. 2013, 47, 117–122. [Google Scholar] [CrossRef]
- Mukherjee, A.; Soto, C. Role of calcineurin in neurodegeneration produced by misfolded proteins and endoplasmic reticulum stress. Curr. Opin. Cell Biol. 2011, 23, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Song, M.; Yao, C. Calcineurin in development and disease. Genes Dis. 2021, 9, 915–927. [Google Scholar] [CrossRef] [PubMed]
- DeGiosio, R.A.; Grubisha, M.J.; MacDonald, M.L.; McKinney, B.C.; Camacho, C.J.; Sweet, R.A. More than a marker: Potential pathogenic functions of MAP2. Front. Mol. Neurosci. 2022, 15, 974890. [Google Scholar] [CrossRef] [PubMed]
- Kounakis, K.; Tavernarakis, N. The Cytoskeleton as a Modulator of Aging and Neurodegeneration. Adv. Exp. Med. Biol. 2019, 1178, 227–245. [Google Scholar] [CrossRef] [PubMed]
- Kowall, N.W.; Quigley, B.J., Jr.; Krause, J.E.; Lu, F.; Kosofsky, B.E.; Ferrante, R.J. Substance P and substance P receptor histochemistry in human neurodegenerative diseases. Regul. Pept. 1993, 46, 174–185. [Google Scholar] [CrossRef]
- Jyoti Dutta, B.; Singh, S.; Seksaria, S.; Das Gupta, G.; Bodakhe, S.H.; Singh, A. Potential role of IP3/Ca2+ signaling and phosphodiesterases: Relevance to neurodegeneration in Alzheimer’s disease and possible therapeutic strategies. Biochem. Pharmacol. 2022, 201, 115071. [Google Scholar] [CrossRef]
- Mackay, J.P.; Nassrallah, W.B.; Raymond, L.A. Cause or compensation? Altered neuronal Ca2+ handling in Huntington’s disease. CNS Neurosci. Ther. 2018, 24, 301–310. [Google Scholar] [CrossRef]
- Wang, Q.; Chu, C.H.; Qian, L.; Chen, S.H.; Wilson, B.; Oyarzabal, E.; Jiang, L.; Ali, S.; Robinson, B.; Kim, H.C.; et al. Substance P exacerbates dopaminergic neurodegeneration through neurokinin-1 receptor-independent activation of microglial NADPH oxidase. J. Neurosci. 2014, 34, 12490–12503. [Google Scholar] [CrossRef]
- Gupta, K.K.; Singh, S.K. Cdk5: A main culprit in neurodegeneration. Int. J. Neurosci. 2019, 129, 1192–1197. [Google Scholar] [CrossRef]
- Shah, K.; Rossie, S. Tale of the Good and the Bad Cdk5: Remodeling of the Actin Cytoskeleton in the Brain. Mol. Neurobiol. 2018, 55, 3426–3438. [Google Scholar] [CrossRef]
- Jin, H.; Komita, M.; Aoe, T. The Role of BiP Retrieval by the KDEL Receptor in the Early Secretory Pathway and its Effect on Protein Quality Control and Neurodegeneration. Front. Mol. Neurosci. 2017, 10, 222. [Google Scholar] [CrossRef]
- Sou, S.N.; Ilieva, K.M.; Polizzi, K.M. Binding of human BiP to the ER stress transducers IRE1 and PERK requires ATP. Biochem. Biophys. Res. Commun. 2012, 420, 473–478. [Google Scholar] [CrossRef]
- Lee, S.H.; Meilandt, W.J.; Xie, L.; Gandham, V.D.; Ngu, H.; Barck, K.H.; Rezzonico, M.G.; Imperio, J.; Lalehzadeh, G.; Huntley, M.A.; et al. Trem2 restrains the enhancement of tau accumulation and neurodegeneration by β-amyloid pathology. Neuron 2021, 109, 1283–1301.e6. [Google Scholar] [CrossRef]
- Guo, Y.; Wei, X.; Yan, H.; Qin, Y.; Yan, S.; Liu, J.; Zhao, Y.; Jiang, F.; Lou, H. TREM2 deficiency aggravates α-synuclein-induced neurodegeneration and neuroinflammation in Parkinson’s disease models. FASEB J. 2019, 33, 12164–12174. [Google Scholar] [CrossRef]
- Iurlaro, R.; Muñoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016, 283, 2640–2652. [Google Scholar] [CrossRef]
- Scheper, W.; Hoozemans, J.J. The unfolded protein response in neurodegenerative diseases: A neuropathological perspective. Acta Neuropathol. 2015, 130, 315–331. [Google Scholar] [CrossRef]
- Fischer, R.; Maier, O. Interrelation of oxidative stress and inflammation in neurodegenerative disease: Role of TNF. Oxid. Med. Cell. Longev. 2015, 2015, 610813. [Google Scholar] [CrossRef]
- Brambilla, L.; Guidotti, G.; Martorana, F.; Iyer, A.M.; Aronica, E.; Valori, C.F.; Rossi, D. Disruption of the astrocytic TNFR1-GDNF axis accelerates motor neuron degeneration and disease progression in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2016, 25, 3080–3095. [Google Scholar] [CrossRef][Green Version]
- Klimaschewski, L.; Claus, P. Fibroblast Growth Factor Signalling in the Diseased Nervous System. Mol. Neurobiol. 2021, 58, 3884–3902. [Google Scholar] [CrossRef]
- Pehar, M.; Vargas, M.R.; Cassina, P.; Barbeito, A.G.; Beckman, J.S.; Barbeito, L. Complexity of astrocyte-motor neuron interactions in amyotrophic lateral sclerosis. Neurodegener. Dis. 2005, 2, 139–146. [Google Scholar] [CrossRef]
- Stankiewicz, T.R.; Linseman, D.A. Rho family GTPases: Key players in neuronal development, neuronal survival, and neurodegeneration. Front. Cell. Neurosci. 2014, 8, 314. [Google Scholar] [CrossRef] [PubMed]
- Arrazola Sastre, A.; Luque Montoro, M.; Gálvez-Martín, P.; Lacerda, H.M.; Lucia, A.M.; Llavero, F.; Zugaza, J.L. Small GTPases of the Ras and Rho Families Switch on/off Signaling Pathways in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 6312. [Google Scholar] [CrossRef] [PubMed]
- Habib, R.; Noureen, N.; Nadeem, N. Decoding Common Features of Neurodegenerative Disorders: From Differentially Expressed Genes to Pathways. Curr. Genomics 2018, 19, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Musilli, M.; Ciotti, M.T.; Pieri, M.; Martino, A.; Borrelli, S.; Dinallo, V.; Diana, G. Therapeutic effects of the Rho GTPase modulator CNF1 in a model of Parkinson’s disease. Neuropharmacology 2016, 109, 357–365. [Google Scholar] [CrossRef]
- Reddy, J.M.; Raut, N.G.R.; Seifert, J.L.; Hynds, D.L. Regulation of Small GTPase Prenylation in the Nervous System. Mol. Neurobiol. 2020, 57, 2220–2231. [Google Scholar] [CrossRef]
- Roser, A.E.; Tönges, L.; Lingor, P. Modulation of Microglial Activity by Rho-Kinase (ROCK) Inhibition as Therapeutic Strategy in Parkinson’s Disease and Amyotrophic Lateral Sclerosis. Front. Aging Neurosci. 2017, 9, 94. [Google Scholar] [CrossRef]
- Hughes, D.; Mallucci, G.R. The unfolded protein response in neurodegenerative disorders—Therapeutic modulation of the PERK pathway. FEBS J. 2019, 286, 342–355. [Google Scholar] [CrossRef]
- Taalab, Y.M.; Ibrahim, N.; Maher, A.; Hassan, M.; Mohamed, W.; Moustafa, A.A.; Salama, M.; Johar, D.; Bernstein, L. Mechanisms of disordered neurodegenerative function: Concepts and facts about the different roles of the protein kinase RNA-like endoplasmic reticulum kinase (PERK). Rev. Neurosci. 2018, 29, 387–415. [Google Scholar] [CrossRef]
- Ghemrawi, R.; Khair, M. Endoplasmic Reticulum Stress and Unfolded Protein Response in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 6127. [Google Scholar] [CrossRef]
- Vidal, R.L.; Sepulveda, D.; Troncoso-Escudero, P.; Garcia-Huerta, P.; Gonzalez, C.; Plate, L.; Jerez, C.; Canovas, J.; Rivera, C.A.; Castillo, V.; et al. Enforced dimerization between XBP1s and ATF6f enhances the protective effects of the UPR in models of neurodegeneration. Mol. Ther. 2021, 29, 1862–1882. [Google Scholar] [CrossRef]
- Tziortzouda, P.; van den Bosch, L.; Hirth, F. Triad of TDP43 control in neurodegeneration: Autoregulation, localization and aggregation. Nat. Rev. Neurosci. 2021, 22, 197–208. [Google Scholar] [CrossRef]
- Flores, B.N.; Li, X.; Malik, A.M.; Martinez, J.; Beg, A.A.; Barmada, S.J. An Intramolecular Salt Bridge Linking TDP43 RNA Binding, Protein Stability, and TDP43-Dependent Neurodegeneration. Cell Rep. 2019, 27, 1133–1150.e8. [Google Scholar] [CrossRef]
- Lai, W.F.; Wong, W.T. Roles of the actin cytoskeleton in aging and age-associated diseases. Ageing Res. Rev. 2020, 58, 101021. [Google Scholar] [CrossRef]
- Sen, S.; Lagas, S.; Roy, A.; Kumar, H. Cytoskeleton saga: Its regulation in normal physiology and modulation in neurodegenerative disorders. Eur. J. Pharmacol. 2022, 925, 175001. [Google Scholar] [CrossRef]
- Ross, O.A.; Rutherford, N.J.; Baker, M.; Soto-Ortolaza, A.I.; Carrasquillo, M.M.; DeJesus-Hernandez, M.; Adamson, J.; Li, M.; Volkening, K.; Finger, E.; et al. Ataxin-2 repeat-length variation and neurodegeneration. Hum. Mol. Genet. 2011, 20, 3207–3212. [Google Scholar] [CrossRef]
- Xue, Y.C.; Ng, C.S.; Xiang, P.; Liu, H.; Zhang, K.; Mohamud, Y.; Luo, H. Dysregulation of RNA-Binding Proteins in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2020, 13, 78. [Google Scholar] [CrossRef]
- Blažeković, A.; Jerčić, K.G.; Borovečki, F. SNCA 3′ UTR Genetic Variants in Patients with Parkinson’s Disease. Biomolecules 2021, 11, 1799. [Google Scholar] [CrossRef]
- Wang, Q.; Tian, Q.; Song, X.; Liu, Y.; Li, W. SNCA Gene Polymorphism may Contribute to an Increased Risk of Alzheimer’s Disease. J. Clin. Lab. Anal. 2016, 30, 1092–1099. [Google Scholar] [CrossRef]
- Vilariño-Güell, C.; Wider, C.; Soto-Ortolaza, A.I.; Cobb, S.A.; Kachergus, J.M.; Keeling, B.H.; Dachsel, J.C.; Hulihan, M.M.; Dickson, D.W.; Wszolek, Z.K.; et al. Characterization of DCTN1 genetic variability in neurodegeneration. Neurology 2009, 72, 2024–2028. [Google Scholar] [CrossRef]
- Xu, H.; Jia, J. Immune-Related Hub Genes and the Competitive Endogenous RNA Network in Alzheimer’s Disease. J. Alzheimers Dis. 2020, 77, 1255–1265. [Google Scholar] [CrossRef]
- Drobny, A.; Prieto Huarcaya, S.; Dobert, J.; Kluge, A.; Bunk, J.; Schlothauer, T.; Zunke, F. The role of lysosomal cathepsins in neurodegeneration: Mechanistic insights, diagnostic potential and therapeutic approaches. Biochim. Biophys. Acta Mol. Cell Res. 2022, 1869, 119243. [Google Scholar] [CrossRef] [PubMed]
- Vidoni, C.; Follo, C.; Savino, M.; Melone, M.A.; Isidoro, C. The Role of Cathepsin D in the Pathogenesis of Human Neurodegenerative Disorders. Med. Res. Rev. 2016, 36, 845–870. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Cha, S.J.; Lee, J.W.; Kim, H.J.; Kim, K. Recent Advances on the Role of GSK3β in the Pathogenesis of Amyotrophic Lateral Sclerosis. Brain Sci. 2020, 10, 675. [Google Scholar] [CrossRef] [PubMed]
- Duda, P.; Wiśniewski, J.; Wójtowicz, T.; Wójcicka, O.; Jaśkiewicz, M.; Drulis-Fajdasz, D.; Rakus, D.; McCubrey, J.A.; Gizak, A. Targeting GSK3 signaling as a potential therapy of neurodegenerative diseases and aging. Expert Opin. Ther. Targets 2018, 22, 833–848. [Google Scholar] [CrossRef]
- Hu, X.; Zhou, X.; He, W.; Yang, J.; Xiong, W.; Wong, P.; Wilson, C.G.; Yan, R. BACE1 deficiency causes altered neuronal activity and neurodegeneration. J. Neurosci. 2010, 30, 8819–8829. [Google Scholar] [CrossRef]
- Spadoni, O.; Crestini, A.; Piscopo, P.; Malvezzi-Campeggi, L.; Carunchio, I.; Pieri, M.; Zona, C.; Confaloni, A. Gene expression profiles of APP and BACE1 in Tg SOD1G93A cortical cells. Cell. Mol. Neurobiol. 2009, 29, 635–641. [Google Scholar] [CrossRef]
- Gerson, J.E.; Sandoval-Pistorius, S.; Welday, J.P.; Rodriguez, A.; Gregory, J.D.; Liggans, N.; Schache, K.; Li, X.; Trzeciakiewicz, H.; Barmada, S.; et al. Disrupting the Balance of Protein Quality Control Protein UBQLN2 Accelerates Tau Proteinopathy. J. Neurosci. 2022, 42, 1845–1863. [Google Scholar] [CrossRef]
- Lee, Y.; Lee, B.H.; Yip, W.; Chou, P.; Yip, B.S. Neurofilament Proteins as Prognostic Biomarkers in Neurological Disorders. Curr. Pharm. Des. 2020, 25, 4560–4569. [Google Scholar] [CrossRef]
- De Los Reyes Corrales, T.; Losada-Pérez, M.; Casas-Tintó, S. JNK Pathway in CNS Pathologies. Int. J. Mol. Sci. 2021, 22, 3883. [Google Scholar] [CrossRef]
- Huang, Q.; Du, X.; He, X.; Yu, Q.; Hu, K.; Breitwieser, W.; Shen, Q.; Ma, S.; Li, M. JNK-mediated activation of ATF2 contributes to dopaminergic neurodegeneration in the MPTP mouse model of Parkinson’s disease. Exp. Neurol. 2016, 277, 296–304. [Google Scholar] [CrossRef]
- Zheng, C.; Zhou, X.W.; Wang, J.Z. The dual roles of cytokines in Alzheimer’s disease: Update on interleukins, TNF-α, TGF-β and IFN-γ. Transl. Neurodegener. 2016, 5, 7. [Google Scholar] [CrossRef]
- Ejlerskov, P.; Hultberg, J.G.; Wang, J.; Carlsson, R.; Ambjørn, M.; Kuss, M.; Liu, Y.; Porcu, G.; Kolkova, K.; Friis Rundsten, C.; et al. Lack of Neuronal IFN-β-IFNAR Causes Lewy Body- and Parkinson’s Disease-like Dementia. Cell 2015, 163, 324–339. [Google Scholar] [CrossRef]
- Wilhelmus, M.M.; Otte-Höller, I.; Wesseling, P.; de Waal, R.M.; Boelens, W.C.; Verbeek, M.M. Specific association of small heat shock proteins with the pathological hallmarks of Alzheimer’s disease brains. Neuropathol. Appl. Neurobiol. 2006, 32, 119–130. [Google Scholar] [CrossRef]
- Narayan, M.; Seeley, K.W.; Jinwal, U.K. Identification of Apo B48 and other novel biomarkers in amyotrophic lateral sclerosis patient fibroblasts. Biomark. Med. 2016, 10, 453–462. [Google Scholar] [CrossRef]
- Criado-Marrero, M.; Blazier, D.M.; Gould, L.A.; Gebru, N.T.; Rodriguez Ospina, S.; Armendariz, D.S.; Darling, A.L.; Beaulieu-Abdelahad, D.; Blair, L.J. Evidence against a contribution of the CCAAT-enhancer binding protein homologous protein (CHOP) in mediating neurotoxicity in rTg4510 mice. Sci. Rep. 2022, 12, 7372. [Google Scholar] [CrossRef]
- Gavilán, M.P.; Pintado, C.; Gavilán, E.; Jiménez, S.; Ríos, R.M.; Vitorica, J.; Castaño, A.; Ruano, D. Dysfunction of the unfolded protein response increases neurodegeneration in aged rat hippocampus following proteasome inhibition. Aging Cell 2009, 8, 654–665. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Das, S.; Hyman, B.T. APOE and Alzheimer’s disease: Advances in genetics, pathophysiology, and therapeutic approaches. Lancet Neurol. 2021, 20, 68–80. [Google Scholar] [CrossRef]
- Rozpędek-Kamińska, W.; Siwecka, N.; Wawrzynkiewicz, A.; Wojtczak, R.; Pytel, D.; Diehl, J.A.; Majsterek, I. The PERK-Dependent Molecular Mechanisms as a Novel Therapeutic Target for Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 2108. [Google Scholar] [CrossRef]
- Bruni, A.C.; Bernardi, L.; Gabelli, C. From beta amyloid to altered proteostasis in Alzheimer’s disease. Ageing Res. Rev. 2020, 64, 101126. [Google Scholar] [CrossRef]
- Kim, B.; Choi, Y.; Kim, H.S.; Im, H.I. Methyl-CpG Binding Protein 2 in Alzheimer Dementia. Int. Neurourol. J. 2019, 23 (Suppl. S2), S72–S81. [Google Scholar] [CrossRef]
- Del-Aguila, J.L.; Koboldt, D.C.; Black, K.; Chasse, R.; Norton, J.; Wilson, R.K.; Cruchaga, C. Alzheimer’s disease: Rare variants with large effect sizes. Curr. Opin. Genet. Dev. 2015, 33, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Dias, V.; Junn, E.; Mouradian, M.M. The role of oxidative stress in Parkinson’s disease. J. Parkinsons Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Vélez, G.E.; Zoghbi, H.Y. Parkinson’s Disease Genetics and Pathophysiology. Annu. Rev. Neurosci. 2021, 44, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.S.; Geng, W.S.; Jia, J.J.; Chen, L.; Zhang, P.P. Cellular and Molecular Basis of Neurodegeneration in Parkinson Disease. Front. Aging Neurosci. 2018, 10, 109. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Meng, L.; He, M.; Zhang, Z. Tau in the Pathophysiology of Parkinson’s Disease. J. Mol. Neurosci. 2021, 71, 2179–2191. [Google Scholar] [CrossRef]
- Cardona, F.; Perez-Tur, J. Other Proteins Involved in Parkinson’s Disease and Related Disorders. Curr. Protein Pept. Sci. 2017, 18, 765–778. [Google Scholar] [CrossRef]
- Cacabelos, R. Parkinson’s Disease: From Pathogenesis to Pharmacogenomics. Int. J. Mol. Sci. 2017, 18, 551. [Google Scholar] [CrossRef]
- Puschmann, A. New Genes Causing Hereditary Parkinson’s Disease or Parkinsonism. Curr. Neurol. Neurosci. Rep. 2017, 17, 66. [Google Scholar] [CrossRef]
- Chen, C.M.; Chen, Y.C.; Chiang, M.C.; Fung, H.C.; Chang, K.H.; Lee-Chen, G.J.; Wu, Y.R. Association of GCH1 and MIR4697, but not SIPA1L2 and VPS13C polymorphisms, with Parkinson’s disease in Taiwan. Neurobiol. Aging 2016, 39, 221.e1–221.e5. [Google Scholar] [CrossRef]
- Feldman, E.L.; Goutman, S.A.; Petri, S.; Mazzini, L.; Savelieff, M.G.; Shaw, P.J.; Sobue, G. Amyotrophic lateral sclerosis. Lancet 2022, 400, 1363–1380. [Google Scholar] [CrossRef]
- Kiskinis, E.; Sandoe, J.; Williams, L.A.; Boulting, G.L.; Moccia, R.; Wainger, B.J.; Han, S.; Peng, T.; Thams, S.; Mikkilineni, S.; et al. Pathways disrupted in human ALS motor neurons identified by genetic correction of mutant SOD1. Cell Stem Cell 2014, 14, 781–795. [Google Scholar] [CrossRef]
- Chen, J.; Bassot, A.; Giuliani, F.; Simmen, T. Amyotrophic Lateral Sclerosis (ALS): Stressed by Dys-functional Mitochondria-Endoplasmic Reticulum Contacts (MERCs). Cells 2021, 10, 1789. [Google Scholar] [CrossRef]
- Kaus, A.; Sareen, D. ALS Patient Stem Cells for Unveiling Disease Signatures of Motoneuron Susceptibility: Perspectives on the Deadly Mitochondria, ER Stress and Calcium Triad. Front. Cell Neurosci. 2015, 9, 448. [Google Scholar] [CrossRef]
- Kirola, L.; Mukherjee, A.; Mutsuddi, M. Recent Updates on the Genetics of Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Mol. Neurobiol. 2022, 59, 5673–5694. [Google Scholar] [CrossRef]
- Maurel, C.; Dangoumau, A.; Marouillat, S.; Brulard, C.; Chami, A.; Hergesheimer, R.; Corcia, P.; Blasco, H.; Andres, C.R.; Vourc’h, P. Causative Genes in Amyotrophic Lateral Sclerosis and Protein Degradation Pathways: A Link to Neurodegeneration. Mol. Neurobiol. 2018, 55, 6480–6499. [Google Scholar] [CrossRef]
- Lambert-Smith, I.A.; Saunders, D.N.; Yerbury, J.J. Proteostasis impairment and ALS. Prog. Biophys. Mol. Biol. 2022, 174, 3–27. [Google Scholar] [CrossRef]
- Schymick, J.C.; Talbot, K.; Traynor, B.J. Genetics of sporadic amyotrophic lateral sclerosis. Hum. Mol. Genet. 2007, 16, R233–R242. [Google Scholar] [CrossRef]
- Rayner, S.L.; Cheng, F.; Hogan, A.L.; Grima, N.; Yang, S.; Ke, Y.D.; Au, C.G.; Morsch, M.; de Luca, A.; Davidson, J.M.; et al. ALS/FTD-causing mutation in cyclin F causes the dysregulation of SFPQ. Hum. Mol. Genet. 2021, 30, 971–984. [Google Scholar] [CrossRef]
- Yerbury, J.J.; Farrawell, N.E.; McAlary, L. Proteome Homeostasis Dysfunction: A Unifying Principle in ALS Pathogenesis. Trends Neurosci. 2020, 43, 274–284. [Google Scholar] [CrossRef]
- Johnson, R.T. Prion diseases. Lancet Neurol. 2005, 4, 635–642. [Google Scholar] [CrossRef]
- Otero, A.; Betancor, M.; Eraña, H.; Borges, N.F.; Lucas, J.J.; Badiola, J.J.; Castilla, J.; Bolea, R. Prion-Associated Neurodegeneration Causes Both Endoplasmic Reticulum Stress and Proteasome Impairment in a Murine Model of Spontaneous Disease. Int. J. Mol. Sci. 2021, 22, 465. [Google Scholar] [CrossRef] [PubMed]
- Torres, M.; Castillo, K.; Armisén, R.; Stutzin, A.; Soto, C.; Hetz, C. Prion protein misfolding affects calcium homeostasis and sensitizes cells to endoplasmic reticulum stress. PLoS ONE 2010, 5, e15658. [Google Scholar] [CrossRef] [PubMed]
- Apetri, A.C.; Surewicz, K.; Surewicz, W.K. The effect of disease-associated mutations on the folding pathway of human prion protein. J. Biol. Chem. 2004, 279, 18008–18014. [Google Scholar] [CrossRef] [PubMed]
- Majer, A.; Medina, S.J.; Sorensen, D.; Martin, M.J.; Frost, K.L.; Phillipson, C.; Manguiat, K.; Booth, S.A. The cell type resolved mouse transcriptome in neuron-enriched brain tissues from the hippocampus and cerebellum during prion disease. Sci. Rep. 2019, 9, 1099. [Google Scholar] [CrossRef]
- Lashuel, H.A. Rethinking protein aggregation and drug discovery in neurodegenerative diseases: Why we need to embrace complexity? Curr. Opin. Chem. Biol. 2021, 64, 67–75. [Google Scholar] [CrossRef]
- Brehme, M.; Voisine, C.; Rolland, T.; Wachi, S.; Soper, J.H.; Zhu, Y.; Orton, K.; Villella, A.; Garza, D.; Vidal, M.; et al. A Chaperome Sub-Network Safeguards Proteostasis in Aging and Neuro-degenerative Disease. Cell Rep. 2014, 9, 1135–1150. [Google Scholar] [CrossRef]
- Klim, J.R.; Pintacuda, G.; Nash, L.A.; Guerra San Juan, I.; Eggan, K. Connecting TDP-43 Pathology with Neuropathy. Trends Neurosci. 2021, 44, 424–440. [Google Scholar] [CrossRef]
- Shih, Y.T.; Hsueh, Y.P. The involvement of endoplasmic reticulum formation and protein synthesis efficiency in VCP- and ATL1-related neurological dis orders. J. Biomed. Sci. 2018, 25, 2. [Google Scholar] [CrossRef]
- Hetz, C.; Saxena, S. ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 2017, 13, 477–491. [Google Scholar] [CrossRef]
- Zhu, B.; Jiang, L.; Huang, T.; Zhao, Y.; Liu, T.; Zhong, Y.; Li, X.; Campos, A.; Pomeroy, K.; Masliah, E.; et al. ER-associated degradation regulates Alzheimer’s amyloid pathology and memory function by modulating γ-secretase activity. Nat. Commun. 2017, 8, 1472. [Google Scholar] [CrossRef]
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019, 20, 421–435. [Google Scholar] [CrossRef]
- Guo, T.; Zhang, D.; Zeng, Y.; Huang, T.Y.; Xu, H.; Zhao, Y. Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Mol. Neurodegener. 2020, 15, 40. [Google Scholar] [CrossRef]
- Asai, M.; Kinjo, A.; Kimura, S.; Mori, R.; Kawakubo, T.; Shirotani, K.; Yagishita, S.; Maruyama, K.; Iwata, N. Perturbed Calcineurin-NFAT Signaling Is Associated with the Development of Alzheimer’s Disease. Biol. Pharm. Bull. 2016, 39, 1646–1652. [Google Scholar] [CrossRef]
- Song, S.; Miranda, C.J.; Braun, L.; Meyer, K.; Frakes, A.E.; Ferraiuolo, L.; Likhite, S.; Bevan, A.K.; Foust, K.D.; McConnell, M.J.; et al. Major histocompatibility complex class I molecules protect motor neurons from astrocyte-induced toxicity in amyotrophic lateral sclerosis. Nat. Med. 2016, 22, 397–403. [Google Scholar] [CrossRef]
- Brini, M.; Calì, T.; Ottolini, D.; Carafoli, E. Neuronal calcium signaling: Function and dysfunction. Cell. Mol. Life Sci. 2014, 71, 2787–2814. [Google Scholar] [CrossRef]
- Karagas, N.E.; Venkatachalam, K. Roles for the Endoplasmic Reticulum in Regulation of Neuronal Calcium Homeostasis. Cells 2019, 8, 1232. [Google Scholar] [CrossRef]
- Chanaday, N.L.; Nosyreva, E.; Shin, O.H.; Zhang, H.; Aklan, I.; Atasoy, D.; Bezprozvanny, I.; Kavalali, E.T. Presynaptic store-operated Ca2+ entry drives excitatory spontaneous neurotransmission and augments endoplasmic reticulum stress. Neuron 2021, 109, 1314–1332.e5. [Google Scholar] [CrossRef]
- Britzolaki, A.; Saurine, J.; Klocke, B.; Pitychoutis, P.M. A Role for SERCA Pumps in the Neurobiology of Neuropsychiatric and Neurodegenerative Disorders. Adv. Exp. Med. Biol. 2020, 1131, 131–161. [Google Scholar] [CrossRef]
- Sicari, D.; Delaunay-Moisan, A.; Combettes, L.; Chevet, E.; Igbaria, A. A guide to assessing endoplasmic reticulum homeostasis and stress in mammalian systems. FEBS J. 2020, 287, 27–42. [Google Scholar] [CrossRef]
- Paschen, W.; Mengesdorf, T. Endoplasmic reticulum stress response and neurodegeneration. Cell Calcium 2005, 38, 409–415. [Google Scholar] [CrossRef]
- Mammucari, C.; Raffaello, A.; Vecellio Reane, D.; Gherardi, G.; de Mario, A.; Rizzuto, R. Mitochondrial calcium uptake in organ physiology: From molecular mechanism to animal models. Pflug. Arch. 2018, 470, 1165–1179. [Google Scholar] [CrossRef] [PubMed]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011, 476, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Raturi, A.; Simmen, T. Where the endoplasmic reticulum and the mitochondrion tie the knot: The mitochondria-associated membrane (MAM). Biochim. Biophys. Acta 2013, 1833, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Pchitskaya, E.; Popugaeva, E.; Bezprozvanny, I. Calcium signaling and molecular mechanisms underlying neurodegenerative diseases. Cell Calcium 2018, 70, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Jadiya, P.; Garbincius, J.F.; Elrod, J.W. Reappraisal of metabolic dysfunction in neurodegeneration: Focus on mitochondrial function and calcium signaling. Acta Neuropathol. Commun. 2021, 9, 124. [Google Scholar] [CrossRef]
- Puri, B.K. Calcium Signaling and Gene Expression. Adv. Exp. Med. Biol. 2020, 1131, 537–545. [Google Scholar] [CrossRef]
- Esteras, N.; Abramov, A.Y. Mitochondrial Calcium Deregulation in the Mechanism of Beta-Amyloid and Tau Pathology. Cells 2020, 9, 2135. [Google Scholar] [CrossRef]
- Verkhratsky, A. The endoplasmic reticulum and neuronal calcium signaling. Cell Calcium 2002, 32, 393–404. [Google Scholar] [CrossRef]
- Wang, J.H.; Kelly, P.T. Postsynaptic calcineurin activity downregulates synaptic transmission by weakening intracellular Ca2+ signaling mechanisms in hippocampal CA1 neurons. J. Neurosci. 1997, 17, 4600–4611. [Google Scholar] [CrossRef]
- Benkert, J.; Hess, S.; Roy, S.; Beccano-Kelly, D.; Wiederspohn, N.; Duda, J.; Simons, C.; Patil, K.; Gaifullina, A.; Mannal, N.; et al. Cav2.3 channels contribute to dopaminergic neuron loss in a model of Parkinson’s disease. Nat Commun. 2019, 10, 5094. [Google Scholar] [CrossRef]
- Leandrou, E.; Emmanouilidou, E.; Vekrellis, K. Voltage-Gated Calcium Channels and alpha-Synuclein: Implications in Parkin-son’s Disease. Front. Mol. Neurosci. 2019, 12, 237. [Google Scholar] [CrossRef]
- Sun, W.Y.; Tyurin, V.A.; Mikulska-Ruminska, K.; Shrivastava, I.H.; Anthonymuthu, T.S.; Zhai, Y.J.; Pan, M.H.; Gong, H.B.; Lu, D.H.; Sun, J.; et al. Phospholipase iPLA2β averts ferroptosis by eliminating a redox lipid death signal. Nat. Chem. Biol. 2021, 17, 465–476. [Google Scholar] [CrossRef]
- Yamada, T.; McGeer, P.L.; Baimbridge, K.G.; McGeer, E.G. Relative sparing in Parkinson’s disease of substantia nigra dopamine neurons containing calbindin-D28K. Brain Res. 1990, 526, 303–307. [Google Scholar] [CrossRef]
- Knörle, R. Neuromelanin in Parkinson’s Disease: From Fenton Reaction to Calcium Signaling. Neurotox. Res. 2018, 33, 515–522. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, R.; Wang, G. Impact of Dopamine Oxidation on Dopaminergic Neurodegeneration. ACS Chem. Neurosci. 2019, 10, 945–953. [Google Scholar] [CrossRef]
- Parent, A.; Fortin, M.; Côté, P.Y.; Cicchetti, F. Calcium-binding proteins in primate basal ganglia. Neurosci. Res. 1996, 25, 309–334. [Google Scholar] [CrossRef]
- Czeredys, M. Dysregulation of Neuronal Calcium Signaling via Store-Operated Channels in Huntington’s Disease. Front. Cell. Dev. Biol. 2020, 8, 611735. [Google Scholar] [CrossRef]
- Giacomello, M.; Oliveros, J.C.; Naranjo, J.R.; Carafoli, E. Neuronal Ca2+ dyshomeostasis in Huntington disease. Prion 2013, 7, 76–84. [Google Scholar] [CrossRef]
- Schmidt, M.E.; Caron, N.S.; Aly, A.E.; Lemarié, F.L.; Dal Cengio, L.; Ko, Y.; Lazic, N.; Anderson, L.; Nguyen, B.; Raymond, L.A.; et al. DAPK1 Promotes Extrasynaptic GluN2B Phos-phorylation and Striatal Spine Instability in the YAC128 Mouse Model of Huntington Disease. Front. Cell. Neurosci. 2020, 14, 590569. [Google Scholar] [CrossRef]
- Ueda, M.; Li, S.; Itoh, M.; Hayakawa-Yano, Y.; Wang, M.X.; Hayakawa, M.; Hasebe-Matsubara, R.; Ohta, K.; Ohta, E.; Mizuno, A.; et al. Polyglutamine expansion disturbs the endoplasmic reticulum formation, leading to caspase-7 activation through Bax. Biochem. Biophys. Res. Commun. 2014, 443, 1232–1238. [Google Scholar] [CrossRef]
- Giacomello, M.; Hudec, R.; Lopreiato, R. Huntington’s disease, calcium, and mitochondria. Biofactors 2011, 37, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Rozas, P.; Bargsted, L.; Martínez, F.; Hetz, C.; Medinas, D.B. The ER proteostasis network in ALS: Determining the differential mo-toneuron vulnerability. Neurosci. Lett. 2017, 636, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, H.; Manfredi, G. Mitochondrial dysfunction and intracellular calcium dysregulation in ALS. Mech. Ageing Dev. 2010, 131, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Gregory, J.M.; Livesey, M.R.; McDade, K.; Selvaraj, B.T.; Barton, S.K.; Chandran, S.; Smith, C. Dysregulation of AMPA receptor subunit expression in sporadic ALS post-mortem brain. J. Pathol. 2020, 250, 67–78. [Google Scholar] [CrossRef]
- Alexianu, M.E.; Ho, B.K.; Mohamed, A.H.; La Bella, V.; Smith, R.G.; Appel, S.H. The role of calcium-binding proteins in selective moto-neuron vulnerability in amyotrophic lateral sclerosis. Ann. Neurol. 1994, 36, 846–858. [Google Scholar] [CrossRef]
- Coussee, E.; de Smet, P.; Bogaert, E.; Elens, I.; van Damme, P.; Willems, P.; Koopman, W.; van den Bosch, L.; Callewaert, G. G37R SOD1 mutant alters mitochondrial complex I activity, Ca(2+) uptake and ATP production. Cell Calcium 2011, 49, 217–225. [Google Scholar] [CrossRef]
- Hong, J.M.; Moon, J.H.; Park, S.Y. Human prion protein-mediated calcineurin activation induces neuron cell death via AMPK and autophagy pathway. Int. J. Biochem. Cell Biol. 2020, 119, 105680. [Google Scholar] [CrossRef]
- Bertoli, A.; Sorgato, M.C. Neuronal pathophysiology featuring PrPC and its control over Ca2+ metabolism. Prion 2018, 12, 28–33. [Google Scholar] [CrossRef]
- Double, K.L.; Reyes, S.; Werry, E.L.; Halliday, G.M. Selective cell death in neurodegeneration: Why are some neurons spared in vulnerable regions? Prog. Neurobiol. 2010, 92, 316–329. [Google Scholar] [CrossRef]
- Subramaniam, S. Selective Neuronal Death in Neurodegenerative Diseases: The Ongoing Mystery. Yale J. Biol. Med. 2019, 92, 695–705. [Google Scholar]
- Saxena, S.; Caroni, P. Selective neuronal vulnerability in neurodegenerative diseases: From stressor thresholds to degeneration. Neuron 2011, 71, 35–48. [Google Scholar] [CrossRef]
- Michel, P.P.; Hirsch, E.C.; Hunot, S. Understanding Dopaminergic Cell Death Pathways in Parkinson Disease. Neuron 2016, 90, 675–691. [Google Scholar] [CrossRef]
- Kweon, J.H.; Kim, S.; Lee, S.B. The cellular basis of dendrite pathology in neurodegenerative diseases. BMB Rep. 2017, 50, 5–11. [Google Scholar] [CrossRef]
- Higley, M.J.; Sabatini, B.L. Calcium signaling in dendrites and spines: Practical and functional considerations. Neuron 2008, 59, 902–913. [Google Scholar] [CrossRef]
- Konur, S.; Ghosh, A. Calcium signaling and the control of dendritic development. Neuron 2005, 46, 401–405. [Google Scholar] [CrossRef]
- Vizard, T.N.; O’Keeffe, G.W.; Gutierrez, H.; Kos, C.H.; Riccardi, D.; Davies, A.M. Regulation of axonal and dendritic growth by the extracellular calcium-sensing receptor. Nat. Neurosci. 2008, 11, 285–291. [Google Scholar] [CrossRef]
- Arendt, T.; Brückner, M.K.; Bigl, V.; Marcova, L. Dendritic reorganisation in the basal forebrain under degenerative conditions and its defects in Alzheimer’s disease. II. Ageing, Korsakoff’s disease, Parkinson’s disease, and Alzheimer’s disease. J. Comp. Neurol. 1995, 351, 189–222. [Google Scholar] [CrossRef]
- Mrdjen, D.; Fox, E.J.; Bukhari, S.A.; Montine, K.S.; Bendall, S.C.; Montine, T.J. The basis of cellular and regional vulnerability in Alzheimer’s disease. Acta Neuropathol. 2019, 138, 729–749. [Google Scholar] [CrossRef]
- Morrison, B.M.; Hof, P.R.; Morrison, J.H. Determinants of neuronal vulnerability in neurodegenerative diseases. Ann. Neurol. 1998, 44 (Suppl. S1), S32–S44. [Google Scholar] [CrossRef]
- Surmeier, D.J.; Obeso, J.A.; Halliday, G.M. Selective neuronal vulnerability in Parkinson disease. Nat. Rev. Neurosci. 2017, 18, 101–113. [Google Scholar] [CrossRef]
- Han, I.; You, Y.; Kordower, J.H.; Brady, S.T.; Morfini, G.A. Differential vulnerability of neurons in Huntington’s disease: The role of cell type-specific features. J. Neurochem. 2010, 113, 1073–1091. [Google Scholar] [CrossRef] [PubMed]
- Luebke, J.I.; Weaver, C.M.; Rocher, A.B.; Rodriguez, A.; Crimins, J.L.; Dickstein, D.L.; Wearne, S.L.; Hof, P.R. Dendritic vulnerability in neurodegenerative disease: Insights from analyses of cortical pyramidal neurons in transgenic mouse models. Brain Struct. Funct. 2010, 214, 181–199. [Google Scholar] [CrossRef] [PubMed]
- Wojda, U.; Salinska, E.; Kuznicki, J. Calcium ions in neuronal degeneration. IUBMB Life 2008, 60, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Gleichmann, M.; Cheng, A. Mitochondria in neuroplasticity and neurological disorders. Neuron 2008, 60, 748–766. [Google Scholar] [CrossRef]
- Rossoll, W.; Bassell, G.J. Crosstalk of Local Translation and Mitochondria: Powering Plasticity in Axons and Dendrites. Neuron 2019, 101, 204–206. [Google Scholar] [CrossRef]
- Caracci, M.O.; Fuentealba, L.M.; Marzolo, M.P. Golgi Complex Dynamics and Its Implication in Prevalent Neurological Disorders. Front. Cell Dev. Biol. 2019, 7, 75. [Google Scholar] [CrossRef]
- Chung, C.G.; Kwon, M.J.; Jeon, K.H.; Hyeon, D.Y.; Han, M.H.; Park, J.H.; Cha, I.J.; Cho, J.H.; Kim, K.; Rho, S.; et al. Golgi Outpost Synthesis Impaired by Toxic Polyglutamine Proteins Contributes to Dendritic Pathology in Neurons. Cell Rep. 2017, 20, 356–369. [Google Scholar] [CrossRef]
- Mattson, M.P.; Magnus, T. Ageing and neuronal vulnerability. Nat. Rev. Neurosci. 2006, 7, 278–294. [Google Scholar] [CrossRef]
- Fu, H.; Hardy, J.; Duff, K.E. Selective vulnerability in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1350–1358. [Google Scholar] [CrossRef]
- Diack, A.B.; Head, M.W.; McCutcheon, S.; Boyle, A.; Knight, R.; Ironside, J.W.; Manson, J.C.; Will, R.G. Variant CJD. 18 years of research and surveillance. Prion 2014, 8, 286–295. [Google Scholar] [CrossRef]
- Harris, D.A.; Heather, L. New Insights into Prion Structure and Toxicity. Neuron 2006, 50, 353–357. [Google Scholar] [CrossRef]
- Chételat, G.; La Joie, R.; Villain, N.; Perrotin, A.; de La Sayette, V.; Eustache, F.; Vandenberghe, R. Amyloid imaging in cognitively normal individuals, at-risk populations and preclinical Alzheimer’s disease. Neuroimage Clin. 2013, 2, 356–365. [Google Scholar] [CrossRef]
- Nijssen, J.; Comley, L.H.; Hedlund, E. Motor neuron vulnerability and resistance in amyotrophic lateral sclerosis. Acta Neuropathol. 2017, 133, 863–885. [Google Scholar] [CrossRef]
- Tong, B.C.; Wu, A.J.; Li, M.; Cheung, K.H. Calcium signaling in Alzheimer’s disease & therapies. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1745–1760. [Google Scholar] [CrossRef]
- Tradewell, M.L.; Cooper, L.A.; Minotti, S.; Durham, H.D. Calcium dysregulation, mitochondrial pathology and protein aggregation in a culture model of amyotrophic lateral sclerosis: Mechanistic relationship and differential sensitivity to intervention. Neurobiol. Dis. 2011, 42, 265–275. [Google Scholar] [CrossRef]
- Lim, D.; Dematteis, G.; Tapella, L.; Genazzani, A.A.; Calì, T.; Brini, M.; Verkhratsky, A. Ca2+ handling at the mitochondria-ER contact sites in neurodegeneration. Cell Calcium 2021, 98, 102453. [Google Scholar] [CrossRef]
- Kulkarni, V.A.; Firestein, B.L. The dendritic tree and brain disorders. Mol. Cell. Neurosci. 2012, 50, 10–20. [Google Scholar] [CrossRef]
- Emoto, K. Dendrite remodeling in development and disease. Dev. Growth Differ. 2011, 53, 277–286. [Google Scholar] [CrossRef]
- Jagadha, V.; Becker, L.E. Dendritic pathology: An overview of Golgi studies in man. Can. J. Neurol. Sci. 1989, 16, 41–50. [Google Scholar] [CrossRef]
- McAllister, A.K. Neurotrophins and neuronal differentiation in the central nervous system. Cell. Mol. Life Sci. 2001, 58, 1054–1060. [Google Scholar] [CrossRef]
- Ng, T.; Ryu, J.R.; Sohn, J.H.; Tan, T.; Song, H.; Ming, G.L.; Goh, E.L. Class 3 semaphorin mediates dendrite growth in adult newborn neurons through Cdk5/FAK pathway. PLoS ONE 2013, 8, e65572. [Google Scholar] [CrossRef] [PubMed]
- Keeler, A.B.; Molumby, M.J.; Weiner, J.A. Protocadherins branch out: Multiple roles in dendrite development. Cell Adhes. Migr. 2015, 9, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Quach, T.T.; Massicotte, G.; Belin, M.F.; Honnorat, J.; Glasper, E.R.; Devries, A.C.; Jakeman, L.B.; Baudry, M.; Duchemin, A.M.; Kolattukudy, P.E. CRMP3 is required for hippocampal CA1 dendritic organization and plasticity. FASEB J. 2008, 22, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Quach, T.T.; Auvergnon, N.; Khanna, R.; Belin, M.F.; Kolattukudy, P.E.; Honnorat, J.; Duchemin, A.M. Opposing Morphogenetic Defects on Dendrites and Mossy Fibers of Dentate Granular Neurons in CRMP3-Deficient Mice. Brain Sci. 2018, 8, 196. [Google Scholar] [CrossRef] [PubMed]
- Quach, T.T.; Wilson, S.M.; Rogemond, V.; Chounlamountri, N.; Kolattukudy, P.E.; Martinez, S.; Khanna, M.; Belin, M.F.; Khanna, R.; Honnorat, J.; et al. Mapping CRMP3 domains involved in dendrite morphogenesis and voltage-gated calcium channel regulation. J. Cell Sci. 2013, 126, 4262–4273. [Google Scholar] [CrossRef]
- Quach, T.T.; Stratton, H.J.; Khanna, R.; Kolattukudy, P.E.; Honnorat, J.; Meyer, K.; Duchemin, A.M. Intellectual disability: Dendritic anomalies and emerging genetic perspectives. Acta Neuropathol. 2021, 141, 139–158. [Google Scholar] [CrossRef]
- Brittain, J.M.; Duarte, D.B.; Wilson, S.M.; Zhu, W.; Ballard, C.; Johnson, P.L.; Liu, N.; Xiong, W.; Ripsch, M.S.; Wang, Y.; et al. Suppression of inflammatory and neuropathic pain by uncoupling CRMP-2 from the presynaptic Ca2+ channel complex. Nat. Med. 2011, 17, 822–829. [Google Scholar] [CrossRef]
- Jeanne, M.; Demory, H.; Moutal, A.; Khanna, R.; Dobyns, W.B.; Bézieau, S.; Lohkamp, B.; Toutain, A.; Laumonnier, F.; Honnorat, J.; et al. Missense variants in DPYSL5 cause a neurodevelopmental disorder with corpus callosum agenesis and cerebellar abnormalities. Am. J. Hum. Genet. 2021, 108, 951–961. [Google Scholar] [CrossRef]
- Graus, F.; Titulaer, M.J.; Balu, R.; Benseler, S.; Bien, C.G.; Cellucci, T.; Cortese, I.; Dale, R.C.; Gelfand, J.M.; Geschwind, M.; et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol. 2016, 15, 391–404. [Google Scholar] [CrossRef]
- Quach, T.T.; Moutal, A.; Khanna, R.; Deems, N.P.; Duchemin, A.M.; Barrientos, R.M. Collapsin Response Mediator Proteins: Novel Targets for Alzheimer’s Disease. J. Alzheimers Dis. 2020, 77, 949–960. [Google Scholar] [CrossRef]
- Khanna, R.; Wilson, S.M.; Brittain, J.M.; Weimer, J.; Sultana, R.; Butterfield, A.; Hensley, K. Opening Pandora’s jar: A primer on the putative roles of CRMP2 in a panoply of neurodegenerative, sensory and motor neuron, and central disorders. Future Neurol. 2012, 7, 749–771. [Google Scholar] [CrossRef]
- Moutal, A.; White, K.A.; Chefdeville, A.; Laufmann, R.N.; Vitiello, P.F.; Feinstein, D.; Weimer, J.M.; Khanna, R. Dysregulation of CRMP2 Post-Translational Modifications Drive Its Pathological Functions. Mol. Neurobiol. 2019, 56, 6736–6755. [Google Scholar] [CrossRef]
- Knudsen, A.; Bredholt, G.; Storstein, A.; Oltedal, L.; Davanger, S.; Krossnes, B.; Honnorat, J.; Vedeler, C.A. Antibodies to CRMP3-4 associated with limbic encephalitis and thymoma. Clin. Exp. Immunol. 2007, 149, 16–22. [Google Scholar] [CrossRef]
- Yang, Y.; Zhao, B.; Ji, Z.; Zhang, G.; Zhang, J.; Li, S.; Guo, G.; Lin, H. CRMPs colocalize and interact with cytoskeleton in hippocampal neurons. Int. J. Clin. Exp. Med. 2015, 8, 22337–22344. [Google Scholar]
- Deo, R.C.; Schmidt, E.F.; Elhabazi, A.; Togashi, H.; Burley, S.K.; Strittmatter, S.M. Structural bases for CRMP function in plexin-dependent semaphorin3A signaling. EMBO J. 2004, 23, 9–22. [Google Scholar] [CrossRef]
- Ji, Z.S.; Li, J.P.; Fu, C.H.; Luo, J.X.; Yang, H.; Zhang, G.W.; Wu, W.; Lin, H.S. Spastin interacts with collapsin response mediator protein 3 to regulate neurite growth and branching. Neural Regen. Res. 2021, 16, 2549–2556. [Google Scholar] [CrossRef]
- Heo, S.; Diering, G.H.; Na, C.H.; Nirujogi, R.S.; Bachman, J.L.; Pandey, A.; Huganir, R.L. Identification of long-lived synaptic proteins by proteomic analysis of synaptosome protein turnover. Proc. Natl. Acad. Sci. USA 2018, 115, E3827–E3836. [Google Scholar] [CrossRef]
- Expression Atlas. Available online: https://www.ebi.ac.uk/gxa/home (accessed on 15 September 2021).
- Iqbal, J.; Zhang, K.; Jin, N.; Zhao, Y.; Liu, Q.; Ni, J.; Shen, L. Effect of Sodium Selenate on Hippocampal Proteome of 3xTg-AD Mice-Exploring the Antioxidant Dogma of Selenium against Alzheimer’s Disease. ACS Chem. Neurosci. 2018, 9, 1637–1651. [Google Scholar] [CrossRef]
- Jahrling, J.B.; Hernandez, C.M.; Denner, L.; Dineley, K.T. PPARgamma recruitment to active ERK during memory consolidation is required for Alzheimer’s disease-related cognitive enhancement. J. Neurosci. 2014, 34, 4054–4063. [Google Scholar] [CrossRef]
- Denner, L.A.; Rodriguez-Rivera, J.; Haidacher, S.J.; Jahrling, J.B.; Carmical, J.R.; Hernandez, C.M.; Zhao, Y.; Sadygov, R.G.; Starkey, J.M.; Spratt, H.; et al. Cognitive enhancement with rosiglitazone links the hippocampal PPAR and ERK MAPK signaling pathways. J. Neurosci. 2012, 32, 16725–16735. [Google Scholar] [CrossRef]
- Madrid, L.; Moreno-Grau, S.; Ahmad, S.; González-Pérez, A.; de Rojas, I.; Xia, R.; Martino Adami, P.V.; García-González, P.; Kleineidam, L.; Yang, Q.; et al. Multiomics integrative analysis identifies APOE allele-specific blood biomarkers associated to Alzheimer’s disease etiopathogenesis. Aging 2021, 13, 9277–9329. [Google Scholar] [CrossRef] [PubMed]
- Yagensky, O.; Kohansal-Nodehi, M.; Gunaseelan, S.; Rabe, T.; Zafar, S.; Zerr, I.; Härtig, W.; Urlaub, H.; Chua, J.J. Increased expression of heme-binding protein 1 early in Alzheimer’s disease is linked to neurotoxicity. eLife 2019, 8, e47498. [Google Scholar] [CrossRef] [PubMed]
- Goriounova, N.A.; Heyer, D.B.; Wilbers, R.; Verhoog, M.B.; Giugliano, M.; Verbist, C.; Obermayer, J.; Kerkhofs, A.; Smeding, H.; Verberne, M.; et al. Large and fast human pyramidal neurons associate with intelligence. eLife 2018, 7, e41714. [Google Scholar] [CrossRef] [PubMed]
- Savage, J.E.; Jansen, P.R.; Stringer, S.; Watanabe, K.; Bryois, J.; de Leeuw, C.A.; Nagel, M.; Awasthi, S.; Barr, P.B.; Coleman, J.R.; et al. Genome-wide association meta-analysis in 269,867 individuals identifies new genetic and functional links to intelligence. Nat. Genet. 2018, 51, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Zabaneh, D.; Krapohl, E.; Gaspar, H.A.; Curtis, C.; Lee, S.H.; Patel, H.; Newhouse, S.; Wu, H.M.; Simpson, M.A.; Putallaz, M.; et al. A genome-wide association study for extremely high intelligence. Mol. Psychiatry 2018, 23, 1226–1232. [Google Scholar] [CrossRef]
- Liu-Seifert, H.; Siemers, E.; Price, K.; Han, B.; Selzler, K.J.; Henley, D.; Sundell, K.; Aisen, P.; Cummings, J.; Raskin, J.; et al. Cognitive Impairment Precedes and Predicts Functional Impairment in Mild Alzheimer’s Disease. J. Alzheimers Dis. 2015, 47, 205–214. [Google Scholar] [CrossRef]
- Huang, L.; Wan, J.; Wu, Y.; Tian, Y.; Yao, Y.; Yao, S.; Ji, X.; Wang, S.; Su, Z.; Xu, H. Challenges in adeno-associated virus-based treatment of central nervous system diseases through systemic injection. Life Sci. 2021, 270, 119142. [Google Scholar] [CrossRef]
- Nectow, A.R.; Nestler, E.J. Viral tools for neuroscience. Nat. Rev. Neurosci. 2020, 21, 669–681. [Google Scholar] [CrossRef]
- Vincent, K.A.; Piraino, S.T.; Wadsworth, S.C. Analysis of recombinant adeno-associated virus packaging and requirements for rep and cap gene products. J. Virol. 1997, 71, 1897–1905. [Google Scholar] [CrossRef]
- Kotterman, M.A.; Schaffer, D.V. Engineering adeno-associated viruses for clinical gene therapy. Nat. Rev. Genet. 2014, 15, 445–451. [Google Scholar] [CrossRef]
- Tenenbaum, L.; Chtarto, A.; Lehtonen, E.; Velu, T.; Brotchi, J.; Levivier, M.J. Recombinant AAV-mediated gene delivery to the central nervous system. Gene Med. 2004, 6 (Suppl. S1), S212–S222. [Google Scholar] [CrossRef]
- Blackburn, S.D.; Steadman, R.A.; Johnson, F.B. Attachment of adeno-associated virus type 3H to fibroblast growth factor receptor 1. Arch. Virol. 2006, 151, 617–623. [Google Scholar] [CrossRef]
- Mietzsch, M.; Eddington, C.; Jose, A.; Hsi, J.; Chipman, P.; Henley, T.; Choudhry, M.; McKenna, R.; Agbandje-McKenna, M. Improved Genome Packaging Efficiency of Adeno-associated Virus Vectors Using Rep Hybrids. J. Virol. 2021, 95, e0077321. [Google Scholar] [CrossRef]
- Powell, S.K.; Rivera-Soto, R.; Gray, S.J. Viral expression cassette elements to enhance transgene target specificity and expression in gene therapy. Discov. Med. 2015, 19, 49–57. [Google Scholar]
- Herzog, C.D.; Bishop, K.M.; Brown, L.; Wilson, A.; Kordower, J.H.; Bartus, R.T. Gene transfer provides a practical means for safe, long-term, targeted delivery of biologically active neurotrophic factor proteins for neurodegenerative diseases. Drug Deliv. Transl. Res. 2011, 1, 361–382. [Google Scholar] [CrossRef]
- Ramaswamy, S.; McBride, J.L.; Herzog, C.D.; Brandon, E.; Gasmi, M.; Bartus, R.T.; Kordower, J.H. Neurturin gene therapy improves motor function and prevents death of striatal neurons in a 3-nitropropionic acid rat model of Huntington’s disease. Neurobiol. Dis. 2007, 26, 375–384. [Google Scholar] [CrossRef]
- Kiyota, T.; Zhang, G.; Morrison, C.M.; Bosch, M.E.; Weir, R.A.; Lu, Y.; Dong, W.; Gendelman, H.E. AAV2/1 CD74 Gene Transfer Reduces β-amyloidosis and Improves Learning and Memory in a Mouse Model of Alzheimer’s Disease. Mol. Ther. 2015, 23, 1712–1721. [Google Scholar] [CrossRef]
- Mochizuki, H.; Hayakawa, H.; Migita, M.; Shibata, M.; Tanaka, R.; Suzuki, A.; Shimo-Nakanishi, Y.; Urabe, T.; Yamada, M.; Tamayose, K.; et al. An AAV-derived Apaf-1 dominant negative inhibitor prevents MPTP toxicity as antiapoptotic gene therapy for Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2001, 98, 10918–10923. [Google Scholar] [CrossRef]
- Miniarikova, J.; Zimmer, V.; Martier, R.; Brouwers, C.C.; Pythoud, C.; Richetin, K.; Rey, M.; Lubelski, J.; Evers, M.M.; van Deventer, S.J.; et al. AAV5-miHTT gene therapy demonstrates suppression of mutant huntingtin aggregation and neuronal dysfunction in a rat model of Huntington’s disease. Gene Ther. 2017, 24, 630–639. [Google Scholar] [CrossRef]
- Duan, W.; Guo, M.; Yi, L.; Liu, Y.; Li, Z.; Ma, Y.; Zhang, G.; Liu, Y.; Bu, H.; Song, X.; et al. The deletion of mutant SOD1 via CRISPR/Cas9/sgRNA prolongs survival in an amyotrophic lateral sclerosis mouse model. Gene Ther. 2020, 27, 157–169. [Google Scholar] [CrossRef]
- Foust, K.D.; Salazar, D.L.; Likhite, S.; Ferraiuolo, L.; Ditsworth, D.; Ilieva, H.; Meyer, K.; Schmelzer, L.; Braun, L.; Cleveland, D.W.; et al. Therapeutic AAV9-mediated suppression of mutant SOD1 slows disease progression and extends survival in models of inherited ALS. Mol. Ther. 2013, 21, 2148–2159. [Google Scholar] [CrossRef] [PubMed]
- Brandon, E.F.; Hermsen, H.P.; van Eijkeren, J.C.; Tiesjema, B. Effect of administration route on the biodistribution and shedding of replication-deficient AAV2: A qualitative modelling approach. Curr. Gene Ther. 2010, 10, 91–106. [Google Scholar] [CrossRef]
- Drouin, L.M.; Agbandje-McKenna, M. Adeno-associated virus structural biology as a tool in vector development. Future Virol. 2013, 8, 1183–1199. [Google Scholar] [CrossRef] [PubMed]
- Chu, W.S.; Ng, J. Immunomodulation in Administration of rAAV: Preclinical and Clinical Adjuvant Pharmacotherapies. Front. Immunol. 2021, 12, 658038. [Google Scholar] [CrossRef]
- O’Connor, D.M.; Boulis, N.M. Gene therapy for neurodegenerative diseases. Trends Mol. Med. 2015, 21, 504–512. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, K.; Riyad, J.M.; Weber, T. Expressing Transgenes That Exceed the Packaging Capacity of Adeno-Associated Virus Capsids. Hum. Gene Ther. Methods 2016, 27, 1–12. [Google Scholar] [CrossRef]
- Kumar, N.; Stanford, W.; de Solis, C.; Aradhana Abraham, N.D.; Dao, T.J.; Thaseen, S.; Sairavi, A.; Gonzalez, C.U.; Ploski, J.E. The Development of an AAV-Based CRISPR SaCas9 Genome Editing System That Can Be Delivered to Neurons in vivo and Regulated via Doxycycline and Cre-Recombinase. Front. Mol. Neurosci. 2018, 11, 413. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ND | Clinical Signs and Symptoms | Key Protein Candidates | Other Involved Proteins | |
|---|---|---|---|---|
| Specificity | Commonality • | |||
| HD | Chorea; dystonia; impaired posture, balance and speech; cognitive disorders; learning difficulty. | HUNTINGTIN [8,9,10] | HD71Q [11]; ATN1 [12]; HIP-1 [13]; DMPK [14]; FOXP2 [15]; PRNP [16] | MAP1 [17,18]; Calbindin [19,20]; Calcineurin [21,22]; MAP2 [23,24]; SUBSTANCE P [25,26]; IP3 [27,28]; CDK5 [29,30]; BIP [31,32]; TREM2 [33,34]; IRE1 [35,36]; TNFR1 [37,38]; FGF [39,40]; RAC [41,42]; CDC42 [43,44]; RHO [45,46]; PERK [47,48]; ATF6 [49,50];TDP-43 [51,52]; ACTIN [53,54]; ATXN2 [55,56]; SNCA [57,58]; DCTN1 [59,60]; CTSD [61,62]; GSK3 [63,64]; BACE1 [65,66]; UBQLN2 [67,68]; JNK [69,70]; IFN [71,72]; HSP20 [73,74]; CHOP [75,76]. |
| AD | Memory loss; repeat statements; depression; apathy; irritability; delusions; aggressiveness. | [APP, TAU] [77,78] | [PSEN1, PSEN2, CR1, APOE, PICALM, BIN1, TREM2, ABCA7, PLD3] [79]; [ADAM10, MEF2C] [80]; [UNC5C, AKAP9] [81] | |
| PD | Motor deficit; often appear asymmetrical; resting tremor; rigidity; dystonia; cognitive disorders. | ALPHA- SYNUCLEIN [82,83,84] | [TAU, LRRK2, SNCA] [85]; [DNAJC6, VPS35] [86]; [VPS13C, PARK2, COMT] [87]; RAB39B [88]; MIR4697 [89] | |
| ALS | Difficulty walking; weakness in legs and hands; slurred speech; inappropriate laughing; cognitive dysfunction. | SUPEROXIDE DISMUTASE [90,91,92,93,94] | [C9orf72, FUS, VCP, ALS2, SQSTM1, TARDBF] [95]; [KIFA, SETX, OPTN] [96]; [VEGF, ANG] [97]; [CYCLINE F] [98]; [GRN, PRPH] [99] | |
| CJD | Memory loss; impaired thinking; insomnia; personality changes; jerky movements. | PRION [100,101,102,103] | [COX6, FZD9, RXRG, SOX11] [104] | |
| ND | Neuron Vulnerability | Neuron Resistance | ||
|---|---|---|---|---|
| Brain Areas Neuron Types | Molecular Mechanisms Prevalence | Brain Areas Neuron Types | Molecular Mechanisms Prevalence | |
| AD | Frontal lobe; Entorhinal cortex; Hippocampus ; Subiculum; Amygdala; Locus Coeruleus; Cingulate gyrus; Nucleus basalis magnocellularis. Pn. Cn. Ln. RIn. | Sensitive to oxygen, glucose, energy deprivation, and excitotoxicity. Increased Ca++ entry. Low CBP. MisfP | Dentate Gyrus. Cerebellum. Gn. PurKn expressing PcP4. Sn. CI. | High expression of Y1, calbindin, High CBP as compared to Pn. |
| PD | Striatum; Substantia nigra; Raphe nucleus; Locus nucleus; Limbic cortex; Inferior olivary nucleus; Medulla oblongata; Subiculum. DAn. Ssn. basal forebrain Cn. | Neuromelanin, Dp itself. α-Syn. Sensitive to TFs. Mitochondrial-ER dysfunction. Ca++ oscillation. Low CBP. Expression of GIRK2. Important CaV1 transcripts. | Ventral Tegmental Area. Pedunculopontin nucleus. DAn. GABAn. GluTn. | High expression of calbindin and other CBP. High mitochondrial mass. Undetectable GIRK2 expression. |
| ALS | Spinal cord; Motor neurons network; Brain stem; Corpus callosum; Dentate Gyrus; Entorhinal Cortex; Extrapyramidal alteration. Cn. ffMn with large axon expressing MMP9. Mn. | Ca++ dysh. Low CBP. Atypical AMPAR. Mutant SOD1 depletes Hsp70/75. EAAT2 high expression. Mitochondrial abnormalities. | Spinal cord SrMn is more resistant (as compared to ffMn); On and neurons in Onuf’s nuclei. | SrMN expresses Os which binds to αxβ2 integrin (CD11c), its receptor. EAAT2 expression reduced. High expression of calbindin, CBP, and parvalbumin |
| HD | Cerebral cortex; Thalamus; Striatum; Hypothalamus; Cerebellum; Amygdala; Globus pallidus; Putamen; Hippocampus. EnKn. ParVin. Ssn. SPn. MSn. | Decreased GDNF, CNTF, BDNF, and CBP. Sensitive to AMPA, NMDA. Increased (Ca++)ic. Reduction in ATP. | Cerebral cortex. Striatum. Cn. AsN. SomaTn. | High expression of parvalbumin, calbindin, and other Ca++buffering proteins |
| CJD | Cerebellum; Cerebral cortex; Striatum; Putamen; Thalamus; Corpus callosum. ParVin. GABAn. ILB4n. | Increased AMPA, Calcineurin activity, Ca++ permeability. Interaction with the α2δ-1 subunit of VGCC. Damage in mitochondria. | Hippocampus. Cerebellum. Pn, Gn in both areas. | High expression of calbindin and other CBP. |
| Disease | Clinicaltrial.gov ID | Vectors | Transgenes | Injection Sites |
|---|---|---|---|---|
| Alzheimer | NCT 05040217 | AAV2 | BDNF cDNA | Stereotaxic injection into the brain |
| NCT03634007 | AAVrh.10 | APOE2 cDNA | Intrathecal Injection | |
| Parkinson | NCT01621581 NCT04167540 | AAV2 | GDNF cDNA | Stereotaxic injection into the putamen |
| NCT00985517 | AAV2 | Neurturin cDNA | Substantia nigra and putamen | |
| NCT01973543 NCT00229736 NCT03562494 | AAV2 | AADC, Dopa Decarboxylase cDNA | Stereotaxic injection into the striatum | |
| Huntington | NCT05243017 | AAV5 | HTTmiRNA | Stereotaxic injection into the striatum |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quach, T.T.; Stratton, H.J.; Khanna, R.; Mackey-Alfonso, S.; Deems, N.; Honnorat, J.; Meyer, K.; Duchemin, A.-M. Neurodegenerative Diseases: From Dysproteostasis, Altered Calcium Signalosome to Selective Neuronal Vulnerability to AAV-Mediated Gene Therapy. Int. J. Mol. Sci. 2022, 23, 14188. https://doi.org/10.3390/ijms232214188
Quach TT, Stratton HJ, Khanna R, Mackey-Alfonso S, Deems N, Honnorat J, Meyer K, Duchemin A-M. Neurodegenerative Diseases: From Dysproteostasis, Altered Calcium Signalosome to Selective Neuronal Vulnerability to AAV-Mediated Gene Therapy. International Journal of Molecular Sciences. 2022; 23(22):14188. https://doi.org/10.3390/ijms232214188
Chicago/Turabian StyleQuach, Tam T., Harrison J. Stratton, Rajesh Khanna, Sabrina Mackey-Alfonso, Nicolas Deems, Jérome Honnorat, Kathrin Meyer, and Anne-Marie Duchemin. 2022. "Neurodegenerative Diseases: From Dysproteostasis, Altered Calcium Signalosome to Selective Neuronal Vulnerability to AAV-Mediated Gene Therapy" International Journal of Molecular Sciences 23, no. 22: 14188. https://doi.org/10.3390/ijms232214188
APA StyleQuach, T. T., Stratton, H. J., Khanna, R., Mackey-Alfonso, S., Deems, N., Honnorat, J., Meyer, K., & Duchemin, A.-M. (2022). Neurodegenerative Diseases: From Dysproteostasis, Altered Calcium Signalosome to Selective Neuronal Vulnerability to AAV-Mediated Gene Therapy. International Journal of Molecular Sciences, 23(22), 14188. https://doi.org/10.3390/ijms232214188