
Comparative Genomics and Functional Studies of Putative m6A Methyltransferase (METTL) Genes in Cotton

 , , ,
, , ,
Abstract
1. Introduction
2. Results
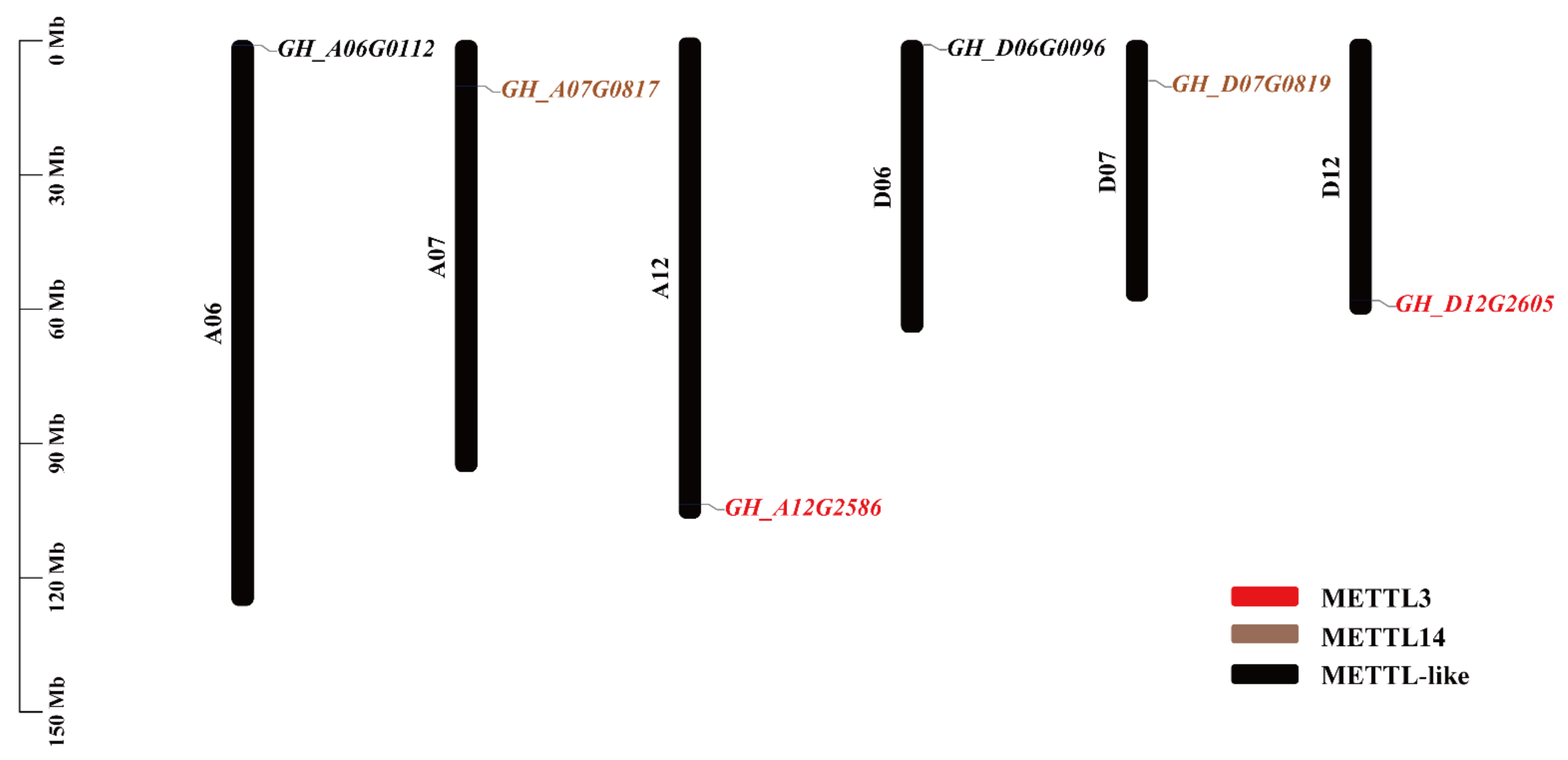
2.1. Identification and Chromosomal Location of RNA Methyltransferase Genes in G. hirsutum
2.2. Structural Organization of GhMETTL Genes
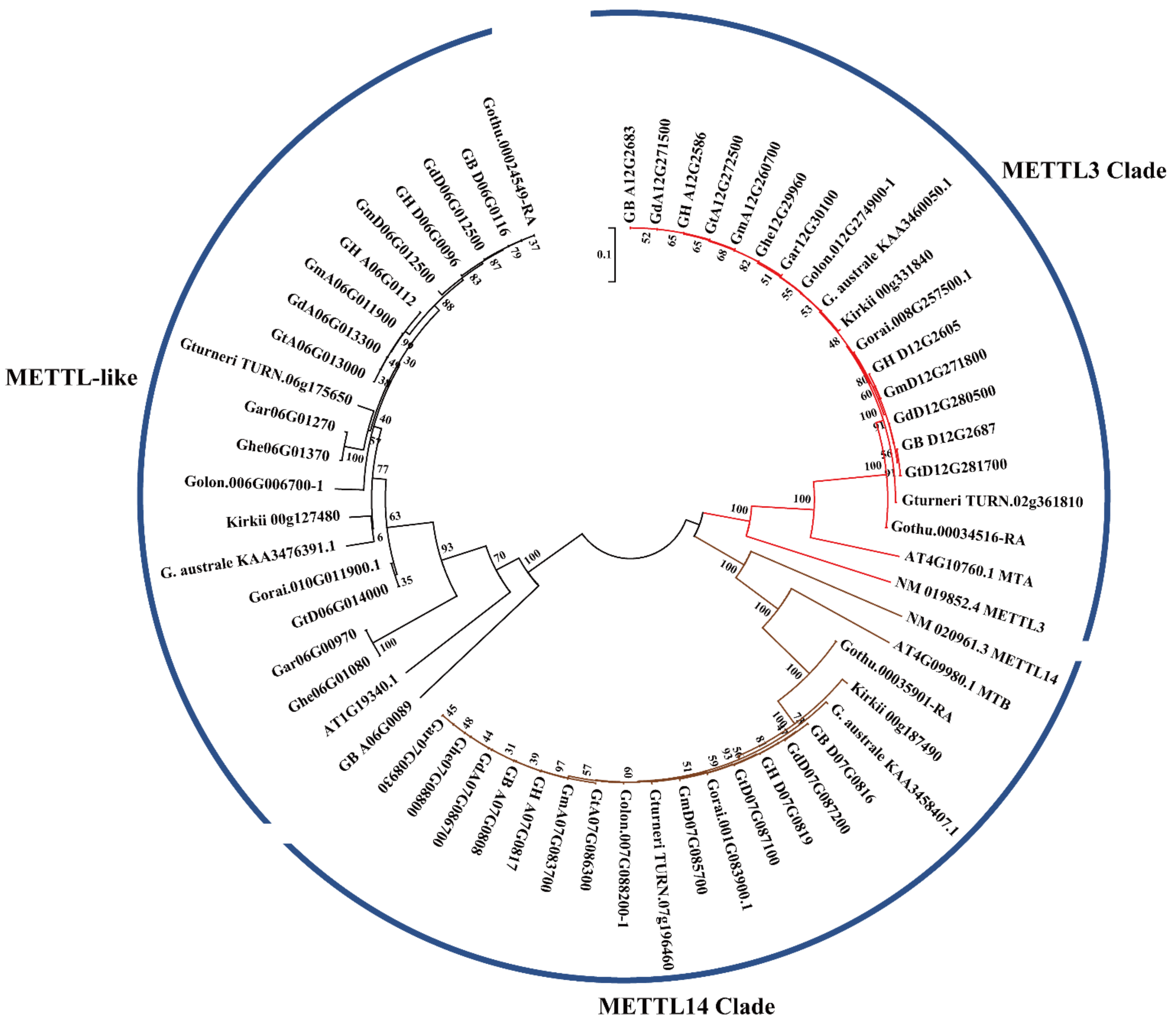
2.3. Phylogenetic Analysis of METTL Genes in Gossypium
2.4. Cis-Element Analysis of METTL Genes in G. Hirsutum
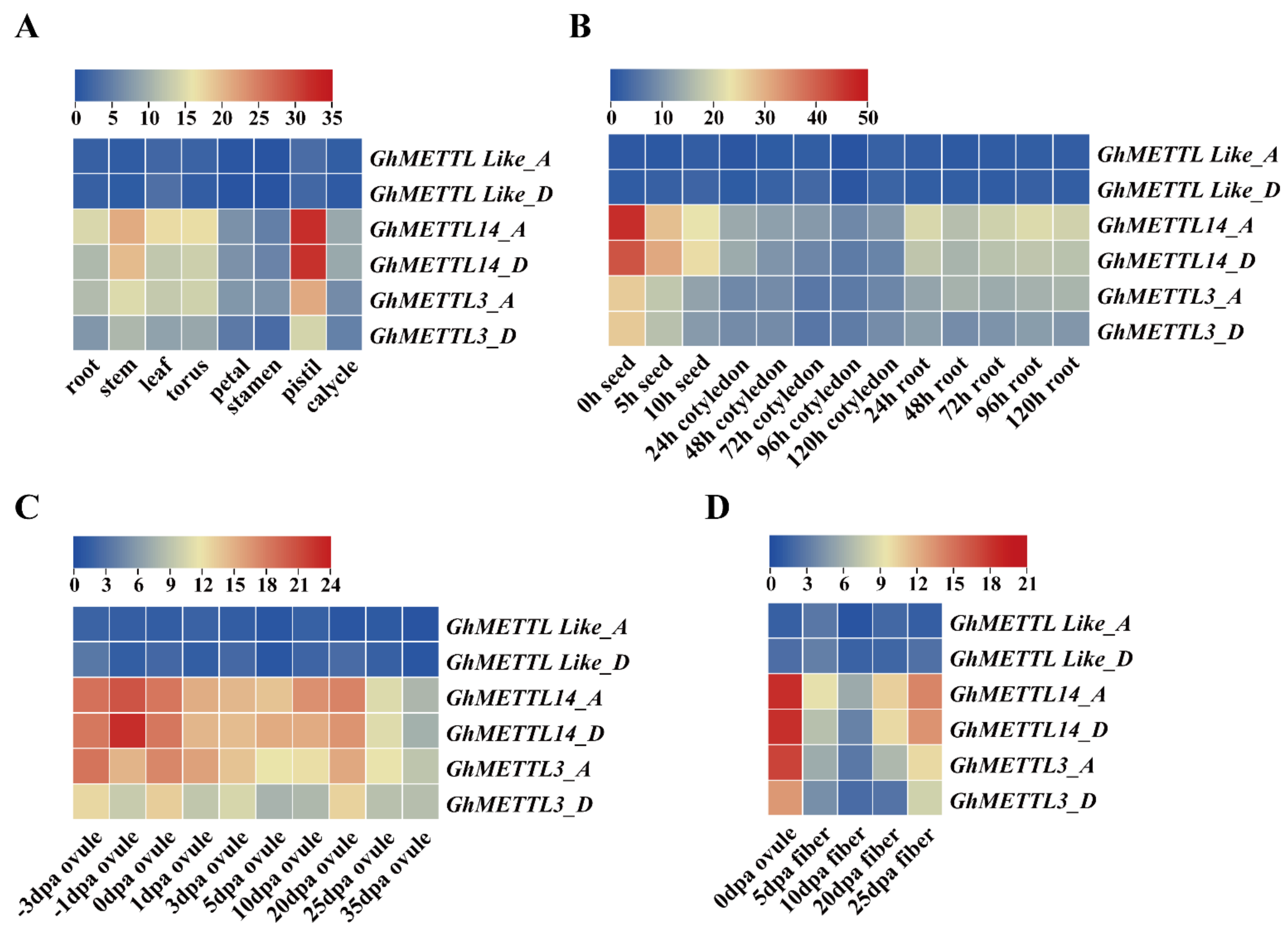
2.5. Expression Patterns of GhMETTL Genes in Different G. hirsutum Tissues
2.6. Expression Changes of GhMETTL Genes in G. Hirsutum under Different Stresses
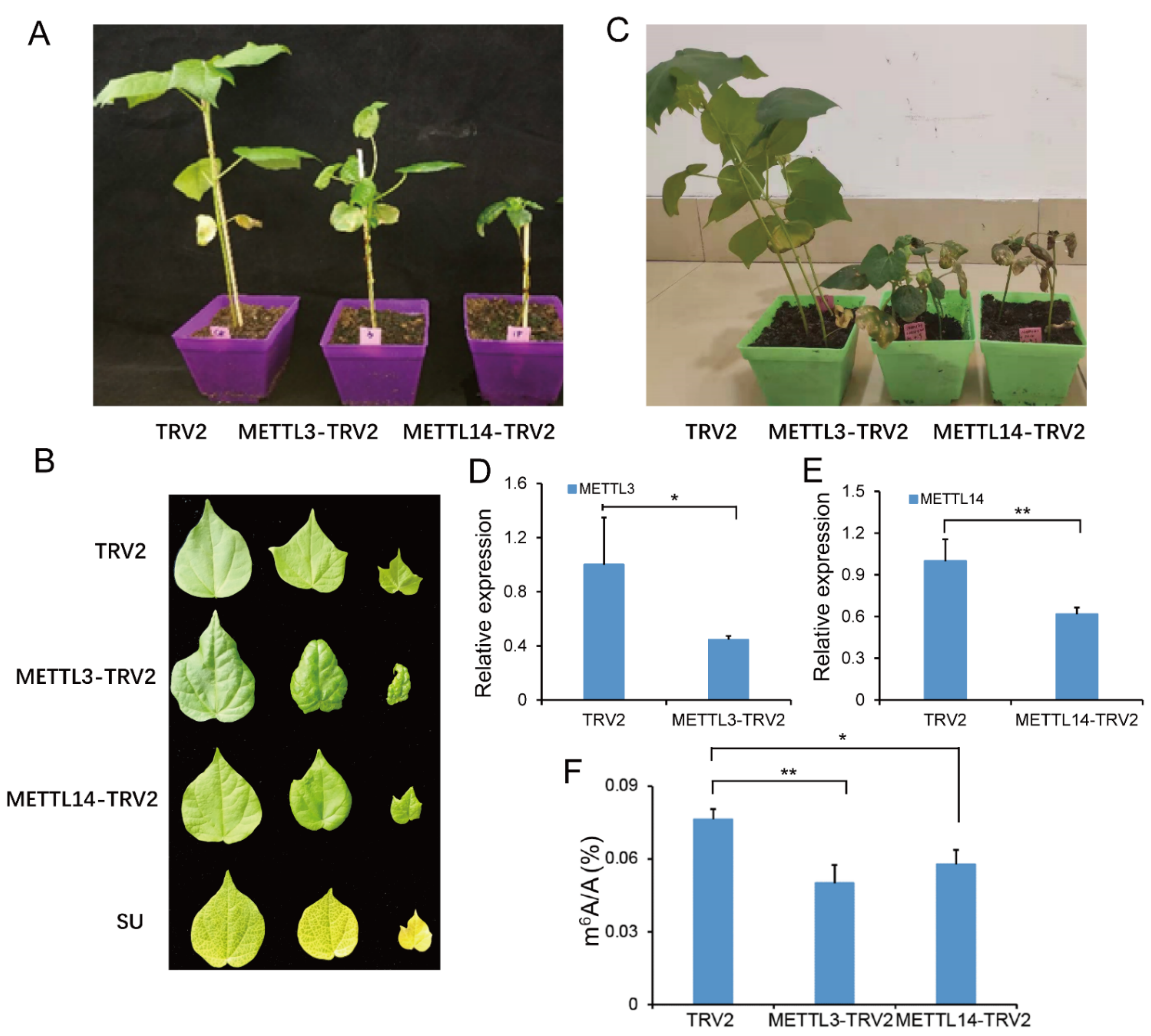
2.7. Suppressing GhMETTL3 and GhMETTL14 Caused Growth Arrest in G. hirsutum
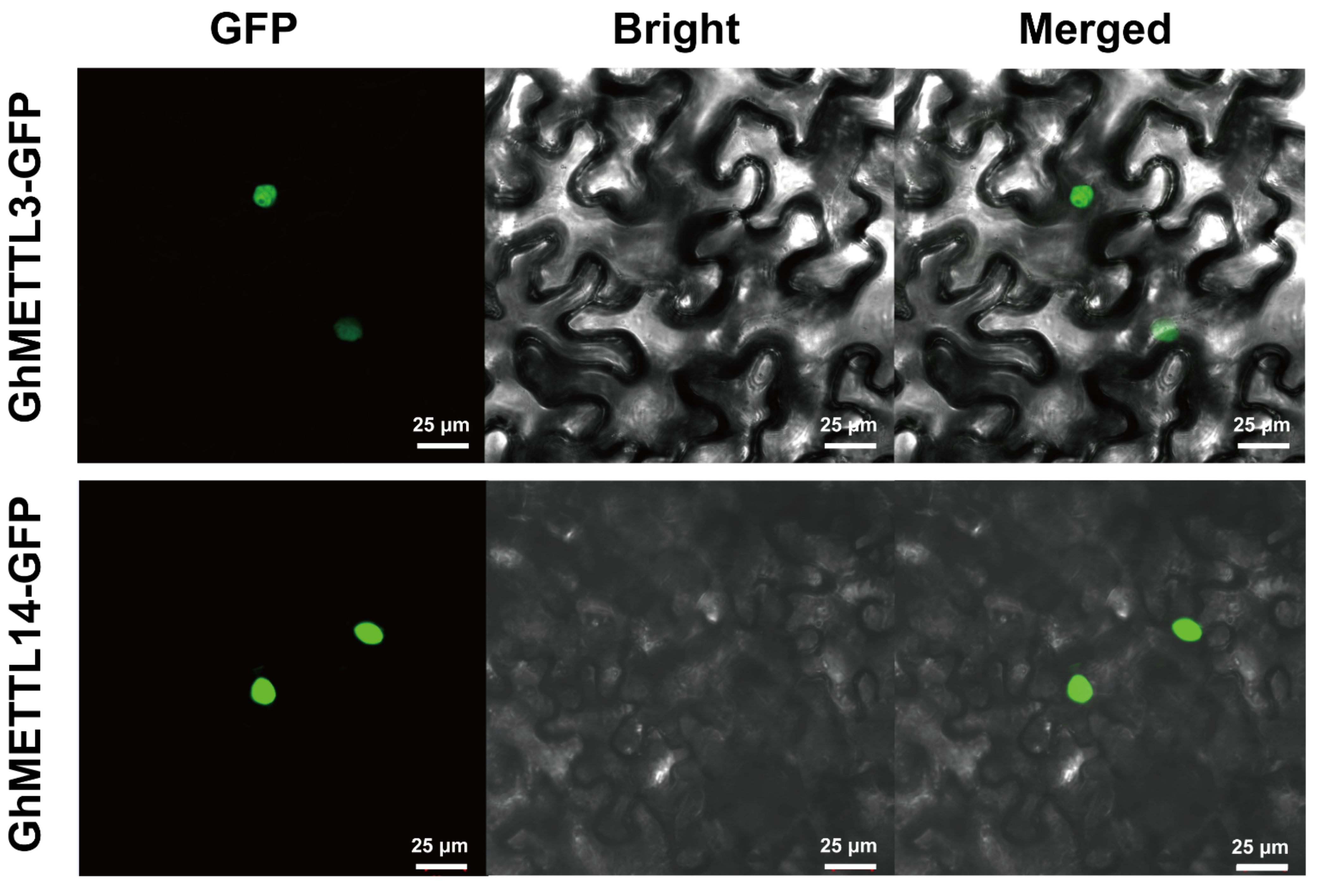
2.8. Subcellular Localization of GhMETTL3 and GhMETTL14 Proteins
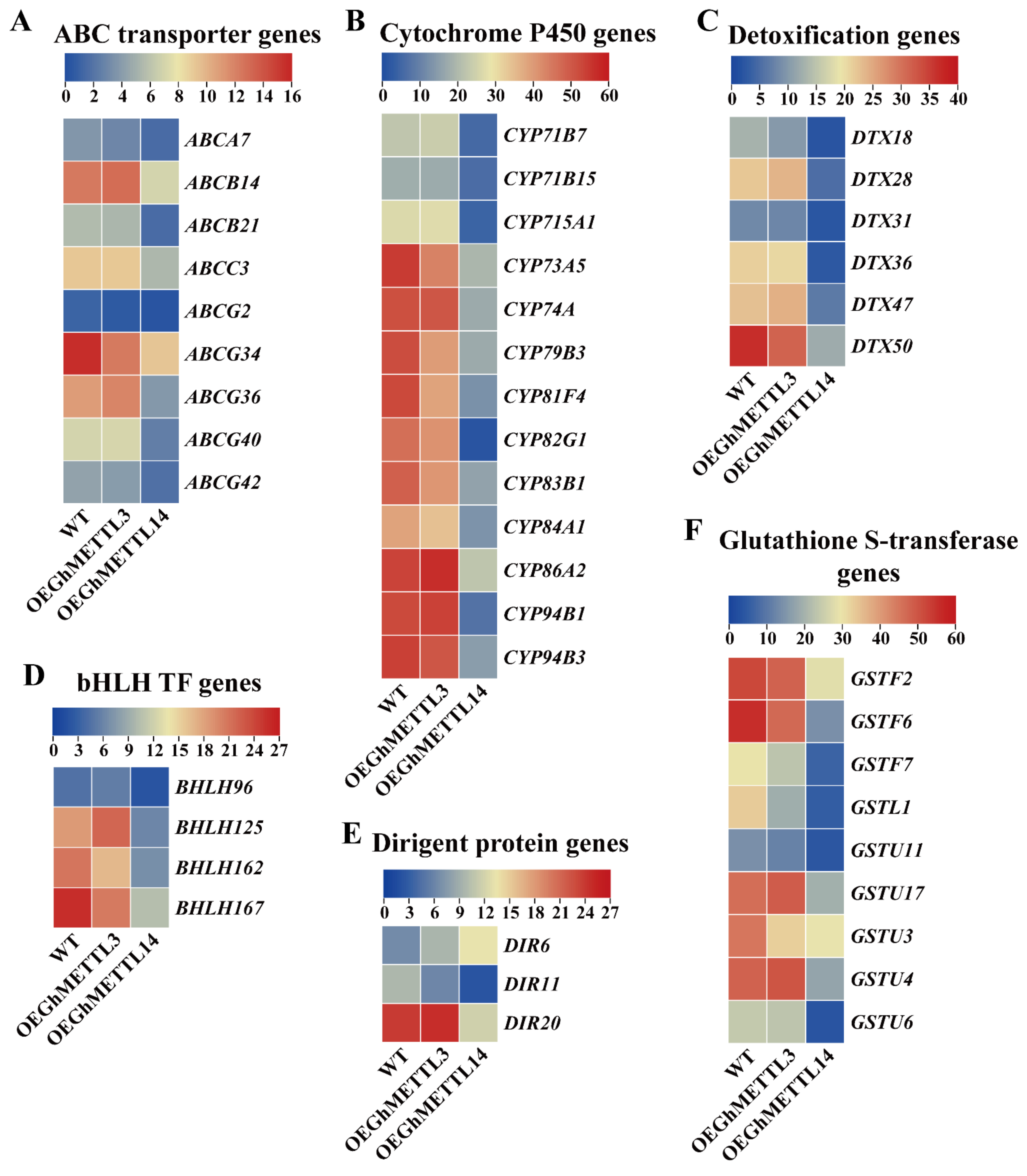
2.9. Overexpression of GhMETTL3 and GhMETTL14 Produced Distinct Differentially Expressed Genes (DEGs) in A. Thaliana
2.10. GhMETTL3 and GhMETTL14 Exhibited Divergent Functions in Transgenic Arabidopsis
3. Discussion
4. Materials and Methods
4.1. Identification of METTL Family Genes and METTL Proteins in Diploid and Tetraploid Gossypium Species
4.2. Chromosomal Location and Gene Structure Analyses
4.3. Sequence Alignment and Phylogenetic Tree Construction
4.4. Analysis of Cis-Acting Element in Promoter Regions of GhMETTLs
4.5. Expression Patterns of GhMETTLs in Different Tissues and Stress Conditions
4.6. Virus-Induced Gene Silencing (VIGS) Assays
4.7. RNA Extraction, cDNA Synthesis, and RT-qPCR Expression Analyses
4.8. Quantitative Analysis of m6A Level in Cotton Leaf Tissue
4.9. Subcellular Localization of GhMETTL3 and GhMETTL14 Proteins and Microscopic Observation
4.10. Arabidopsis Transformation
4.11. RNA Sequencing of OEGhMETTL3, OEGhMETTL14, and Wild Type A. Thaliana Plants
4.12. Read Data Quality Control, Mapping, and Calculations of the Differentially Expressed Genes
4.13. Gene Annotation and Enrichment Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Liu, N.; Pan, T. N6-methyladenosine–encoded epitranscriptomics. Nat. Struct. Mol. Biol. 2016, 23, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Gao, G.; Tang, R.; Wang, W.; Wang, Y.; Tian, S.; Qin, G. m6 A-mediated regulation of crop development and stress responses. Plant Biotechnol. J. 2022, 20, 1447–1455. [Google Scholar] [CrossRef]
- Yue, H.; Nie, X.; Yan, Z.; Weining, S. N6-methyladenosine regulatory machinery in plants: Composition, function and evolution. Plant Biotechnol J. 2019, 17, 1194–1208. [Google Scholar] [CrossRef] [PubMed]
- Bokar, J.A.; Shambaugh, M.E.; Polayes, D.; Matera, A.G.; Rottman, F.M. Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. RNA 1997, 3, 1233–1247. [Google Scholar] [PubMed]
- Bujnicki, J.M.; Feder, M.; Radlinska, M.; Blumenthal, R.M. Structure prediction and phylogenetic analysis of a functionally diverse family of proteins homologous to the MT-A70 subunit of the human mRNA:m(6)A methyltransferase. J. Mol. Evol. 2002, 55, 431–444. [Google Scholar] [CrossRef]
- Liu, J.; Yue, Y.; Han, D.; Wang, X.; Fu, Y.; Zhang, L.; Jia, G.; Yu, M.; Lu, Z.; Deng, X.; et al. A METTL3–METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 2013, 10, 93–95. [Google Scholar] [CrossRef]
- Liu, J.; Li, K.; Cai, J.; Zhang, M.; Zhang, X.; Xiong, X.; Meng, H.; Xu, X.; Huang, Z.; Peng, J.; et al. Landscape and Regulation of m6A and m6Am Methylome across Human and Mouse Tissues. Mol. Cell 2019, 77, 426–440. [Google Scholar] [CrossRef]
- Śledź, P.; Jinek, M. Structural insights into the molecular mechanism of the m(6)A writer complex. eLife 2016, 5, e18434. [Google Scholar]
- Wang, P.; Doxtader, K.A.; Nam, Y. Structural Basis for Cooperative Function of Mettl3 and Mettl14 Methyltransferases. Mol. Cell 2016, 63, 306–317. [Google Scholar] [CrossRef]
- Wang, X.; Feng, J.; Xue, Y.; Guan, Z.; Zhang, D.; Liu, Z.; Gong, Z.; Huang, J.; Tang, C.; Zou, T.; et al. Structural basis of N6-adenosine methylation by the METTL3–METTL14 complex. Nature 2016, 534, 575–578. [Google Scholar] [CrossRef]
- Ping, X.-L.; Sun, B.-F.; Wang, L.; Xiao, W.; Yang, X.; Wang, W.-J.; Adhikari, S.; Shi, Y.; Lv, Y.; Chen, Y.-S.; et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014, 24, 177–189. [Google Scholar] [CrossRef]
- Zhong, S.; Li, H.; Bodi, Z.; Button, J.; Vespa, L.; Herzog, M.; Fray, R.G. MTA is an Arabidopsis messenger RNA adenosine methylase and interacts with a homolog of a sex-specific splicing factor. Plant Cell 2008, 20, 1278–1288. [Google Scholar] [CrossRef]
- Lu, L.; Zhang, Y.; He, Q.; Qi, Z.; Zhang, G.; Xu, W.; Yi, T.; Wu, G.; Li, R. MTA, an RNA m6A methyltransferase, enhances drought tolerance by regulating the development of trichomes and roots in poplar. Int. J. Mol. Sci. 2020, 21, 2462. [Google Scholar] [CrossRef]
- Arribas-Hernández, L.; Bressendorff, S.; Hansen, M.H.; Poulsen, C.; Erdmann, S.; Brodersen, P. An m6A-YTH module controls developmental timing and morphogenesis in arabidopsis. Plant Cell 2018, 30, 952–967. [Google Scholar] [CrossRef]
- He, Y.; Li, Y.; Yao, Y.; Zhang, H.; Wang, Y.; Gao, J.; Fan, M. Overexpression of watermelon m6A methyltransferase ClMTB enhances drought tolerance in tobacco by mitigating oxidative stress and photosynthesis inhibition and modulating stress-responsive gene expression. Plant Physiol. Biochem. 2021, 168, 340–352. [Google Scholar] [CrossRef]
- Wendel, F.J.; Grover, C. Taxonomy and Evolution of the Cotton Genus, Gossypium, 2nd ed.; American Society of Agronomy Inc.: Madison, WI, USA; Crop Science Society of America Inc.: Madison, WI, USA; Soil Science Society of America Inc.: Madison, WI, USA, 2015. [Google Scholar]
- Wang, K.; Wendel, J.F.; Hua, J. Designations for individual genomes and chromosomes in Gossypium. J. Cotton Res. 2018, 1, 3. [Google Scholar] [CrossRef]
- Chen, Z.; Grover, C.E.; Li, P.; Wang, Y.; Nie, H.; Zhao, Y.; Wang, M.; Liu, F.; Zhou, Z.; Wang, X.; et al. Molecular evolution of the plastid genome during diversification of the cotton genus. Mol. Phylogenetics Evol. 2017, 112, 268–276. [Google Scholar] [CrossRef]
- Huang, G.; Huang, J.-Q.; Chen, X.-Y.; Zhu, Y.-X. Recent advances and future perspectives in cotton research. Annu. Rev. Plant Biol. 2021, 72, 437–462. [Google Scholar] [CrossRef]
- Paterson, A.H.; Wendel, J.F.; Gundlach, H.; Guo, H.; Jenkins, J.; Jin, D.; Llewellyn, D.; Showmaker, K.C.; Shu, S.; Udall, J.; et al. Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibres. Nature 2012, 492, 423–427. [Google Scholar] [CrossRef]
- Wang, K.; Wang, Z.; Li, F.; Ye, W.; Wang, J.; Song, G.; Yue, Z.; Cong, L.; Shang, H.; Zhu, S.; et al. The draft genome of a diploid cotton Gossypium raimondii. Nat. Genet. 2012, 44, 1098–1103. [Google Scholar] [CrossRef]
- Udall, J.A.; Long, E.; Hanson, C.; Yuan, D.; Ramaraj, T.; Conover, J.L.; Gong, L.; Arick, M.A.; Grover, C.E.; Peterson, D.G.; et al. De novo genome sequence assemblies of Gossypium raimondii and Gossypium turneri. G3 Genes Genomes Genet. 2019, 9, 3079–3085. [Google Scholar] [CrossRef] [PubMed]
- Grover, C.E.; Arick, M.A., II; Thrash, A.; Conover, J.L.; Sanders, W.S.; Peterson, D.G.; Frelichowski, J.E.; Scheffler, J.A.; Scheffler, B.E.; Wendel, J.F. Insights into the evolution of the New World diploid cottons (Gossypium, Subgenus Houzingenia) based on genome sequencing. Genome Biol. Evol. 2018, 11, 53–71. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Fan, G.; Wang, K.; Sun, F.; Yuan, Y.; Song, G.; Li, Q.; Ma, Z.; Lu, C.; Zou, C.; et al. Genome sequence of the cultivated cotton Gossypium arboreum. Nat. Genet. 2014, 46, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Huang, G.; He, S.; Yang, Z.; Sun, G.; Ma, X.; Li, N.; Zhang, X.; Sun, J.; Liu, M.; et al. Resequencing of 243 diploid cotton accessions based on an updated A genome identifies the genetic basis of key agronomic traits. Nat. Genet. 2018, 50, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Wu, Z.; Percy, R.G.; Bai, M.; Li, Y.; Frelichowski, J.E.; Hu, J.; Wang, K.; Yu, J.Z.; Zhu, Y. Genome sequence of Gossypium herbaceum and genome updates of Gossypium arboreum and Gossypium hirsutum provide insights into cotton A-genome evolution. Nat. Genet. 2020, 52, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Grover, C.E.; Pan, M.; Yuan, D.; Arick, M.A.; Hu, G.; Brase, L.; Stelly, D.M.; Lu, Z.; Schmitz, R.J.; Peterson, D.G.; et al. The Gossypium longicalyx Genome as a Resource for Cotton Breeding and Evolution. G3 Genes|Genomes|Genetics 2020, 10, 1457–1467. [Google Scholar] [CrossRef]
- Sheng, K.; Sun, Y.; Liu, M.; Cao, Y.; Han, Y.; Li, C.; Muhammad, U.; Daud, M.K.; Wang, W.; Li, H.; et al. A reference-grade genome assembly for Gossypium bickii and insights into its genome evolution and formation of pigment glands and gossypol. Plant Commun. 2022, 100421. [Google Scholar] [CrossRef]
- Cai, Y.; Cai, X.; Wang, Q.; Wang, P.; Zhang, Y.; Cai, C.; Xu, Y.; Wang, K.; Zhou, Z.; Wang, C.; et al. Genome sequencing of the Australian wild diploid species Gossypium australe highlights disease resistance and delayed gland morphogenesis. Plant Biotechnol. J. 2019, 18, 814–828. [Google Scholar] [CrossRef]
- Zhang, T.; Hu, Y.; Jiang, W.; Fang, L.; Guan, X.; Chen, J.; Zhang, J.; Saski, C.A.; Scheffler, B.E.; Stelly, D.M.; et al. Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 2015, 33, 531–537. [Google Scholar] [CrossRef]
- Chen, Z.J.; Sreedasyam, A.; Ando, A.; Song, Q.; De Santiago, L.M.; Hulse-Kemp, A.M.; Ding, M.; Ye, W.; Kirkbride, R.C.; Jenkins, J.; et al. Genomic diversifications of five Gossypium allopolyploid species and their impact on cotton improvement. Nat. Genet. 2020, 52, 525–533. [Google Scholar] [CrossRef]
- Li, F.; Fan, G.; Lu, C.; Xiao, G.; Zou, C.; Kohel, R.J.; Ma, Z.; Shang, H.; Ma, X.; Wu, J.; et al. Genome sequence of cultivated Upland cotton (Gossypium hirsutum TM-1) provides insights into genome evolution. Nat. Biotechnol. 2015, 33, 524–530. [Google Scholar] [CrossRef]
- Liu, X.; Zhao, B.; Zheng, H.-J.; Hu, Y.; Lu, G.; Yang, C.; Chen, J.-D.; Chen, J.-J.; Chen, D.-Y.; Zhang, L.; et al. Gossypium barbadense genome sequence provides insight into the evolution of extra-long staple fiber and specialized metabolites. Sci. Rep. 2015, 5, 14139. [Google Scholar] [CrossRef]
- Yuan, D.; Tang, Z.; Wang, M.; Gao, W.; Tu, L.; Jin, X.; Chen, L.; He, Y.; Zhang, L.; Zhu, L.; et al. The genome sequence of Sea-Island cotton (Gossypium barbadense) provides insights into the allopolyploidization and development of superior spinnable fibres. Sci. Rep. 2015, 5, 17662. [Google Scholar] [CrossRef]
- Wang, M.; Tu, L.; Yuan, D.; Zhu, D.; Shen, C.; Li, J.; Liu, F.; Pei, L.; Wang, P.; Zhao, G.; et al. Reference genome sequences of two cultivated allotetraploid cottons, Gossypium hirsutum and Gossypium barbadense. Nat. Genet. 2019, 51, 224–229. [Google Scholar] [CrossRef]
- Hu, Y.; Chen, J.; Fang, L.; Zhang, Z.; Ma, W.; Niu, Y.; Ju, L.; Deng, J.; Zhao, T.; Lian, J.; et al. Gossypium barbadense and Gossypium hirsutum genomes provide insights into the origin and evolution of allotetraploid cotton. Nat. Genet. 2019, 51, 739–748. [Google Scholar] [CrossRef]
- Yang, Z.; Ge, X.; Yang, Z.; Qin, W.; Sun, G.; Wang, Z.; Li, Z.; Liu, J.; Wu, J.; Wang, Y.; et al. Extensive intraspecific gene order and gene structural variations in upland cotton cultivars. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Udall, J.A.; Long, E.; Ramaraj, T.; Conover, J.L.; Yuan, D.; Grover, C.E.; Gong, L.; Arick, M.A., 2nd; Masonbrink, R.E.; Peterson, D.G.; et al. The genome sequence of Gossypioides kirkii illustrates a descending dysploidy in plants. Front. Plant Sci. 2019, 10, 1541. [Google Scholar] [CrossRef]
- Yu, J.; Jung, S.; Cheng, C.-H.; Lee, T.; Zheng, P.; Buble, K.; Crabb, J.; Humann, J.; Hough, H.; Jones, D.; et al. CottonGen: The community database for cotton genomics, genetics, and breeding research. Plants 2021, 10, 2805. [Google Scholar] [CrossRef]
- Poole, R.L. The TAIR database. Methods Mol. Biol. 2007, 406, 179–212. [Google Scholar]
- Hernandez-Garcia, C.M.; Finer, J.J. Identification and validation of promoters and cis-acting regulatory elements. Plant Sci. 2014, 217, 109–119. [Google Scholar] [CrossRef]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lu, Z.; Gomez, A.; Hon, G.C.; Yue, Y.; Han, D.; Fu, Y.; Parisien, M.; Dai, Q.; Jia, G.; et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 2013, 505, 117–120. [Google Scholar] [CrossRef]
- Zhao, X.; Yang, Y.; Sun, B.-F.; Shi, Y.; Yang, X.; Xiao, W.; Hao, Y.-J.; Ping, X.-L.; Chen, Y.-S.; Wang, W.-J.; et al. FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res. 2014, 24, 1403–1419. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Wang, X.; Lu, Z.; Zhao, B.S.; Ma, H.; Hsu, P.J.; Liu, C.; He, C. YTHDF3 facilitates translation and decay of N6-methyladenosine-modified RNA. Cell Res. 2017, 27, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Visvanathan, A.; Somasundaram, K. mRNA traffic control reviewed: N6-methyladenosine (m(6) A) takes the driver’s seat. Bioessays 2018, 40, 1700093. [Google Scholar] [CrossRef]
- Bodi, Z.; Zhong, S.; Mehra, S.; Song, J.; Graham, N.; Li, H.; May, S.; Fray, R.G. Adenosine Methylation in Arabidopsis mRNA is Associated with the 3′ End and Reduced Levels Cause Developmental Defects. Front. Plant Sci. 2012, 3, 48. [Google Scholar] [CrossRef]
- Růžička, K.; Zhang, M.; Campilho, A.; Bodi, Z.; Kashif, M.; Saleh, M.; Eeckhout, D.; El-Showk, S.; Li, H.; Zhong, S.; et al. Identification of factors required for m6 A mRNA methylation in Arabidopsis reveals a role for the conserved E3 ubiquitin ligase HAKAI. New Phytol. 2017, 215, 157–172. [Google Scholar] [CrossRef]
- Zhang, F.; Zhang, Y.-C.; Liao, J.-Y.; Yu, Y.; Zhou, Y.-F.; Feng, Y.-Z.; Yang, Y.-W.; Lei, M.-Q.; Bai, M.; Wu, H.; et al. The subunit of RNA N6-methyladenosine methyltransferase OsFIP regulates early degeneration of microspores in rice. PLoS Genet. 2019, 15, e1008120. [Google Scholar] [CrossRef]
- Sharma, P.; Chatterjee, M.; Burman, N.; Khurana, J.P. Cryptochrome 1 regulates growth and development in Brassica through alteration in the expression of genes involved in light, phytohormone and stress signalling. Plant Cell Env. 2014, 37, 961–977. [Google Scholar] [CrossRef]
- Lopez, L.; Carbone, F.; Bianco, L.; Giuliano, G.; Facella, P.; Perrotta, G. Tomato plants overexpressing cryptochrome 2 reveal altered expression of energy and stress-related gene products in response to diurnal cues. Plant Cell Environ. 2011, 35, 994–1012. [Google Scholar] [CrossRef]
- D’Amico-Damião, V.; Carvalho, R.F. Cryptochrome-related abiotic stress responses in plants. Front. Plant Sci. 2018, 9, 1897. [Google Scholar] [CrossRef]
- D’Amico-Damião, V.; Lúcio, J.C.B.; Oliveira, R.; Gaion, L.A.; Barreto, R.F.; Carvalho, R.F. Cryptochrome 1a depends on blue light fluence rate to mediate osmotic stress responses in tomato. J. Plant Physiol. 2021, 258–259, 153374. [Google Scholar] [CrossRef]
- Meyer, K.D.; Patil, D.P.; Zhou, J.; Zinoviev, A.; Skabkin, M.A.; Elemento, O.; Pestova, T.V.; Qian, S.-B.; Jaffrey, S.R. 5′ UTR m6A promotes cap-independent translation. Cell 2015, 163, 999–1010. [Google Scholar] [CrossRef]
- Hu, J.; Cai, J.; Park, S.J.; Lee, K.; Li, Y.; Chen, Y.; Yun, J.Y.; Xu, T.; Kang, H. N(6) -Methyladenosine mRNA methylation is important for salt stress tolerance in Arabidopsis. Plant J. 2021, 106, 1759–1775. [Google Scholar] [CrossRef]
- Li, Z.; Shi, J.; Yu, L.; Zhao, X.; Ran, L.; Hu, D.; Song, B. N 6 -methyl-adenosine level in Nicotiana tabacum is associated with tobacco mosaic virus. Virol. J. 2018, 15, 87. [Google Scholar] [CrossRef]
- Miao, Z.; Zhang, T.; Qi, Y.; Song, J.; Han, Z.; Ma, C. Evolution of the RNA N (6)-Methyladenosine Methylome Mediated by Genomic Duplication. Plant Physiol. 2020, 182, 345–360. [Google Scholar] [CrossRef]
- Wendel, J.F. The wondrous cycles of polyploidy in plants. Am. J. Bot. 2015, 102, 1753–1756. [Google Scholar] [CrossRef]
- Chen, Z.; Cao, J.-F.; Zhang, X.-F.; Shangguan, X.-X.; Mao, Y.-B.; Wang, L.-J.; Chen, X.-Y. Cotton genome: Challenge into the polyploidy. Sci. Bull. 2017, 62, 1622–1623. [Google Scholar] [CrossRef]
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive Analysis of mRNA Methylation Reveals Enrichment in 3′ UTRs and near Stop Codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef]
- Meyer, K.D.; Jaffrey, S.R. Rethinking m6A readers, writers, and erasers. Annu. Rev. Cell Dev. Biol. 2017, 33, 319–342. [Google Scholar] [CrossRef]
- Luo, G.-Z.; MacQueen, A.; Zheng, G.; Duan, H.; Dore, L.C.; Lu, Z.; Liu, J.; Chen, K.; Jia, G.; Bergelson, J.; et al. Unique features of the m6A methylome in Arabidopsis thaliana. Nat. Commun. 2014, 5, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Jung, S.; Cheng, C.-H.; Ficklin, S.P.; Lee, T.; Zheng, P.; Jones, D.; Percy, R.G.; Main, D. CottonGen: A genomics, genetics and breeding database for cotton research. Nucleic Acids Res. 2013, 42, D1229–D1236. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.C.J.; Schlechter, R.; Vannozzi, A.; Höll, J.; Hmmam, I.; Bogs, J.; Tornielli, G.B.; Castellarin, S.D.; Matus, J.T. A systems-oriented analysis of the grapevine R2R3-MYB transcription factor family uncovers new insights into the regulation of stilbene accumulation. DNA Res. 2016, 23, 451–466. [Google Scholar] [CrossRef]
- Mistry, J.; Finn, R.D.; Eddy, S.R.; Bateman, A.; Punta, M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013, 41, e121. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Mistry, J.; Schuster-Böckler, B.; Griffiths-Jones, S.; Hollich, V.; Lassmann, T.; Moxon, S.; Marshall, M.; Khanna, A.; Durbin, R.; et al. Pfam: Clans, web tools and services. Nucleic Acids Res. 2006, 34, D247–D251. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, T.J.; Eddy, S.R. nhmmer: DNA homology search with profile HMMs. Bioinformatics 2013, 29, 2487–2489. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 2017, 46, D493–D496. [Google Scholar] [CrossRef]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef]
- Chen, C.J.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.H.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Hu, B.; Jin, J.; Guo, A.-Y.; Zhang, H.; Luo, J.; Gao, G. GSDS 2.0: An upgraded gene feature visualization server. Bioinformatics 2015, 31, 1296–1297. [Google Scholar] [CrossRef]
- Wilkins, M.R.; Gasteiger, E.; Bairoch, A.; Sanchez, J.C.; Williams, K.L.; Appel, R.D.; Hochstrasser, D.F. Protein Identification and Analysis Tools in the ExPASy Server. In 2-D Proteome Analysis Protocols. Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 1999; Volume 112, pp. 531–552. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Zhao, B.; Cao, J.F.; Hu, G.J.; Chen, Z.W.; Wang, L.Y.; Shangguan, X.X.; Wang, L.J.; Mao, Y.B.; Zhang, T.Z.; Wendel, J.F.; et al. Core cis-element variation confers subgenome-biased expression of a transcription factor that functions in cotton fiber elongation. New Phytol. 2018, 218, 1061–1075. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Risso, D.; Ngai, J.; Speed, T.P.; Dudoit, S. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol. 2014, 32, 896–902. [Google Scholar] [CrossRef]
- Wang, M.; Wang, Q.; Zhang, B. Evaluation and selection of reliable reference genes for gene expression under abiotic stress in cotton (Gossypium hirsutum L.). Gene 2013, 530, 44–50. [Google Scholar] [CrossRef]
- Zhang, Y.; Parmigiani, G.; Johnson, W.E. Combat-seq: Batch effect adjustment for RNA-seq count data. NAR Genom. Bioinform. 2020, 2, lqaa078. [Google Scholar] [CrossRef]
- You, Q.; Xu, W.Y.; Zhang, K.; Zhang, L.W.; Yi, X.; Yao, D.X.; Wang, C.C.; Zhang, X.Y.; Zhao, X.H.; Provart, N.J.; et al. ccNET: Database of co-expression networks with functional modules for diploid and polyploid Gossypium. Nucleic Acids Res. 2017, 45, 5625–5626. [Google Scholar] [CrossRef]
- Huang, C.; Li, P.; Cao, J.; Zheng, Z.; Huang, J.; Zhang, X.; Shangguan, X.; Wang, L.; Chen, Z. Comprehensive identification and expression analysis of CRY gene family in Gossypium. BMC Genom. 2022, 23, 1–17. [Google Scholar] [CrossRef]
- Zhu, J.; Chen, J.; Gao, F.; Xu, C.; Wu, H.; Chen, K.; Si, Z.; Yan, H.; Zhang, T. Rapid mapping and cloning of the virescent-1 gene in cotton by bulked segregant analysis–next generation sequencing and virus-induced gene silencing strategies. J. Exp. Bot. 2017, 68, 4125–4135. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Ruan, J.-X.; Huang, J.-Q.; Yang, C.-Q.; Fang, X.; Chen, Z.-W.; Hong, H.; Wang, L.-J.; Mao, Y.-B.; Lu, S.; et al. Characterization of gossypol biosynthetic pathway. Proc. Natl. Acad. Sci. USA 2018, 115, E5410–E5418. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.-Q.; Fang, X.; Tian, X.; Chen, P.; Lin, J.-L.; Guo, X.-X.; Li, J.-X.; Fan, Z.; Song, W.-M.; Chen, F.-Y.; et al. Aromatization of natural products by a specialized detoxification enzyme. Nat. Chem. Biol. 2020, 16, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.-F.; Huang, J.-Q.; Liu, X.; Huang, C.-C.; Zheng, Z.-S.; Zhang, X.-F.; Shangguan, X.-X.; Wang, L.-J.; Zhang, Y.-G.; Wendel, J.F.; et al. Genome-wide characterization of the GRF family and their roles in response to salt stress in Gossypium. BMC Genom. 2020, 21, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Cao, J.; Huang, C.; Zheng, Z.; Liu, X.; Shangguan, X.; Wang, L.; Zhang, Y.; Chen, Z. Characterization of cotton ARF factors and the role of GhARF2b in fiber development. BMC Genom. 2021, 22, 1–15. [Google Scholar] [CrossRef]
- Cui, W.; Chen, Z.; Shangguan, X.; Li, T.; Wang, L.; Xue, X.; Cao, J. TRY intron2 determined its expression in inflorescence activated by SPL9 and MADS-box genes in Arabidopsis. Plant Sci. 2022, 321, 111311. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(-Delta Delta C) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Cao, J.-F.; Zhao, B.; Huang, C.-C.; Chen, Z.-W.; Zhao, T.; Liu, H.-R.; Hu, G.-J.; Shangguan, X.-X.; Shan, C.-M.; Wang, L.-J.; et al. The miR319-targeted GhTCP4 promotes the transition from cell elongation to wall thickening in cotton fiber. Mol. Plant 2020, 13, 1063–1077. [Google Scholar] [CrossRef]
- Bent, A. Arabidopsis thaliana Floral Dip Transformation Method. Methods Mol. Biol. 2006, 343, 87–104. [Google Scholar] [CrossRef]
- Shangguan, X.; Yang, Q.; Wu, X.; Cao, J. Function analysis of a cotton R2R3 MYB transcription factor GhMYB3 in regulating plant trichome development. Plant Biol. 2021, 23, 1118–1127. [Google Scholar] [CrossRef]
- Chen, Z.; Zhao, J.; Song, J.; Han, S.; Du, Y.; Qiao, Y.; Liu, Z.; Qiao, J.; Li, W.; Li, J.; et al. Influence of graphene on the multiple metabolic pathways of Zea mays roots based on transcriptome analysis. PLoS ONE 2021, 16, e0244856. [Google Scholar] [CrossRef]
- Chen, Z.; Zhao, J.; Qiao, J.; Li, W.; Guan, Z.; Liu, Z.; Bai, X.; Xing, B.; Zhang, J.; Li, J.; et al. Graphene-Mediated Antioxidant Enzyme Activity and Respiration in Plant Roots. ACS Agric. Sci. Technol. 2022, 2, 646–660. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014, 15, 550. [Google Scholar] [CrossRef]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Locus Name | Chr | Genomics Position | CDS | No. of Introns | Size (aa) | MW | pI |
|---|---|---|---|---|---|---|---|---|
| GhMETTL3_A | GH_A12G2586 | A12 | 104,206,704-104,211,250 | 2106 | 6 | 701 | 78.50 | 5.98 |
| GhMETTL3_D | GH_D12G2605 | D12 | 58,367,589-58,370,785 | 2106 | 6 | 701 | 78.42 | 5.92 |
| GhMETTL14_A | GH_A07G0817 | A07 | 10,130,789-10,135,646 | 3558 | 5 | 1185 | 132.59 | 7.10 |
| GhMETTL14_D | GH_D07G0819 | D07 | 8,927,821-8,932,500 | 3555 | 5 | 1184 | 132.49 | 6.92 |
| GhMETTL-Like_A | GH_A06G0112 | A06 | 1,007,934-1,011,291 | 1269 | 8 | 422 | 48.45 | 7.18 |
| GhMETTL-Like_D | GH_D06G0096 | D06 | 890,591-893,931 | 1269 | 8 | 422 | 48.47 | 6.88 |
| Species | METTL Number | METTL3 Homologs | METTL14 Homologs | METTL-Like Homologs |
|---|---|---|---|---|
| Gossypium hirsutum | 6 | GH_A12G2586/GH_D12G2605 | GH_A07G0817/GH_D07G0819 | GH_A06G0112/GH_D06G0096 |
| Gossypium barbadense | 6 | GB_A12G2683/GB_D12G2687 | GB_A07G0808/GB_D07G0816 | GB_A06G0089/GB_D06G0116 |
| Gossypium tomentosum | 6 | GtA12G272500/GtD12G281700 | GtA07G086300/GtD07G087100 | GtA06G013000/GtD06G014000 |
| Gossypium mustelinum | 6 | GmA12G260700/GmD12G271800 | GmA07G083700/GmD07G085700 | GmA06G011900/GmD06G012500 |
| Gossypium darwinii | 6 | GdA12G271500/GdD12G280500 | GdA07G086700/GdD07G087200 | GdA06G013300/GdD06G012500 |
| Gossypium herbaceum | 4 | Ghe12G29960 | Ghe07G08800 | Ghe06G01080/Ghe06G01370 |
| Gossypium arboreum | 4 | Gar12G30100 | Gar07G08930 | Gar06G00970/Gar06G01270 |
| Gossypium longicalyx | 3 | Golon.012G274900-1 | Golon.007G088200-1 | Golon.006G006700-1 |
| Gossypium thurberi | 3 | Gothu.00034516-RA | Gothu.00035901-RA | Gothu.00024549-RA |
| Gossypium raimondii | 3 | Gorai.008G257500.1 | Gorai.001G083900.1 | Gorai.010G011900.1 |
| Gossypium turneri | 3 | Gossypium turneri_TURN.02g361810 | Gossypium turneri_TURN.07g196460 | Gossypium turneri_TURN.06g175650 |
| Gossypium australe | 3 | G. australe_KAA3460050.1 | G. australe_KAA3458407.1 | G. australe_KAA3476391.1 |
| Gossypioides kirkii | 3 | Kirkii_Version3_Juiced.00g331840.m01.polypeptide | Kirkii_Version3_Juiced.00g187490.m01.polypeptide | Kirkii_Version3_Juiced.00g127480.m01.polypeptide |
| Arabidopsis thaliana | 3 | AT4G10760.1_MTA | AT4G09980.1_MTB | AT1G19340.1 |
| Homo sapiens | 2 | NM_019852.4_METTL3 | NM_020961.3_METTL14 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, J.; Huang, C.; Liu, J.; Li, C.; Liu, X.; Zheng, Z.; Hou, L.; Huang, J.; Wang, L.; Zhang, Y.; et al. Comparative Genomics and Functional Studies of Putative m6A Methyltransferase (METTL) Genes in Cotton. Int. J. Mol. Sci. 2022, 23, 14111. https://doi.org/10.3390/ijms232214111
Cao J, Huang C, Liu J, Li C, Liu X, Zheng Z, Hou L, Huang J, Wang L, Zhang Y, et al. Comparative Genomics and Functional Studies of Putative m6A Methyltransferase (METTL) Genes in Cotton. International Journal of Molecular Sciences. 2022; 23(22):14111. https://doi.org/10.3390/ijms232214111
Chicago/Turabian StyleCao, Junfeng, Chaochen Huang, Jun’e Liu, Chenyi Li, Xia Liu, Zishou Zheng, Lipan Hou, Jinquan Huang, Lingjian Wang, Yugao Zhang, and et al. 2022. "Comparative Genomics and Functional Studies of Putative m6A Methyltransferase (METTL) Genes in Cotton" International Journal of Molecular Sciences 23, no. 22: 14111. https://doi.org/10.3390/ijms232214111
APA StyleCao, J., Huang, C., Liu, J., Li, C., Liu, X., Zheng, Z., Hou, L., Huang, J., Wang, L., Zhang, Y., Shangguan, X., & Chen, Z. (2022). Comparative Genomics and Functional Studies of Putative m6A Methyltransferase (METTL) Genes in Cotton. International Journal of Molecular Sciences, 23(22), 14111. https://doi.org/10.3390/ijms232214111