
Exploring the Molecular Mechanism of Sepal Formation in the Decorative Flowers of Hydrangea macrophylla ′Endless Summer′ Based on the ABCDE Model

Abstract
1. Introduction
2. Results
2.1. Phenotypic Observation
2.2. Quality Analysis of Transcriptome Sequencing
2.3. Functional Annotation and Expression Analysis of Transcriptome Unigenes
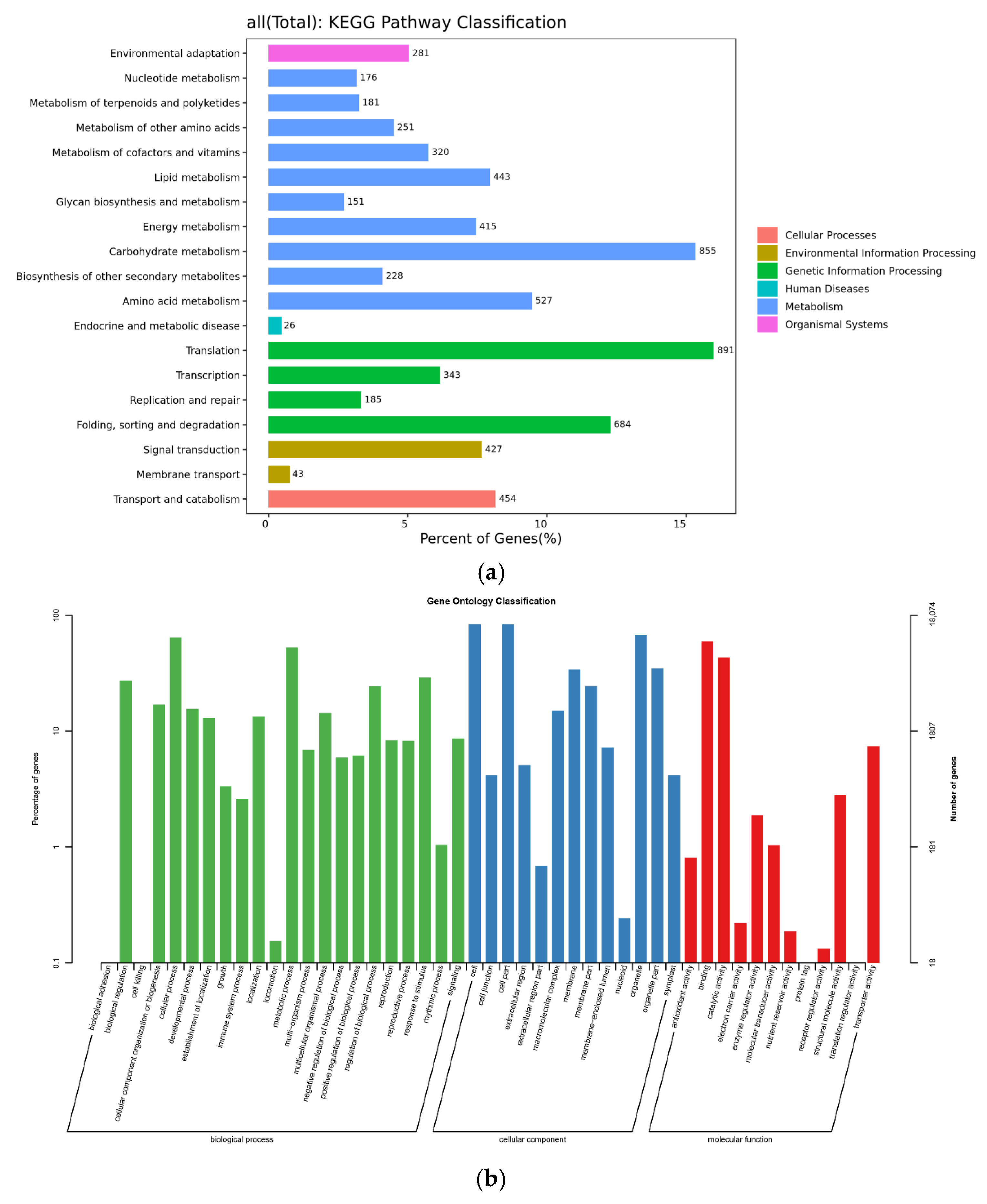
2.3.1. Analysis of KEGG Metabolic Pathway
2.3.2. GO Function Classification
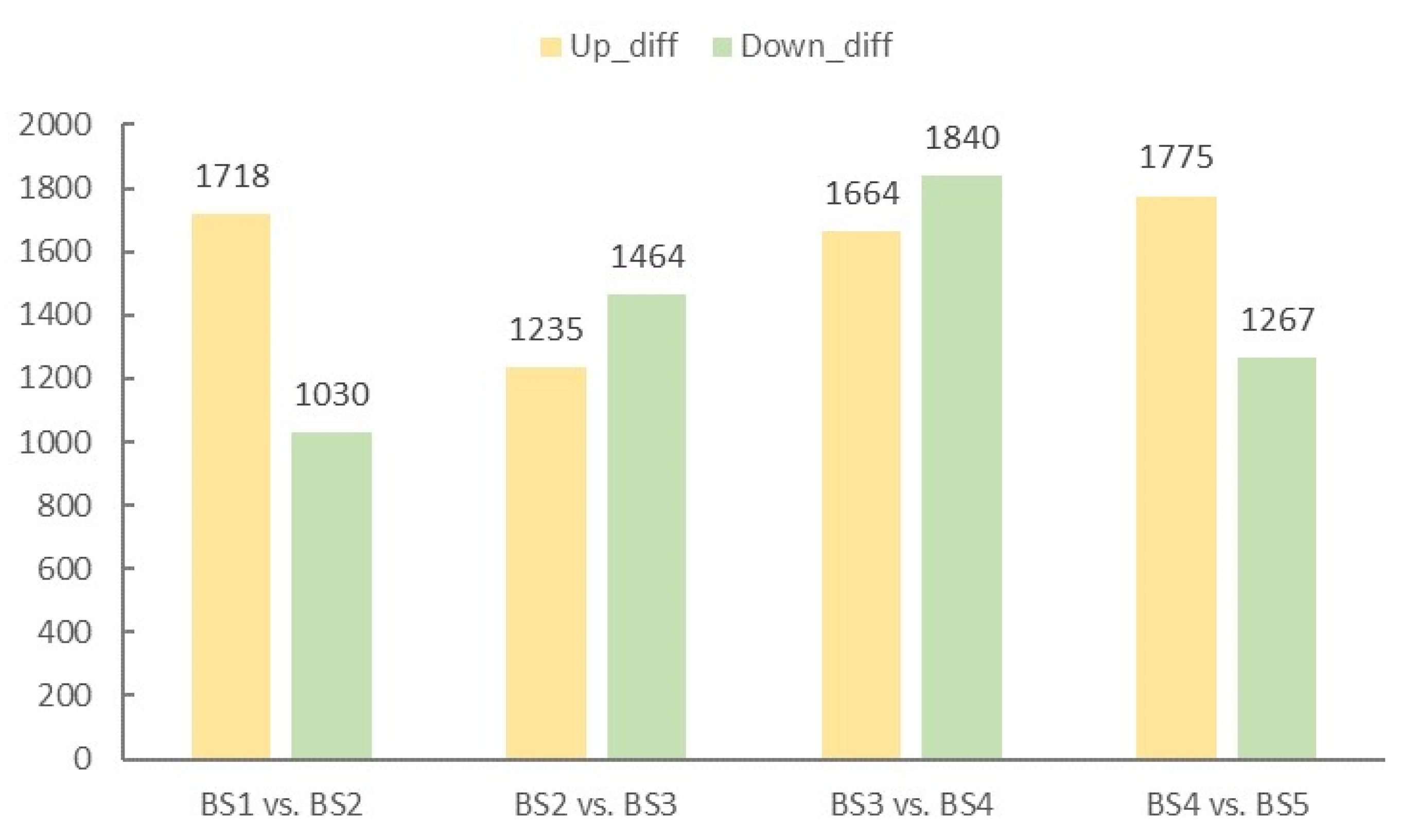
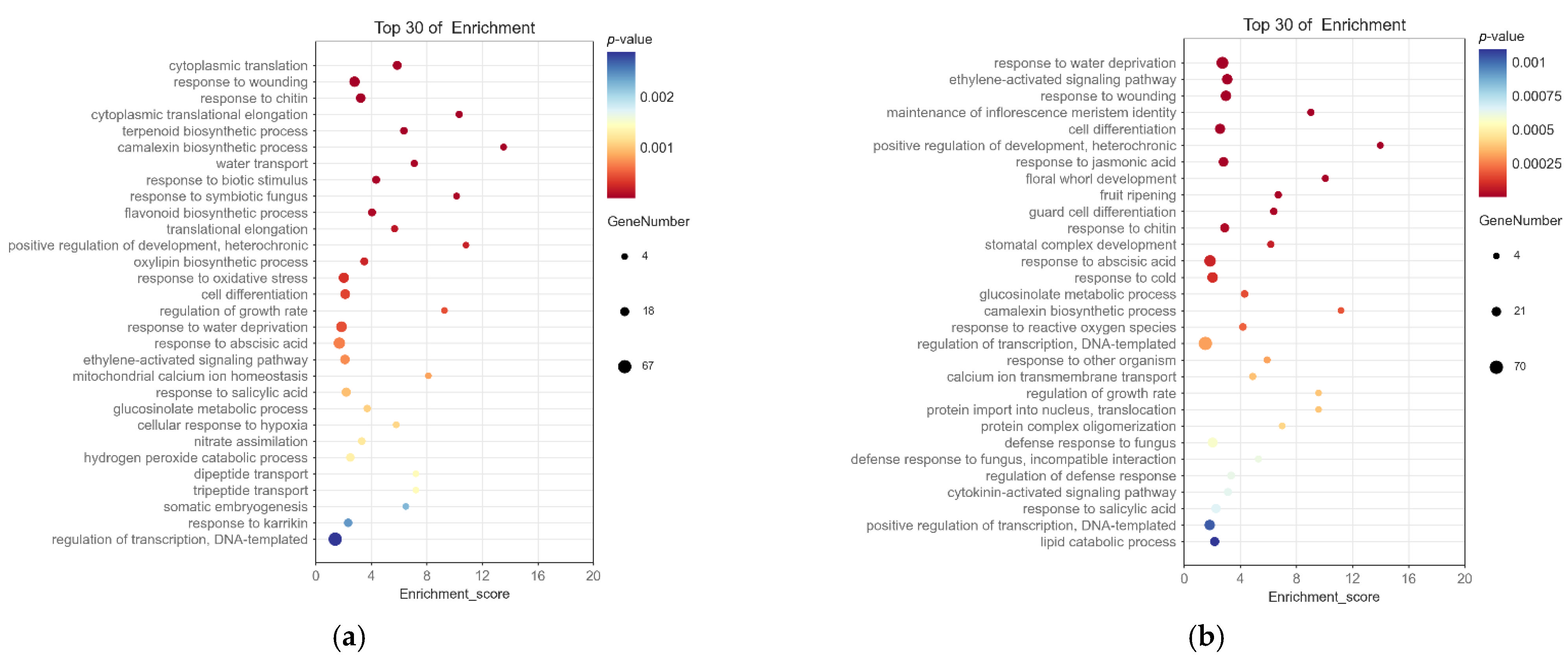
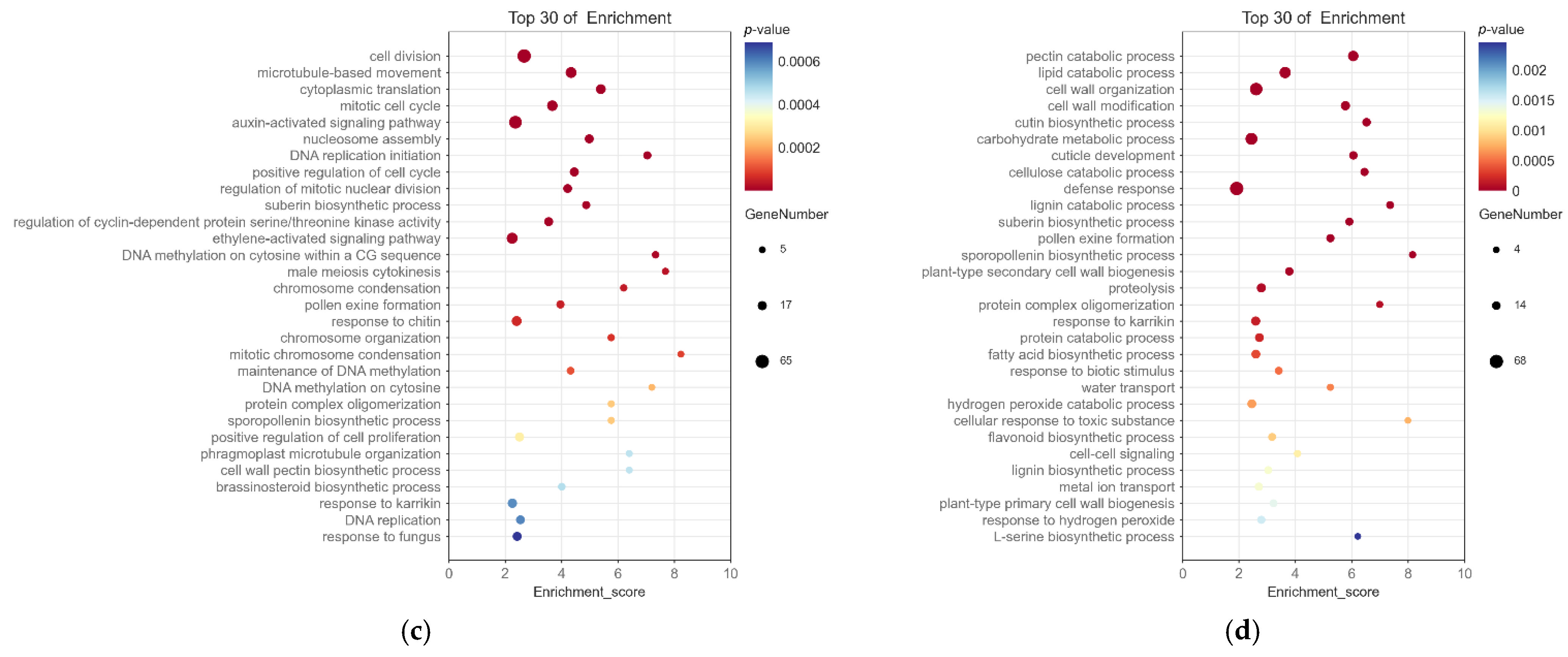
2.4. Screening and Enrichment Analysis of Differentially Expressed Genes
2.5. Analysis of Differentially Expressed Genes Related to the Development of Decorative Flowers
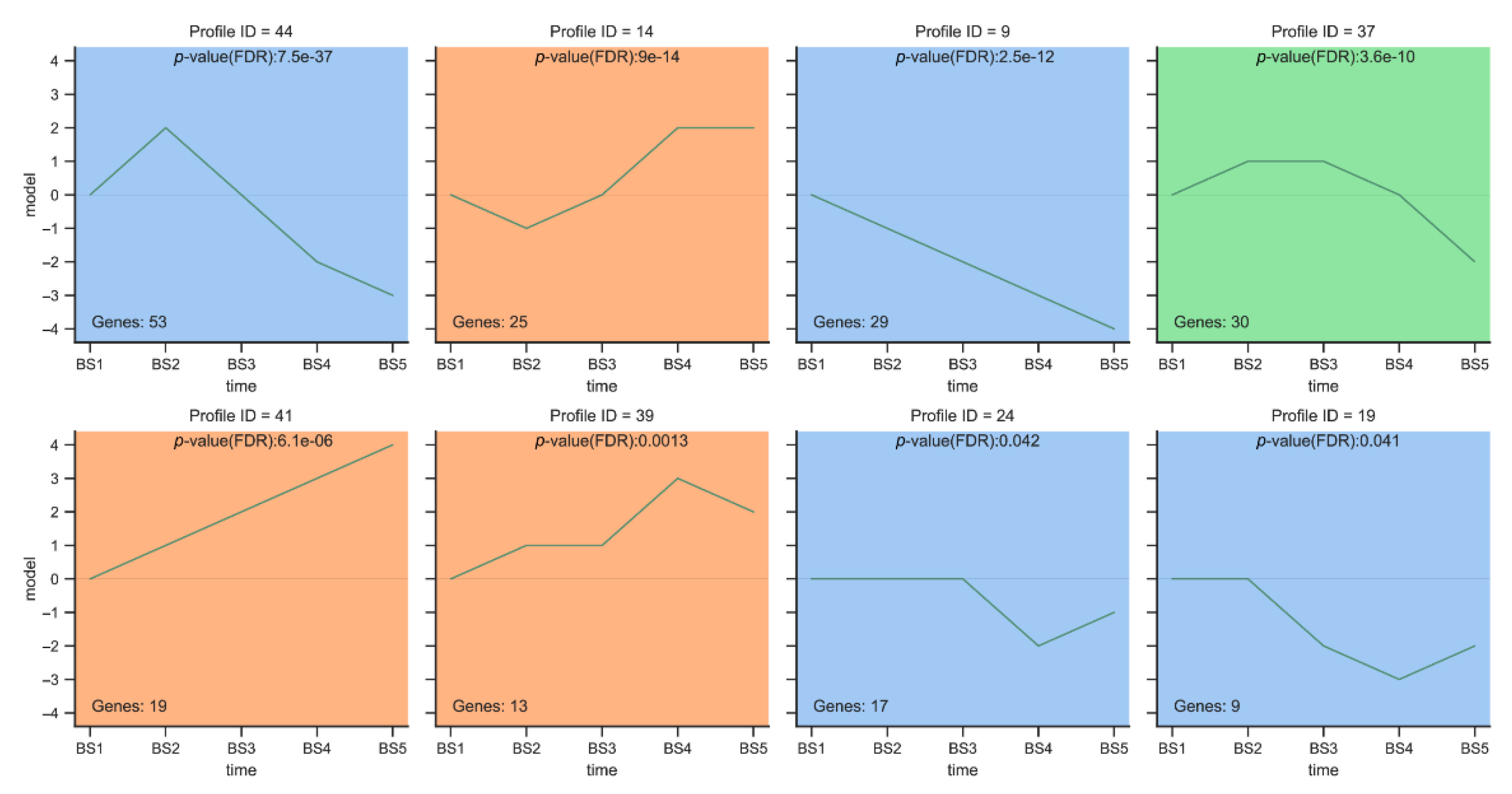
2.5.1. Trend Analysis
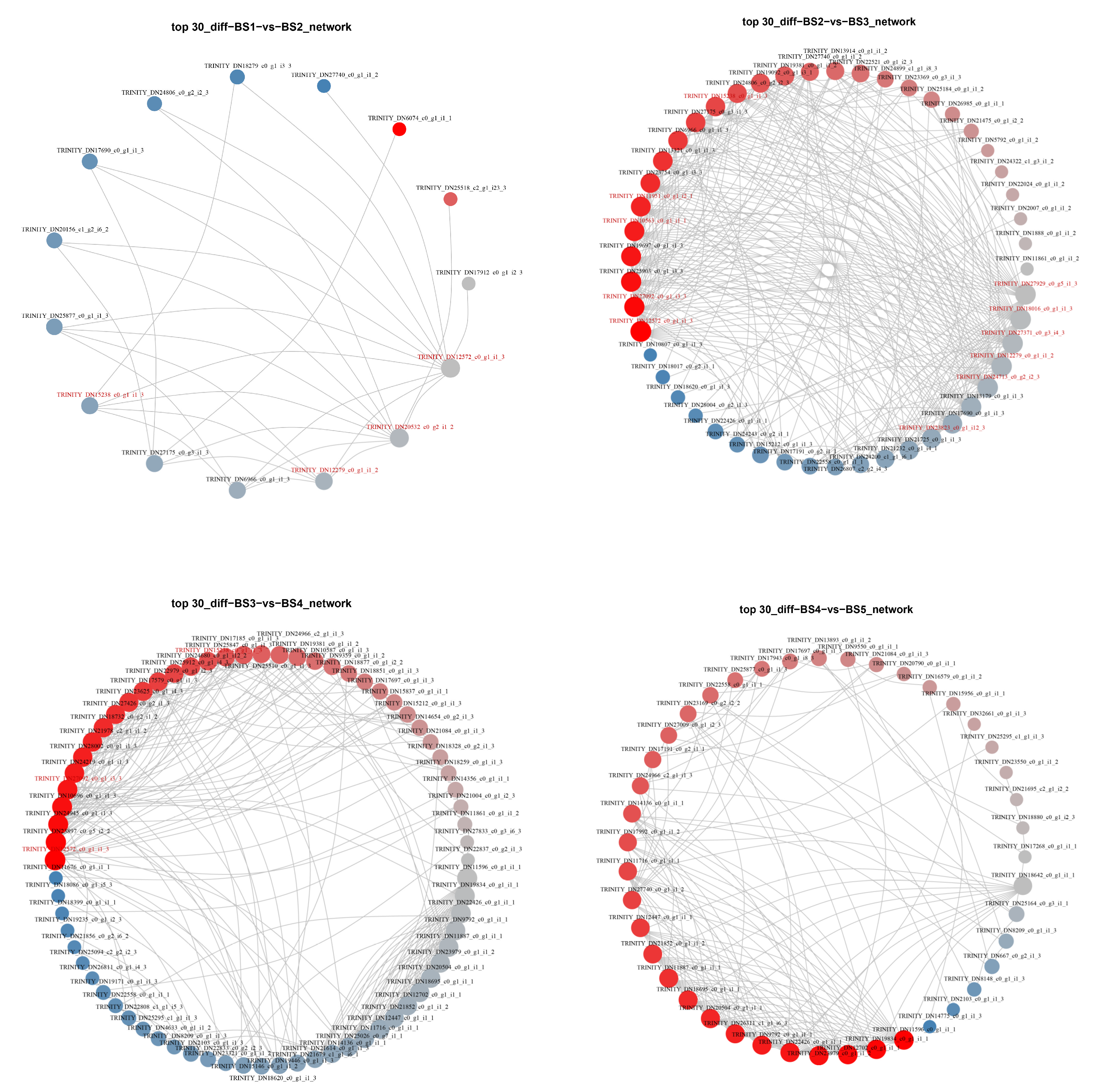
2.5.2. Protein–Protein Interaction (PPI) Analysis
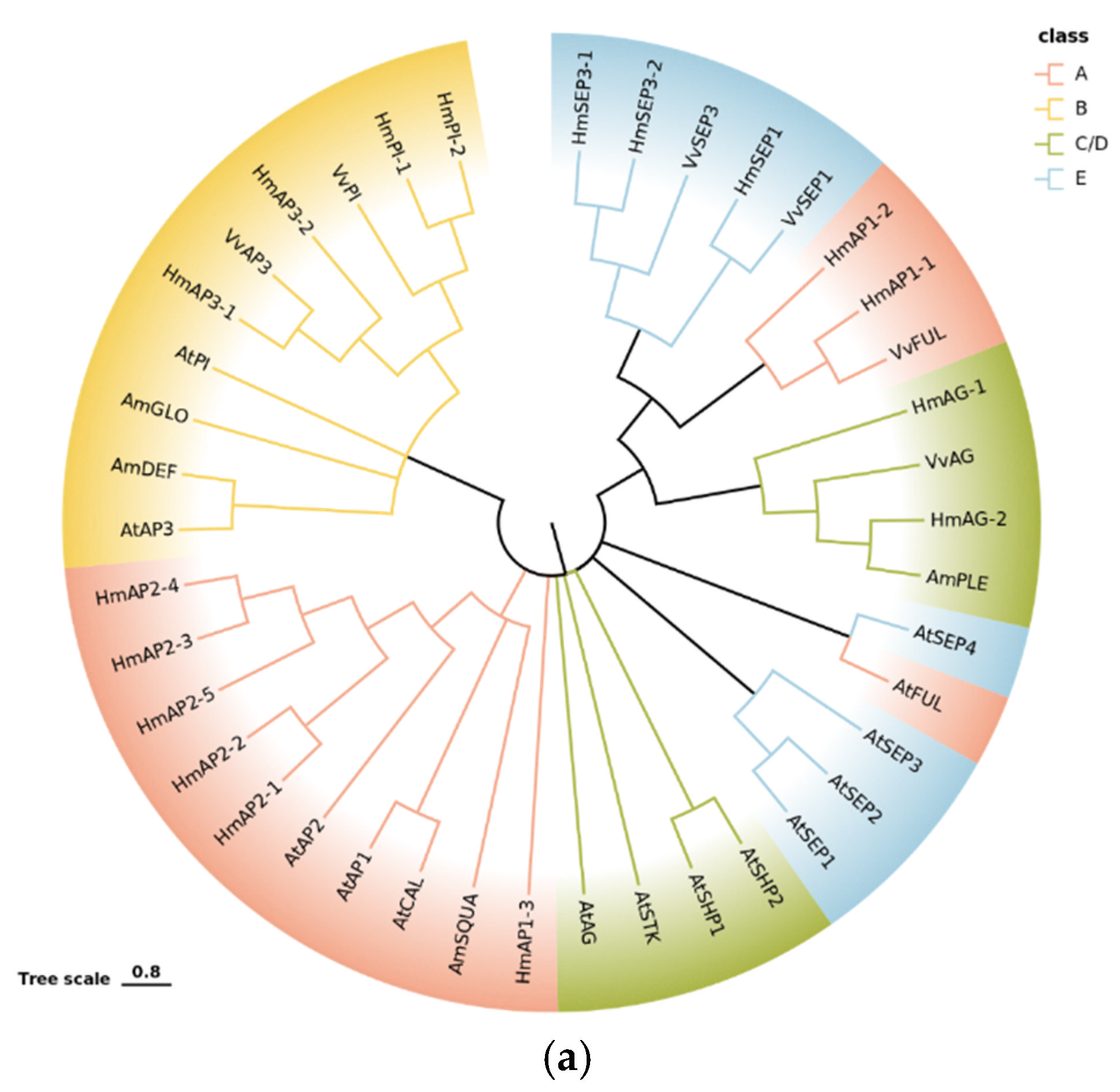
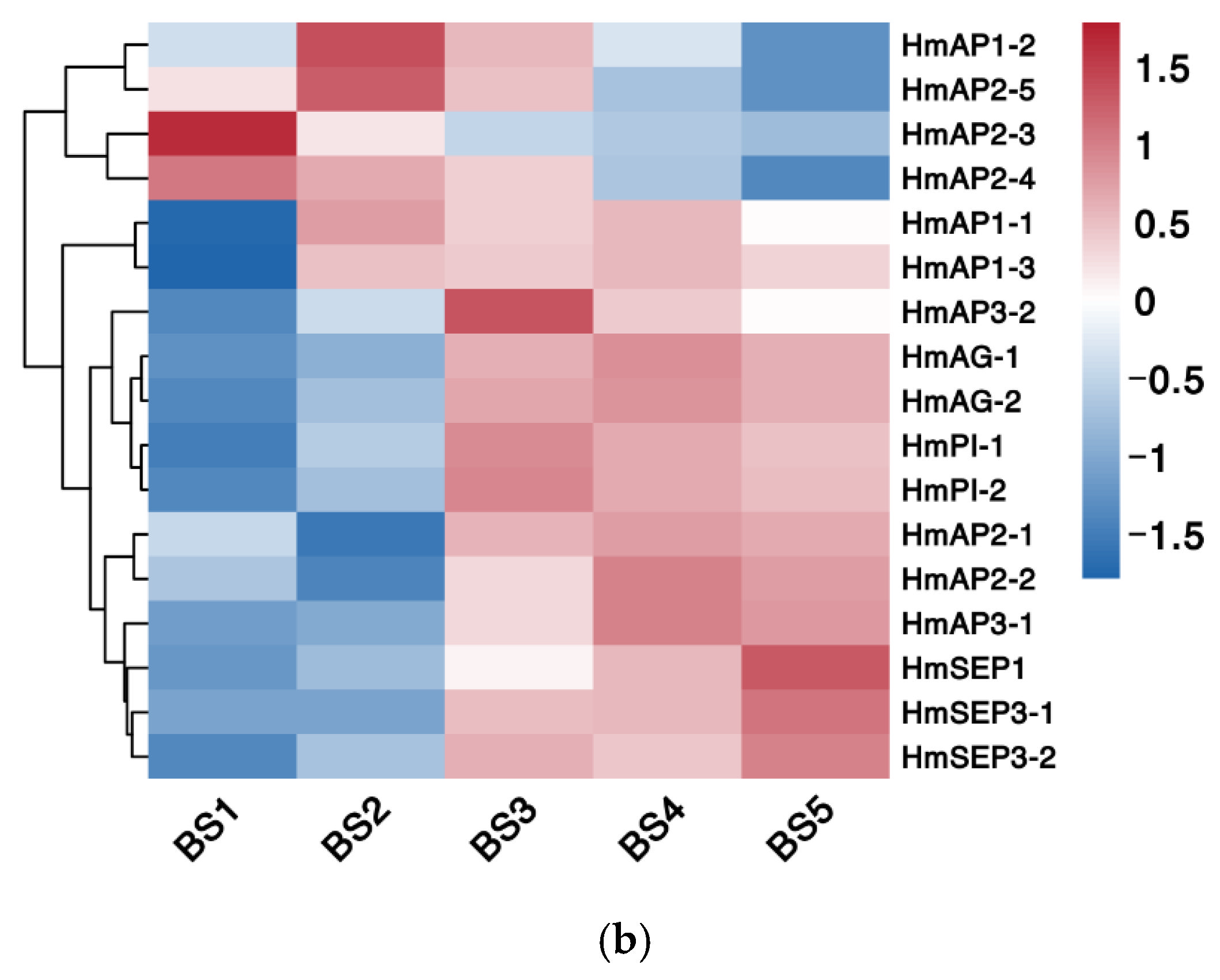
2.5.3. Phylogenetic and Expression Pattern Analysis
2.6. Real-Time Quantitative PCR (RT-qPCR) Verification of Differentially Expressed Genes
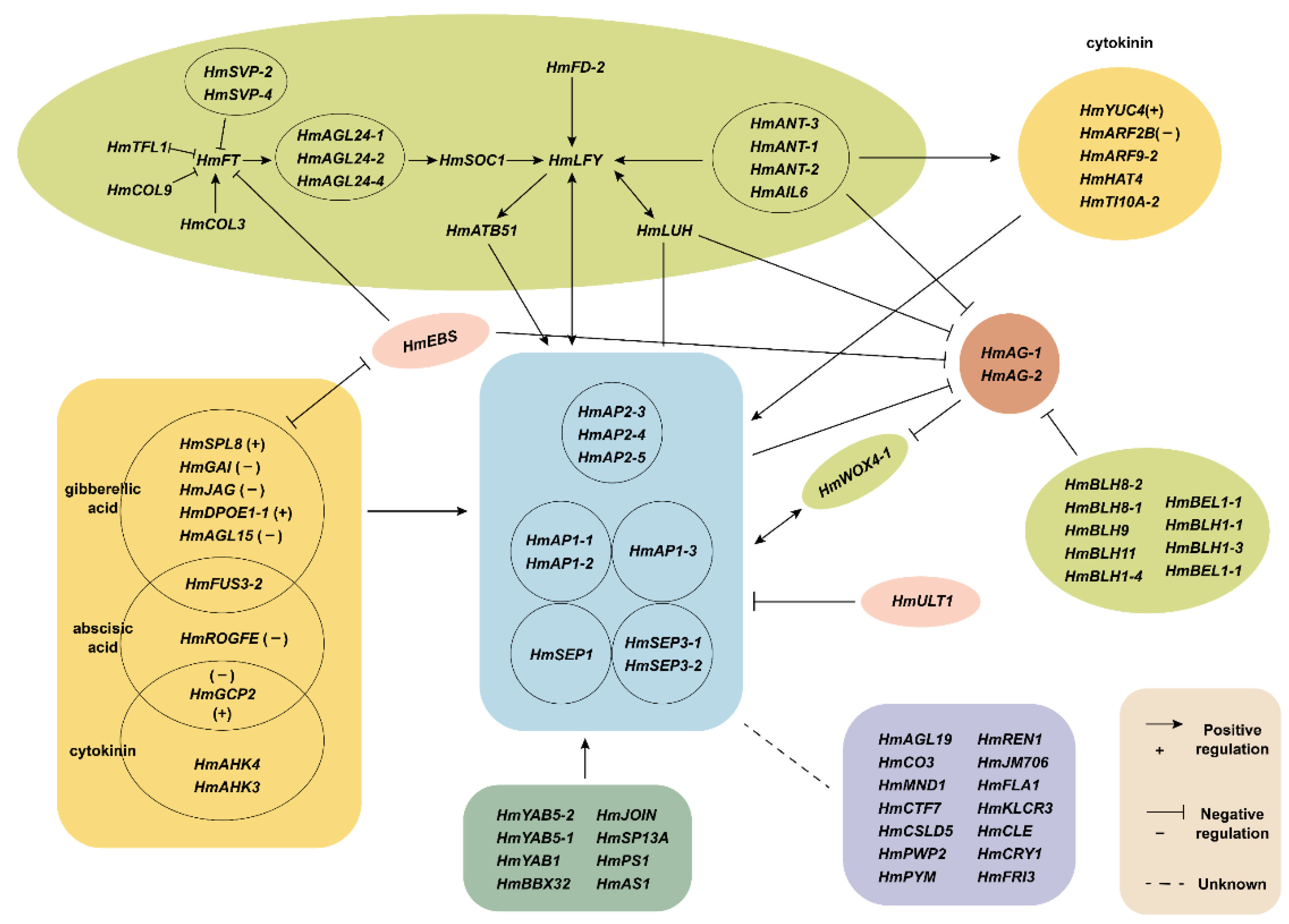
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Transcriptome Sequencing
4.2.1. Extraction and Detection of Total RNA from Transcriptome Sequencing Samples
4.2.2. Construction of cDNA Library and Transcriptome Sequencing
4.2.3. Sequencing Data Quality Monitoring and Sequence Assembly
4.3. Bioinformatics Analysis and Screening of Differentially Expressed Genes
4.4. RT-qPCR Verification
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kitamura, Y.; Hosokawa, M.; Uemachi, T.; Yazawa, S. Increase in the number of decorative florets in the inflorescence of Hydrangea through phytoplasma infection. Sci. Hortic. 2010, 124, 134–138. [Google Scholar] [CrossRef]
- Krizek, B.A.; Fletcher, J.C. Molecular mechanisms of flower development: An armchair guide. Nat. Rev. Genet. 2005, 6, 688–698. [Google Scholar] [CrossRef]
- Tang, L.; Song, Y.; Li, J. Advances on the molecular mechanism of floral organ development. J. Northwest Bot. 2013, 33, 1063–1070. [Google Scholar]
- Jing, D.; Guo, Q.; Chen, W.; Xia, Y.; Wu, D.; Dang, J.; He, Q.; Liang, G. Model evolution and molecular regulation of floral organ development in angiosperms. Plant Physiol. J. 2018, 54, 355–362. [Google Scholar]
- Cong, N.; Cheng, Z.; Wan, J. The ABCDE model of floral organ development. Chin. Agric. Sci. Bull. 2007, 23, 124–128. [Google Scholar]
- Bowman, J.L.; Smyth, D.R.; Meyerowitz, E.M. Genetic Interactions among floral homeotic genes of Arabidopsis. Development 1991, 112, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Coen, E.S.; Meyerowitz, E.M. The war of the whorls: Genetic interactions controlling flower development. Nature 1991, 353, 31–37. [Google Scholar] [CrossRef]
- Pinyopich, A.; Ditta, G.S.; Savidge, B.; Liljegren, S.J.; Baumann, E.; Wisman, E.; Yanofsky, M.F. Assessing the redundancy of MADS-box genes during carpel and ovule development. Nature 2003, 424, 85–88. [Google Scholar] [CrossRef]
- Theißen, G. Development of floral organ identity: Stories from the MADS house. Curr. Opin. Plant Biol. 2001, 4, 75–85. [Google Scholar] [CrossRef]
- Wang, D.; Guo, A.; Yang, C.; Li, D.; Tian, J. Progress on molecular mechanism of floral organ development. J. Northwest AF Univ. Nat. Sci. Ed. 2007, 35, 203–209. [Google Scholar]
- Luan, Y. Analysis of MADS-box gene family in different plants and the flower transcriptome of Carya cathayensis Sarg. Master’s Thesis, Zhejiang A&F University, Lin’an, China, 2019. [Google Scholar]
- Ma, H.; Yanofsky, M.F.; Meyerowitz, E.M. AGL1-AGL6, an Arabidopsis gene family with similarity to floral homeotic and transcription factor genes. Genes Dev. 1991, 5, 484–495. [Google Scholar] [CrossRef] [PubMed]
- Arora, R.; Agarwal, P.; Ray, S.; Singh, A.K.; Singh, V.P.; Tyagi, A.K.; Kapoor, S. MADS-box gene family in rice: Genome-wide identification, organization and expression profiling during reproductive development and stress. BMC Genom. 2007, 8, 242. [Google Scholar] [CrossRef]
- Díaz-Riquelme, J.; Lijavetzky, D.; Martínez-Zapater, J.M.; Carmona, M.a.J. Genome-wide analysis of MIKCC-type MADS box genes in grapevine. Plant Physiol. 2009, 149, 354–369. [Google Scholar] [CrossRef] [PubMed]
- Münster, T.; Dirosa, A.; Kim, J.T.; Martin, W.; Saedler, H.; Theißen, G.; Pahnke, J. Floral homeotic genes were recruited from homologous MADS-box genes preexisting in the common ancestor of ferns and seedplants. Proc. Natl. Acad. Sci. USA 1997, 94, 2415–2420. [Google Scholar] [CrossRef] [PubMed]
- Becker, A.; Theißen, G. The major clades of MADS-box genes and their role in the development and evolution of flowering plants. Mol. Phylogenet. Evol. 2003, 29, 464–489. [Google Scholar] [CrossRef]
- Uemachi, T.; Kurokawa, M.; Nishio, T. Comparison of inflorescence composition and development in the lacecap and its sport, hortensia Hydrangea macrophylla (Thunb.) Ser. J.-Jpn. Soc. Hortic. Sci. 2006, 75, 154–160. [Google Scholar] [CrossRef][Green Version]
- Kanno, A.; Saeki, H.; Kameya, T.; Saedler, H.; Theissen, G. Heterotopic expression of class B floral homeotic genes supports a modified ABC model for tulip (Tulipa gesneriana). Plant Mol. Biol. 2003, 52, 831–841. [Google Scholar] [CrossRef]
- Nakamura, T.; Fukuda, T.; Nakano, M.; Hasebe, M.; Kameya, T.; Kanno, A. The modified ABC model explains the development of the petaloid perianth of Agapanthus praecox ssp. orientalis (Agapanthaceae) flowers. Plant Mol. Biol. 2005, 58, 435–445. [Google Scholar] [CrossRef]
- Kramer, E.M.; Elena, M.; Stilio; Di, V.S.; Schlüter; Philipp, M. Complex patterns of gene duplication in the APETALA3 and PISTILLATA lineages of the Ranunculaceae. Int. J. Plant Sci. 2003, 164, 803–808. [Google Scholar] [CrossRef]
- Kitamura, Y.; Hosokawa, M.; Uemachi, T.; Yazawa, S. Selection of ABC genes for candidate genes of morphological changes in hydrangea floral organs induced by phytoplasma infection. Sci. Hortic. 2009, 122, 603–609. [Google Scholar] [CrossRef]
- Uemachi, T.; Kato, Y.; Nishio, T. Comparison of decorative and non-decorative flowers in Hydrangea macrophylla (Thunb.) Ser. Sci. Hortic. 2004, 102, 325–334. [Google Scholar] [CrossRef]
- Theißen, G.; Melzer, R.; Rümpler, F. MADS-domain transcription factors and the floral quartet model of flower development: Linking plant development and evolution. Development 2016, 143, 3259–3271. [Google Scholar] [CrossRef] [PubMed]
- Ditta, G.; Pinyopich, A.; Robles, P.; Pelaz, S.; Yanofsky, M.F. The SEP4 Gene of Arabidopsis thaliana functions in floral organ and meristem identity. Curr. Biol. 2004, 14, 1935–1940. [Google Scholar] [CrossRef] [PubMed]
- Shan, H.; Zhan, N.; Liu, C.; Xu, G.; Zhang, J.; Chen, Z.; Kong, H. Patterns of gene duplication and functional diversification during the evolution of the AP1/SQUA subfamily of plant MADS-box genes. Mol. Phylogenet. Evol. 2007, 44, 26–41. [Google Scholar] [CrossRef]
- Mandel, M.A.; Gustafson-Brown, C.; Savidge, B.; Yanofsky, M.F. Molecular characterization of the Arabidopsis floral homeotic gene APETALA1. Nature 1992, 360, 273–277. [Google Scholar] [CrossRef]
- Busch, M.A.; Bomblies, K.; Weigel, D. Activation of a floral homeotic Gene in Arabidopsis. Science 1999, 285, 585–587. [Google Scholar] [CrossRef]
- Kater, M.M.; Dreni, L.; Colombo, L. Functional conservation of MADS-box factors controlling floral organ identity in rice and Arabidopsis. J. Exp. Bot. 2006, 57, 3433–3444. [Google Scholar] [CrossRef]
- Kempin, S.A.; Savidge, B.; Yanofsky, M.F. Molecular basis of the cauliflower phenotype in Arabidopsis. Science 1995, 267, 522–525. [Google Scholar] [CrossRef]
- Theißen, G. Shattering developments. Nature 2000, 404, 711–713. [Google Scholar] [CrossRef]
- Theißen, G.; Becker, A.; Rosa, A.D.; Kanno, A.; Kim, J.T.; Münster, T.; Winter, K.U.; Saedler, H. A short history of MADS-box genes in plants. Plant Mol. Biol. 2000, 42, 115–149. [Google Scholar] [CrossRef]
- Cheng, Z. Identification of MADS-box genes and their function in floral organ development of Phyllostachys edulis. Ph.D. Thesis, Chinese Academy of Forestry, Beijing, China, 2016. [Google Scholar]
- Nashima, K.; Shirasawa, K.; Ghelfi, A.; Hirakawa, H.; Isobe, S.; Suyama, T.; Wada, T.; Kurokura, T.; Uemachi, T.; Azuma, M.; et al. Genome sequence of Hydrangea macrophylla and its application in analysis of the double flower phenotype. DNA Res. 2020, 28, dsaa026. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Wang, T.; Wei, X. Function of AP2 gene during floral organs development in higher plant: Review. Chin. J. Trop. Agric. 2005, 25, 50–56. [Google Scholar]
- Wen, K.; Liu, X. The important role of AP2 functional genes in plant floral development. Biotechnol. Bull. 2010, 2, 1–7. [Google Scholar]
- Parcy, F.; Nilsson, O.; Busch, M.A.; Lee, I.; Weigel, D. A genetic framework for floral patterning. Nature 1998, 395, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Piñeiro, M.; Coupland, G. The control of flowering time and floral identity in Arabidopsis. Plant Physiol. 1998, 117, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Jofuku, K.D.; Boer, B.G.d.; Montagu, M.V.; KOkamuro, J. Control of Arabidopsis Flower and Seed Development by the Homeotic Gene APETALA2. Plant Cell 1994, 6, 1211–1225. [Google Scholar] [PubMed]
- Chen, X. A microRNA as a translational repressor of APETALA2 in Arabidopsis flower development. Science 2004, 303, 2022–2025. [Google Scholar] [CrossRef] [PubMed]
- Elliott, R.C.; Betzner, A.S.; Huttner, E.; Oakes, M.P.; Tucker, W.Q.; Gerentes, D.; Perez, P.; Smyth, D.R. AINTEGUMENTA, an APETALA2-like gene of Arabidopsis with pleiotropic roles in ovule development and floral organ growth. Plant Cell 1996, 8, 155–168. [Google Scholar]
- Klucher, K.M.; Chow, H.; Fischer, R.R.L. The AINTEGUMENTA gene of Arabidopsis required for ovule and female gametophyte development is related to the floral homeotic gene APETALA2. Plant Cell 1996, 8, 137–153. [Google Scholar]
- Tsai, W.C.; Pan, Z.J.; Hsiao, Y.Y.; Chen, L.J.; Liu, Z.J. Evolution and function of MADS-box genes involved in orchid floral development. J. Syst. Evol. 2013, 52, 397–410. [Google Scholar] [CrossRef]
- Pelaz, S.; Ditta, G.S.; Baumann, E.; Wisman, E.; Yanofsky, M.F. B and C function floral organ identity functions require SEPALLATA MADS-box genes. Nature 2000, 405, 200–203. [Google Scholar] [CrossRef] [PubMed]
- Pelaz, S.; Tapia-López, R.; Alvarez-Buylla, E.R.; Yanofsky, M.F. Conversion of leaves into petals in Arabidopsis. Curr. Biol. 2001, 11, 182–184. [Google Scholar] [CrossRef]
- Takashi, H.; Koji, G. Complexes of MADS-box proteins are sufficient to convert leaves into floral organs. Nature 2001, 409, 525–529. [Google Scholar]
- Zhang, G. Cloning and Expression Analysis of the pH-Regulating Gene NHX1 in Sepals of Hydrangea macrophylla Vacuole. Master’s Thesis, Chinese Academy of Agricultural Sciences, Beijing, China, 2021. [Google Scholar]
- Livak, K.J.; Schmittgen, T. Analysis of relative gene expression data using real-time quantitative PCR and the 2-DDCt method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Reads | Raw Bases | Clean Reads | Clean Bases | Valid Bases | Q30 | GC |
|---|---|---|---|---|---|---|---|
| BS1 | 51.67M | 7.75G | 50.52M | 7.36G | 94.96% | 88.66% | 45.55% |
| BS2 | 47.87M | 7.18G | 46.82M | 6.84G | 95.22% | 91.14% | 45.52% |
| BS3 | 50.73M | 7.61G | 49.60M | 7.25G | 95.34% | 90.85% | 45.37% |
| BS4 | 49.45M | 7.42G | 48.24M | 7.06G | 95.14% | 91.86% | 45.60% |
| BS5 | 47.49M | 7.12G | 46.46M | 6.79G | 95.31% | 91.69% | 45.38% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Q.; Lyu, T.; Lyu, Y. Exploring the Molecular Mechanism of Sepal Formation in the Decorative Flowers of Hydrangea macrophylla ′Endless Summer′ Based on the ABCDE Model. Int. J. Mol. Sci. 2022, 23, 14112. https://doi.org/10.3390/ijms232214112
Wang Q, Lyu T, Lyu Y. Exploring the Molecular Mechanism of Sepal Formation in the Decorative Flowers of Hydrangea macrophylla ′Endless Summer′ Based on the ABCDE Model. International Journal of Molecular Sciences. 2022; 23(22):14112. https://doi.org/10.3390/ijms232214112
Chicago/Turabian StyleWang, Qi, Tong Lyu, and Yingmin Lyu. 2022. "Exploring the Molecular Mechanism of Sepal Formation in the Decorative Flowers of Hydrangea macrophylla ′Endless Summer′ Based on the ABCDE Model" International Journal of Molecular Sciences 23, no. 22: 14112. https://doi.org/10.3390/ijms232214112
APA StyleWang, Q., Lyu, T., & Lyu, Y. (2022). Exploring the Molecular Mechanism of Sepal Formation in the Decorative Flowers of Hydrangea macrophylla ′Endless Summer′ Based on the ABCDE Model. International Journal of Molecular Sciences, 23(22), 14112. https://doi.org/10.3390/ijms232214112