
Receptor Tyrosine Kinase Inhibitors for the Treatment of Recurrent and Unresectable Bone Sarcomas

 , , , , , and
, , , , , and
Abstract
1. Bone Sarcomas: Introduction and Background
1.1. Epidemiology
1.2. Principles of Treatment
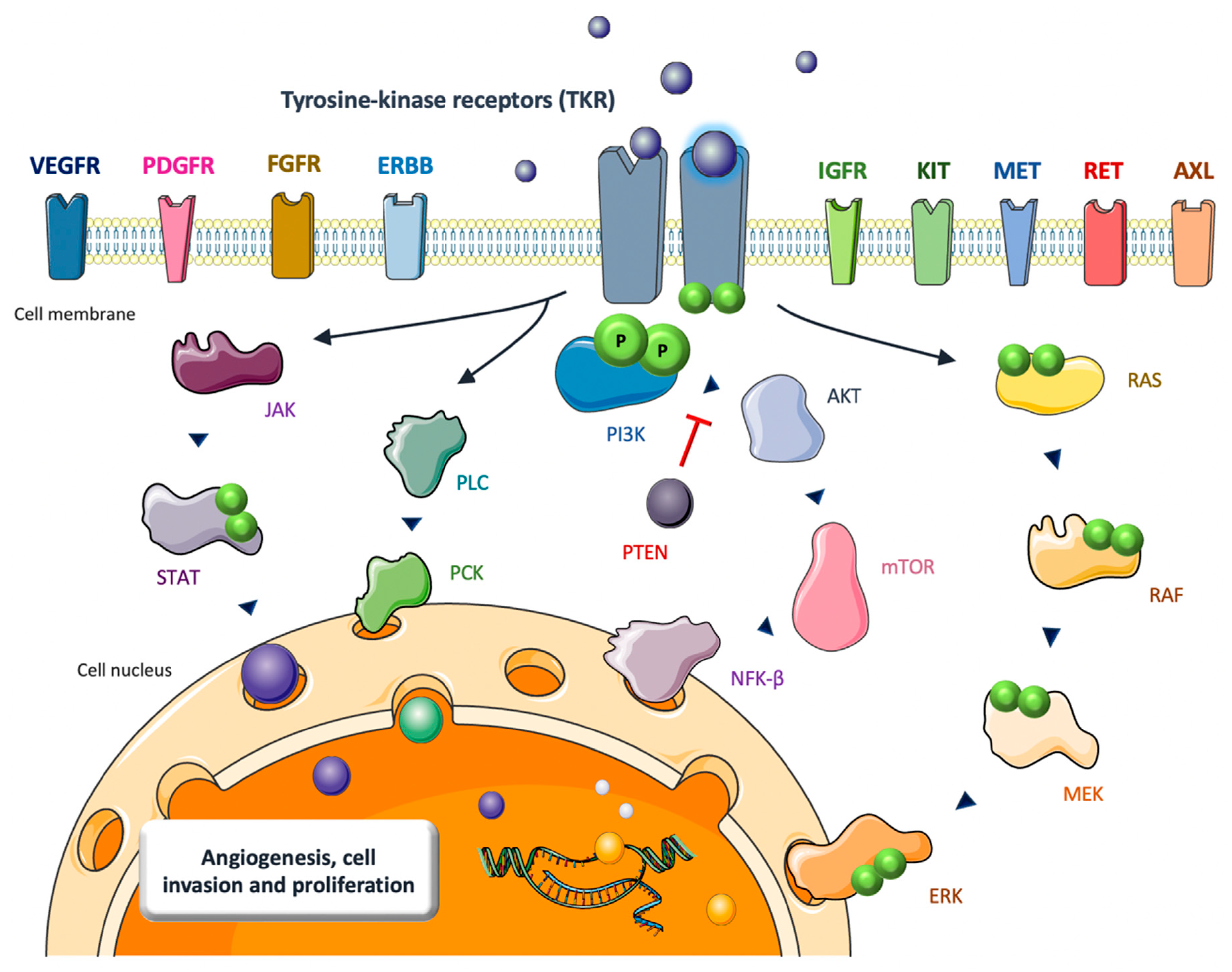
2. TKIs in Bone Sarcomas: Biological Rationale
2.1. Osteosarcoma
2.2. Ewing Sarcoma
2.3. Other Bone Sarcomas
3. TKIs in Bone Sarcomas: Clinical Results
3.1. Osteosarcoma
3.2. Ewing Sarcoma
3.3. Other Bone Sarcomas
3.4. Optimizing the Use of TKIs in Bone Sarcomas
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stiller, C.A.; Trama, A.; Serraino, D.; Rossi, S.; Navarro, C.; Chirlaque, M.D.; Casali, P.G. Rarecare Working Group Descriptive epidemiology of sarcomas in Europe: Report from the RARECARE project. Eur. J. Cancer 2013, 49, 684–695. [Google Scholar] [CrossRef] [PubMed]
- Valery, P.C.; Laversanne, M.; Bray, F. Bone cancer incidence by morphological subtype: A global assessment. Cancer Causes Control 2015, 26, 1127–1139. [Google Scholar] [CrossRef]
- Casali, P.G.; Bielack, S.; Abecassis, N.; Aro, H.T.; Bauer, S.; Biagini, R.; Bonvalot, S.; Boukovinas, I.; Bovee, J.V.M.G.; Brennan, B.; et al. Bone sarcomas: ESMO–PaedCan–EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv79–iv95. [Google Scholar] [CrossRef] [PubMed]
- Smeland, S.; Bielack, S.S.; Whelan, J.; Bernstein, M.; Hogendoorn, P.; Krailo, M.D.; Gorlick, R.; Janeway, K.A.; Ingleby, F.C.; Anninga, J.; et al. Survival and prognosis with osteosarcoma: Outcomes in more than 2000 patients in the EURAMOS-1 (European and American Osteosarcoma Study) cohort. Eur. J. Cancer 2019, 109, 36–50. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S.; Briccoli, A.; Mercuri, M.; Bertoni, F.; Picci, P.; Tienghi, A.; Del Prever, A.B.; Fagioli, F.; Comandone, A.; Bacci, G. Postrelapse survival in osteosarcoma of the extremities: Prognostic factors for long-term survival. J. Clin. Oncol. 2003, 21, 710–715. [Google Scholar] [CrossRef]
- Palmerini, E.; Torricelli, E.; Cascinu, S.; Pierini, M.; De Paolis, M.; Donati, D.; Cesari, M.; Longhi, A.; Abate, M.; Paioli, A.; et al. Is there a role for chemotherapy after local relapse in high-grade osteosarcoma? Pediatr. Blood Cancer 2019, 66, e27792. [Google Scholar] [CrossRef]
- Womer, R.B.; West, D.C.; Krailo, M.D.; Dickman, P.S.; Pawel, B.R.; Grier, H.E.; Marcus, K.; Sailer, S.; Healey, J.H.; Dormans, J.P.; et al. Randomized controlled trial of interval-compressed chemotherapy for the treatment of localized Ewing sarcoma: A report from the Children’s Oncology Group. J. Clin. Oncol. 2012, 30, 4148–4154. [Google Scholar] [CrossRef]
- Cotterill, S.J.; Ahrens, S.; Paulussen, M.; Jürgens, H.F.; Voûte, P.A.; Gadner, H.; Craft, A.W. Prognostic factors in Ewing’s tumor of bone: Analysis of 975 patients from the European Intergroup Cooperative Ewing’s Sarcoma Study Group. J. Clin. Oncol. 2000, 18, 3108–3114. [Google Scholar] [CrossRef]
- Felix, A.; Berlanga, P.; Toulmonde, M.; Landman-Parker, J.; Dumont, S.; Vassal, G.; Le Deley, M.-C.; Gaspar, N. Systematic review of phase-I/II trials enrolling refractory and recurrent Ewing sarcoma: Actual knowledge and future directions to optimize the research. Cancer Med. 2021, 10, 1589–1604. [Google Scholar] [CrossRef]
- Stacchiotti, S.; Dagrada, G.P.; Sanfilippo, R.; Negri, T.; Vittimberga, I.; Ferrari, S.; Grosso, F.; Apice, G.; Tricomi, M.; Colombo, C.; et al. Anthracycline-based chemotherapy in extraskeletal myxoid chondrosarcoma: A retrospective study. Clin. Sarcoma Res. 2013, 3, 16. [Google Scholar] [CrossRef]
- Stacchiotti, S.; Gronchi, A.; Fossati, P.; Akiyama, T.; Alapetite, C.; Baumann, M.; Blay, J.Y.; Bolle, S.; Boriani, S.; Bruzzi, P.; et al. Best practices for the management of local-regional recurrent chordoma: A position paper by the Chordoma Global Consensus Group. Ann. Oncol. 2017, 28, 1230–1242. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, A.; Simeone, N.; Guzzo, M.; Maniezzo, M.; Collini, P.; Morosi, C.; Greco, F.G.; Frezza, A.M.; Casali, P.G.; Stacchiotti, S. Rechallenge of denosumab in jaw osteonecrosis of patients with unresectable giant cell tumour of bone: A case series analysis and literature review. ESMO Open 2020, 5, e000663. [Google Scholar] [CrossRef] [PubMed]
- Nooij, M.A.; Whelan, J.; Bramwell, V.H.C.; Taminiau, A.T.; Cannon, S.; Hogendoorn, P.C.W.; Pringle, J.; Uscinska, B.M.; Weeden, S.; Kirkpatrick, A.; et al. Doxorubicin and cisplatin chemotherapy in high-grade spindle cell sarcomas of the bone, other than osteosarcoma or malignant fibrous histiocytoma: A European Osteosarcoma Intergroup Study. Eur. J. Cancer 2005, 41, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Ottaviani, G.; Jaffe, N. The epidemiology of osteosarcoma. Cancer Treat. Res. 2009, 152, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Kansara, M.; Teng, M.W.; Smyth, M.J.; Thomas, D.M. Translational biology of osteosarcoma. Nat. Rev. Cancer 2014, 14, 722–735. [Google Scholar] [CrossRef] [PubMed]
- Mutsaers, A.J.; Walkley, C.R. Cells of origin in osteosarcoma: Mesenchymal stem cells or osteoblast committed cells? Bone 2014, 62, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Bahrami, A.; Pappo, A.; Easton, J.; Dalton, J.; Hedlund, E.; Ellison, D.; Shurtleff, S.; Wu, G.; Wei, L.; et al. Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep. 2014, 7, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Sayles, L.C.; Breese, M.R.; Koehne, A.L.; Leung, S.G.; Lee, A.G.; Liu, H.-Y.; Spillinger, A.; Shah, A.T.; Tanasa, B.; Straessler, K.; et al. Genome-Informed Targeted Therapy for Osteosarcoma. Cancer Discov. 2019, 9, 46–63. [Google Scholar] [CrossRef]
- Perry, J.A.; Kiezun, A.; Tonzi, P.; Van Allen, E.M.; Carter, S.L.; Baca, S.C.; Cowley, G.S.; Bhatt, A.S.; Rheinbay, E.; Pedamallu, C.S.; et al. Complementary genomic approaches highlight the PI3K/mTOR pathway as a common vulnerability in osteosarcoma. Proc. Natl. Acad. Sci. USA 2014, 111, E5564–E5573. [Google Scholar] [CrossRef]
- Paul, M.K.; Mukhopadhyay, A.K. Tyrosine kinase—Role and significance in Cancer. Int. J. Med. Sci. 2004, 1, 101–115. [Google Scholar] [CrossRef]
- Quan, G.M.Y.; Choong, P.F.M. Anti-angiogenic therapy for osteosarcoma. Cancer Metastasis Rev. 2006, 25, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Zhang, Y.-J.; Zhu, K.; Wang, W.-C. A systematic review of vascular endothelial growth factor expression as a biomarker of prognosis in patients with osteosarcoma. Tumor Biol. 2013, 34, 1895–1899. [Google Scholar] [CrossRef] [PubMed]
- Navid, F.; Santana, V.M.; Neel, M.; McCarville, M.B.; Shulkin, B.L.; Wu, J.; Billups, C.A.; Mao, S.; Daryani, V.M.; Stewart, C.F.; et al. A phase II trial evaluating the feasibility of adding bevacizumab to standard osteosarcoma therapy. Int. J. Cancer 2017, 141, 1469–1477. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Niu, X.; Yao, W. Receptor Tyrosine Kinases in Osteosarcoma Treatment: Which Is the Key Target? Front. Oncol. 2020, 10, 1642. [Google Scholar] [CrossRef]
- Bowles, D.W.; Kessler, E.R.; Jimeno, A. Multi-targeted tyrosine kinase inhibitors in clinical development: Focus on XL-184 (cabozantinib). Drugs Today (Barc) 2011, 47, 857–868. [Google Scholar] [CrossRef]
- Li, A.Y.; McCusker, M.G.; Russo, A.; Scilla, K.A.; Gittens, A.; Arensmeyer, K.; Mehra, R.; Adamo, V.; Rolfo, C. RET fusions in solid tumors. Cancer Treat. Rev. 2019, 81, 101911. [Google Scholar] [CrossRef]
- Rettew, A.N.; Young, E.D.; Lev, D.C.; Kleinerman, E.S.; Abdul-Karim, F.W.; Getty, P.J.; Greenfield, E.M. Multiple receptor tyrosine kinases promote the in vitro phenotype of metastatic human osteosarcoma cell lines. Oncogenesis 2012, 1, e34. [Google Scholar] [CrossRef]
- Luo, J.; Xia, Y.; Yin, Y.; Luo, J.; Liu, M.; Zhang, H.; Zhang, C.; Zhao, Y.; Yang, L.; Kong, L. ATF4 destabilizes RET through nonclassical GRP78 inhibition to enhance chemosensitivity to bortezomib in human osteosarcoma. Theranostics 2019, 9, 6334–6353. [Google Scholar] [CrossRef]
- Kim, M.; Jung, J.-Y.; Choi, S.; Lee, H.; Morales, L.D.; Koh, J.-T.; Kim, S.H.; Choi, Y.-D.; Choi, C.; Slaga, T.J.; et al. GFRA1 promotes cisplatin-induced chemoresistance in osteosarcoma by inducing autophagy. Autophagy 2017, 13, 149–168. [Google Scholar] [CrossRef]
- Zhang, D.; Cui, G.; Sun, C.; Fan, R.; Lei, L.; Williamson, R.A.; Wang, Y.; Zhang, J.; Chen, P.; Wang, A.; et al. Hypoxia promotes osteosarcoma cell proliferation and migration through enhancing platelet-derived growth factor-BB/platelet-derived growth factor receptor-β axis. Biochem. Biophys. Res. Commun. 2019, 512, 360–366. [Google Scholar] [CrossRef]
- Takagi, S.; Takemoto, A.; Takami, M.; Oh-Hara, T.; Fujita, N. Platelets promote osteosarcoma cell growth through activation of the platelet-derived growth factor receptor-Akt signaling axis. Cancer Sci. 2014, 105, 983–988. [Google Scholar] [CrossRef] [PubMed]
- Fernanda Amary, M.; Ye, H.; Berisha, F.; Khatri, B.; Forbes, G.; Lehovsky, K.; Frezza, A.M.; Behjati, S.; Tarpey, P.; Pillay, N.; et al. Fibroblastic growth factor receptor 1 amplification in osteosarcoma is associated with poor response to neo-adjuvant chemotherapy. Cancer Med. 2014, 3, 980–987. [Google Scholar] [CrossRef] [PubMed]
- Weekes, D.; Kashima, T.G.; Zandueta, C.; Perurena, N.; Thomas, D.P.; Sunters, A.; Vuillier, C.; Bozec, A.; El-Emir, E.; Miletich, I.; et al. Regulation of osteosarcoma cell lung metastasis by the c-Fos/AP-1 target FGFR1. Oncogene 2016, 35, 2852–2861. [Google Scholar] [CrossRef] [PubMed]
- Miiji, L.N.O.; Petrilli, A.S.; Di Cesare, S.; Odashiro, A.N.; Burnier, M.N.; de Toledo, S.R.; Garcia, R.J.; Alves, M.T.S. C-kit expression in human osteosarcoma and in vitro assays. Int. J. Clin. Exp. Pathol. 2011, 4, 775–781. [Google Scholar]
- Patanè, S.; Avnet, S.; Coltella, N.; Costa, B.; Sponza, S.; Olivero, M.; Vigna, E.; Naldini, L.; Baldini, N.; Ferracini, R.; et al. MET overexpression turns human primary osteoblasts into osteosarcomas. Cancer Res. 2006, 66, 4750–4757. [Google Scholar] [CrossRef]
- Wang, Y.-H.; Han, X.-D.; Qiu, Y.; Xiong, J.; Yu, Y.; Wang, B.; Zhu, Z.-Z.; Qian, B.-P.; Chen, Y.-X.; Wang, S.-F.; et al. Increased expression of insulin-like growth factor-1 receptor is correlated with tumor metastasis and prognosis in patients with osteosarcoma. J. Surg. Oncol. 2012, 105, 235–243. [Google Scholar] [CrossRef]
- Rettew, A.N.; Getty, P.J.; Greenfield, E.M. Receptor tyrosine kinases in osteosarcoma: Not just the usual suspects. Adv. Exp. Med. Biol. 2014, 804, 47–66. [Google Scholar] [CrossRef]
- Jedlicka, P. Ewing Sarcoma, an enigmatic malignancy of likely progenitor cell origin, driven by transcription factor oncogenic fusions. Int. J. Clin. Exp. Pathol. 2010, 3, 338–347. [Google Scholar]
- Riggi, N.; Knoechel, B.; Gillespie, S.M.; Rheinbay, E.; Boulay, G.; Suvà, M.L.; Rossetti, N.E.; Boonseng, W.E.; Oksuz, O.; Cook, E.B.; et al. EWS-FLI1 utilizes divergent chromatin remodeling mechanisms to directly activate or repress enhancer elements in Ewing sarcoma. Cancer Cell 2014, 26, 668–681. [Google Scholar] [CrossRef]
- Grünewald, T.G.P.; Cidre-Aranaz, F.; Surdez, D.; Tomazou, E.M.; de Álava, E.; Kovar, H.; Sorensen, P.H.; Delattre, O.; Dirksen, U. Ewing sarcoma. Nat. Rev. Dis. Prim. 2018, 4, 5. [Google Scholar] [CrossRef]
- Brohl, A.S.; Solomon, D.A.; Chang, W.; Wang, J.; Song, Y.; Sindiri, S.; Patidar, R.; Hurd, L.; Chen, L.; Shern, J.F.; et al. The genomic landscape of the Ewing Sarcoma family of tumors reveals recurrent STAG2 mutation. PLoS Genet 2014, 10, e1004475. [Google Scholar] [CrossRef] [PubMed]
- Mora, J.; Rodríguez, E.; de Torres, C.; Cardesa, T.; Ríos, J.; Hernández, T.; Cardesa, A.; de Alava, E. Activated growth signaling pathway expression in Ewing sarcoma and clinical outcome. Pediatr. Blood Cancer 2012, 58, 532–538. [Google Scholar] [CrossRef]
- Scotlandi, K.; Benini, S.; Sarti, M.; Serra, M.; Lollini, P.L.; Maurici, D.; Picci, P.; Manara, M.C.; Baldini, N. Insulin-like growth factor I receptor-mediated circuit in Ewing’s sarcoma/peripheral neuroectodermal tumor: A possible therapeutic target. Cancer Res. 1996, 56, 4570–4574. [Google Scholar] [PubMed]
- Asmane, I.; Watkin, E.; Alberti, L.; Duc, A.; Marec-Berard, P.; Ray-Coquard, I.; Cassier, P.; Decouvelaere, A.-V.; Ranchère, D.; Kurtz, J.-E.; et al. Insulin-like growth factor type 1 receptor (IGF-1R) exclusive nuclear staining: A predictive biomarker for IGF-1R monoclonal antibody (Ab) therapy in sarcomas. Eur. J. Cancer 2012, 48, 3027–3035. [Google Scholar] [CrossRef]
- Toretsky, J.A.; Thakar, M.; Eskenazi, A.E.; Frantz, C.N. Phosphoinositide 3-hydroxide kinase blockade enhances apoptosis in the Ewing’s sarcoma family of tumors. Cancer Res. 1999, 59, 5745–5750. [Google Scholar] [PubMed]
- Lee, T.H.; Bolontrade, M.F.; Worth, L.L.; Guan, H.; Ellis, L.M.; Kleinerman, E.S. Production of VEGF165 by Ewing’s sarcoma cells induces vasculogenesis and the incorporation of CD34+ stem cells into the expanding tumor vasculature. Int. J. Cancer 2006, 119, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Strammiello, R.; Benini, S.; Manara, M.C.; Perdichizzi, S.; Serra, M.; Spisni, E.; Picci, P.; Scotlandi, K. Impact of IGF-I/IGF-IR circuit on the angiogenetic properties of Ewing’s sarcoma cells. Horm. Metab. Res. 2003, 35, 675–684. [Google Scholar] [CrossRef]
- Babina, I.S.; Turner, N.C. Advances and challenges in targeting FGFR signalling in cancer. Nat. Rev. Cancer 2017, 17, 318–332. [Google Scholar] [CrossRef]
- Cidre-Aranaz, F.; Grünewald, T.G.P.; Surdez, D.; García-García, L.; Carlos Lázaro, J.; Kirchner, T.; González-González, L.; Sastre, A.; García-Miguel, P.; López-Pérez, S.E.; et al. EWS-FLI1-mediated suppression of the RAS-antagonist Sprouty 1 (SPRY1) confers aggressiveness to Ewing sarcoma. Oncogene 2017, 36, 766–776. [Google Scholar] [CrossRef]
- Lew, E.D.; Furdui, C.M.; Anderson, K.S.; Schlessinger, J. The precise sequence of FGF receptor autophosphorylation is kinetically driven and is disrupted by oncogenic mutations. Sci. Signal. 2009, 2, ra6. [Google Scholar] [CrossRef]
- Helsten, T.; Elkin, S.; Arthur, E.; Tomson, B.N.; Carter, J.; Kurzrock, R. The FGFR Landscape in Cancer: Analysis of 4853 Tumors by Next-Generation Sequencing. Clin. Cancer Res. 2016, 22, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Agelopoulos, K.; Richter, G.H.S.; Schmidt, E.; Dirksen, U.; von Heyking, K.; Moser, B.; Klein, H.-U.; Kontny, U.; Dugas, M.; Poos, K.; et al. Deep Sequencing in Conjunction with Expression and Functional Analyses Reveals Activation of FGFR1 in Ewing Sarcoma. Clin. Cancer Res. 2015, 21, 4935–4946. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Jia, S.F.; Hung, M.C.; Kleinerman, E.S. E1A sensitizes HER2/neu-overexpressing Ewing’s sarcoma cells to topoisomerase II-targeting anticancer drugs. Cancer Res. 2001, 61, 3394–3398. [Google Scholar]
- Mendoza-Naranjo, A.; Wai, D.H.; Farrar, J.E.; Zhu, Q.; Mistry, P.; Lazic, N.; Ayala, F.R.R.; da Cunha, I.W.; Arceci, R.J.; Soares, F.A.; et al. Abstract 431: ErbB4 is a novel driver of metastasis and anoikis resistance in Ewing’s sarcoma. Cancer Res. 2012, 72, 431. [Google Scholar] [CrossRef]
- Uren, A.; Merchant, M.S.; Sun, C.J.; Vitolo, M.I.; Sun, Y.; Tsokos, M.; Illei, P.B.; Ladanyi, M.; Passaniti, A.; Mackall, C.; et al. Beta-platelet-derived growth factor receptor mediates motility and growth of Ewing’s sarcoma cells. Oncogene 2003, 22, 2334–2342. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, Y.X.; Mandal, D.; Wang, S.; Hughes, D.; Pollock, R.E.; Lev, D.; Kleinerman, E.; Hayes-Jordan, A. Inhibiting platelet-derived growth factor beta reduces Ewing’s sarcoma growth and metastasis in a novel orthotopic human xenograft model. In Vivo 2009, 23, 903–909. [Google Scholar]
- Fleuren, E.D.G.; Hillebrandt-Roeffen, M.H.S.; Flucke, U.E.; Te Loo, D.M.W.M.; Boerman, O.C.; van der Graaf, W.T.A.; Versleijen-Jonkers, Y.M.H. The role of AXL and the in vitro activity of the receptor tyrosine kinase inhibitor BGB324 in Ewing sarcoma. Oncotarget 2014, 5, 12753–12768. [Google Scholar] [CrossRef]
- Fleuren, E.D.G.; Roeffen, M.H.S.; Leenders, W.P.; Flucke, U.E.; Vlenterie, M.; Schreuder, H.W.; Boerman, O.C.; van der Graaf, W.T.A.; Versleijen-Jonkers, Y.M.H. Expression and clinical relevance of MET and ALK in Ewing sarcomas. Int. J. Cancer 2013, 133, 427–436. [Google Scholar] [CrossRef]
- Do, I.; Araujo, E.S.; Kalil, R.K.; Bacchini, P.; Bertoni, F.; Unni, K.K.; Park, Y.-K. Protein expression of KIT and gene mutation of c-kit and PDGFRs in Ewing sarcomas. Pathol. Res. Pract. 2007, 203, 127–134. [Google Scholar] [CrossRef]
- Sancéau, J.; Poupon, M.-F.; Delattre, O.; Sastre-Garau, X.; Wietzerbin, J. Strong inhibition of Ewing tumor xenograft growth by combination of human interferon-alpha or interferon-beta with ifosfamide. Oncogene 2002, 21, 7700–7709. [Google Scholar] [CrossRef]
- Zhang, Y.-X.; van Oosterwijk, J.G.; Sicinska, E.; Moss, S.; Remillard, S.P.; van Wezel, T.; Bühnemann, C.; Hassan, A.B.; Demetri, G.D.; Bovée, J.V.M.G.; et al. Functional profiling of receptor tyrosine kinases and downstream signaling in human chondrosarcomas identifies pathways for rational targeted therapy. Clin. Cancer Res. 2013, 19, 3796–3807. [Google Scholar] [CrossRef]
- Polychronidou, G.; Karavasilis, V.; Pollack, S.M.; Huang, P.H.; Lee, A.; Jones, R.L. Novel therapeutic approaches in chondrosarcoma. Future Oncol. 2017, 13, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Schrage, Y.M.; Briaire-de Bruijn, I.H.; de Miranda, N.F.C.C.; van Oosterwijk, J.; Taminiau, A.H.M.; van Wezel, T.; Hogendoorn, P.C.W.; Bovée, J.V.M.G. Kinome profiling of chondrosarcoma reveals SRC-pathway activity and dasatinib as option for treatment. Cancer Res. 2009, 69, 6216–6222. [Google Scholar] [CrossRef] [PubMed]
- Ayala, G.; Liu, C.; Nicosia, R.; Horowitz, S.; Lackman, R. Microvasculature and VEGF expression in cartilaginous tumors. Hum. Pathol. 2000, 31, 341–346. [Google Scholar] [CrossRef]
- Klenke, F.M.; Abdollahi, A.; Bertl, E.; Gebhard, M.-M.; Ewerbeck, V.; Huber, P.E.; Sckell, A. Tyrosine kinase inhibitor SU6668 represses chondrosarcoma growth via antiangiogenesis in vivo. BMC Cancer 2007, 7, 49. [Google Scholar] [CrossRef] [PubMed]
- Shalaby, A.; Presneau, N.; Ye, H.; Halai, D.; Berisha, F.; Idowu, B.; Leithner, A.; Liegl, B.; Briggs, T.R.W.; Bacsi, K.; et al. The role of epidermal growth factor receptor in chordoma pathogenesis: A potential therapeutic target. J. Pathol. 2011, 223, 336–346. [Google Scholar] [CrossRef]
- Tamborini, E.; Virdis, E.; Negri, T.; Orsenigo, M.; Brich, S.; Conca, E.; Gronchi, A.; Stacchiotti, S.; Manenti, G.; Casali, P.G.; et al. Analysis of receptor tyrosine kinases (RTKs) and downstream pathways in chordomas. Neuro Oncol. 2010, 12, 776–789. [Google Scholar] [CrossRef]
- Tamborini, E.; Miselli, F.; Negri, T.; Lagonigro, M.S.; Staurengo, S.; Dagrada, G.P.; Stacchiotti, S.; Pastore, E.; Gronchi, A.; Perrone, F.; et al. Molecular and biochemical analyses of platelet-derived growth factor receptor (PDGFR) B, PDGFRA, and KIT receptors in chordomas. Clin. Cancer Res. 2006, 12, 6920–6928. [Google Scholar] [CrossRef]
- Weinberger, P.M.; Yu, Z.; Kowalski, D.; Joe, J.; Manger, P.; Psyrri, A.; Sasaki, C.T. Differential expression of epidermal growth factor receptor, c-Met, and HER2/neu in chordoma compared with 17 other malignancies. Arch. Otolaryngol. Head Neck Surg. 2005, 131, 707–711. [Google Scholar] [CrossRef]
- Presneau, N.; Shalaby, A.; Idowu, B.; Gikas, P.; Cannon, S.R.; Gout, I.; Diss, T.; Tirabosco, R.; Flanagan, A.M. Potential therapeutic targets for chordoma: PI3K/AKT/TSC1/TSC2/mTOR pathway. Br. J. Cancer 2009, 100, 1406–1414. [Google Scholar] [CrossRef]
- Hu, Y.; Mintz, A.; Shah, S.R.; Quinones-Hinojosa, A.; Hsu, W. The FGFR/MEK/ERK/brachyury pathway is critical for chordoma cell growth and survival. Carcinogenesis 2014, 35, 1491–1499. [Google Scholar] [CrossRef] [PubMed]
- De Vita, A.; Vanni, S.; Miserocchi, G.; Fausti, V.; Pieri, F.; Spadazzi, C.; Cocchi, C.; Liverani, C.; Calabrese, C.; Casadei, R.; et al. A Rationale for the Activity of Bone Target Therapy and Tyrosine Kinase Inhibitor Combination in Giant Cell Tumor of Bone and Desmoplastic Fibroma: Translational Evidences. Biomedicines 2022, 10, 372. [Google Scholar] [CrossRef] [PubMed]
- Mahdal, M.; Neradil, J.; Mudry, P.; Paukovcekova, S.; Staniczkova Zambo, I.; Urban, J.; Macsek, P.; Pazourek, L.; Tomas, T.; Veselska, R. New Target for Precision Medicine Treatment of Giant-Cell Tumor of Bone: Sunitinib Is Effective in the Treatment of Neoplastic Stromal Cells with Activated PDGFRβ Signaling. Cancers 2021, 13, 3543. [Google Scholar] [CrossRef] [PubMed]
- Toulmonde, M.; Lucchesi, C.; Verbeke, S.; Crombe, A.; Adam, J.; Geneste, D.; Chaire, V.; Laroche-Clary, A.; Perret, R.; Bertucci, F.; et al. High throughput profiling of undifferentiated pleomorphic sarcomas identifies two main subgroups with distinct immune profile, clinical outcome and sensitivity to targeted therapies. EBioMedicine 2020, 62, 103131. [Google Scholar] [CrossRef] [PubMed]
- Duffaud, F.; Mir, O.; Boudou-Rouquette, P.; Piperno-Neumann, S.; Penel, N.; Bompas, E.; Delcambre, C.; Kalbacher, E.; Italiano, A.; Collard, O.; et al. Efficacy and safety of regorafenib in adult patients with metastatic osteosarcoma: A non-comparative, randomised, double-blind, placebo-controlled, phase 2 study. Lancet Oncol. 2019, 20, 120–133. [Google Scholar] [CrossRef]
- Duffaud, F.; Blay, J.-Y.; Mir, O.; Chevreau, C.M.; Rouquette, P.B.; Kalbacher, E.; Penel, N.; Perrin, C.; Laurence, V.; Bompas, E.; et al. LBA68 Results of the randomized, placebo (PL)-controlled phase II study evaluating the efficacy and safety of regorafenib (REG) in patients (pts) with metastatic relapsed Ewing sarcoma (ES), on behalf of the French Sarcoma Group (FSG) and UNICANCER. Ann. Oncol. 2020, 31, S1199. [Google Scholar] [CrossRef]
- Davis, L.E.; Bolejack, V.; Ryan, C.W.; Ganjoo, K.N.; Loggers, E.T.; Chawla, S.; Agulnik, M.; Livingston, M.B.; Reed, D.; Keedy, V.; et al. Randomized Double-Blind Phase II Study of Regorafenib in Patients With Metastatic Osteosarcoma. J. Clin. Oncol. 2019, 37, 1424–1431. [Google Scholar] [CrossRef]
- Attia, S.; Bolejack, V.; Ganjoo, K.N.; George, S.; Agulnik, M.; Rushing, D.A.; Loggers, E.T.; Livingston, M.B.; Wright, J.A.; Chawla, S.P.; et al. A phase II trial of regorafenib (REGO) in patients (pts) with advanced Ewing sarcoma and related tumors (EWS) of soft tissue and bone: SARC024 trial results. J. Clin. Oncol. 2017, 35, 11005. [Google Scholar] [CrossRef]
- Italiano, A.; Mir, O.; Mathoulin-Pelissier, S.; Penel, N.; Piperno-Neumann, S.; Bompas, E.; Chevreau, C.; Duffaud, F.; Entz-Werlé, N.; Saada, E.; et al. Cabozantinib in patients with advanced Ewing sarcoma or osteosarcoma (CABONE): A multicentre, single-arm, phase 2 trial. Lancet Oncol. 2020, 21, 446–455. [Google Scholar] [CrossRef]
- Grignani, G.; Palmerini, E.; Dileo, P.; Asaftei, S.D.; D’Ambrosio, L.; Pignochino, Y.; Mercuri, M.; Picci, P.; Fagioli, F.; Casali, P.G.; et al. A phase II trial of sorafenib in relapsed and unresectable high-grade osteosarcoma after failure of standard multimodal therapy: An Italian Sarcoma Group study. Ann. Oncol. 2012, 23, 508–516. [Google Scholar] [CrossRef]
- Grignani, G.; Palmerini, E.; Ferraresi, V.; D’Ambrosio, L.; Bertulli, R.; Asaftei, S.D.; Tamburini, A.; Pignochino, Y.; Sangiolo, D.; Marchesi, E.; et al. Sorafenib and everolimus for patients with unresectable high-grade osteosarcoma progressing after standard treatment: A non-randomised phase 2 clinical trial. Lancet Oncol. 2015, 16, 98–107. [Google Scholar] [CrossRef]
- Gaspar, N.; Campbell-Hewson, Q.; Gallego Melcon, S.; Locatelli, F.; Venkatramani, R.; Hecker-Nolting, S.; Gambart, M.; Bautista, F.; Thebaud, E.; Aerts, I.; et al. Phase I/II study of single-agent lenvatinib in children and adolescents with refractory or relapsed solid malignancies and young adults with osteosarcoma (ITCC-050)☆. ESMO Open 2021, 6, 100250. [Google Scholar] [CrossRef]
- Gaspar, N.; Venkatramani, R.; Hecker-Nolting, S.; Melcon, S.G.; Locatelli, F.; Bautista, F.; Longhi, A.; Lervat, C.; Entz-Werle, N.; Casanova, M.; et al. Lenvatinib with etoposide plus ifosfamide in patients with refractory or relapsed osteosarcoma (ITCC-050): A multicentre, open-label, multicohort, phase 1/2 study. Lancet Oncol. 2021, 22, 1312–1321. [Google Scholar] [CrossRef]
- Chugh, R.; Wathen, J.K.; Maki, R.G.; Benjamin, R.S.; Patel, S.R.; Meyers, P.A.; Myers, P.A.; Priebat, D.A.; Reinke, D.K.; Thomas, D.G.; et al. Phase II multicenter trial of imatinib in 10 histologic subtypes of sarcoma using a bayesian hierarchical statistical model. J. Clin. Oncol. 2009, 27, 3148–3153. [Google Scholar] [CrossRef]
- Palmerini, E.; Lopez-Pousa, A.; Grignani, G.; Redondo, A.; Hindi, N.; Stacchiotti, S.; Sebio, A.; Lopez-Martin, J.A.; Valverde Morales, C.M.; Martinez-Trufero, J.; et al. IMMUNOSARC: A collaborative Spanish (GEIS) and Italian (ISG) sarcoma groups phase I/II trial of sunitinib and nivolumab in advanced soft tissue and bone sarcoma: Results from the phase II part, bone sarcoma cohort. J. Clin. Oncol. 2020, 38, 11522. [Google Scholar] [CrossRef]
- Xie, L.; Xu, J.; Sun, X.; Guo, W.; Gu, J.; Liu, K.; Zheng, B.; Ren, T.; Huang, Y.; Tang, X.; et al. Apatinib plus camrelizumab (anti-PD1 therapy, SHR-1210) for advanced osteosarcoma (APFAO) progressing after chemotherapy: A single-arm, open-label, phase 2 trial. J. Immunother. Cancer 2020, 8, e000798. [Google Scholar] [CrossRef] [PubMed]
- Schuetze, S.M.; Bolejack, V.; Choy, E.; Ganjoo, K.N.; Staddon, A.P.; Chow, W.A.; Tawbi, H.A.; Samuels, B.L.; Patel, S.R.; von Mehren, M.; et al. Phase 2 study of dasatinib in patients with alveolar soft part sarcoma, chondrosarcoma, chordoma, epithelioid sarcoma, or solitary fibrous tumor: Dasatinib Treatment of Indolent Sarcomas. Cancer 2017, 123, 90–97. [Google Scholar] [CrossRef]
- Stacchiotti, S.; Longhi, A.; Ferraresi, V.; Grignani, G.; Comandone, A.; Stupp, R.; Bertuzzi, A.; Tamborini, E.; Pilotti, S.; Messina, A.; et al. Phase II study of imatinib in advanced chordoma. J. Clin. Oncol. 2012, 30, 914–920. [Google Scholar] [CrossRef]
- Stacchiotti, S.; Morosi, C.; Lo Vullo, S.; Casale, A.; Palassini, E.; Frezza, A.M.; Dinoi, G.; Messina, A.; Gronchi, A.; Cavalleri, A.; et al. Imatinib and everolimus in patients with progressing advanced chordoma: A phase 2 clinical study. Cancer 2018, 124, 4056–4063. [Google Scholar] [CrossRef]
- Stacchiotti, S.; Tamborini, E.; Lo Vullo, S.; Bozzi, F.; Messina, A.; Morosi, C.; Casale, A.; Crippa, F.; Conca, E.; Negri, T.; et al. Phase II study on lapatinib in advanced EGFR-positive chordoma. Ann. Oncol. 2013, 24, 1931–1936. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Dumas, J.; Adnane, L.; Lynch, M.; Carter, C.A.; Schütz, G.; Thierauch, K.-H.; Zopf, D. Regorafenib (BAY 73-4506): A new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int. J. Cancer 2011, 129, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Pan, P.-J.; Liu, Y.-C.; Hsu, F.-T. Protein Kinase B and Extracellular Signal-Regulated Kinase Inactivation is Associated with Regorafenib-Induced Inhibition of Osteosarcoma Progression In Vitro and In Vivo. J. Clin. Med. 2019, 8, E900. [Google Scholar] [CrossRef] [PubMed]
- Fioramonti, M.; Fausti, V.; Pantano, F.; Iuliani, M.; Ribelli, G.; Lotti, F.; Pignochino, Y.; Grignani, G.; Santini, D.; Tonini, G.; et al. Cabozantinib Affects Osteosarcoma Growth Through A Direct Effect On Tumor Cells and Modifications In Bone Microenvironment. Sci. Rep. 2018, 8, 4177. [Google Scholar] [CrossRef]
- Kokkali-Zervos, S.; Ardavanis, A.; Panousieris, M.; Duran, J.; Boukovinas, I. 1645P Real-world data on cabozantinib in advanced osteosarcoma and Ewing sarcoma—A study of the Hellenic Group of Sarcoma and Rare Cancers. Ann. Oncol. 2020, 31, S984. [Google Scholar] [CrossRef]
- Zheng, B.; Ren, T.; Huang, Y.; Guo, W. Apatinib inhibits migration and invasion as well as PD-L1 expression in osteosarcoma by targeting STAT3. Biochem. Biophys. Res. Commun. 2018, 495, 1695–1701. [Google Scholar] [CrossRef]
- Xie, L.; Xu, J.; Sun, X.; Tang, X.; Yan, T.; Yang, R.; Guo, W. Apatinib for Advanced Osteosarcoma after Failure of Standard Multimodal Therapy: An Open Label Phase II Clinical Trial. Oncologist 2019, 24, e542–e550. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Gu, Z.; Wang, X.; Liu, Z.; Yao, W.; Wang, J.; Zhang, P.; Cai, Q.; Ge, H. Efficacy and safety of apatinib in treatment of osteosarcoma after failed standard multimodal therapy: An observational study. Medicine (Baltimore) 2019, 98, e15650. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Li, J.; Xie, Q.; Diao, L.; Gai, L.; Yang, W. Efficacy and safety of apatinib monotherapy in advanced bone and soft tissue sarcoma: An observational study. Cancer Biol. Ther. 2018, 19, 198–204. [Google Scholar] [CrossRef]
- Mao, W.-F.; Shao, M.-H.; Gao, P.-T.; Ma, J.; Li, H.-J.; Li, G.-L.; Han, B.-H.; Yuan, C.-G. The important roles of RET, VEGFR2 and the RAF/MEK/ERK pathway in cancer treatment with sorafenib. Acta Pharmacol. Sin. 2012, 33, 1311–1318. [Google Scholar] [CrossRef]
- Hussein, Z.; Mizuo, H.; Hayato, S.; Namiki, M.; Shumaker, R. Clinical Pharmacokinetic and Pharmacodynamic Profile of Lenvatinib, an Orally Active, Small-Molecule, Multitargeted Tyrosine Kinase Inhibitor. Eur. J. Drug Metab. Pharm. 2017, 42, 903–914. [Google Scholar] [CrossRef]
- Pignochino, Y.; Grignani, G.; Cavalloni, G.; Motta, M.; Tapparo, M.; Bruno, S.; Bottos, A.; Gammaitoni, L.; Migliardi, G.; Camussi, G.; et al. Sorafenib blocks tumour growth, angiogenesis and metastatic potential in preclinical models of osteosarcoma through a mechanism potentially involving the inhibition of ERK1/2, MCL-1 and ezrin pathways. Mol. Cancer 2009, 8, 118. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K. Axitinib, a novel anti-angiogenic drug with promising activity in various solid tumors. Curr. Opin. Investig. Drugs 2008, 9, 658–671. [Google Scholar] [PubMed]
- Wedge, S.R.; Kendrew, J.; Hennequin, L.F.; Valentine, P.J.; Barry, S.T.; Brave, S.R.; Smith, N.R.; James, N.H.; Dukes, M.; Curwen, J.O.; et al. AZD2171: A highly potent, orally bioavailable, vascular endothelial growth factor receptor-2 tyrosine kinase inhibitor for the treatment of cancer. Cancer Res. 2005, 65, 4389–4400. [Google Scholar] [CrossRef] [PubMed]
- Geller, J.I.; Fox, E.; Turpin, B.K.; Goldstein, S.L.; Liu, X.; Minard, C.G.; Kudgus, R.A.; Reid, J.M.; Berg, S.L.; Weigel, B.J. A study of axitinib, a VEGF receptor tyrosine kinase inhibitor, in children and adolescents with recurrent or refractory solid tumors: A Children’s Oncology Group phase 1 and pilot consortium trial (ADVL1315). Cancer 2018, 124, 4548–4555. [Google Scholar] [CrossRef]
- Fox, E.; Aplenc, R.; Bagatell, R.; Chuk, M.K.; Dombi, E.; Goodspeed, W.; Goodwin, A.; Kromplewski, M.; Jayaprakash, N.; Marotti, M.; et al. A phase 1 trial and pharmacokinetic study of cediranib, an orally bioavailable pan-vascular endothelial growth factor receptor inhibitor, in children and adolescents with refractory solid tumors. J. Clin. Oncol. 2010, 28, 5174–5181. [Google Scholar] [CrossRef]
- Waller, C.F. Imatinib Mesylate. Recent Results Cancer Res. 2018, 212, 1–27. [Google Scholar] [CrossRef]
- Gobin, B.; Moriceau, G.; Ory, B.; Charrier, C.; Brion, R.; Blanchard, F.; Redini, F.; Heymann, D. Imatinib mesylate exerts anti-proliferative effects on osteosarcoma cells and inhibits the tumour growth in immunocompetent murine models. PLoS ONE 2014, 9, e90795. [Google Scholar] [CrossRef]
- Longhi, A.; Paioli, A.; Palmerini, E.; Cesari, M.; Abate, M.E.; Setola, E.; Spinnato, P.; Donati, D.; Hompland, I.; Boye, K. Pazopanib in relapsed osteosarcoma patients: Report on 15 cases. Acta Oncol. 2019, 58, 124–128. [Google Scholar] [CrossRef]
- Raciborska, A.; Bilska, K. Sorafenib in patients with progressed and refractory bone tumors. Med. Oncol. 2018, 35, 126. [Google Scholar] [CrossRef]
- Xie, L.; Guo, W.; Wang, Y.; Yan, T.; Ji, T.; Xu, J. Apatinib for advanced sarcoma: Results from multiple institutions’ off-label use in China. BMC Cancer 2018, 18, 396. [Google Scholar] [CrossRef]
- Wang, Y.; Min, L.; Zhou, Y.; Luo, Y.; Duan, H.; Tu, C. The efficacy and safety of apatinib in Ewing’s sarcoma: A retrospective analysis in one institution. Cancer Manag. Res. 2018, 10, 6835–6842. [Google Scholar] [CrossRef] [PubMed]
- Merchant, M.S. Potential Use of Imatinib in Ewing’s Sarcoma: Evidence for In Vitro and In Vivo Activity. Cancer Spectr. Knowl. Environ. 2002, 94, 1673–1679. [Google Scholar] [CrossRef] [PubMed]
- Chao, J.; Budd, G.T.; Chu, P.; Frankel, P.; Garcia, D.; Junqueira, M.; Loera, S.; Somlo, G.; Sato, J.; Chow, W.A. Phase II clinical trial of imatinib mesylate in therapy of KIT and/or PDGFRalpha-expressing Ewing sarcoma family of tumors and desmoplastic small round cell tumors. Anticancer Res. 2010, 30, 547–552. [Google Scholar] [PubMed]
- Lalchandani, U.R.; Sahai, V.; Hersberger, K.; Francis, I.R.; Wasnik, A.P. A Radiologist’s Guide to Response Evaluation Criteria in Solid Tumors. Curr. Probl. Diagn. Radiol. 2019, 48, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Mullin, R.J.; Keith, B.R.; Liu, L.-H.; Ma, H.; Rusnak, D.W.; Owens, G.; Alligood, K.J.; Spector, N.L. Anti-tumor activity of GW572016: A dual tyrosine kinase inhibitor blocks EGF activation of EGFR/erbB2 and downstream Erk1/2 and AKT pathways. Oncogene 2002, 21, 6255–6263. [Google Scholar] [CrossRef]
- Vora, T.S.; Peretz Soroka, H.; Noujaim, J.C.; Marcoux, N.; Alcindor, T.; Karachiwala, H.; Alvi, S.; Shaikh, F.; Abdul Razak, A.R.; Gupta, A.A. Real-world experience of tyrosine kinase inhibitors in patients (pt) with recurrent bone tumours (BT): A CanSaRCC study. J. Clin. Oncol. 2022, 40, 11530. [Google Scholar] [CrossRef]
- Xie, L.; Xu, J.; Sun, X.; Li, K.; Li, X.; He, F.; Gu, J.; Lv, Z.; Tang, X.; Sun, K.; et al. 1646P Apatinib for treatment of inoperable metastatic or locally advanced chondrosarcoma: What we can learn about the biological behavior of chondrosarcoma from a multicenter study. Ann. Oncol. 2020, 31, S984. [Google Scholar] [CrossRef]
- Liu, Z.; Gao, S.; Zhu, L.; Wang, J.; Zhang, P.; Li, P.; Zhang, F.; Yao, W. Efficacy and safety of anlotinib in patients with unresectable or metastatic bone sarcoma: A retrospective multiple institution study. Cancer Med. 2021, 10, 7593–7600. [Google Scholar] [CrossRef]
- George, S.; Merriam, P.; Maki, R.G.; Van den Abbeele, A.D.; Yap, J.T.; Akhurst, T.; Harmon, D.C.; Bhuchar, G.; O’Mara, M.M.; D’Adamo, D.R.; et al. Multicenter Phase II Trial of Sunitinib in the Treatment of Nongastrointestinal Stromal Tumor Sarcomas. J. Clin. Oncol. 2009, 27, 3154–3160. [Google Scholar] [CrossRef]
- Li, J.; Zhou, J.; Liu, Y.; Sun, X.; Song, W. Comprehensive treatment for multicentric giant cell tumors of the pelvis and spine using apatinib: A case report and literature review. J. Cancer Res. Ther. 2020, 16, 1020. [Google Scholar] [CrossRef]
- de Jonge, M.J.A.; Hamberg, P.; Verweij, J.; Savage, S.; Suttle, A.B.; Hodge, J.; Arumugham, T.; Pandite, L.N.; Hurwitz, H.I. Phase I and pharmacokinetic study of pazopanib and lapatinib combination therapy in patients with advanced solid tumors. Investig. New Drugs 2013, 31, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Fleuren, E.D.G.; Versleijen-Jonkers, Y.M.H.; Boerman, O.C.; van der Graaf, W.T.A. Targeting receptor tyrosine kinases in osteosarcoma and Ewing sarcoma: Current hurdles and future perspectives. Biochim. Biophys. Acta 2014, 1845, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Hurlburt, W.; Greer, A.; Reeves, K.A.; Hillerman, S.; Chang, H.; Fargnoli, J.; Graf Finckenstein, F.; Gottardis, M.M.; Carboni, J.M. Differential mechanisms of acquired resistance to insulin-like growth factor-i receptor antibody therapy or to a small-molecule inhibitor, BMS-754807, in a human rhabdomyosarcoma model. Cancer Res. 2010, 70, 7221–7231. [Google Scholar] [CrossRef] [PubMed]
- Pignochino, Y.; Dell’Aglio, C.; Basiricò, M.; Capozzi, F.; Soster, M.; Marchiò, S.; Bruno, S.; Gammaitoni, L.; Sangiolo, D.; Torchiaro, E.; et al. The Combination of Sorafenib and Everolimus Abrogates mTORC1 and mTORC2 upregulation in osteosarcoma preclinical models. Clin. Cancer Res. 2013, 19, 2117–2131. [Google Scholar] [CrossRef]
- Stacchiotti, S.; Marrari, A.; Tamborini, E.; Palassini, E.; Virdis, E.; Messina, A.; Crippa, F.; Morosi, C.; Gronchi, A.; Pilotti, S.; et al. Response to imatinib plus sirolimus in advanced chordoma. Ann. Oncol. 2009, 20, 1886–1894. [Google Scholar] [CrossRef]
- Casanova, M.; Bautista, F.; Campbell Hewson, Q.; Makin, G.; Marshall, L.V.; Verschuur, A.; Canete, A.; Corradini, N.; Ploeger, B.; Mueller, U.; et al. Phase I study of regorafenib in combination with vincristine and irinotecan in pediatric patients with recurrent or refractory solid tumors. J. Clin. Oncol. 2020, 38, 10507. [Google Scholar] [CrossRef]
- Brennan, R.C.; Furman, W.; Mao, S.; Wu, J.; Turner, D.C.; Stewart, C.F.; Santana, V.; McGregor, L.M. Phase I dose escalation and pharmacokinetic study of oral gefitinib and irinotecan in children with refractory solid tumors. Cancer Chemother. Pharmacol. 2014, 74, 1191–1198. [Google Scholar] [CrossRef]
- Jakacki, R.I.; Hamilton, M.; Gilbertson, R.J.; Blaney, S.M.; Tersak, J.; Krailo, M.D.; Ingle, A.M.; Voss, S.D.; Dancey, J.E.; Adamson, P.C. Pediatric phase I and pharmacokinetic study of erlotinib followed by the combination of erlotinib and temozolomide: A Children’s Oncology Group Phase I Consortium Study. J. Clin. Oncol. 2008, 26, 4921–4927. [Google Scholar] [CrossRef]
- Pollard, J.A.; Alonzo, T.A.; Gerbing, R.; Brown, P.; Fox, E.; Choi, J.; Fisher, B.; Hirsch, B.; Kahwash, S.; Getz, K.; et al. Sorafenib in Combination With Standard Chemotherapy for Children With High Allelic Ratio FLT3/ITD+ Acute Myeloid Leukemia: A Report From the Children’s Oncology Group Protocol AAML1031. J. Clin. Oncol. 2022, 40, 2023–2035. [Google Scholar] [CrossRef]
- Fleuren, E.D.G.; Terry, R.L.; Meyran, D.; Omer, N.; Trapani, J.A.; Haber, M.; Neeson, P.J.; Ekert, P.G. Enhancing the Potential of Immunotherapy in Paediatric Sarcomas: Breaking the Immunosuppressive Barrier with Receptor Tyrosine Kinase Inhibitors. Biomedicines 2021, 9, 1798. [Google Scholar] [CrossRef]
- Wilky, B.A.; Trucco, M.M.; Subhawong, T.K.; Florou, V.; Park, W.; Kwon, D.; Wieder, E.D.; Kolonias, D.; Rosenberg, A.E.; Kerr, D.A.; et al. Axitinib plus pembrolizumab in patients with advanced sarcomas including alveolar soft-part sarcoma: A single-centre, single-arm, phase 2 trial. Lancet Oncol. 2019, 20, 837–848. [Google Scholar] [CrossRef]
- Paoluzzi, L.; Cacavio, A.; Ghesani, M.; Karambelkar, A.; Rapkiewicz, A.; Weber, J.; Rosen, G. Response to anti-PD1 therapy with nivolumab in metastatic sarcomas. Clin. Sarcoma Res. 2016, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.; Rastogi, S.; Shamim, S.A.; Barwad, A.; Sethi, M. Good and sustained response to pembrolizumab and pazopanib in advanced undifferentiated pleomorphic sarcoma: A case report. Clin. Sarcoma Res. 2020, 10, 10. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Clinical Trial (Phase) | Agent | Tumor | N | Age Range (Years) | Outcomes | G3/G4 Adverse Events | |
|---|---|---|---|---|---|---|---|
| Duffaud et al. (REGOBONE) (phase II multi-cohort) | Regorafenib (vs. placebo) | OS [75] | 38 | 21–50 | DCR: 17/26 (65%; 95% CI 47–95) (vs. 0% in placebo); mPFS: 16 w (95% CI 8.0–27.3) vs. 4 w (95% CI 3.0–5.7) | Hypertension (24%), PPS (10%), fatigue (10%), hypophosphatemia (10%), chest pain (10%) | |
| ES [76] | 41 | 16–59 | PR: 5/36 (21.7%) (vs. 0% in placebo); mPFS: 11.4 w (95% CI 4.6–22.9) vs. 3.9 w (95% CI 3.3–7.3) | Diarrhea (13%), PPS (13%), thrombocytopenia (9%), fatigue (9%), mucositis (9%), febrile neutropenia (9%) | |||
| Davis et al. (SARC024) (phase II) [77] | Regorafenib (vs. placebo) | OS | 42 | 18–76 | mPFS: 3.6 m (95% CI 2.0–7.6) vs. 1.7 m (95% CI 1.2–1.8) (HR 0.42; 95% CI 0.21–0.85; p = 0.017) | Hypertension (14%), rash (9%), hypophosphatemia (9%), extremity pain (9%), thrombocytopenia (9%), PPS (5%) | |
| Attia et al. (REGO) (phase II) [78] | Regorafenib | ES | 30 | 19–65 | PR: 3/28 (10.7%); SD: 18/28 (64.3%); mPFS: 3.6 m (95% CI 2.8–3.8) | Hypophosphatemia (20%), hypertension (6.7%), ALT increase (6.7%), fatigue (3.3%), abdominal pain (3.3%), diarrhea (3.3%), hypokalemia (3.3%), oral mucositis (3.3%), neutropenia (3.3%), rash (3.3%) | |
| Italiano et al. (CABONE) (phase II) [79] | Cabozantinib | OS/ES | 90 | 20–53 | OS (n: 42) | PR: 5/42 (12%; 95% CI 4–26), SD: 14/42 (33%; 95% CI 20–50); mPFS: 7.2 m PR (95% CI 4.7–10.9); 4.5 m SD (95% CI 1.8–9.5); 1.8 m PD (95% CI 0.8–1.9) | Hypophosphatemia (9%), neutropenia (7%), AST increase (6%), PPS (6%), pneumothorax (6%) |
| ES (n: 39) | PR: 10/39 (26%; 95% CI 13–42); mPFS: 4.4 m (95% CI 3.7–5.6) | ||||||
| Grignani et al. (phase II) [80] | Sorafenib | OS | 35 | 15–62 | ORR: 14% (95% CI 2–26) DCR: 49% (95% CI 31–67) mPFS: 4 m (95% CI 2–5) | PPS (9%), anemia (6%), thrombocytopenia (6%), CK elevation (6%), leucopenia (3%), rash (3%), mucositis (3%), nausea (3%), fatigue (3%), lipase elevation (3%), pneumothorax (3%), bleeding (3%) | |
| Grignani et al. (phase II) [81] | Sorafenib + everolimus | OS | 38 | 18–64 | ORR: 10% (95% CI 0.3–21); DCR: 63% PFS at 6 m: 45% (95% CI 28–61) | Hypophosphatemia (16%), lymphopenia (16%), PPS (13%), thrombocytopenia (11%), fatigue (5%), mucositis (5%), diarrhea (5%), anemia (5%), pneumothorax (3%) | |
| Gaspar et al. (phase I/II) [82] | Lenvatinib | OS | 31 | 9–22 | ORR: 7% (95% CI 0.8–22.1) PFS at 4 m: 29% (95% CI 14–48); mPFS: 3 m (95% CI 1.8–5.4) | Hypertension (3%), diarrhea (3%), proteinuria (3%), decreased weight (3%), abdominal pain (3%) | |
| Gaspar et al. (phase I/II) [83] | Lenvatinib + IF/VP16 | OS | 35 | 2–25 | PFS at 4 m: 51% (95% CI 34–69); | Neutropenia (77%), thrombocytopenia (71%), anemia (54%), leukopenia (54%) | |
| Chugh et al. (phase II) [84] | Imatinib | OS/ES/ others | 185 | 14–83 | OS (n: 27) | PR: 0/27 (0%); SD: 5/27 (19%) | No G3/G4 adverse events reported |
| ES (n: 13) | PR/SD: 0/13 (0%) | ||||||
| Palmerini et al. (IMMUNO-SARC) (phase I/II) [85] | Sunitinib + nivolumab | OS/ES/ others | 40 | 21–74 | DCR: 24/40 (60%) (1 CR, 1 PR, 22 SD) mPFS 3.7 m (95% CI 3.4–4) mOS 14.2 m (95% CI 7.1–21.3) | Neutropenia (10%), anemia (10%), ALT/AST increase (7.5%), fatigue (5%), oral mucositis (5%), thrombocytopenia (2.5%), dysphagia (2.5%), gastric hemorrhage (2.5%), malaise (2.5%), thromboembolism (2.5%), pneumonitis (2.5%) | |
| Xie et al. (APFAO) (phase II) [86] | Apatinib + camrelizumab | OS | 43 | 11–43 | ORR: 9/43 (20.9%) PFS at 6 m: 50.9% (95% CI 34.6–65.0) | Wound dehiscence (14%), ALP increase (9.3%), AST/ALT increase (9.3%), blood bilirubin increase (9.3%), hypertriglyceridemia (7.0%), anorexia (7.0%), weight loss (7.0%), pneumothorax (7.0%), platelet count decrease (4.7%), diarrhea (4.7%), PPS (4.7%), limb pain (4.7%), leukopenia (4.7%), rash (4.7%), oral mucositis (4.7%), hypertension (4.7%), toothache (4.7%), nausea (4.7%), non-cardiac chest pain (4.7%), hypothyroidism (2.3%), blood LDH increase (2.3%), proteinuria (2.3%), cough (2.3%), hemorrhoidal hemorrhage (2.3%), fatigue (2.3%), peripheral neuroinflammation (2.3%) | |
| Schuetze et al. (phase II) [87] | Dasatinib | CS | 33 | 22–87 | ORR: 6/33 (18.2%); mPFS: 5.5 m | Pain (17%), dyspnea (11%), pleural effusion (6%), diarrhea (5%), anemia (3%), thrombocytopenia (2%), neutropenia (<1%), lymphopenia (<1%) | |
| Chordoma | 32 | ORR: 6/32 (18.8%); mPFS: 6.3 m | |||||
| Stacchiotti et al. (phase II) [88] | Imatinib | Chordoma | 50 | 24–86 | PR: 1/50 (2%); SD 35/50 (70%); ORR: 2% (95% CI 0–5.3); mPFS: 9.2 m | Fluid retention (29%) | |
| Stacchiotti et al. (phase II) [89] | Imatinib + everolimus | Chordoma | 40 | 49–70 | PR: 9/40 (20.9%); SD 24/40 (55.8%) mPFS: 11.5 m (95% CI 4.6–17.6) | Infection (16%), fatigue (9%), anemia (2%), leukopenia (2%), febrile neutropenia (2%), thrombocytopenia (2%), cardiac ischemia (2%) | |
| Stacchiotti et al. (phase II) [90] | Lapatinib | Chordoma (EGFR) | 18 | 35–75 | PR: 6/18 (33.3%); SD: 7/18 (38.9%) mPFS: 6 m (95% CI 3–8) | Anemia (5.6%), rash (5.6%), thromboembolism (5.6%) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Albarrán, V.; Villamayor, M.L.; Chamorro, J.; Rosero, D.I.; Pozas, J.; San Román, M.; Calvo, J.C.; Pérez de Aguado, P.; Moreno, J.; Guerrero, P.; et al. Receptor Tyrosine Kinase Inhibitors for the Treatment of Recurrent and Unresectable Bone Sarcomas. Int. J. Mol. Sci. 2022, 23, 13784. https://doi.org/10.3390/ijms232213784
Albarrán V, Villamayor ML, Chamorro J, Rosero DI, Pozas J, San Román M, Calvo JC, Pérez de Aguado P, Moreno J, Guerrero P, et al. Receptor Tyrosine Kinase Inhibitors for the Treatment of Recurrent and Unresectable Bone Sarcomas. International Journal of Molecular Sciences. 2022; 23(22):13784. https://doi.org/10.3390/ijms232213784
Chicago/Turabian StyleAlbarrán, Víctor, María Luisa Villamayor, Jesús Chamorro, Diana Isabel Rosero, Javier Pozas, María San Román, Juan Carlos Calvo, Patricia Pérez de Aguado, Jaime Moreno, Patricia Guerrero, and et al. 2022. "Receptor Tyrosine Kinase Inhibitors for the Treatment of Recurrent and Unresectable Bone Sarcomas" International Journal of Molecular Sciences 23, no. 22: 13784. https://doi.org/10.3390/ijms232213784
APA StyleAlbarrán, V., Villamayor, M. L., Chamorro, J., Rosero, D. I., Pozas, J., San Román, M., Calvo, J. C., Pérez de Aguado, P., Moreno, J., Guerrero, P., González, C., García de Quevedo, C., Álvarez-Ballesteros, P., & Vaz, M. Á. (2022). Receptor Tyrosine Kinase Inhibitors for the Treatment of Recurrent and Unresectable Bone Sarcomas. International Journal of Molecular Sciences, 23(22), 13784. https://doi.org/10.3390/ijms232213784