
The IL-33/ST2 Pathway in Cerebral Malaria


Abstract
1. Introduction
1.1. Cerebral Malaria (CM)
1.2. Immunopathological Mechanisms Associated to CM
1.3. Blood–Brain Barrier in Cerebral Malaria
2. IL-33
2.1. Nuclear and Extracellular IL-33
2.2. Post-Translational Regulation of IL-33
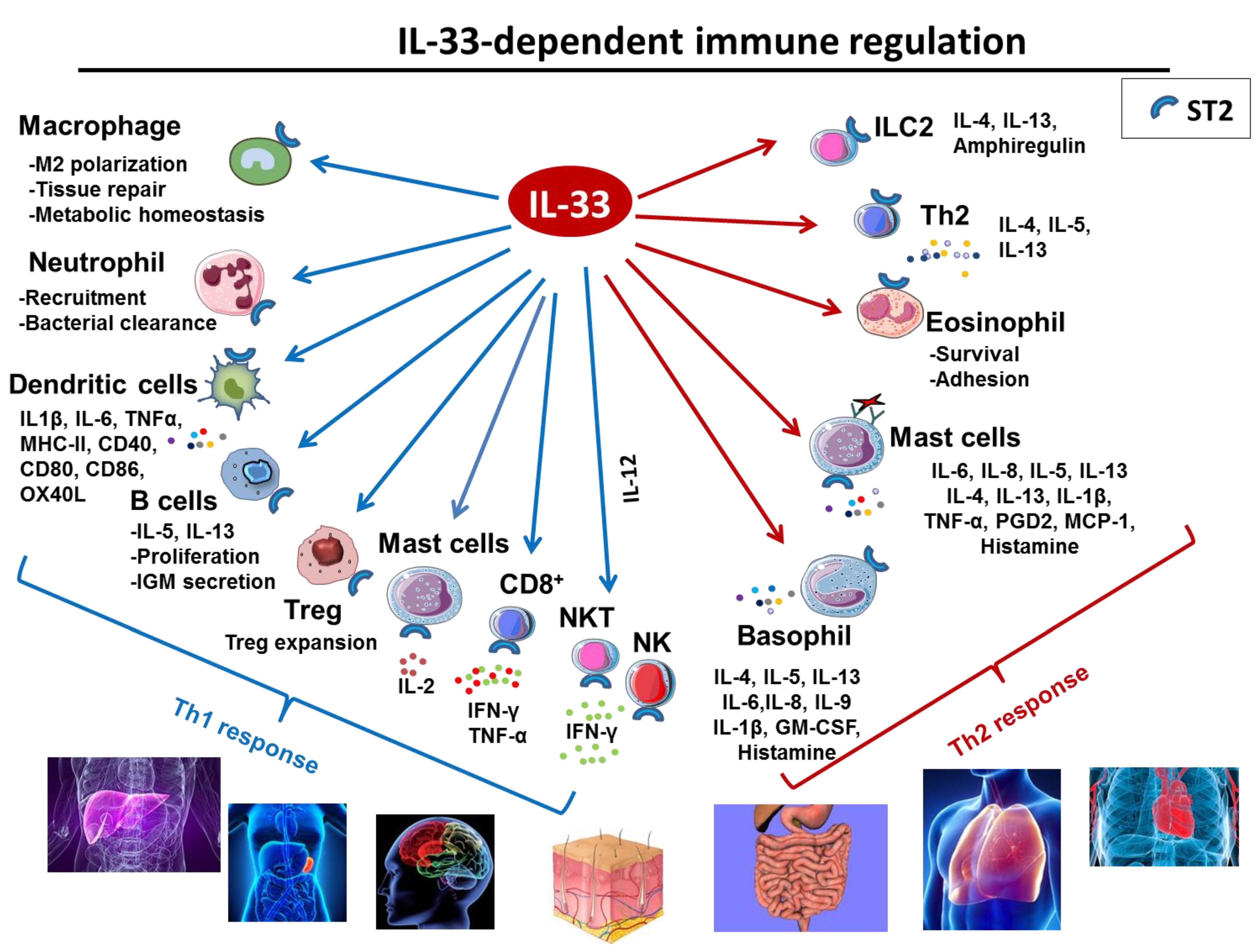
2.3. IL-33 in Immune Responses
2.4. IL-33 in the Central Nervous System
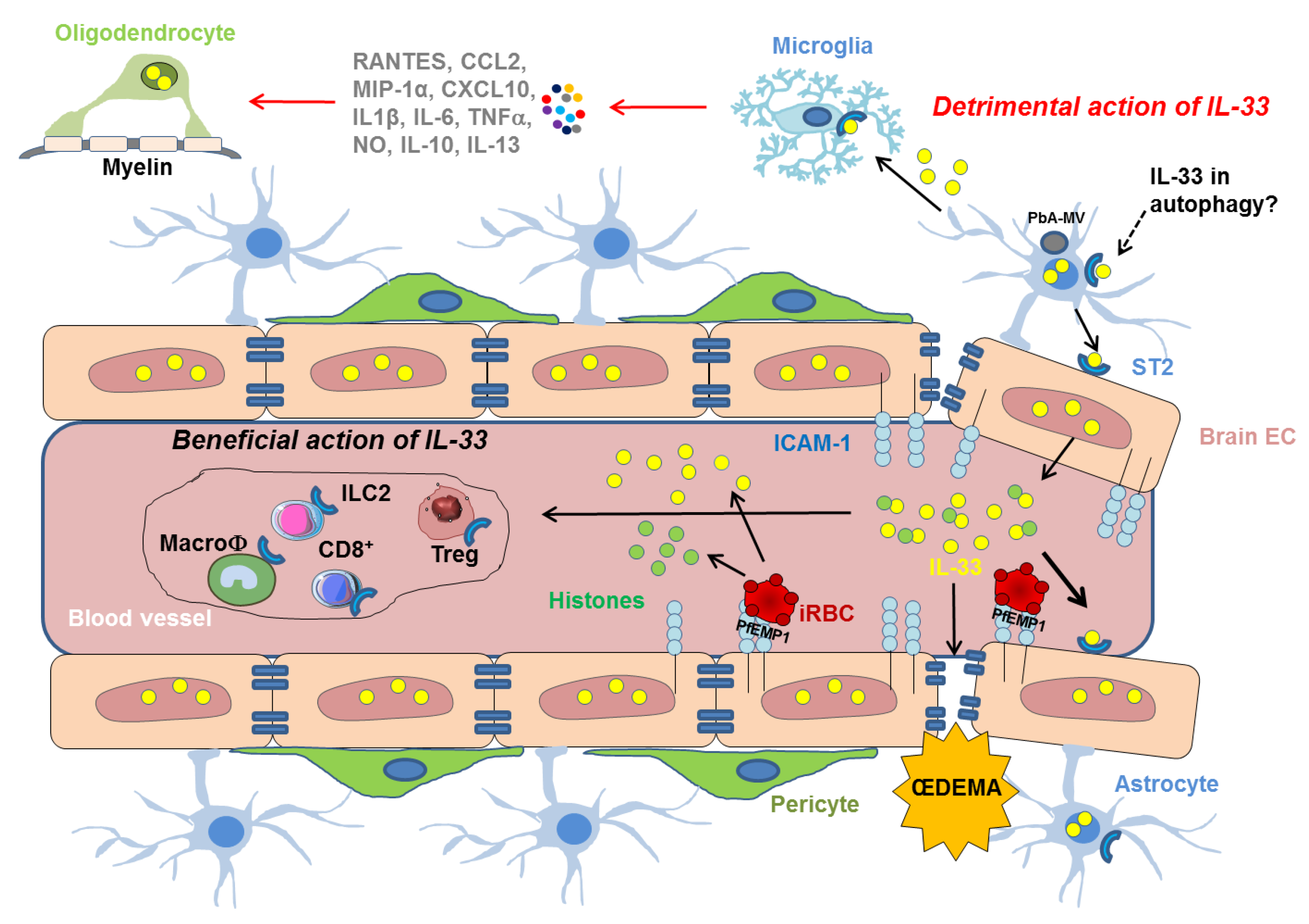
3. IL-33 in Cerebral Malaria
3.1. IL-33 Immune Regulation in Cerebral Malaria
3.2. IL-33, Autophagy and Cerebral Malaria
3.3. Proposed Role of IL-33 in CM-Associated Endothelial Disruption
3.4. IL-33 in Experimental Models of Cerebral Malaria
4. Conclusions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| BBB | blood–brain barrier |
| B-CSF | Blood–cerebrospinal fluid |
| CM | cerebral malaria |
| CNS | central nervous system |
| DAMPs | damage-associated molecular pattern |
| DC | dendritic cell |
| EC | endothelial cell |
| ECM | experimental cerebral malaria |
| FL-IL-33 | full length Interleukin-33 |
| HCM | human cerebral malaria |
| ICH | intracranial hemorrhage |
| ILC2 | type-2 innate lymphoid cell |
| iRBC | infected red blood cell |
| LAP | LC3-Associated Phagocytosis |
| LC3 | Microtubule-associated protein 1A/1B-light chain 3 |
| MC | mast cells |
| MHC-I | major histocompatibility complex class I |
| mIL-33 | mature Interleukin-33 |
| MS | multiple sclerosis |
| PbA | Plasmodium berghei ANKA |
| PbA-MVs | parasite microvesicules |
| PfEMP1 | Plasmodium erythrocyte membrane protein 1 |
| TLRs | Toll-like-receptors |
| Tregs | regulatory T cells |
References
- Idro, R.; Marsh, K.; John, C.C.; Newton, C.R.J. Cerebral Malaria: Mechanisms of Brain Injury and Strategies for Improved Neurocognitive Outcome. Pediatr. Res. 2010, 68, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Hunt, N.H.; Golenser, J.; Chan-Ling, T.; Parekh, S.; Rae, C.; Potter, S.; Medana, I.M.; Miu, J.; Ball, H.J. Immunopathogenesis of Cerebral Malaria. Int. J. Parasitol. 2006, 36, 569–582. [Google Scholar] [CrossRef] [PubMed]
- Medana, I.M.; Turner, G.D.H. Human Cerebral Malaria and the Blood-Brain Barrier. Int. J. Parasitol. 2006, 36, 555–568. [Google Scholar] [CrossRef] [PubMed]
- van der Heyde, H.C.; Nolan, J.; Combes, V.; Gramaglia, I.; Grau, G.E. A Unified Hypothesis for the Genesis of Cerebral Malaria: Sequestration, Inflammation and Hemostasis Leading to Microcirculatory Dysfunction. Trends Parasitol. 2006, 22, 503–508. [Google Scholar] [CrossRef]
- Bagot, S.; Nogueira, F.; Collette, A.; do Rosario, V.; Lemonier, F.; Cazenave, P.-A.; Pied, S. Comparative Study of Brain CD8+ T Cells Induced by Sporozoites and Those Induced by Blood-Stage Plasmodium Berghei ANKA Involved in the Development of Cerebral Malaria. Infect. Immun. 2004, 72, 2817–2826. [Google Scholar] [CrossRef]
- Blanc, A.-L.; Keswani, T.; Gorgette, O.; Bandeira, A.; Malissen, B.; Cazenave, P.-A.; Pied, S. Suppression of CD4+ Effector Responses by Naturally Occurring CD4+ CD25+ Foxp3+ Regulatory T Cells Contributes to Experimental Cerebral Malaria. Infect. Immun. 2016, 84, 329–338. [Google Scholar] [CrossRef]
- Dalko, E.; Genete, D.; Auger, F.; Dovergne, C.; Lambert, C.; Herbert, F.; Cazenave, P.-A.; Roland, J.; Pield, S. Heme Dampens T-Cell Sequestration by Modulating Glial Cell Responses during Rodent Cerebral Malaria. Brain Behav. Immun. 2016, 58, 280–290. [Google Scholar] [CrossRef]
- Barrera, V.; Haley, M.J.; Strangward, P.; Attree, E.; Kamiza, S.; Seydel, K.B.; Taylor, T.E.; Milner, D.A.; Craig, A.G.; Couper, K.N. Comparison of CD8+ T Cell Accumulation in the Brain During Human and Murine Cerebral Malaria. Front. Immunol. 2019, 10, 1747. [Google Scholar] [CrossRef]
- Huggins, M.A.; Johnson, H.L.; Jin, F.; N′Songo, A.; Hanson, L.M.; LaFrance, S.J.; Butler, N.S.; Harty, J.T.; Johnson, A.J. Perforin Expression by CD8 T Cells Is Sufficient To Cause Fatal Brain Œdema during Experimental Cerebral Malaria. Infect. Immun. 2017, 85, e00985-16. [Google Scholar] [CrossRef]
- Obermeier, B.; Verma, A.; Ransohoff, R.M. The Blood-Brain Barrier. Handb. Clin. Neurol. 2016, 133, 39–59. [Google Scholar] [CrossRef]
- Ngo-Thanh, H.; Sasaki, T.; Suzue, K.; Yokoo, H.; Isoda, K.; Kamitani, W.; Shimokawa, C.; Hisaeda, H.; Imai, T. Blood–Cerebrospinal Fluid Barrier: Another Site Disrupted during Experimental Cerebral Malaria Caused by Plasmodium Berghei ANKA. Int. J. Parasitol. 2020, 50, 1167–1175. [Google Scholar] [CrossRef] [PubMed]
- Hunt, N.H.; Grau, G.E. Cytokines: Accelerators and Brakes in the Pathogenesis of Cerebral Malaria. Trends Immunol. 2003, 24, 491–499. [Google Scholar] [CrossRef]
- Bush, M.A.; Anstey, N.M.; Yeo, T.W.; Florence, S.M.; Granger, D.L.; Mwaikambo, E.D.; Weinberg, J.B. Vascular Dysfunction in Malaria: Understanding the Role of the Endothelial Glycocalyx. Front. Cell Dev. Biol. 2021, 9, 751251. [Google Scholar] [CrossRef] [PubMed]
- Touré, F.S.; Ouwe-Missi-Oukem-Boyer, O.; Bisvigou, U.; Moussa, O.; Rogier, C.; Pino, P.; Mazier, D.; Bisser, S. Apoptosis: A Potential Triggering Mechanism of Neurological Manifestation in Plasmodium Falciparum Malaria. Parasite Immunol. 2008, 30, 47–51. [Google Scholar] [CrossRef]
- Pino, P.; Vouldoukis, I.; Kolb, J.P.; Mahmoudi, N.; Desportes-Livage, I.; Bricaire, F.; Danis, M.; Dugas, B.; Mazier, D. Plasmodium Falciparum-Infected Erythrocyte Adhesion Induces Caspase Activation and Apoptosis in Human Endothelial Cells. J. Infect. Dis. 2003, 187, 1283–1290. [Google Scholar] [CrossRef]
- N’Dilimabaka, N.; Taoufiq, Z.; Zougbédé, S.; Bonnefoy, S.; Lorthiois, A.; Couraud, P.O.; Rebollo, A.; Snounou, G.; Mazier, D.; Moreno Sabater, A.P. Falciparum Isolate-Specific Distinct Patterns of Induced Apoptosis in Pulmonary and Brain Endothelial Cells. PLoS ONE 2014, 9, e90692. [Google Scholar] [CrossRef]
- Brown, H.; Rogerson, S.; Taylor, T.; Tembo, M.; Mwenechanya, J.; Molyneux, M.; Turner, G. Blood-Brain Barrier Function in Cerebral Malaria in Malawian Children. Am. J. Trop. Med. Hyg. 2001, 64, 207–213. [Google Scholar] [CrossRef]
- Wilson, N.O.; Huang, M.-B.; Anderson, W.; Bond, V.; Powell, M.; Thompson, W.E.; Armah, H.B.; Adjei, A.A.; Gyasi, R.; Tettey, Y.; et al. Soluble Factors from Plasmodium Falciparum-Infected Erythrocytes Induce Apoptosis in Human Brain Vascular Endothelial and Neuroglia Cells. Mol. Biochem. Parasitol. 2008, 162, 172–176. [Google Scholar] [CrossRef]
- Shaw, T.N.; Stewart-Hutchinson, P.J.; Strangward, P.; Dandamudi, D.B.; Coles, J.A.; Villegas-Mendez, A.; Gallego-Delgado, J.; van Rooijen, N.; Zindy, E.; Rodriguez, A.; et al. Perivascular Arrest of CD8+ T Cells Is a Signature of Experimental Cerebral Malaria. PLoS Pathog. 2015, 11, e1005210. [Google Scholar] [CrossRef]
- Tripathi, A.K.; Sullivan, D.J.; Stins, M.F. Plasmodium Falciparum-Infected Erythrocytes Decrease the Integrity of Human Blood-Brain Barrier Endothelial Cell Monolayers. J. Infect. Dis. 2007, 195, 942–950. [Google Scholar] [CrossRef]
- Chakravorty, S.J.; Carret, C.; Nash, G.B.; Ivens, A.; Szestak, T.; Craig, A.G. Altered Phenotype and Gene Transcription in Endothelial Cells, Induced by Plasmodium Falciparum-Infected Red Blood Cells: Pathogenic or Protective? Int. J. Parasitol. 2007, 37, 975–987. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tripathi, A.K.; Sha, W.; Shulaev, V.; Stins, M.F.; Sullivan, D.J. Plasmodium Falciparum–Infected Erythrocytes Induce NF-ΚB Regulated Inflammatory Pathways in Human Cerebral Endothelium. Blood 2009, 114, 4243–4252. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.; White, N.J. Molecular Mechanisms of Cytoadherence in Malaria. Am. J. Physiol. 1999, 276, C1231–C1242. [Google Scholar] [CrossRef] [PubMed]
- Eugenin, E.A.; Martiney, J.A.; Berman, J.W. The Malaria Toxin Hemozoin Induces Apoptosis in Human Neurons and Astrocytes: Potential Role in the Pathogenesis of Cerebral Malaria. Brain Res. 2019, 1720, 146317. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, J.; Owyang, A.; Oldham, E.; Song, Y.; Murphy, E.; McClanahan, T.K.; Zurawski, G.; Moshrefi, M.; Qin, J.; Li, X.; et al. IL-33, an Interleukin-1-like Cytokine That Signals via the IL-1 Receptor-Related Protein ST2 and Induces T Helper Type 2-Associated Cytokines. Immunity 2005, 23, 479–490. [Google Scholar] [CrossRef]
- McCarthy, P.C.; Phair, I.R.; Greger, C.; Pardali, K.; McGuire, V.A.; Clark, A.R.; Gaestel, M.; Arthur, J.S.C. IL-33 Regulates Cytokine Production and Neutrophil Recruitment via the P38 MAPK-Activated Kinases MK2/3. Immunol. Cell Biol. 2019, 97, 54–71. [Google Scholar] [CrossRef]
- Nakajima, S.; Ishimaru, K.; Kobayashi, A.; Yu, G.; Nakamura, Y.; Oh-oka, K.; Suzuki-Inoue, K.; Kono, K.; Nakao, A. Resveratrol Inhibits IL-33–Mediated Mast Cell Activation by Targeting the MK2/3–PI3K/Akt Axis. Sci. Rep. 2019, 9, 18423. [Google Scholar] [CrossRef]
- Nishizaki, T. IL-33 Suppresses GSK-3β Activation through an ST2-Independent MyD88/TRAF6/RIP/PI3K/Akt Pathway. Heliyon 2018, 4, e00971. [Google Scholar] [CrossRef]
- Duez, C.; Gross, B.; Marquillies, P.; Ledroit, V.; Ryffel, B.; Glineur, C. Glineur Corine Regulation of IL (Interleukin)-33 Production in Endothelial Cells via Kinase Activation and Fas/CD95 Upregulation. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2619–2631. [Google Scholar] [CrossRef]
- Dohi, E.; Choi, E.Y.; Rose, I.V.L.; Murata, A.S.; Chow, S.; Niwa, M.; Kano, S. Behavioral Changes in Mice Lacking Interleukin-33. eNeuro 2017, 4, ENEURO.0147-17.2017. [Google Scholar] [CrossRef]
- Pichery, M.; Mirey, E.; Mercier, P.; Lefrancais, E.; Dujardin, A.; Ortega, N.; Girard, J.-P. Endogenous IL-33 Is Highly Expressed in Mouse Epithelial Barrier Tissues, Lymphoid Organs, Brain, Embryos, and Inflamed Tissues: In Situ Analysis Using a Novel Il-33–LacZ Gene Trap Reporter Strain. J. Immunol. 2012, 188, 3488–3495. [Google Scholar] [CrossRef] [PubMed]
- Moussion, C.; Ortega, N.; Girard, J.-P. The IL-1-like Cytokine IL-33 Is Constitutively Expressed in the Nucleus of Endothelial Cells and Epithelial Cells in Vivo: A Novel “Alarmin”? PLoS ONE 2008, 3, e3331. [Google Scholar] [CrossRef] [PubMed]
- Küchler, A.M.; Pollheimer, J.; Balogh, J.; Sponheim, J.; Manley, L.; Sorensen, D.R.; De Angelis, P.M.; Scott, H.; Haraldsen, G. Nuclear Interleukin-33 Is Generally Expressed in Resting Endothelium but Rapidly Lost upon Angiogenic or Proinflammatory Activation. Am. J. Pathol. 2008, 173, 1229–1242. [Google Scholar] [CrossRef]
- Hardman, C.S.; Panova, V.; McKenzie, A.N.J. IL-33 Citrine Reporter Mice Reveal the Temporal and Spatial Expression of IL-33 during Allergic Lung Inflammation. Eur. J. Immunol. 2013, 43, 488–498. [Google Scholar] [CrossRef] [PubMed]
- Carriere, V. IL-33, the IL-1-like Cytokine Ligand for ST2 Receptor, Is a Chromat...—PubMed—NCBI. Available online: http://www.ncbi.nlm.nih.gov.gate2.inist.fr/pubmed/?term=carriere+V+2007 (accessed on 9 March 2015).
- Roussel, L.; Erard, M.; Cayrol, C.; Girard, J.-P. Molecular Mimicry between IL-33 and KSHV for Attachment to Chromatin through the H2A–H2B Acidic Pocket. EMBO Rep. 2008, 9, 1006–1012. [Google Scholar] [CrossRef]
- Travers, J.; Rochman, M.; Miracle, C.E.; Habel, J.E.; Brusilovsky, M.; Caldwell, J.M.; Rymer, J.K.; Rothenberg, M.E. Chromatin Regulates IL-33 Release and Extracellular Cytokine Activity. Nat. Commun. 2018, 9, 3244. [Google Scholar] [CrossRef] [PubMed]
- Gautier, V.; Cayrol, C.; Farache, D.; Roga, S.; Monsarrat, B.; Burlet-Schiltz, O.; Gonzalez de Peredo, A.; Girard, J.-P. Extracellular IL-33 Cytokine, but Not Endogenous Nuclear IL-33, Regulates Protein Expression in Endothelial Cells. Sci. Rep. 2016, 6, 34255. [Google Scholar] [CrossRef]
- Marsman, G.; Zeerleder, S.; Luken, B.M. Extracellular Histones, Cell-Free DNA, or Nucleosomes: Differences in Immunostimulation. Cell Death Dis. 2016, 7, e2518. [Google Scholar] [CrossRef]
- Luzina, I.G.; Pickering, E.M.; Kopach, P.; Kang, P.H.; Lockatell, V.; Todd, N.W.; Papadimitriou, J.C.; McKenzie, A.N.J.; Atamas, S.P. Full-Length IL-33 Promotes Inflammation but Not Th2 Response In Vivo in an ST2-Independent Fashion. J. Immunol. 2012, 189, 403–410. [Google Scholar] [CrossRef]
- Cayrol, C.; Girard, J.-P. The IL-1-like Cytokine IL-33 Is Inactivated after Maturation by Caspase-1. Proc. Natl. Acad. Sci. USA 2009, 106, 9021–9026. [Google Scholar] [CrossRef]
- Ali, S.; Nguyen, D.Q.; Falk, W.; Martin, M.U. Caspase 3 Inactivates Biologically Active Full Length Interleukin-33 as a Classical Cytokine but Does Not Prohibit Nuclear Translocation. Biochem. Biophys. Res. Commun. 2010, 391, 1512–1516. [Google Scholar] [CrossRef] [PubMed]
- Lüthi, A.U.; Cullen, S.P.; McNeela, E.A.; Duriez, P.J.; Afonina, I.S.; Sheridan, C.; Brumatti, G.; Taylor, R.C.; Kersse, K.; Vandenabeele, P.; et al. Suppression of Interleukin-33 Bioactivity through Proteolysis by Apoptotic Caspases. Immunity 2009, 31, 84–98. [Google Scholar] [CrossRef] [PubMed]
- Madouri, F.; Guillou, N.; Fauconnier, L.; Marchiol, T.; Rouxel, N.; Chenuet, P.; Ledru, A.; Apetoh, L.; Ghiringhelli, F.; Chamaillard, M.; et al. Caspase-1 Activation by NLRP3 Inflammasome Dampens IL-33-Dependent House Dust Mite-Induced Allergic Lung Inflammation. J. Mol. Cell Biol. 2015, 7, 351–365. [Google Scholar] [CrossRef] [PubMed]
- Cayrol, C.; Duval, A.; Schmitt, P.; Roga, S.; Camus, M.; Stella, A.; Burlet-Schiltz, O.; Gonzalez-de-Peredo, A.; Girard, J.-P. Environmental Allergens Induce Allergic Inflammation through Proteolytic Maturation of IL-33. Nat. Immunol. 2018, 19, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Lefrançais, E.; Duval, A.; Mirey, E.; Roga, S.; Espinosa, E.; Cayrol, C.; Girard, J.-P. Central Domain of IL-33 Is Cleaved by Mast Cell Proteases for Potent Activation of Group-2 Innate Lymphoid Cells. Proc. Natl. Acad. Sci. USA 2014, 111, 15502–15507. [Google Scholar] [CrossRef]
- Lefrançais, E.; Roga, S.; Gautier, V.; Gonzalez-de-Peredo, A.; Monsarrat, B.; Girard, J.-P.; Cayrol, C. IL-33 Is Processed into Mature Bioactive Forms by Neutrophil Elastase and Cathepsin G. Proc. Natl. Acad. Sci. USA 2012, 109, 1673–1678. [Google Scholar] [CrossRef]
- Cohen, E.S.; Scott, I.C.; Majithiya, J.B.; Rapley, L.; Kemp, B.P.; England, E.; Rees, D.G.; Overed-Sayer, C.L.; Woods, J.; Bond, N.J.; et al. Oxidation of the Alarmin IL-33 Regulates ST2-Dependent Inflammation. Nat. Commun. 2015, 6, 8327. [Google Scholar] [CrossRef]
- Griesenauer, B.; Paczesny, S. The ST2/IL-33 Axis in Immune Cells during Inflammatory Diseases. Front. Immunol. 2017, 8, 475. [Google Scholar] [CrossRef]
- Molofsky, A.B.; Savage, A.; Locksley, R.M. Interleukin-33 in Tissue Homeostasis, Injury and Inflammation. Immunity 2015, 42, 1005–1019. [Google Scholar] [CrossRef]
- Cayrol, C.; Girard, J.-P. Interleukin-33 (IL-33): A Nuclear Cytokine from the IL-1 Family. Immunol. Rev. 2018, 281, 154–168. [Google Scholar] [CrossRef]
- Liew, F.Y.; Girard, J.-P.; Turnquist, H.R. Interleukin-33 in Health and Disease. Nat. Rev. Immunol. 2016, 16, 676–689. [Google Scholar] [CrossRef] [PubMed]
- Bonilla, W.V.; Fröhlich, A.; Senn, K.; Kallert, S.; Fernandez, M.; Johnson, S.; Kreutzfeldt, M.; Hegazy, A.N.; Schrick, C.; Fallon, P.G.; et al. The Alarmin Interleukin-33 Drives Protective Antiviral CD8+ T Cell Responses. Science 2012, 335, 984–989. [Google Scholar] [CrossRef] [PubMed]
- Komai-Koma, M.; Wang, E.; Kurowska-Stolarska, M.; Li, D.; McSharry, C.; Xu, D. Interleukin-33 Promoting Th1 Lymphocyte Differentiation Dependents on IL-12. Immunobiology 2016, 221, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Pascual-Reguant, A.; Bayat Sarmadi, J.; Baumann, C.; Noster, R.; Cirera-Salinas, D.; Curato, C.; Pelczar, P.; Huber, S.; Zielinski, C.E.; Löhning, M.; et al. TH17 Cells Express ST2 and Are Controlled by the Alarmin IL-33 in the Small Intestine. Mucosal Immunol. 2017, 10, 1431–1442. [Google Scholar] [CrossRef]
- Wohlfert, E.A.; Grainger, J.R.; Bouladoux, N.; Konkel, J.E.; Oldenhove, G.; Ribeiro, C.H.; Hall, J.A.; Yagi, R.; Naik, S.; Bhairavabhotla, R.; et al. GATA3 Controls Foxp3+ Regulatory T Cell Fate during Inflammation in Mice. J. Clin. Investg. 2011, 121, 4503–4515. [Google Scholar] [CrossRef]
- Yang, Q.; Li, G.; Zhu, Y.; Liu, L.; Chen, E.; Turnquist, H.; Zhang, X.; Finn, O.J.; Chen, X.; Lu, B. IL-33 Synergizes with TCR and IL-12 Signaling to Promote the Effector Function of CD8+ T Cells. Eur. J. Immunol. 2011, 41, 3351–3360. [Google Scholar] [CrossRef]
- Gao, K.; Li, X.; Zhang, L.; Bai, L.; Dong, W.; Gao, K.; Shi, G.; Xia, X.; Wu, L.; Zhang, L. Transgenic Expression of IL-33 Activates CD8+ T Cells and NK Cells and Inhibits Tumor Growth and Metastasis in Mice. Cancer Lett. 2013, 335, 463–471. [Google Scholar] [CrossRef]
- Hung, L.-Y.; Tanaka, Y.; Herbine, K.; Pastore, C.; Singh, B.; Ferguson, A.; Vora, N.; Douglas, B.; Zullo, K.; Behrens, E.; et al. Cellular Context of IL-33 Expression Dictates Impact on Anti-Helminth Immunity. Sci. Immunol. 2020, 5, eabc6259. [Google Scholar] [CrossRef]
- Dinarello, C.A. Introduction to the Interleukin-1 Family of Cytokines and Receptors: Drivers of Innate Inflammation and Acquired Immunity. Immunol. Rev. 2018, 281, 5–7. [Google Scholar] [CrossRef]
- Komai-Koma, M.; Gilchrist, D.S.; McKenzie, A.N.J.; Goodyear, C.S.; Xu, D.; Liew, F.Y. IL-33 Activates B1 Cells and Exacerbates Contact Sensitivity. J. Immunol. 2011, 186, 2584–2591. [Google Scholar] [CrossRef] [PubMed]
- Sattler, S.; Ling, G.-S.; Xu, D.; Hussaarts, L.; Romaine, A.; Zhao, H.; Fossati-Jimack, L.; Malik, T.; Cook, H.T.; Botto, M.; et al. IL-10-Producing Regulatory B Cells Induced by IL-33 (BregIL-33) Effectively Attenuate Mucosal Inflammatory Responses in the Gut. J. Autoimmun. 2014, 50, 107–122. [Google Scholar] [CrossRef]
- Mu, R.; Huang, H.-Q.; Li, Y.-H.; Li, C.; Ye, H.; Li, Z.-G. Elevated Serum Interleukin 33 Is Associated with Autoantibody Production in Patients with Rheumatoid Arthritis. J. Rheumatol. 2010, 37, 2006–2013. [Google Scholar] [CrossRef] [PubMed]
- Kerr, A.L.; Cheng, S.-Y.; Jones, T.A. Experience-Dependent Neural Plasticity in the Adult Damaged Brain. J. Commun. Disord. 2011, 44, 538–548. [Google Scholar] [CrossRef] [PubMed]
- Gadani, S.P.; Walsh, J.T.; Smirnov, I.; Zheng, J.; Kipnis, J. The Glia-Derived Alarmin IL-33 Orchestrates the Immune Response and Promotes Recovery Following CNS Injury. Neuron 2015, 85, 703–709. [Google Scholar] [CrossRef] [PubMed]
- Abd Rachman Isnadi, M.F.; Chin, V.K.; Abd Majid, R.; Lee, T.Y.; Atmadini Abdullah, M.; Bello Omenesa, R.; Osamah Ibraheem, Z.; Basir, R. Critical Roles of IL-33/ST2 Pathway in Neurological Disorders. Mediat. Inflamm. 2018, 2018, 5346413. [Google Scholar] [CrossRef] [PubMed]
- Yndart, A.; Kaushik, A.; Agudelo, M.; Raymond, A.; Atluri, V.S.; Saxena, S.K.; Nair, M. Investigation of Neuropathogenesis in HIV-1 Clade B and C Infection Associated with IL-33 and ST2 Regulation. ACS Chem. Neurosci. 2015, 6, 1600–1612. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Z.; Thangavel, R.; Kempuraj, D.; Yang, E.; Zaheer, S.; Zaheer, A. Alzheimer’s Disease: Evidence for the Expression of Interleukin-33 and Its Receptor ST2 in the Brain. J. Alzheimers Dis. 2014, 40, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Jafarzadeh, A.; Mahdavi, R.; Jamali, M.; Hajghani, H.; Nemati, M.; Ebrahimi, H.-A. Increased Concentrations of Interleukin-33 in the Serum and Cerebrospinal Fluid of Patients with Multiple Sclerosis. Oman Med. J. 2016, 31, 40–45. [Google Scholar] [CrossRef]
- Xiao, Y.; Lai, L.; Chen, H.; Shi, J.; Zeng, F.; Li, J.; Feng, H.; Mao, J.; Zhang, F.; Wu, N.; et al. Interleukin-33 Deficiency Exacerbated Experimental Autoimmune Encephalomyelitis with an Influence on Immune Cells and Glia Cells. Mol. Immunol. 2018, 101, 550–563. [Google Scholar] [CrossRef]
- Besnard, A.-G.; Guabiraba, R.; Niedbala, W.; Palomo, J.; Reverchon, F.; Shaw, T.N.; Couper, K.N.; Ryffel, B.; Liew, F.Y. IL-33-Mediated Protection against Experimental Cerebral Malaria Is Linked to Induction of Type 2 Innate Lymphoid Cells, M2 Macrophages and Regulatory T Cells. PLoS Pathog. 2015, 11, e1004607. [Google Scholar] [CrossRef]
- Longhi-Balbinot, D.T.; Rossaneis, A.C.; Pinho-Ribeiro, F.A.; Bertozzi, M.M.; Cunha, F.Q.; Alves-Filho, J.C.; Cunha, T.M.; Peron, J.P.S.; Miranda, K.M.; Casagrande, R.; et al. The Nitroxyl Donor, Angeli’s Salt, Reduces Chronic Constriction Injury-Induced Neuropathic Pain. Chem. Biol. Interact. 2016, 256, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Ma, L.; Luo, C.-L.; Wang, T.; Zhang, M.-Y.; Shen, X.; Meng, H.-H.; Ji, M.-M.; Wang, Z.-F.; Chen, X.-P.; et al. IL-33 Exerts Neuroprotective Effect in Mice Intracerebral Hemorrhage Model Through Suppressing Inflammation/Apoptotic/Autophagic Pathway. Mol. Neurobiol. 2017, 54, 3879–3892. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Wen, Y.; Wang, L.; Wen, L.; You, W.; Wei, S.; Mao, L.; Wang, H.; Chen, Z.; Yang, X. Therapeutic Opportunities of Interleukin-33 in the Central Nervous System. Front. Immunol. 2021, 12, 654626. [Google Scholar] [CrossRef] [PubMed]
- Fairlie-Clarke, K.; Barbour, M.; Wilson, C.; Hridi, S.U.; Allan, D.; Jiang, H.-R. Expression and Function of IL-33/ST2 Axis in the Central Nervous System Under Normal and Diseased Conditions. Front. Immunol. 2018, 9, 2596. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Zhou, Y.; Xiao, W.; Liang, Z.; Dai, J.; Weng, X.; Wu, X. Interleukin-33 Ameliorates Ischemic Brain Injury in Experimental Stroke through Promoting Th2 Response and Suppressing Th17 Response. Brain Res. 2015, 1597, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Yasuoka, S.; Kawanokuchi, J.; Parajuli, B.; Jin, S.; Doi, Y.; Noda, M.; Sonobe, Y.; Takeuchi, H.; Mizuno, T.; Suzumura, A. Production and Functions of IL-33 in the Central Nervous System. Brain Res. 2011, 1385, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Allan, D.; Fairlie-Clarke, K.J.; Elliott, C.; Schuh, C.; Barnett, S.C.; Lassmann, H.; Linnington, C.; Jiang, H.-R. Role of IL-33 and ST2 Signalling Pathway in Multiple Sclerosis: Expression by Oligodendrocytes and Inhibition of Myelination in Central Nervous System. Acta Neuropathol. Commun. 2016, 4, 75. [Google Scholar] [CrossRef] [PubMed]
- Jiao, M.; Li, X.; Chen, L.; Wang, X.; Yuan, B.; Liu, T.; Dong, Q.; Mei, H.; Yin, H. Neuroprotective Effect of Astrocyte-Derived IL-33 in Neonatal Hypoxic-Ischemic Brain Injury. J. Neuroinflamm. 2020, 17, 251. [Google Scholar] [CrossRef] [PubMed]
- Vainchtein, I.D.; Chin, G.; Cho, F.S.; Kelley, K.W.; Miller, J.G.; Chien, E.C.; Liddelow, S.A.; Nguyen, P.T.; Nakao-Inoue, H.; Dorman, L.C.; et al. Astrocyte-Derived Interleukin-33 Promotes Microglial Synapse Engulfment and Neural Circuit Development. Science 2018, 359, 1269–1273. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.T.; Dorman, L.C.; Pan, S.; Vainchtein, I.D.; Han, R.T.; Nakao-Inoue, H.; Taloma, S.E.; Barron, J.J.; Molofsky, A.B.; Kheirbek, M.A.; et al. Microglial Remodeling of the Extracellular Matrix Promotes Synapse Plasticity. Cell 2020, 182, 388–403. [Google Scholar] [CrossRef]
- Ayimba, E.; Hegewald, J.; Ségbéna, A.Y.; Gantin, R.G.; Lechner, C.J.; Agosssou, A.; Banla, M.; Soboslay, P.T. Proinflammatory and Regulatory Cytokines and Chemokines in Infants with Uncomplicated and Severe Plasmodium Falciparum Malaria. Clin. Exp. Immunol. 2011, 166, 218–226. [Google Scholar] [CrossRef]
- Fernander, E.M.; Adogamhe, P.; Datta, D.; Bond, C.; Zhao, Y.; Bangirana, P.; Conroy, A.L.; Opoka, R.O.; John, C.C. Elevated Plasma Soluble ST2 Levels Are Associated With Neuronal Injury and Neurocognitive Impairment in Children with Cerebral Malaria. Pathog. Immun. 2022, 7, 60–80. [Google Scholar] [CrossRef] [PubMed]
- Palomo, J.; Reverchon, F.; Piotet, J.; Besnard, A.-G.; Couturier-Maillard, A.; Maillet, I.; Tefit, M.; Erard, F.; Mazier, D.; Ryffel, B.; et al. Critical Role of IL-33 Receptor ST2 in Experimental Cerebral Malaria Development. Eur. J. Immunol. 2015, 45, 1354–1365. [Google Scholar] [CrossRef] [PubMed]
- Ghazanfari, N.; Mueller, S.N.; Heath, W.R. Cerebral Malaria in Mouse and Man. Front. Immunol. 2018, 9, 2016. [Google Scholar] [CrossRef] [PubMed]
- Avril, M.; Bernabeu, M.; Benjamin, M.; Brazier, A.J.; Smith, J.D. Interaction between Endothelial Protein C Receptor and Intercellular Adhesion Molecule 1 to Mediate Binding of Plasmodium Falciparum-Infected Erythrocytes to Endothelial Cells. mBio 2016, 7, e00615-16. [Google Scholar] [CrossRef]
- Wei, J.; Zhao, J.; Schrott, V.; Zhang, Y.; Gladwin, M.; Bullock, G.; Zhao, Y. Red Blood Cells Store and Release Interleukin-33. J. Investig. Med. 2015, 63, 806–810. [Google Scholar] [CrossRef]
- Demyanets, S.; Konya, V.; Kastl, S.P.; Kaun, C.; Rauscher, S.; Niessner, A.; Pentz, R.; Pfaffenberger, S.; Rychli, K.; Lemberger, C.E.; et al. Interleukin-33 Induces Expression of Adhesion Molecules and Inflammatory Activation in Human Endothelial Cells and in Human Atherosclerotic Plaques. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2080–2089. [Google Scholar] [CrossRef]
- Boubou, M.I.; Collette, A.; Voegtlé, D.; Mazier, D.; Cazenave, P.-A.; Pied, S. T Cell Response in Malaria Pathogenesis: Selective Increase in T Cells Carrying the TCR Vβ8 during Experimental Cerebral Malaria. Int. Immunol. 1999, 11, 1553–1562. [Google Scholar] [CrossRef]
- Shrivastava, S.K.; Dalko, E.; Delcroix-Genete, D.; Herbert, F.; Cazenave, P.-A.; Pied, S. Uptake of Parasite-Derived Vesicles by Astrocytes and Microglial Phagocytosis of Infected Erythrocytes May Drive Neuroinflammation in Cerebral Malaria. Glia 2017, 65, 75–92. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Bento, C.F.; Deretic, V. Therapeutic Targeting of Autophagy in Neurodegenerative and Infectious Diseases. J. Exp. Med. 2015, 212, 979–990. [Google Scholar] [CrossRef]
- Deretic, V.; Saitoh, T.; Akira, S. Autophagy in Infection, Inflammation and Immunity. Nat. Rev. Immunol. 2013, 13, 722–737. [Google Scholar] [CrossRef] [PubMed]
- Leleu, I.; Genete, D.; Desnoulez, S.S.; Saidi, N.; Brodin, P.; Lafont, F.; Tomavo, S.; Pied, S. A Noncanonical Autophagy Is Involved in the Transfer of Plasmodium-Microvesicles to Astrocytes. Autophagy 2021, 18, 1583–1598. [Google Scholar] [CrossRef] [PubMed]
- Collette, A.; Bagot, S.; Ferrandiz, M.E.; Cazenave, P.-A.; Six, A.; Pied, S. A Profound Alteration of Blood TCRB Repertoire Allows Prediction of Cerebral Malaria. J. Immunol. 2004, 173, 4568–4575. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Shi, L.; Hua, S.; Qi, C.; Fang, M. IL-33 Ameliorates Experimental Colitis Involving Regulation of Autophagy of Macrophages in Mice. Cell Biosci. 2019, 9, 10. [Google Scholar] [CrossRef]
- Wang, Z.; Wu, L.; Pan, B.; Chen, Y.; Zhang, T.; Tang, N. Interleukin 33 Mediates Hepatocyte Autophagy and Innate Immune Response in the Early Phase of Acetaminophen-Induced Acute Liver Injury. Toxicology 2021, 456, 152788. [Google Scholar] [CrossRef]
- Wirth, C.C.; Glushakova, S.; Scheuermayer, M.; Repnik, U.; Garg, S.; Schaack, D.; Kachman, M.M.; Weißbach, T.; Zimmerberg, J.; Dandekar, T.; et al. Perforin-like Protein PPLP2 Permeabilizes the Red Blood Cell Membrane during Egress of Plasmodium Falciparum Gametocytes. Cell. Microbiol. 2014, 16, 709–733. [Google Scholar] [CrossRef]
- Gillrie, M.R.; Lee, K.; Gowda, D.C.; Davis, S.P.; Monestier, M.; Cui, L.; Hien, T.T.; Day, N.P.J.; Ho, M. Plasmodium Falciparum Histones Induce Endothelial Proinflammatory Response and Barrier Dysfunction. Am. J. Pathol. 2012, 180, 1028–1039. [Google Scholar] [CrossRef]
- Moxon, C.A.; Alhamdi, Y.; Storm, J.; Toh, J.M.H.; McGuinness, D.; Ko, J.Y.; Murphy, G.; Lane, S.; Taylor, T.E.; Seydel, K.B.; et al. Parasite Histones Are Toxic to Brain Endothelium and Link Blood Barrier Breakdown and Thrombosis in Cerebral Malaria. Blood Adv. 2020, 4, 2851–2864. [Google Scholar] [CrossRef]
- Shibui, A.; Takamori, A.; Tolba, M.E.M.; Nambu, A.; Shimura, E.; Yamaguchi, S.; Sanjoba, C.; Suto, H.; Sudo, K.; Okumura, K.; et al. IL-25, IL-33 and TSLP Receptor Are Not Critical for Development of Experimental Murine Malaria. Biochem. Biophys. Rep. 2016, 5, 191–195. [Google Scholar] [CrossRef][Green Version]
- Reverchon, F.; Mortaud, S.; Sivoyon, M.; Maillet, I.; Laugeray, A.; Palomo, J.; Montécot, C.; Herzine, A.; Meme, S.; Meme, W.; et al. IL-33 Receptor ST2 Regulates the Cognitive Impairments Associated with Experimental Cerebral Malaria. PLoS Pathog. 2017, 13, e1006322. [Google Scholar] [CrossRef]
- Morita, H.; Arae, K.; Unno, H.; Miyauchi, K.; Toyama, S.; Nambu, A.; Oboki, K.; Ohno, T.; Motomura, K.; Matsuda, A.; et al. An Interleukin-33-Mast Cell-Interleukin-2 Axis Suppresses Papain-Induced Allergic Inflammation by Promoting Regulatory T Cell Numbers. Immunity 2015, 43, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Huang, L.; Zhang, X.; Zhang, M.; Wang, Q.; Lin, H.; Yu, Z.; Li, X.; Liu, X.B.; Wu, Q.; et al. Mast Cells-Derived Exosomes Worsen the Development of Experimental Cerebral Malaria. Acta Trop. 2021, 224, 106145. [Google Scholar] [CrossRef] [PubMed]
- Strangward, P.; Haley, M.J.; Albornoz, M.G.; Barrington, J.; Shaw, T.; Dookie, R.; Zeef, L.; Baker, S.M.; Winter, E.; Tzeng, T.-C.; et al. Targeting the IL33-NLRP3 Axis Improves Therapy for Experimental Cerebral Malaria. Proc. Natl. Acad. Sci. USA 2018, 115, 7404–7409. [Google Scholar] [CrossRef]
- Prakash, D.; Fesel, C.; Jain, R.; Cazenave, P.-A.; Mishra, G.C.; Pied, S. Clusters of Cytokines Determine Malaria Severity in Plasmodium Falciparum–Infected Patients from Endemic Areas of Central India. J. Infect. Dis. 2006, 194, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Brown, H.; Turner, G.; Rogerson, S.; Tembo, M.; Mwenechanya, J.; Molyneux, M.; Taylor, T. Cytokine Expression in the Brain in Human Cerebral Malaria. J. Infect. Dis. 1999, 180, 1742–1746. [Google Scholar] [CrossRef] [PubMed]
- Tiemi Shio, M.; Eisenbarth, S.C.; Savaria, M.; Vinet, A.F.; Bellemare, M.-J.; Harder, K.W.; Sutterwala, F.S.; Bohle, D.S.; Descoteaux, A.; Flavell, R.A.; et al. Malarial Hemozoin Activates the NLRP3 Inflammasome through Lyn and Syk Kinases. PLoS Pathog. 2009, 5, e1000559. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Mice | Role of IL-33 | References |
|---|---|---|
| WT | Microglia activation/proliferation induced by IL-33 in CNS inflammation | [65,77] |
| Effect of IL-33 mediated by systemic ILC-2, Th2, M2 and Treg | [71] | |
| In the brain during ECM, IL-33 mRNA expression not altered but IL-33 protein doubled | [84] | |
| Nuclear IL-33 detected by immunostaining in the brain during ECM. | [84] | |
| No circulating IL-33 detected in serum of PbA-infected mice | [84] | |
| IL-33 increased in the spleen and lung after sporozoite infection | [84] | |
| Contribution of IL-33 to cognitive defects associated in ECM | [101] | |
| Early and direct role of IL-33/ST2 pathway in the exacerbated neuroinflammation during ECM | [101] | |
| IL-1β induced by IL-33/ST2 pathway triggers IL-33 expression in oligodendrocytes | [101] | |
| Exacerbated IL-33 expression by astrocytes and oligodendrocytes after PbA-infection | [101] | |
| Decrease of IL-33 mRNA and upregulation of IL-33 protein in hippocampus, SVZ and frontal cortex in ECM | [101] | |
| ST2-deficient | Reduction of cerebral inflammation | [84] |
| Significant reduction of ECM symptoms | [84] | |
| Resistant to PbA-induced neuropathology and improved survival | [84] | |
| Reduced brain sequestration of CD4+ T cells and CD8+ T cells | [84] | |
| Local expression of ICAM-1, CXCR3, and LT-α strongly reduced | [84] | |
| Parasitemia and brain parasite load similar to WT mice | [84] | |
| IFN-γ, TNFα and CXCL10 mRNA expression not altered in the brain | [84] | |
| No cognitive defect post-PbA infection | [101] | |
| CXCL9, CXCL10, IL-1β drastically reduced | [101] | |
| Microglia activation after PbA-infection | [101] | |
| IL-33-deficient | Similar survival and parasitemia than WT mice | [100] |
| Reduced anxiety-like behavior | [30] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Glineur, C.; Leleu, I.; Pied, S. The IL-33/ST2 Pathway in Cerebral Malaria. Int. J. Mol. Sci. 2022, 23, 13457. https://doi.org/10.3390/ijms232113457
Glineur C, Leleu I, Pied S. The IL-33/ST2 Pathway in Cerebral Malaria. International Journal of Molecular Sciences. 2022; 23(21):13457. https://doi.org/10.3390/ijms232113457
Chicago/Turabian StyleGlineur, Corine, Inès Leleu, and Sylviane Pied. 2022. "The IL-33/ST2 Pathway in Cerebral Malaria" International Journal of Molecular Sciences 23, no. 21: 13457. https://doi.org/10.3390/ijms232113457
APA StyleGlineur, C., Leleu, I., & Pied, S. (2022). The IL-33/ST2 Pathway in Cerebral Malaria. International Journal of Molecular Sciences, 23(21), 13457. https://doi.org/10.3390/ijms232113457