Abstract
Caffeine is the most frequently used substance with a central nervous system stimulant effect, but its consumption is most often due to the intake of foods and drinks that contain it (coffee, tea, chocolate, food supplements with plant extracts of Guarana, Mate herba, Cola nuts). Due to its innocuity, caffeine is a safe xanthine alkaloid for human consumption in a wide range of doses, being used for its central nervous stimulating effect, lipolytic and diuresis-enhancing properties, but also as a permitted ergogenic compound in athletes. In addition to the mechanisms that explain the effects of caffeine on the targeted organ, there are many proposed mechanisms by which this substance would have antioxidant effects. As such, its consumption prevents the occurrence/progression of certain neurodegenerative diseases as well as other medical conditions associated with increased levels of reactive oxygen or nitrogen species. However, most studies that have assessed the beneficial effects of caffeine have used pure caffeine. The question, therefore, arises whether the daily intake of caffeine from food or drink has similar benefits, considering that in foods or drinks with a high caffeine content, there are other substances that could interfere with this action, either by potentiating or decreasing its antioxidant capacity. Natural sources of caffeine often combine plant polyphenols (phenol-carboxylic acids, catechins) with known antioxidant effects; however, stimulant drinks and dietary supplements often contain sugars or artificial sweeteners that can significantly reduce the effects of caffeine on oxidative stress. The objective of this review is to clarify the effects of caffeine in modulating oxidative stress and assess these benefits, considering the source and the dose administered.
1. Introduction
Caffeine is the most used substance with a centrally excitatory effect since it is a common ingredient in beverages and foods. According to data published by the EFSA (European Food Safety Authority), the average daily consumption of caffeine in young adults (18–65 years old) is 37–319 mg and derives mainly from products based on coffee and cocoa beans, tea leaves, guarana berries, and kola nuts [1]. The consumption of caffeine begins in childhood and resides in sweets and juices, but in conditions of moderate consumption, the doses are low and do not raise health concerns [2]. After oral administration, caffeine is rapidly absorbed with a bioavailability of approximately 99%. It is distributed in most tissues, including the brain, and then it is metabolized in the liver and excreted via the kidneys, mostly in the form of metabolites [3].
Although caffeine has been commonly used since ancient times, it is still a studied and controversial substance. The studies published in the literature regarding this substance convey both the beneficial effects and toxic effects (dose-related) on health. In recent years, however, several studies have attempted to establish the link between caffeine and oxidative status, as many of the conditions in which caffeine has been shown to have a beneficial influence can be attributed to a decrease in oxidative stress (OS).
Oxidative stress is defined as an imbalance between the production of reactive species and the ability of extracellular and intracellular protection systems to neutralize them. A growing number of medical conditions are correlated with the increase in the level of reactive oxygen species (ROS), but the pathophysiological mechanisms in which they are involved are beyond the purpose of our paper, and, in addition, their positive effects should not be excluded [4]. Briefly, these species originate from the oxygen molecule (O2), which in the presence of a free electron, forms the superoxide anion (). The latter, under the action of superoxide dismutase (SOD), is converted to hydrogen peroxide (H2O2), a more stable compound with a half-life longer than . However, both and H2O2, in the presence of transitional metals (Cu, Fe, Zn), generate a highly reactive free radical, the hydroxyl radical (•OH), through the Fenton and Haber–Weiss reactions. In addition, can interact with nitric oxide (NO), forming reactive nitrogen species (RNS), such as peroxynitrite (ONOO−), from which •OH can be obtained [5,6,7].
2. Pharmacokinetic and Pharmacodynamic Properties of Caffeine
2.1. Caffeine: Mechanism of Action
The peripheral and central effects of caffeine in the currently used doses occur as a result of antagonizing P1 adenosine receptors [8]. P1 receptors have four subtypes A1, A2A, A2B, and A3. Xanthine derivatives, such as caffeine, theophylline, and other naturally occurring xanthines, have an affinity for all four receptor subtypes. A1 and A3 receptors are negatively coupled to adenylate cyclase (AC) and decrease cyclic adenosine monophosphate (cAMP) concentration, while A2A and A2B receptors stimulate AC activity and increase intracellular cAMP levels [9]. Since the localization of these receptors is different, the consequent effects of their stimulation or blocking are complex. For example, the density of A1 receptors is increased in the heart so that antagonists of these receptors, such as caffeine, stimulate the heart. The A2A subtype is predominantly found in coronary arteries; as a consequence, adenosine has coronary dilator effects. [10].
Adenosine receptors are also located in the kidney and are involved in the control of the glomerular filtration rate (GFR) and the hydro-electrolytic balance. More specifically, through A1 receptors, adenosine produces afferent arteriolar constriction and decreases GFR, which implies that antagonists of these receptors increase GFR and diuresis, specifically natriuresis [11].
In the CNS, adenosine favors the release of neurotransmitters through A2A receptors and inhibits it through A1 receptors, modulating sleep and vigilance [12]. The central stimulating action of caffeine is explained by antagonizing adenosine receptors [13]. A2A agonists have been shown to have pro-nociceptive effects, the intensity of the pain being directly proportional to the increase in cAMP level [14]. Moreover, the stimulation of A2A receptors located in the pial vessel causes vasodilatation [15], which can contribute to the occurrence of headaches and migraines. Therefore, the use of caffeine in migraine treatment is due to antagonizing these effects [14].
Caffeine is a non-selective inhibitor of phosphodiesterase (PDE) enzymes involved in the inactivation of cAMP and cyclic guanosine monophosphate (cGMP) with the formation of 5′-AMP and 5′-GMP. This increases the level of cAMP and cGMP [16]. Mammalian PDEs, divided into 11 subfamilies (PDE1 to PDE11), have different specificities for cyclic nucleotides. PDEs 4, 7, and 8 selectively hydrolyze cAMP; PDEs 5, 6, and 9 selectively hydrolyze cGMP; and PDEs 1, 2, 3, 10, and 11 possess an affinity for both cAMP and cGMP. The tissue distribution of these isoforms is variable, and modulating their activity has different consequences [17]. The effects of caffeine that can be explained as a result of the inhibition of PDEs include the (weak) bronchodilator effect and the lipolytic effect [8].
In high doses, caffeine, as an agonist of ryanodine receptors, favors the contraction of striated muscles by releasing calcium ions from the sarcoplasmic reticulum (see Figure 1) [18].
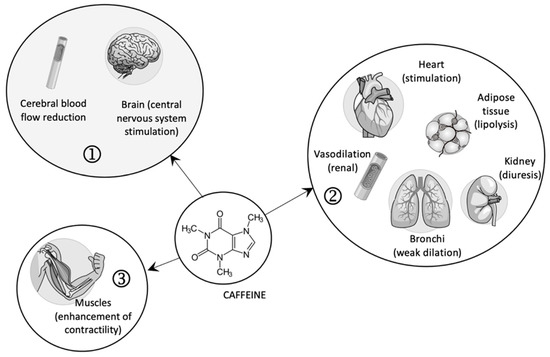
Figure 1.
The pharmacological effects of caffeine (1—central effects; 2—peripheral effects; 3—contraction of skeletal muscles).
Although the complex effects of caffeine are explained in specialized literature based on the three mechanisms presented above, the effects are different depending on the dose used for the antagonism of adenosine receptors, the inhibition of PDE enzymes, and the mobilization of calcium from the endoplasmic reticulum. For example, the affinity for adenosine receptors was observed only in low doses and not high ones [13,19], while the consequent effects of PDEs inhibition are especially visible in doses several times higher than those currently used due to the low affinity of caffeine on PDEs [13].
Since these effects of caffeine are well known and intensively discussed in the literature, only the mechanisms involved in the modulation of oxidative stress will be detailed in this paper.
Although the beneficial effect of caffeine to reduce OS is well documented and demonstrated in both in vivo and in vitro studies, the way it does so has not been fully elucidated.
2.2. Caffeine: The Metabolic Pathways Involved in Biotransformation and Oxidative Stress
As a natural xanthine derivative, structurally similar to endogenous purine nitrogenous bases, caffeine is metabolized almost entirely and involves both microsomal (especially CYP1A2) and non-microsomal enzyme xanthine oxidase (XO)—see Figure 2.
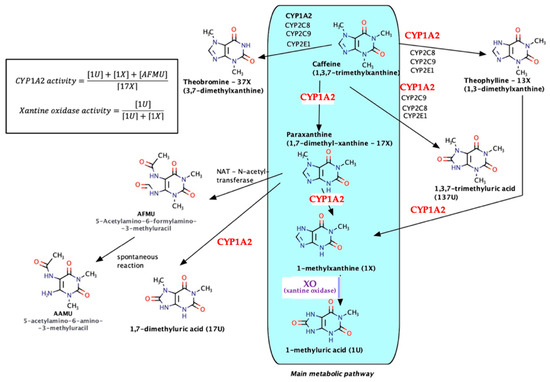
Figure 2.
Caffeine metabolic pathways.
The main metabolic pathway is the formation of paraxanthine (17-X) followed by N7-demethylation, reactions via CYP1A2 (80% of the total amount), and then oxidation under the action of XO with the formation of 1-methyluric acid (1U). Secondary metabolites (theobromine, theophylline, trimethyluric acid, etc.) each occur in much lower percentages (6–8%). As a result, taking into account the relative innocuity of caffeine, the urinary metabolites/caffeine can be used in pharmacogenetics for the determination of the CYP1A2 phenotype (molar ratio of 7-demethylated compounds–AFMU, 1-methylxanthine, 1-methyluric acid, and the immediate precursor paraxanthine), as for the quantitative evaluation of the catalytic activity of XO (molar ratio of 1-methyluric acid to total 1-methyluric acid + 1-methylxanthine [20,21,22,23,24].
As a substrate of XO, caffeine and its metabolites can competitively inhibit uric acid formation. The latter compound has dual characteristics—at the extracellular level, it acts as a powerful antioxidant, but inside the cell, it acts as a promoter of the formation of ROS; at the intracellular level, uric acid is a compound that generates endothelial dysfunction by favoring the formation of cytokines and lipid peroxidation, reducing the amount of endothelial nitric oxide, and favoring the pro-inflammatory cascade ERK/p38 MAPK [25,26].
Mammalian XO comes from hepatic xanthine dehydrogenase (XDR), which has an oxidizing action on some biological substrates via NAD+ with the consequent formation of NADH; after its release into plasma, XDR, following partial proteolysis, will produce XO which, concomitantly with the formation of uric acid, generates the formation of ROS [27,28].
In many pathological processes in which OS has an essential role, increased XO activity is involved: type 1 diabetes mellitus, in which the production of mitochondrial superoxide is not modified, and the generation of ROS thus becomes XO-dependent [29], myocardial ischemia and reperfusion lesions [30], atherosclerosis [25], and neoplastic processes [31].
Recent studies point out that longevity itself is negatively correlated with the level of OS, although antioxidant medication brings only modest benefits, except for ischemia-reperfusion syndrome in which scavenger and trapping drugs are ineffective, while the inhibition of XO can be life saving [31]. A recent retrospective clinical cohort study in the Irish population by Ruiz F et al. confirmed that hyperuricemia is an independent marker of mortality in both sexes [32].
The effects of caffeine on uricemia are still controversial. As a result of the structure and metabolites that form, caffeine is a substrate for XO, competitively interfering with purine endogenous compounds that represent the physiological substrate of the enzyme (xanthine, hypoxanthine). However, the effects of exogenous caffeine intake on uric acid levels remain questionable. Conversely, if the caffeine intake comes from natural sources (tea, coffee), the present phytocompounds may also be XO inhibitors (flavonoids, polyphenols). Thus, in an in vitro study on human immortalized normal liver cell line HL-7702, Wu D et al. showed that tea and its components inhibited XO and decreased the mRNA expression of XDR, although this effect was not attributed to caffeine but to catechins and gallic acid [33]. A meta-analysis conducted by Peluso I et al. on the effect of tea extracts in patients with asymptomatic hyperuricemia did not reach a clear conclusion due to the lack of bioavailability data of flavanol-type compounds and the small number of randomized studies used in the statistical analysis [34]. Another meta-analysis based on nine clinical studies demonstrated that daily coffee intake decreases the plasma level of uric acid, the effect being gender specific (more obvious in men than in women despite an equivalent dose) [35]. A clinical study conducted on the Korean population aimed at assessing the consumption of pure caffeine and beverages containing caffeine (tea, coffee) on the catabolism of endogenous purines and serum uric acid level; the authors concluded that regardless of the source, the moderate intake of caffeine does not influence the plasma level of uric acid in any way [36]. The contradictory results obtained by various researchers (decrease, increase, or lack of influence on uricemia) may be due to the difference in the source of caffeine (pure caffeine, extracts of tea, coffee, Mathe folium, guarana, etc.), but also by the polymorphism of CYP1A2 and XO. The variable activity of the two enzymes could explain gender and racial differences in caffeine metabolism with consequences in uric acid production [37]. Previous studies showed a higher CYP1A2 activity in men than in women, in Europeans than in South Africans, as well as in Swedes than in Koreans [38]. Other phytochemical compounds (catechins, flavonoids, etc.) present in natural extracts can influence the biological effect through multiple mechanisms (antioxidant effect, inhibition of XO).
2.3. The Main Mechanisms of Action of Caffeine in Correlation with Oxidative Stress
2.3.1. Caffeine and the Purinergic Signaling Pathway
The purinergic signaling pathway is mediated by extracellular adenosine, adenosine diphosphate (ADP), and adenosine triphosphate (ATP). Nucleotides and nucleosides are purinergic receptor agonists. Nucleotides stimulate the P2X (receptor-gated ion channels) and P2Y (G protein-coupled receptors) receptors, while adenosine is a P1 receptor agonist (P1R-G-coupled receptors) [39].
If we refer to the effects of caffeine in relation to the type of receptor antagonized, the effects are dependent on the type of consumption. Although caffeine has an affinity for both A2A and A1 receptors, in the case of acute administration, the effects are specific to blocking A1 receptors. In the case of chronic consumption, however, the establishment of tolerance of A1 receptors to caffeine was observed, and the effects were specific to A2A receptor antagonism [40]. Taking into account the fact that the stimulation of both receptor subtypes changes the concentration of cAMP, with a decrease in cAMP in the case of the stimulation of A1 receptors and an increase in cAMP in the case of the stimulation of A2A receptors [9], the question arises whether the antioxidant/beneficial action of caffeine could be due to this second messenger.
The way cAMP modulates OS has not been elucidated. The decrease in cAMP in vascular smooth muscle cells was correlated with an increase in the production of the superoxide anion [41]. In the central nervous system, cAMP-activated protein kinase A (PKA) forms at the mitochondrial level a complex with A-kinase anchoring protein 121 (AKAP121), which phosphorylates the fission modulator dynamin-related protein 1 (Drp1) with the increase in the level of reduced glutathione (GSH), but also the activity of mitochondrial SOD2. Thus, cell apoptosis is prevented after exposure to neurotoxic substances [42]. cGMP has beneficial effects, such as protecting cells against redox stress by activating antioxidant gene expression and against chromosomal instability [43,44]. As far as the second action mechanism of caffeine is concerned, PDE inhibition (in high doses) would have the effects described above.
Even if the antioxidant effects of caffeine consumption could not be directly correlated with cAMP and/or cGMP levels, these effects have been demonstrated in experimental studies.
- The importance of purinergic receptors in neurodegenerative disorders
In a study by Leite et al., caffeine and another A2A purinergic receptor antagonist were shown to have similar effects on memory impairment as assessed using the Novel Object Recognition (NOR) and OS generated by aging tests in rats. Administration of both compounds led to a decrease in ROS and RNS, which suggested that the beneficial effect was correlated with the blocking of these receptors [45]. Neurodegeneration and the accumulation of beta-amyloid plaques (amyloid-beta accumulation) (Aβ) from Alzheimer’s disease (AD) have also been shown to be correlated with the release of inflammatory cytokines following A2A receptor stimulation. The beneficial effect of caffeine in reducing memory impairment by regulating the expression of brain-derived neurotrophic factor (BDNF) and tropomyosin receptor kinase B (TrkB) or by regulating the expression of nuclear erythroid 2-related factor (Nrf2) and Toll-like receptor 4 (TLR 4)-induced glial cells-mediated neuronal cell death and neuroinflammation [46].
Due to the complex interrelations between adenosine and nucleotides, the role of adenosine in the pathogenesis of neurodegenerative diseases cannot be separated from its phosphorylate derivatives.
The role of purinergic receptors in the pathogenesis of some degenerative CNS diseases has been intensively studied, and the effects following the stimulation and the blocking of these receptors are extremely complex. For example, OS has been observed to inhibit hydrolases (ecto-nucleotidase, E-NTPDase1/CD39 and ecto-5′-nucleotide/CD73) involved in the conversion of ATP into ADP, ADP into AMP, and AMP into adenosine, thus disrupting the redox homeostasis of the cell. This is because adenosine, via the purinergic P1 receptors, increases the activity of intracellular antioxidant systems, thereby decreasing the production of ROS. Once the activity of these ecto-enzymes is inhibited, the activation of adenosine-controlled antioxidant systems is prevented [39].
In AD, once Aβ-plates accumulate, ATP release increases in the extracellular space that activates the P2X7 receptors. In the CNS, this translates into an increase in the release of cytokines and chemokines and a decrease in α-secretase (proteolytic enzymes that cleave the amyloid precursor). However, the stimulation of P2Y2 receptors has the opposite effect, as it stimulates α-secretase activity with neuroprotective effects. On the other hand, stimulation of the P2Y13 receptors by ADP increases the activity of heme oxygenase-1 (HO-1), an enzyme involved in cytoprotection [47]. The role of P2Y13 receptors in the reduction of OS generated by H2O2 has also been demonstrated on N2A neuroblastoma cells, and it has been observed that the agonists of these receptors activate HO-1. The HO-1 activation is, however, dependent on the presence of the transcription factor Nrf2 [48].
Based on the above, it can be concluded that the harmful effects following the activation of P2Y7 receptors are counteracted by the activation of P2Y2 receptors, P2Y13 receptors, and A2A receptors.
In the pathogenesis of Parkinson’s disease (PD), on the other hand, P2Y6 receptors appear to be involved, and their stimulation causes an increase in cytokine production. This has been demonstrated in vitro on neuronal SH-SY5Y cells (cell cultures used in the study of the molecular and cellular mechanisms of toxic substances contributing to the development of PD), whereby exposure to 1-methyl-4-phenylpyridinium (MPP+), a compound with toxicity to dopaminergic neurons, increases the density of P2Y6 whereas the administration of a receptor agonist enhances its toxicity. At the same time, in the case of the knockdown of P2Y6R cells or following the use of an antagonist of these receptors, the harmful effects of MPP+ are reduced, which is demonstrated by a decrease in the production of ROS [49].
The role of purinergic receptors in the pathogenesis of Alzheimer’s and Parkinson’s diseases, as well as the interrelationships proposed in the literature between purinergic signaling pathways and OS, are schematically presented in Figure 3.
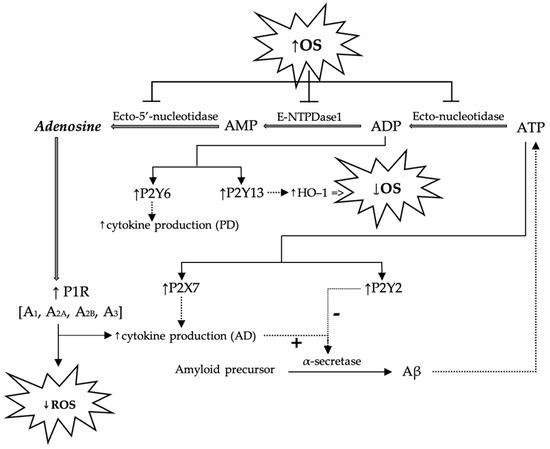
Figure 3.
Schematic presentation of possible mechanisms by which adenosine and purine nucleotides (AMP, ADP, ATP) modulate ROS generation and possible implications in the pathogenesis of Alzheimer’s and Parkinson’s diseases. P1R—purinergic receptor 1; OS—oxidative stress; ROS—reactive oxygen species; Aβ—beta-amyloid; AD—Alzheimer’s disease; PD—Parkinson’s disease.
The role of P2X7 receptors in the modulation of autophagy and inflammation has also been studied in a nonalcoholic steatohepatitis (NASH) model, the authors noting that P2X7 receptor-deleted mice were protected in developing liver inflammation and fibrosis [47].
On the other hand, in vitro and in vivo studies have shown that caffeine potentiates SOD2 activity by inhibiting its inactivation by acetylation. This is possible due to its affinity for sirtuin 3 (SIRT3), a mitochondrial nicotinamide adenine dinucleotide (NAD+)-dependent on acetylase, involved in the deacetylation process of two lysine residues (lysine 68 and 122) of SOD. The binding of caffeine to SIRT3 increases the affinity of deacetylase for the substrate with the increased antioxidant capacity of SOD [50].
The destruction of cholinergic neurons in Alzheimer’s disease and dopaminergic neurons in Parkinson’s disease has been correlated with high levels of ROS and neuroinflammation. Caffeine has often been proposed as an antioxidant agent with beneficial effects in the management of neurodegenerative diseases. Among the proposed mechanisms is the enhancement of the Nrf2 effect and its associated genes and the blocking of A2A receptors [46]. Additionally, in an animal model at menopause (ovariectomized female rats), caffeine has shown a beneficial effect in reducing the anxiety of this period, with the effects demonstrated by a behavioral study (elevated plus maze test). In this study, the effectiveness of caffeine was also explained by its ability to reduce lipid peroxidation and improve the ability of antioxidant systems in the brain to reduce the level of ROS [51].
The suggested mechanisms with which caffeine has neuroprotective effects (e.g., AD and PD) are reshown in Figure 4.
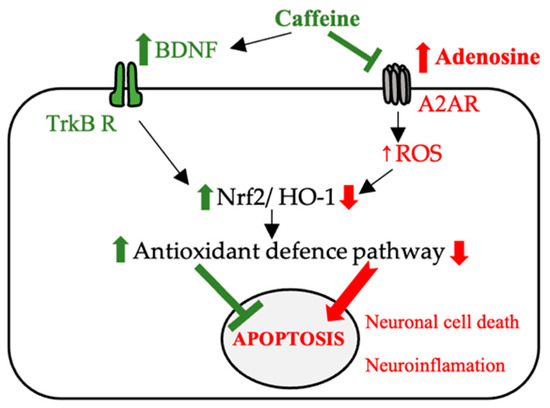
Figure 4.
The possible mechanisms involved in the neuroprotective effect of caffeine (BDNF—brain-derived neurotrophic factor, TrKB R—tropomyosin receptor kinase B, Nrf2—nuclear erythroid 2-related factor, A2AR—adenosine receptor, HO-1—heme oxygenase-1, ROS—reactive oxygen species).
In preclinical studies, caffeine has demonstrated a protective effect against the occurrence of neural damage produced after administration of d-galactose [52], cadmium (Cd) [53], lipopolysaccharides (LPS) [54], or exposure to hyperoxia [55]. The doses of pure caffeine used in these studies were 3 mg/kg/day ip (against d-galactose injuries) [52], 30 mg/kg/day ip (against Cd and LPS injuries) [53,54], and 10 mg/kg/day ip (against hyperoxia injuries) [55]. If the harmful effects of exposure to d-galactose, Cd, or LPS have been studied in adult animals, the animal model of hyperoxia has been used to assess the neuroprotective effects of caffeine at critical periods of brain development in 6-day-old rats. The brain of rats at six days post-partum corresponds, from the point of view of development to that of the human fetus in the 28–32 weeks of gestation, and this animal model can be used to evaluate the sequelae of prematurity triggered by OS and free radical-mediated tissue damage [55].
The neuroprotective effect of caffeine after exposure to neurotoxic compounds has been demonstrated in numerous in-vivo preclinical studies. The results are summarized in Table 1.

Table 1.
The proposed neuroprotective mechanisms of caffeine after in-vivo animal exposure to neurotoxic compounds.
In patients with Parkinson‘s disease, consumption of caffeine for 6 weeks in doses of at least 200 mg/day was associated with an improvement in motor symptoms, and a slowing of the progression of dyskinesia was observed [60,61]. Based on these observations, the benefits of caffeine were evaluated in a placebo-controlled phase 3 trial, with no improvement in motor symptoms observed after 6 months of daily consumption of 400 mg of caffeine [62]. Therefore, even if preclinical studies have shown a neuroprotective effect of caffeine in PD patients, the results of clinical studies are controversial, the results being dependent on the stage of the disease, chronic drug treatment, duration of treatment, and dose of caffeine.
However, epidemiological studies and meta-analyses reinforce the idea of a direct correlation between caffeine/coffee consumption and decreased risk/incidence of neurodegenerative diseases, as shown in Table 2.
Table 2.
The association of caffeine/coffee intake with the risk of neurodegenerative disorders.
- The importance of purinergic receptors for liver function
In the liver, adenosine is released either as a result of hypoxia or as a result of exposure to hepatotoxic compounds. The occurrence of liver fibrosis following chronic alcohol consumption has been explained by increased adenosine levels and the consequent stimulation of A2A receptors on the surface of hepatic stellate cells [69]. This has been correlated with the increased expression of transforming growth factor beta (TGFβ) and connective tissue growth factor (CTGF), which contribute to fibroblast proliferation, stimulation of collagen, and extracellular matrix synthesis [70], while purinergic receptor antagonists have been shown to have an effect on reducing these consequences [69], the proposed mechanisms include both the decrease in inflammatory biomarkers and oxidative stress [71]. The mechanism is shown in Figure 5.
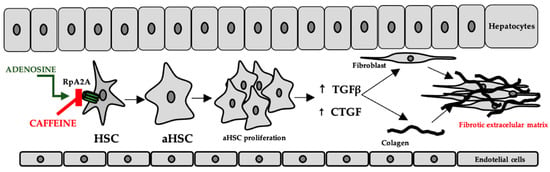
Figure 5.
The role of adenosine and caffeine in the occurrence/prevention of liver fibrosis (HSC—hepatic stellate cells, aHSC—activated hepatic stellate cells, TGFβ—transforming growth factor beta, CTGF—connective tissue growth factor).
Preclinical studies conducted on rats showed that the administration of caffeine in doses of 30 mg/kg/day and 100 mg/kg/day has beneficial effects on oxidative stress leading to a decrease in MDA values (in a dose-dependent manner) and advanced oxidation protein products (AOPP). The decrease in the AOPP level reflects the ability of cells to protect themselves against OS injury [72]. Caffeine has also been shown to be effective in improving biological markers in models of liver damage. Thus, administration of 37.5 mg/kg per day significantly reduced plasma levels of tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and interleukin-6 (IL-6), decreased malondialdehyde (MDA) and increased GSH and hepatic glutathione peroxidase (GPx) in male rats with thioacetamide-induced liver disease. In addition to improving biochemical markers, caffeine has also improved the histological and functional appearance of the liver [71]. The benefits of caffeine consumption in reducing liver bone were also evaluated in an animal model of nonalcoholic fatty liver disease induced by a high-fat diet. In this case, animals treated with caffeine at 20 or 30 mg/kg for 8 weeks had lower plasma levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST) and bilirubin, and increased the level of low albumin. The authors also assessed how caffeine interferes with hepatic lipid metabolism by observing a decrease in hepatic mRNA expression of fatty acid synthase and acetyl-CoA carboxylase along with an increase in hepatic carnitine palmitoyltransferase 1 (CPT1) and proliferation-activated receptor α, which suggested that the administration of caffeine regulates hepatic de novo lipogenesis and β-oxidation [73].
- Caffeine, adenosine receptors, and sport
Caffeine is an ergogenic compound that was included on the list of prohibited substances by the World Anti-Doping Agency between 1984 and 2004, followed by its inclusion in the list of substances monitored exclusively during competitions. In fact, the consumption of caffeine by athletes has also increased after its removal from the list of prohibited substances in sports [74]. The ergogenic effects are primarily due to the antagonism of A1 receptors [75], which are dose dependent and also correlated with the increase in cAMP levels. It should be stated that the increase in cAMP is the consequence of both the blocking of A1 receptors (in low doses) and the inhibition of PDEs (in high doses) [8]. Caffeine stimulates lipolysis, following the increase in cAMP, increasing the availability of free fatty acids that are then oxidized for energy without significantly altering carbohydrate metabolism, although a glycogen-sparing effect has been observed [76].
In addition to its ergogenic effect, caffeine has been shown to have many other advantages in sports.
In an animal model, intense exercise led to increased levels of AST and creatine kinase (CK) as well as SOD and GPx activity in hepatocytes. Caffeine consumption has been shown to prevent the increase of AST, even though it does not influence the plasma CK level while decreasing the activity of oxidative enzymes [77]. It has also been suggested that moderate consumption of caffeine enhances the beneficial effects of low-intensity physical exercise, an effect demonstrated by a decrease in the level of inflammatory markers (IL-1β, IL-6, TNF-α, and interferon-gamma—INF-γ) [78].
In humans, an improvement in psychomotor state and oxidative stress markers (GPX, SOD) has been observed following the ingestion of 5 mg/kg caffeine in athletes participating in an endurance race after 26 h of sleep deprivation [79].
2.3.2. Caffeine and Glutamatergic Neurotransmission
The antagonism of purinergic receptors has much more complex consequences in the context of OS modulation, the literature describing the interrelationships between caffeine administration, glutamatergic neurotransmission, and its influence on the processes involved in neurodegeneration (generation of free radicals, neuroinflammation). Thus, studies in the literature directly link the administration of caffeine and the increased level of glutamate (Glu) [80]. Animal models using caffeine have shown that caffeine increases available Glu in the nucleus accumbens (NAc) and posterior hypothalamus, to some extent, by blocking A1 receptors [80,81,82]. Because caffeine exhibits the same psychostimulant properties as other substances with an addictive effect, caffeine can have similar effects to those produced by Glu as a generator of excitotoxicity. Glu generates ROS by opening Ca2+ channels mediated by excessive stimulation of N-methyl-D-aspartate (NMDA) receptors, resulting in OS, oxidative damage, and apoptosis [83,84]. Another way in which caffeine is able to influence the extracellular level of Glu (by modulating glutamate/aspartate uptake) is represented by blocking A2 receptors [85]. Regarding the A2A receptor subtype, it is widely distributed in microglia and has the role of potentiating inflammatory effects [86]. Associated with this mechanism, we can also mention the negative influence on the recapture of excess Glu by glutamate transporter 1, but also the increase in the activity of phospho-extracellular signal-regulated kinases (pERK 1/2), which has an immediate effect, including the marked increase in the extracellular level of Glu, activation of microglia, and initiation of neuroinflammatory processes [86,87,88]. One interesting thing about the effects of adenosine is that it foregrounds inflammation. Thus, if in peripheral tissues adenosine mediates anti-inflammatory effects through A2A receptors, in the case of neuroinflammation, the activation of these receptors is responsible for neuronal degradation [89] through the influence exerted on glial cells, which involves the stimulation of cyclooxygenase type 2 (COX2), nitric oxide synthase (NOS), the release of prostaglandins, cytokines, and stimulation of Glu efflux from the presynaptic level [90,91,92].
In contrast, a study by Ning et al. demonstrated the protective effect of caffeine on inflammatory processes and the decrease in Glu levels in the case of acute injuries [93]. In PD investigation, multiple preclinical studies have claimed that prior exposure (for different periods of time, from one week to three weeks) of experimental animals (mice, rats) to caffeine reduced the loss of dopaminergic neuronal mass in the striatal area in a dose-dependent manner (30 mg/kg, 10 mg/kg 1 g/L in drinking water), thus conferring the neuroprotective effect [94,95]. This effect of caffeine is due to blocking A2A receptors and reducing Glu release as a result of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) administration. Thus, the binding and automatic blocking of this receptor subtype reduces the activity of protein kinase A (PKA) by decreasing the level of extracellular Ca2+ that can penetrate inside the cell, thus lowering the amount of Glu released, hence reducing neuroinflammation [87,96].
3. From Pure Caffeine to Coffee Products
All the above studies have assessed the antioxidant potential of pure caffeine, which raises the question of whether the active compound from food has the same properties. The phytochemical complexity of natural extracts can influence the effect of caffeine, but we must bear in mind that foods also contain sugars and sweeteners.
Many energy drinks combine taurine with caffeine. Although it is a compound that is endogenously synthesized in the body, antioxidant effects are only visible in higher concentrations. The additional intake could bring benefits correlated with the antioxidant action (stabilization of cell membranes, inhibition of enzymes involved in the generation of ROS, modulation of transcription factors such as Nrf2 and NF-kB) [97].
3.1. The Importance of the Phytocomplex in the Modulation of Oxidative Stress
The main natural caffeine sources are Coffeae semen, Theae folium, Colae semen, Mate folium, Guarana, and small quantities are found in Cacao semen. The content of caffeine in coffee beans is influenced by multiple factors (genetic, environmental, agricultural, etc.) but is usually between 1–1.2% in Coffea arabica and 1.7–2.5% in Coffea canephora [98]. Xanthine alkaloids are usually found in herbal drugs complexed with different compounds such as tannins or chlorogenic acid (CGA) and caffeine chlorogenate. These polyphenolic compounds have high antioxidant capacities, and, overall, they greatly influence the systemic effects of the total extracts/beverages. Of the compounds that are extracted together with caffeine from plant products, the most important are likely catechin derivatives found in green tea. Catechins are a group of flavan-3-ols with multiple phenoxy groups, a chemical characteristic that enhances its antioxidant potential compared to other flavonoids. The most important catechin found in green tea is (−)-epigallocatechin gallate, but small quantities of (+) catechin, (−)-epicatechin, gallocatechin, gallocatechin gallate, and epicatechin gallate have been identified. In green coffee seeds, the amount of CGAs varies significantly between different Coffea species, reaching almost 15% in Coffea canephora [99]. CGA content in coffee is closely correlated with caffeine content, and the major CGA encountered is 5-O-caffeoquinolinic acid.
Often, in weight loss supplements or those intended for athletes, the actual caffeine content is higher than that stated on the label (besides the declared pure caffeine, the extracts of tea or green coffee are highlighted separately, and the total alkaloid content is higher) [100].
The antioxidant effect of CGAs has been assessed in both in vitro and in vivo studies.
For in vitro studies, CGAs have been shown to have the ability to trap free radicals [101,102,103,104] and to decrease MDA levels [105] in a dose-dependent manner, with consumption being likely to have beneficial effects for the body, including the progression of neurodegenerative diseases induced by OS. However, in addition to the direct action of neutralizing free radicals, the antioxidant effect of CGA can also be explained by the fact that it activates Nrf2, thus stimulating the activity of endogenous antioxidant enzymes [104]. In this case, as well, studies have been conducted on pure substances. However, a study assessed the ability of coffee extracts to inhibit the aggregation of pathogenic Aβ, tau fibrillization, and tau aggregation, noting that concentrated extracts, respectively dark roast coffee extract, had a significantly higher effect compared to light roast coffee extract and decaffeinated dark roast coffee, in the case of the last two types of extracts the inhibition potency of the oligomerization of Aβ plates being almost identical. In addition, pure caffeine did not have the ability to inhibit the oligomerization process. Of the compounds studied by Mancini et al. that had a favorable effect in inhibiting the aggregation of pathogenic Aβ was quercetin, while phenylidane had a beneficial effect in reducing the aggregation of both tau protein and Aβ. Chlorogenic acid did not influence these processes. Moreover, in high concentrations, caffeic acid and quercetin stimulated the aggregation of α-synuclein, a protein with implications for the pathogenesis of PD [106]. Similar results were obtained by Baeza et al., who demonstrated that pure caffeine, unlike CGAs, does not provide significant protection against OS [107].
Unfortunately, the results obtained in vitro, although they may provide information on the effect of certain substances, must be confirmed in vivo. If we refer to CGA and caffeic acid, although they are absorbed from the digestive tract, they do not cross the blood-brain barrier (BBB) [108]; therefore, the neuroprotective action of coffee extracts cannot be attributed to these compounds. Since the antioxidant action of CGA should not be neglected despite its low bioavailability, attempts have been made to obtain a CGA-loaded liposome to increase its bioavailability and antioxidant capacity in vivo [109]. Although this formulation improves the transmembrane transfer of the substance, it is not possible to benefit from such complexes in food.
The beneficial effects of caffeine explained on the basis of the antioxidant properties of the phytocomplex, have also been demonstrated in patients with hepatocellular carcinoma, with a reduced risk (in most studies) in the case of coffee-consuming patients [110,111].
Important amounts of caffeine can be found in cocoa and cocoa products. As recently reported, commercial cocoa contains, on average, 0.21% caffeine, sweet chocolate 0.07%, and milk chocolate 0.02% caffeine [112] The consumption of these products has also been correlated with beneficial effects on health, explained by a capacity to combat the effects of OS. Yet again, this is a case of a complex composition of these extracts containing increased amounts of polyphenols as well as theobromine, the main compound with a xanthine structure. Since the antioxidant capacity of products depends on the concentration of these active compounds, Belščak et al. demonstrated that the highest antioxidant protection is attributed to cocoa products containing the highest amount of solid cocoa (e.g., dark chocolate) [113].
The polyphenols contained in cocoa and green tea, especially quercetin, have a well-documented antioxidant action [114]. Even though in vitro studies have shown that quercetin crosses the BBB [115], in vivo studies in rats and pigs have shown that both quercetin and its metabolites are distributed in most internal organs, including the brain (even if in a smaller amount), provided that the animals benefit from a diet high in quercetin for a long period of time; otherwise, they are distributed only in the kidneys and liver [116]. As in the case of CGAs, the antioxidant action of quercetin is attributed to the ability to neutralize ROS, respectively RNS [117], but also by activating the Nrf2-pathway [118,119].
3.2. Influence of Taste Regulators on Oxidative Stress (Sugar and Sweeteners)
For understandable health reasons, the World Health Organization (WHO) recommends that the amount of free sugar intake is below the limit of 5–10% of the total energy consumed in a day [120]. This reduction in carbohydrate consumption is based on the desire to limit the conditions associated with metabolic syndrome (type 2 diabetes, endocrine diseases, ischemic heart disease, coronary heart disease, myocardial infarction) [121,122,123]. In addition, a study by Brown et al. states that each extra-sweetened drink negatively influences blood pressure values, and thus, the systolic arterial pressure increases by 1.6 mmHg, and the diastolic increases by 0.8 mmHg [124].
Since food products contain sugar or sweeteners, the dilemma is whether they might generate ROS and interfere with the antioxidant action of caffeine or phytocomplex.
First, an increase in glucose metabolism results in the production of ROS via mitochondrial glucose oxidation, with the formation of and H2O2. Moreover, it has been observed that in the case of hyperglycemic status, the mitochondrial fission process correlates with the increase in the levels of ROS of mitochondrial origin [125]. Another pathway from which ROS results is represented by the nicotinamide adenindinucleotid phosphate oxidase system (NADPH-oxidase), which can be found in the vascular endothelium, smooth muscle cells, cardiomyocytes, and the immune system (neutrophils and macrophages). Thus, the hyperglycemic status favors the activation of NADPH-oxidase and the generation of ROS [126,127,128].
At the same time, constantly increased glucose levels promote the formation of advanced glycation end products (AGEs). These AGEs arise as a result of a non-enzymatic reaction between sugars and amino groups on proteins, lipids, or nucleic acids. These are compounds of interest due to their pathological potential and ability to generate ROS [129]. Three mechanisms for the formation of AGEs are currently known.
The first described mechanism is the Millard reaction in which the aldehyde group on the glucose attaches to the free amino group on the proteins with the formation of a Schiff base, which, after rearrangement, leads to a stable Amadori compound.
The second mechanism consists of the self-oxidation of glucose along with lipid peroxidation, which leads to the formation of dicarbonyl derivatives. These derivatives (glyoxal, methylglyoxal, and 3-deoxyglucosone) ultimately cause the formation of AGEs [130].
The third mechanism is the polyol pathway. This last mechanism involves the reduction of glucose by the action of aldose-reductase to sorbitol, which is further converted to fructose by the action of sorbitol dehydrogenase. Subsequently, fructose-3-phosphate is transformed into α-oxaldehyde, a compound that has the ability to lead to the formation of AGEs [131].
Once formed, these AGEs can react with the receptor for advanced glycation end products (RAGE), leading to the activation of the nuclear factor kappa-B (NF-κβ). Once activated, NF-κβ stimulates the transcription of cytokines (IL-1, IL-6, IL-8, and TNF-α), but also of NADPH-oxidase, all of which favor the production of ROS. The third mechanism is the polyol pathway. This last mechanism involves the reduction of glucose by the action of aldose-reductase to sorbitol, which is further converted to fructose by the action of sorbitol dehydrogenase. Subsequently, fructose-3-phosphate is transformed into α-oxaldehyde, a compound that has the ability to lead to the formation of AGEs [132,133,134,135]. Additionally, the level of AGEs correlates with the level of high-sensitivity C-reactive protein (hs-CRP), which is known to stimulate the production of ROS by neutrophils [136]. At the same time, another ROS-generating mechanism is suggested, this time via insulin, released from the massive amounts of ingested glucose. Insulin activates the type 4 isoform of NADPH-oxidase through which H2O2 is formed [137,138].
Aspartame is included in this category of sweeteners. It is added to various foods, sweets, beverages, oral hygiene products (chewing gum, breath mints), and even in pharmaceutical products as a result of their advantageous hypocaloric properties [139]. The main metabolites of aspartame are represented by approximately 50% phenylalanine, 40% aspartic acid, and 10% methanol [140]. Regarding the consumption of products containing aspartame, various neurological effects have been reported, including effects such as headaches, insomnia, and even seizures, along with changes in the level of catecholamines. In animal studies, the use of high doses of aspartame was also accompanied by neurochemical changes, with the toxicity being attributed to its pro-oxidant effect. Thus, the ROS generated by this sweetener induced an imbalance between pro-apoptotic (Bax) and anti-apoptotic markers (Bcl-2) in favor of pro-apoptotic ones [141]. During metabolism, aspartame releases methanol, which is then subjected to metabolism via three enzyme systems: alcohol dehydrogenase, catalase, and the microsomal oxidizing systems. The latter is considered directly responsible for the generation of ROS [142,143]. Methanol is also oxidized to formaldehyde which then transforms into formic acid. In addition, a close connection between methanol and corticosteroid levels is observed, as well as the presence of mitochondrial lesions, hence the increased production of ROS in conjunction with the alteration of the antioxidant mechanisms [144].
A decrease in GSH was also observed, which is correlated with increased levels of MDA generated from lipid peroxidation. This decrease in GSH is most likely due to methanol metabolites. In fact, multiple studies have demonstrated this oxidative damage, both at the hepatocellular level and in the kidneys, but, most likely, there are oxidative lesions at the central level as well [142,145,146,147,148,149]. In the same manner, a reduction in protein thiols was observed, which resulted from the oxidation of thiolic groups (-SH), which was the result of the decrease in GSH [142,145]. Another important effect to consider is that of the oxidation of -SH groups. There is a risk of a loss of ionic balance, given that ATP-ases are linked to the membrane by these groups and are susceptible to the action of ROS, being recognized as a phenomenon encountered in cytotoxicity [150,151].
Similar data in direct connection with OS were also observed in the case of other sweeteners such as saccharin, sucralose, and cyclamate, with a similar effect on the level of peroxidized lipids, GSH, but also on 8-hydroxy-20-deoxyguanosine (8-OHdG), without describing a clear mechanism of ROS generation [152,153,154,155].
However, the existence of sweetener alternatives without caloric value and with antioxidant properties should also be mentioned, such as Steviol glycosides or Siraitia grosvenorii fruit extract that have been demonstrated in the literature [156,157,158,159,160].
In conclusion, it is considered that the acceleration of the oxidative processes is due to the methanol obtained from aspartame, and the most important matter is that the most affected are diabetic patients, who have differently affected functions, including the antioxidant capacity.
4. Conclusions
The effects of caffeine on the CNS and the body systems are extremely complex in usual doses, explained by the antagonism of adenosine receptors. The blockade of these receptors has been associated with the potential antioxidant action of caffeine, but many of the effects may be the consequence of influencing intracellular transduction mechanisms, a certain effect based on only one of the two mechanisms being very difficult to explain. As far as the antioxidant action of pure caffeine is concerned, the properties of caffeine are intensively studied, and it has been shown to have beneficial effects on health (e.g., neurodegenerative disorders, liver damage, and physical activity). However, these results cannot be fully extrapolated in cases in which the intake of caffeine comes from products intended for human consumption, such as coffee, green/black tea, and cocoa products (sweets and beverages), for two reasons. First, these products also contain other compounds that contribute to the antioxidant capacity, in particular by the content of polyphenolic compounds, namely chlorogenic acids and quercetin. Of these, studies have shown that only quercetin, also in high doses (repeated exposure), reaches sufficient concentrations in the CNS to explain the neuroprotective effects, not only the peripheral antioxidant ones. Moreover, in the case of coffee, the degree of roasting of the beans is also important. Second, sugar and artificial sweeteners can antagonize the antioxidant action of caffeine due to their ability to induce ROS generation. However, natural sweeteners Steviol glycosides or Siraitia grosvenorii fruit extract also have antioxidant properties. It is, therefore, difficult to assess to what extent food products containing caffeine modulate the oxidative status of differences depending on the source of caffeine and the dose ingested, despite the fact that epidemiological studies report a strong link between the consumption of coffee/caffeine and lowering the risk/incidence of neurodegenerative diseases.
Author Contributions
Conceptualization, B.-E.Ő.; writing—original draft preparation, B.-E.Ő., G.J., A.P., and R.-E.Ș.; writing—review and editing, C.-E.V. and A.T.-V. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors would like to thank Adrian Năznean for the English language revision of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Caffeine|EFSA. Available online: https://www.efsa.europa.eu/en/topics/topic/caffeine (accessed on 21 July 2022).
- Ahluwalia, N.; Herrick, K. Caffeine Intake from Food and Beverage Sources and Trends among Children and Adolescents in the United States: Review of National Quantitative Studies from 1999 to 2011. Adv. Nutr. 2015, 6, 102–111. [Google Scholar] [CrossRef] [PubMed]
- dePaula, J.; Farah, A. Caffeine Consumption through Coffee: Content in the Beverage, Metabolism, Health Benefits and Risks. Beverages 2019, 5, 37. [Google Scholar] [CrossRef]
- Jîtcă, G.; Ősz, B.E.; Tero-Vescan, A.; Miklos, A.P.; Rusz, C.-M.; Bătrînu, M.-G.; Vari, C.E. Positive Aspects of Oxidative Stress at Different Levels of the Human Body: A Review. Antioxidants 2022, 11, 572. [Google Scholar] [CrossRef]
- Beckman, J.S.; Beckman, T.W.; Chen, J.; Marshall, P.A.; Freeman, B.A. Apparent Hydroxyl Radical Production by Peroxynitrite: Implications for Endothelial Injury from Nitric Oxide and Superoxide. Proc. Natl. Acad. Sci. USA 1990, 87, 1620–1624. [Google Scholar] [CrossRef]
- Kehrer, J.P. The Haber-Weiss Reaction and Mechanisms of Toxicity. Toxicology 2000, 149, 43–50. [Google Scholar] [CrossRef]
- Radi, R. Oxygen Radicals, Nitric Oxide, and Peroxynitrite: Redox Pathways in Molecular Medicine. Proc. Natl. Acad. Sci. USA 2018, 115, 5839–5848. [Google Scholar] [CrossRef]
- Rodak, K.; Kokot, I.; Kratz, E.M. Caffeine as a Factor Influencing the Functioning of the Human Body-Friend or Foe? Nutrients 2021, 13, 3088. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, K.A.; Balasubramanian, R.; Deflorian, F.; Gao, Z.-G. G Protein-Coupled Adenosine (P1) and P2Y Receptors: Ligand Design and Receptor Interactions. Purinergic Signal 2012, 8, 419–436. [Google Scholar] [CrossRef]
- Mustafa, S.J.; Morrison, R.R.; Teng, B.; Pelleg, A. Adenosine Receptors and the Heart: Role in Regulation of Coronary Blood Flow and Cardiac Electrophysiology. In Adenosine Receptors in Health and Disease. Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2009; pp. 161–188. [Google Scholar] [CrossRef]
- Vallon, V.; Osswald, H. Adenosine Receptors and the Kidney. In Adenosine Receptors in Health and Disease. Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2009; pp. 443–470. [Google Scholar] [CrossRef]
- Marchi, M.; Raiteri, L.; Risso, F.; Vallarino, A.; Bonfanti, A.; Monopoli, A.; Ongini, E.; Raiteri, M. Effects of Adenosine A1 and A2A Receptor Activation on the Evoked Release of Glutamate from Rat Cerebrocortical Synaptosomes. Br. J. Pharmacol. 2002, 136, 434–440. [Google Scholar] [CrossRef] [PubMed]
- Daly, J.W.; Shi, D.; Nikodijevic, O.; Jacobson, K.A. The Role of Adenosine Receptors in the Central Action of Caffeine. Pharmacopsychoecologia 1994, 7, 201–213. [Google Scholar]
- Fried, N.T.; Elliott, M.B.; Oshinsky, M.L. The Role of Adenosine Signaling in Headache: A Review. Brain Sci. 2017, 7, 30. [Google Scholar] [CrossRef] [PubMed]
- Vidyasagar, R.; Greyling, A.; Draijer, R.; Corfield, D.R.; Parkes, L.M. The Effect of Black Tea and Caffeine on Regional Cerebral Blood Flow Measured with Arterial Spin Labeling. J. Cereb. Blood Flow Metab. 2013, 33, 963–968. [Google Scholar] [CrossRef]
- Echeverri, D.; Montes, F.R.; Cabrera, M.; Galán, A.; Prieto, A. Caffeine’s Vascular Mechanisms of Action. Int. J. Vasc. Med. 2010, 2010, 834060. [Google Scholar] [CrossRef]
- Azevedo, M.F.; Faucz, F.R.; Bimpaki, E.; Horvath, A.; Levy, I.; de Alexandre, R.B.; Ahmad, F.; Manganiello, V.; Stratakis, C.A. Clinical and Molecular Genetics of the Phosphodiesterases (PDEs). Endocr. Rev. 2014, 35, 195–233. [Google Scholar] [CrossRef]
- Chirasani, V.R.; Pasek, D.A.; Meissner, G. Structural and Functional Interactions between the Ca2+-, ATP-, and Caffeine-Binding Sites of Skeletal Muscle Ryanodine Receptor (RyR1). J. Biol. Chem. 2021, 297, 101040. [Google Scholar] [CrossRef] [PubMed]
- Fredholm, B.B.; Yang, J.; Wang, Y. Low, but Not High, Dose Caffeine Is a Readily Available Probe for Adenosine Actions. Mol. Asp. Med. 2017, 55, 20–25. [Google Scholar] [CrossRef]
- Tero-Vescan, A.; Osz, B.E.; Maier, A.; Imre, S.; Ormenisan, A.; Hancu, V.; Vari, C.E. Concomitant Quantification of Caffeine, Cotinine and N-Methyl Uric Acid in Urine. Applications for Athlete Monitoring and Pharmacological Screening. Rev. Chim. 2017, 68, 2284–2288. [Google Scholar] [CrossRef]
- Kim, H.J.; Choi, M.S.; Rehman, S.U.; Ji, Y.S.; Yu, J.S.; Nakamura, K.; Yoo, H.H. Determination of Urinary Caffeine Metabolites as Biomarkers for Drug Metabolic Enzyme Activities. Nutrients 2019, 11, 1947. [Google Scholar] [CrossRef]
- Thorn, C.F.; Aklillu, E.; McDonagh, E.M.; Klein, T.E.; Altman, R.B. PharmGKB Summary: Caffeine Pathway. Pharm. Genom. 2012, 22, 389–395. [Google Scholar] [CrossRef]
- Relling, M.V.; Lin, J.; Ayers, G.D.; Evans, W.E. Racial and Gender Differences in N-Acetyltransferase, Xanthine Oxidase, and CYP1A2* Activities. Clin Pharm. Ther. 1992, 52, 643–658. [Google Scholar] [CrossRef]
- Kalow, W.; Tang, B.-K. Use of Caffeine Metabolite Ratios to Explore CYP1A2 and Xanthine Oxidase Activities. Clin Pharm. Ther. 1991, 50, 508–519. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Tsukui, D.; Kono, H. Uric Acid in Inflammation and the Pathogenesis of Atherosclerosis. Int. J. Mol. Sci. 2021, 22, 12394. [Google Scholar] [CrossRef] [PubMed]
- Roumeliotis, S.; Roumeliotis, A.; Dounousi, E.; Eleftheriadis, T.; Liakopoulos, V. Dietary Antioxidant Supplements and Uric Acid in Chronic Kidney Disease: A Review. Nutrients 2019, 11, 1911. [Google Scholar] [CrossRef]
- Nishino, T.; Okamoto, K.; Eger, B.T.; Pai, E.F.; Nishino, T. Mammalian Xanthine Oxidoreductase—Mechanism of Transition from Xanthine Dehydrogenase to Xanthine Oxidase. FEBS J. 2008, 275, 3278–3289. [Google Scholar] [CrossRef] [PubMed]
- Kuppusamy, P.; Zweier, J.L. Characterization of Free Radical Generation by Xanthine Oxidase. Evidence for Hydroxyl Radical Generation. J. Biol. Chem. 1989, 264, 9880–9884. [Google Scholar] [CrossRef]
- Desco, M.-C.; Asensi, M.; Márquez, R.; Martínez-Valls, J.; Vento, M.; Pallardó, F.V.; Sastre, J.; Viña, J. Xanthine Oxidase Is Involved in Free Radical Production in Type 1 Diabetes: Protection by Allopurinol. Diabetes 2002, 51, 1118–1124. [Google Scholar] [CrossRef] [PubMed]
- Chambers, D.E.; Parks, D.A.; Patterson, G.; Roy, R.; McCord, J.M.; Yoshida, S.; Parmley, L.F.; Downey, J.M. Xanthine Oxidase as a Source of Free Radical Damage in Myocardial Ischemia. J. Mol. Cell Cardiol. 1985, 17, 145–152. [Google Scholar] [CrossRef]
- Chen, M.-M.; Meng, L.-H. The Double Faced Role of Xanthine Oxidoreductase in Cancer. Acta Pharm. Sin 2022, 43, 1623–1632. [Google Scholar] [CrossRef]
- Browne, L.D.; Jaouimaa, F.-Z.; Walsh, C.; Perez-Ruiz, F.; Richette, P.; Burke, K.; Stack, A.G. Serum Uric Acid and Mortality Thresholds among Men and Women in the Irish Health System: A Cohort Study. Eur. J. Intern. Med. 2021, 84, 46–55. [Google Scholar] [CrossRef]
- Wu, D.; Chen, R.; Zhang, W.; Lai, X.; Sun, L.; Li, Q.; Zhang, Z.; Cao, J.; Wen, S.; Lai, Z.; et al. Tea and Its Components Reduce the Production of Uric Acid by Inhibiting Xanthine Oxidase. Food Nutr. Res. 2022, 66. [Google Scholar] [CrossRef]
- Peluso, I.; Teichner, A.; Manafikhi, H.; Palmery, M. Camellia Sinensis in Asymptomatic Hyperuricemia: A Meta-Analysis of Tea or Tea Extract Effects on Uric Acid Levels. Crit. Rev. Food Sci. Nutr. 2017, 57, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Park, K.Y.; Kim, H.J.; Ahn, H.S.; Kim, S.H.; Park, E.J.; Yim, S.-Y.; Jun, J.-B. Effects of Coffee Consumption on Serum Uric Acid: Systematic Review and Meta-Analysis. Semin. Arthritis Rheum. 2016, 45, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.; Park, P.S.; Chun, B.-Y.; Choi, B.Y.; Kim, M.K.; Shin, M.-H.; Lee, Y.-H.; Shin, D.H.; Kim, S.-K. The Effect of Coffee, Tea, and Caffeine Consumption on Serum Uric Acid and the Risk of Hyperuricemia in Korean Multi-Rural Communities Cohort. Rheumatol. Int. 2015, 35, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Nehlig, A. Interindividual Differences in Caffeine Metabolism and Factors Driving Caffeine Consumption. Pharm. Rev 2018, 70, 384–411. [Google Scholar] [CrossRef] [PubMed]
- Denden, S.; Bouden, B.; Haj Khelil, A.; Ben Chibani, J.; Hamdaoui, M.H. Gender and Ethnicity Modify the Association between the CYP1A2 Rs762551 Polymorphism and Habitual Coffee Intake: Evidence from a Meta-Analysis. Genet Mol. Res. 2016, 15, gmr.15027487. [Google Scholar] [CrossRef] [PubMed]
- Savio, L.E.B.; Leite-Aguiar, R.; Alves, V.S.; Coutinho-Silva, R.; Wyse, A.T.S. Purinergic Signaling in the Modulation of Redox Biology. Redox. Biol. 2021, 47, 102137. [Google Scholar] [CrossRef]
- Gupta, R.C. Reproductive and Developmental Toxicology; Academic Press: Cambridge, MA, USA, 2011; ISBN 978-0-12-382033-4. [Google Scholar]
- Saha, S.; Li, Y.; Anand-Srivastava, M.B. Reduced Levels of Cyclic AMP Contribute to the Enhanced Oxidative Stress in Vascular Smooth Muscle Cells from Spontaneously Hypertensive Rats. Can. J. Physiol. Pharm. 2008, 86, 190–198. [Google Scholar] [CrossRef]
- Zhang, J.; Feng, J.; Ma, D.; Wang, F.; Wang, Y.; Li, C.; Wang, X.; Yin, X.; Zhang, M.; Dagda, R.K.; et al. Neuroprotective Mitochondrial Remodeling by AKAP121/PKA Protects HT22 Cell from Glutamate-Induced Oxidative Stress. Mol. Neurobiol. 2019, 56, 5586–5607. [Google Scholar] [CrossRef]
- Cui, W.-Q.; Wang, S.-T.; Pan, D.; Chang, B.; Sang, L.-X. Caffeine and Its Main Targets of Colorectal Cancer. World J. Gastrointest. Oncol. 2020, 12, 149–172. [Google Scholar] [CrossRef]
- Rappaport, J.A.; Waldman, S.A. The Guanylate Cyclase C—CGMP Signaling Axis Opposes Intestinal Epithelial Injury and Neoplasia. Front. Oncol. 2018, 8, 299. [Google Scholar] [CrossRef]
- Leite, M.R.; Wilhelm, E.A.; Jesse, C.R.; Brandão, R.; Nogueira, C.W. Protective Effect of Caffeine and a Selective A2A Receptor Antagonist on Impairment of Memory and Oxidative Stress of Aged Rats. Exp. Gerontol. 2011, 46, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Ikram, M.; Park, T.J.; Ali, T.; Kim, M.O. Antioxidant and Neuroprotective Effects of Caffeine against Alzheimer’s and Parkinson’s Disease: Insight into the Role of Nrf-2 and A2AR Signaling. Antioxidants 2020, 9, 902. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Seth, R.K.; Kumar, A.; Kadiiska, M.B.; Michelotti, G.; Diehl, A.M.; Chatterjee, S. Purinergic Receptor X7 Is a Key Modulator of Metabolic Oxidative Stress-Mediated Autophagy and Inflammation in Experimental Nonalcoholic Steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G950–G963. [Google Scholar] [CrossRef]
- Espada, S.; Ortega, F.; Molina-Jijón, E.; Rojo, A.I.; Pérez-Sen, R.; Pedraza-Chaverri, J.; Miras-Portugal, M.T.; Cuadrado, A. The Purinergic P2Y13 Receptor Activates the Nrf2/HO-1 Axis and Protects against Oxidative Stress-Induced Neuronal Death. Free Radic. Biol. Med. 2010, 49, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Xu, S.; Yang, X.; Xiao, Q. Purinergic Receptor P2Y6 Contributes to 1-Methyl-4-Phenylpyridinium-Induced Oxidative Stress and Cell Death in Neuronal SH-SY5Y Cells. J. Neurosci. Res. 2018, 96, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Gan, C.; Gao, Z.; Huang, Y.; Wu, S.; Zhang, D.; Wang, X.; Sheng, J. Caffeine Targets SIRT3 to Enhance SOD2 Activity in Mitochondria. Front. Cell Dev. Biol. 2020, 8, 822. [Google Scholar] [CrossRef]
- Caravan, I.; Sevastre Berghian, A.; Moldovan, R.; Decea, N.; Orasan, R.; Filip, G.A. Modulatory Effects of Caffeine on Oxidative Stress and Anxiety-like Behavior in Ovariectomized Rats. Can. J. Physiol. Pharmacol. 2016, 94, 961–972. [Google Scholar] [CrossRef]
- Ullah, F.; Ali, T.; Ullah, N.; Kim, M.O. Caffeine Prevents D-Galactose-Induced Cognitive Deficits, Oxidative Stress, Neuroinflammation and Neurodegeneration in the Adult Rat Brain. Neurochem. Int. 2015, 90, 114–124. [Google Scholar] [CrossRef]
- Khan, A.; Ikram, M.; Muhammad, T.; Park, J.; Kim, M.O. Caffeine Modulates Cadmium-Induced Oxidative Stress, Neuroinflammation, and Cognitive Impairments by Regulating Nrf-2/HO-1 In Vivo and In Vitro. J. Clin. Med. 2019, 8, 680. [Google Scholar] [CrossRef]
- Badshah, H.; Ikram, M.; Ali, W.; Ahmad, S.; Hahm, J.R.; Kim, M.O. Caffeine May Abrogate LPS-Induced Oxidative Stress and Neuroinflammation by Regulating Nrf2/TLR4 in Adult Mouse Brains. Biomolecules 2019, 9, 719. [Google Scholar] [CrossRef]
- Endesfelder, S.; Weichelt, U.; Strauß, E.; Schlör, A.; Sifringer, M.; Scheuer, T.; Bührer, C.; Schmitz, T. Neuroprotection by Caffeine in Hyperoxia-Induced Neonatal Brain Injury. Int. J. Mol. Sci. 2017, 18, 187. [Google Scholar] [CrossRef] [PubMed]
- Ghoneim, F.M.; Khalaf, H.A.; Elsamanoudy, A.Z.; Abo El-khair, S.M.; Helaly, A.M.; Mahmoud, E.-H.M.; Elshafey, S.H. Protective Effect of Chronic Caffeine Intake on Gene Expression of Brain Derived Neurotrophic Factor Signaling and the Immunoreactivity of Glial Fibrillary Acidic Protein and Ki-67 in Alzheimer’s Disease. Int. J. Clin. Exp. Pathol. 2015, 8, 7710–7728. [Google Scholar] [PubMed]
- Brothers, H.M.; Marchalant, Y.; Wenk, G.L. Caffeine Attenuates Lipopolysaccharide-Induced Neuroinflammation. Neurosci. Lett. 2010, 480, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Bagga, P.; Chugani, A.N.; Patel, A.B. Neuroprotective Effects of Caffeine in MPTP Model of Parkinson’s Disease: A (13)C NMR Study. Neurochem. Int. 2016, 92, 25–34. [Google Scholar] [CrossRef]
- Machado-Filho, J.A.; Correia, A.O.; Montenegro, A.B.A.; Nobre, M.E.P.; Cerqueira, G.S.; Neves, K.R.T.; Naffah-Mazzacoratti, M.d.G.; Cavalheiro, E.A.; de Castro Brito, G.A.; de Barros Viana, G.S. Caffeine Neuroprotective Effects on 6-OHDA-Lesioned Rats Are Mediated by Several Factors, Including pro-Inflammatory Cytokines and Histone Deacetylase Inhibitions. Behav. Brain Res. 2014, 264, 116–125. [Google Scholar] [CrossRef]
- Altman, R.D.; Lang, A.E.; Postuma, R.B. Caffeine in Parkinson’s Disease: A Pilot Open-Label, Dose-Escalation Study. Mov. Disord. 2011, 26, 2427–2431. [Google Scholar] [CrossRef]
- Postuma, R.B.; Lang, A.E.; Munhoz, R.P.; Charland, K.; Pelletier, A.; Moscovich, M.; Filla, L.; Zanatta, D.; Rios Romenets, S.; Altman, R.; et al. Caffeine for Treatment of Parkinson Disease. Neurology 2012, 79, 651–658. [Google Scholar] [CrossRef]
- Postuma, R.B.; Anang, J.; Pelletier, A.; Joseph, L.; Moscovich, M.; Grimes, D.; Furtado, S.; Munhoz, R.P.; Appel-Cresswell, S.; Moro, A.; et al. Caffeine as Symptomatic Treatment for Parkinson Disease (Café-PD). Neurology 2017, 89, 1795–1803. [Google Scholar] [CrossRef]
- Eskelinen, M.H.; Ngandu, T.; Tuomilehto, J.; Soininen, H.; Kivipelto, M. Midlife Coffee and Tea Drinking and the Risk of Late-Life Dementia: A Population-Based CAIDE Study. J. Alzheimers Dis. 2009, 16, 85–91. [Google Scholar] [CrossRef]
- Ross, G.W.; Abbott, R.D.; Petrovitch, H.; Morens, D.M.; Grandinetti, A.; Tung, K.-H.; Tanner, C.M.; Masaki, K.H.; Blanchette, P.L.; Curb, J.D.; et al. Association of Coffee and Caffeine Intake with the Risk of Parkinson Disease. JAMA 2000, 283, 2674–2679. [Google Scholar] [CrossRef]
- Sääksjärvi, K.; Knekt, P.; Rissanen, H.; Laaksonen, M.A.; Reunanen, A.; Männistö, S. Prospective Study of Coffee Consumption and Risk of Parkinson’s Disease. Eur. J. Clin. Nutr. 2008, 62, 908–915. [Google Scholar] [CrossRef] [PubMed]
- Ascherio, A.; Zhang, S.M.; Hernán, M.A.; Kawachi, I.; Colditz, G.A.; Speizer, F.E.; Willett, W.C. Prospective Study of Caffeine Consumption and Risk of Parkinson’s Disease in Men and Women. Ann. Neurol. 2001, 50, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Costa, J.; Lunet, N.; Santos, C.; Santos, J.; Vaz-Carneiro, A. Caffeine Exposure and the Risk of Parkinson’s Disease: A Systematic Review and Meta-Analysis of Observational Studiess. J. Alzheimers Dis. 2010, 20, S221–S238. [Google Scholar] [CrossRef]
- Hong, C.T.; Chan, L.; Bai, C.-H. The Effect of Caffeine on the Risk and Progression of Parkinson’s Disease: A Meta-Analysis. Nutrients 2020, 12, 1860. [Google Scholar] [CrossRef] [PubMed]
- Cronstein, B.N. Caffeine, A Drug for All Seasons. J. Hepatol. 2010, 53, 207–208. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, N.; Chu, H.Y.; Yu, Y.; Zhang, Z.-K.; Zhang, G.; Zhang, B.-T. Connective Tissue Growth Factor: From Molecular Understandings to Drug Discovery. Front. Cell Dev. Biol. 2020, 8, 593269. [Google Scholar] [CrossRef] [PubMed]
- Amer, M.G.; Mazen, N.F.; Mohamed, A.M. Caffeine Intake Decreases Oxidative Stress and Inflammatory Biomarkers in Experimental Liver Diseases Induced by Thioacetamide: Biochemical and Histological Study. Int. J. Immunopathol. Pharm. 2017, 30, 13–24. [Google Scholar] [CrossRef]
- Paşaoğlu, H.; DemiR, F.E.O.; Yilmaz-DemiRtaş, C.; Hussein, A.; Paşaoğlu, Ö.T. The Effect of Caffeine on Oxidative Stress in Liver and Heart Tissues of Rats. Turk. J. Med. Sci. 2011, 41, 665–672. [Google Scholar] [CrossRef]
- Helal, M.; Ayoub, S.; Elkashefand, W.; Ibrahim, T. Caffeine Affects HFD-Induced Hepatic Steatosis by Multifactorial Intervention. Hum. Exp. Toxicol. 2018, 37, 983–990. [Google Scholar] [CrossRef]
- Aguilar-Navarro, M.; Muñoz, G.; Salinero, J.J.; Muñoz-Guerra, J.; Fernández-Álvarez, M.; del Mar Plata, M.; Del Coso, J. Urine Caffeine Concentration in Doping Control Samples from 2004 to 2015. Nutrients 2019, 11, 286. [Google Scholar] [CrossRef]
- Aguiar, A.S.; Speck, A.E.; Canas, P.M.; Cunha, R.A. Neuronal Adenosine A2A Receptors Signal Ergogenic Effects of Caffeine. Sci. Rep. 2020, 10, 13414. [Google Scholar] [CrossRef] [PubMed]
- Graham, T.E. Caffeine and Exercise. Sports Med. 2001, 31, 785–807. [Google Scholar] [CrossRef] [PubMed]
- Barcelos, R.P.; Souza, M.A.; Amaral, G.P.; Stefanello, S.T.; Bresciani, G.; Fighera, M.R.; Soares, F.A.A.; Barbosa, N.V. Caffeine Supplementation Modulates Oxidative Stress Markers in the Liver of Trained Rats. Life Sci. 2014, 96, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Cechella, J.L.; Leite, M.R.; Dobrachinski, F.; da Rocha, J.T.; Carvalho, N.R.; Duarte, M.M.M.F.; Soares, F.A.A.; Bresciani, G.; Royes, L.F.F.; Zeni, G. Moderate Swimming Exercise and Caffeine Supplementation Reduce the Levels of Inflammatory Cytokines without Causing Oxidative Stress in Tissues of Middle-Aged Rats. Amino Acids 2014, 46, 1187–1195. [Google Scholar] [CrossRef] [PubMed]
- Khcharem, A.; Souissi, M.; Atheymen, R.; Souissi, W.; Sahnoun, Z. Effects of Caffeine Ingestion on Psychomotor State and Oxidative Stress Markers after an 8-Km Run Competition in Sleep-Deprived Recreational Runners. Biol. Rhythm Res. 2022, 53, 1334–1346. [Google Scholar] [CrossRef]
- John, J.; Kodama, T.; Siegel, J.M. Caffeine Promotes Glutamate and Histamine Release in the Posterior Hypothalamus. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 307, R704–R710. [Google Scholar] [CrossRef]
- Solinas, M.; Ferré, S.; You, Z.-B.; Karcz-Kubicha, M.; Popoli, P.; Goldberg, S.R. Caffeine Induces Dopamine and Glutamate Release in the Shell of the Nucleus Accumbens. J. Neurosci. 2002, 22, 6321–6324. [Google Scholar] [CrossRef]
- Owolabi, J.O.; Olatunji, S.Y.; Olanrewaju, A.J. Caffeine and Cannabis Effects on Vital Neurotransmitters and Enzymes in the Brain Tissue of Juvenile Experimental Rats. Ann. Neurosci. 2017, 24, 65–73. [Google Scholar] [CrossRef]
- Moghimi, E.; Solomon, J.A.; Gianforcaro, A.; Hamadeh, M.J. Dietary Vitamin D3 Restriction Exacerbates Disease Pathophysiology in the Spinal Cord of the G93A Mouse Model of Amyotrophic Lateral Sclerosis. PLoS ONE 2015, 10, e0126355. [Google Scholar] [CrossRef]
- Jîtcă, G.; Ősz, B.E.; Tero-Vescan, A.; Vari, C.E. Psychoactive Drugs—From Chemical Structure to Oxidative Stress Related to Dopaminergic Neurotransmission. A Review. Antioxidants 2021, 10, 381. [Google Scholar] [CrossRef]
- De Freitas, A.P.; Ferreira, D.D.P.; Fernandes, A.; Martins, R.S.; Borges-Martins, V.P.P.; Sathler, M.F.; dos-Santos-Pereira, M.; Paes-de-Carvalho, R.; Giestal-de-Araujo, E.; de Melo Reis, R.A.; et al. Caffeine Alters Glutamate—Aspartate Transporter Function and Expression in Rat Retina. Neuroscience 2016, 337, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, R.A.; Agha, A.M.; Abdel-Rahman, A.A.; Nassar, N.N. Role of Adenosine A2A Receptor in Cerebral Ischemia Reperfusion Injury: Signaling to Phosphorylated Extracellular Signal-Regulated Protein Kinase (PERK1/2). Neuroscience 2016, 314, 145–159. [Google Scholar] [CrossRef] [PubMed]
- Dall’Igna, O.P.; Porciúncula, L.O.; Souza, D.O.; Cunha, R.A.; Lara, D.R.; Dall’lgna, O.P. Neuroprotection by Caffeine and Adenosine A2A Receptor Blockade of Beta-Amyloid Neurotoxicity. Br. J. Pharm. 2003, 138, 1207–1209. [Google Scholar] [CrossRef] [PubMed]
- Pedata, F.; Pugliese, A.M.; Coppi, E.; Dettori, I.; Maraula, G.; Cellai, L.; Melani, A. Adenosine A2A Receptors Modulate Acute Injury and Neuroinflammation in Brain Ischemia. Mediat. Inflamm. 2014, 2014, 805198. [Google Scholar] [CrossRef] [PubMed]
- Cunha, R.A.; Chen, J.-F.; Sitkovsky, M.V. Opposite Modulation of Peripheral Inflammation and Neuroinflammation by Adenosine A2A Receptors. In Interaction between Neurons and Glia in Aging and Disease; Malva, J.O., Rego, A.C., Cunha, R.A., Oliveira, C.R., Eds.; Springer US: Boston, MA, USA, 2007; pp. 53–79. ISBN 978-0-387-70830-0. [Google Scholar]
- Ingwersen, J.; Wingerath, B.; Graf, J.; Lepka, K.; Hofrichter, M.; Schröter, F.; Wedekind, F.; Bauer, A.; Schrader, J.; Hartung, H.-P.; et al. Dual Roles of the Adenosine A2a Receptor in Autoimmune Neuroinflammation. J. Neuroinflamm. 2016, 13, 48. [Google Scholar] [CrossRef]
- Rajasundaram, S. Adenosine A2A Receptor Signaling in the Immunopathogenesis of Experimental Autoimmune Encephalomyelitis. Front Immunol. 2018, 9, 402. [Google Scholar] [CrossRef]
- Pinna, A.; Serra, M.; Marongiu, J.; Morelli, M. Pharmacological Interactions between Adenosine A2A Receptor Antagonists and Different Neurotransmitter Systems. Parkinsonism Relat. Disord. 2020, 80, S37–S44. [Google Scholar] [CrossRef]
- Ning, Y.-L.; Yang, N.; Chen, X.; Zhao, Z.-A.; Zhang, X.-Z.; Chen, X.-Y.; Li, P.; Zhao, Y.; Zhou, Y.-G. Chronic Caffeine Exposure Attenuates Blast-Induced Memory Deficit in Mice. Chin. J. Traumatol. 2015, 18, 204–211. [Google Scholar] [CrossRef]
- Xu, K.; Xu, Y.; Brown-Jermyn, D.; Chen, J.-F.; Ascherio, A.; Dluzen, D.E.; Schwarzschild, M.A. Estrogen Prevents Neuroprotection by Caffeine in the Mouse 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine Model of Parkinson’s Disease. J. Neurosci. 2006, 26, 535–541. [Google Scholar] [CrossRef]
- Sonsalla, P.K.; Wong, L.-Y.; Harris, S.L.; Richardson, J.R.; Khobahy, I.; Li, W.; Gadad, B.S.; German, D.C. Delayed Caffeine Treatment Prevents Nigral Dopamine Neuron Loss in a Progressive Rat Model of Parkinson’s Disease. Exp. Neurol. 2012, 234, 482–487. [Google Scholar] [CrossRef]
- Köfalvi, A.; Moreno, E.; Cordomí, A.; Cai, N.S.; Fernández-Dueñas, V.; Ferreira, S.G.; Guixà-González, R.; Sánchez-Soto, M.; Yano, H.; Casadó-Anguera, V.; et al. Control of glutamate release by complexes of adenosine and cannabinoid receptors. BMC Biol. 2020, 18, 9. [Google Scholar] [CrossRef] [PubMed]
- Surai, P.F.; Earle-Payne, K.; Kidd, M.T. Taurine as a Natural Antioxidant: From Direct Antioxidant Effects to Protective Action in Various Toxicological Models. Antioxidants 2021, 10, 1876. [Google Scholar] [CrossRef] [PubMed]
- Campa, C.; Doulbeau, S.; Dussert, S.; Hamon, S.; Noirot, M. Diversity in Bean Caffeine Content among Wild Coffea Species: Evidence of a Discontinuous Distribution. Food Chem. 2005, 91, 633–637. [Google Scholar] [CrossRef]
- Campa, C.; Doulbeau, S.; Dussert, S.; Hamon, S.; Noirot, M. Qualitative Relationship between Caffeine and Chlorogenic Acid Contents among Wild Coffea Species. Food Chem. 2005, 93, 135–139. [Google Scholar] [CrossRef]
- Tero-Vescan, A.; Vari, C.-E.; Imre, S.; Hosz, B.-E.; Filip, C.; Hancu, G. Comparative Analysis by HPLC-UV and Capillary Electrophoresis of Dietary Supplements for Weight Loss. Farmacia 2016, 64, 699–705. [Google Scholar]
- Zhang, L.-Y.; Cosma, G.; Gardner, H.; Vallyathan, V.; Castranova, V. Effect of Chlorogenic Acid on Hydroxyl Radical. Mol. Cell Biochem. 2003, 247, 205–210. [Google Scholar] [CrossRef]
- Wu, L. Effect of Chlorogenic Acid on Antioxidant Activity of Flos Lonicerae Extracts. J. Zhejiang Univ.-Sci. B 2007, 8, 673–679. [Google Scholar] [CrossRef]
- Bouayed, J.; Rammal, H.; Dicko, A.; Younos, C.; Soulimani, R. Chlorogenic Acid, a Polyphenol from Prunus Domestica (Mirabelle), with Coupled Anxiolytic and Antioxidant Effects. J. Neurol. Sci. 2007, 262, 77–84. [Google Scholar] [CrossRef]
- Yao, J.; Peng, S.; Xu, J.; Fang, J. Reversing ROS-Mediated Neurotoxicity by Chlorogenic Acid Involves Its Direct Antioxidant Activity and Activation of Nrf2-ARE Signaling Pathway. BioFactors 2019, 45, 616–626. [Google Scholar] [CrossRef]
- Oboh, G.; Agunloye, O.M.; Akinyemi, A.J.; Ademiluyi, A.O.; Adefegha, S.A. Comparative Study on the Inhibitory Effect of Caffeic and Chlorogenic Acids on Key Enzymes Linked to Alzheimer’s Disease and Some Pro-Oxidant Induced Oxidative Stress in Rats’ Brain-In Vitro. Neurochem. Res. 2013, 38, 413–419. [Google Scholar] [CrossRef]
- Mancini, R.S.; Wang, Y.; Weaver, D.F. Phenylindanes in Brewed Coffee Inhibit Amyloid-Beta and Tau Aggregation. Front. Neurosci. 2018, 12, 735. [Google Scholar] [CrossRef] [PubMed]
- Baeza, G.; Amigo-Benavent, M.; Sarriá, B.; Goya, L.; Mateos, R.; Bravo, L. Green Coffee Hydroxycinnamic Acids but Not Caffeine Protect Human HepG2 Cells against Oxidative Stress. Food Res. Int. 2014, 62, 1038–1046. [Google Scholar] [CrossRef]
- Lardeau, A.; Poquet, L. Phenolic Acid Metabolites Derived from Coffee Consumption Are Unlikely to Cross the Blood–Brain Barrier. J. Pharm. Biomed. Anal. 2013, 76, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Sun, C.; Yuan, Y.; Zhu, Y.; Wan, J.; Firempong, C.K.; Omari-Siaw, E.; Xu, Y.; Pu, Z.; Yu, J.; et al. Enhanced Oral Bioavailability and in Vivo Antioxidant Activity of Chlorogenic Acid via Liposomal Formulation. Int. J. Pharm. 2016, 501, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Pelizzaro, F.; Cardin, R.; Sartori, A.; Imondi, A.; Penzo, B.; Farinati, F. Coffee and Hepatocellular Carcinoma: Epidemiologic Evidence and Biologic Mechanisms. Hepatoma Res. 2021, 7, 29. [Google Scholar] [CrossRef]
- Bai, K.; Cai, Q.; Jiang, Y.; Lv, L. Coffee Consumption and Risk of Hepatocellular Carcinoma: A Meta-Analysis of Eleven Epidemiological Studies. Onco Targets Ther. 2016, 9, 4369–4375. [Google Scholar] [CrossRef]
- Zoumas, B.; Kreiser, W.; Martin, R. Theobromine and Caffeine Content of Chocolate Products. J. Food Sci. 2006, 45, 314–316. [Google Scholar] [CrossRef]
- Belščak, A.; Komes, D.; Horžić, D.; Ganić, K.K.; Karlović, D. Comparative Study of Commercially Available Cocoa Products in Terms of Their Bioactive Composition. Food Res. Int. 2009, 42, 707–716. [Google Scholar] [CrossRef]
- Zhang, H.; Tsao, R. Dietary Polyphenols, Oxidative Stress and Antioxidant and Anti-Inflammatory Effects. Curr. Opin. Food Sci. 2016, 8, 33–42. [Google Scholar] [CrossRef]
- Ishisaka, A.; Ichikawa, S.; Sakakibara, H.; Piskula, M.K.; Nakamura, T.; Kato, Y.; Ito, M.; Miyamoto, K.; Tsuji, A.; Kawai, Y.; et al. Accumulation of Orally Administered Quercetin in Brain Tissue and Its Antioxidative Effects in Rats. Free Radic. Biol. Med. 2011, 51, 1329–1336. [Google Scholar] [CrossRef]
- De Boer, V.C.J.; Dihal, A.A.; van der Woude, H.; Arts, I.C.W.; Wolffram, S.; Alink, G.M.; Rietjens, I.M.C.M.; Keijer, J.; Hollman, P.C.H. Tissue Distribution of Quercetin in Rats and Pigs. J. Nutr. 2005, 135, 1718–1725. [Google Scholar] [CrossRef] [PubMed]
- Boots, A.W.; Haenen, G.R.M.M.; Bast, A. Health Effects of Quercetin: From Antioxidant to Nutraceutical. Eur. J. Pharm. 2008, 585, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Arredondo, F.; Echeverry, C.; Abin-Carriquiry, J.A.; Blasina, F.; Antúnez, K.; Jones, D.P.; Go, Y.-M.; Liang, Y.-L.; Dajas, F. After Cellular Internalization, Quercetin Causes Nrf2 Nuclear Translocation, Increases Glutathione Levels, and Prevents Neuronal Death against an Oxidative Insult. Free Radic. Biol. Med. 2010, 49, 738–747. [Google Scholar] [CrossRef] [PubMed]
- Granado-Serrano, A.B.; Martín, M.A.; Bravo, L.; Goya, L.; Ramos, S. Quercetin Modulates Nrf2 and Glutathione-Related Defenses in HepG2 Cells: Involvement of P38. Chem. Biol. Interact. 2012, 195, 154–164. [Google Scholar] [CrossRef]
- Guideline: Sugars Intake for Adults and Children. Available online: https://www.who.int/publications-detail-redirect/9789241549028 (accessed on 13 September 2022).
- Qin, P.; Li, Q.; Zhao, Y.; Chen, Q.; Sun, X.; Liu, Y.; Li, H.; Wang, T.; Chen, X.; Zhou, Q.; et al. Sugar and Artificially Sweetened Beverages and Risk of Obesity, Type 2 Diabetes Mellitus, Hypertension, and All-Cause Mortality: A Dose-Response Meta-Analysis of Prospective Cohort Studies. Eur. J. Epidemiol. 2020, 35, 655–671. [Google Scholar] [CrossRef]
- De Koning, L.; Malik, V.S.; Kellogg, M.D.; Rimm, E.B.; Willett, W.C.; Hu, F.B. Sweetened Beverage Consumption, Incident Coronary Heart Disease, and Biomarkers of Risk in Men. Circulation 2012, 125, 1735–1741. [Google Scholar] [CrossRef]
- Benn, M.; Tybjaerg-Hansen, A.; McCarthy, M.I.; Jensen, G.B.; Grande, P.; Nordestgaard, B.G. Nonfasting Glucose, Ischemic Heart Disease, and Myocardial Infarction: A Mendelian Randomization Study. J. Am. Coll. Cardiol. 2012, 59, 2356–2365. [Google Scholar] [CrossRef]
- Brown, I.J.; Stamler, J.; van Horn, L.; Robertson, C.E.; Chan, Q.; Dyer, A.R.; Huang, C.-C.; Rodriguez, B.L.; Zhao, L.; Daviglus, M.L.; et al. Sugar-Sweetened Beverage, Sugar Intake of Individuals, and Their Blood Pressure. Hypertension 2011, 57, 695–701. [Google Scholar] [CrossRef]
- Yu, T.; Sheu, S.-S.; Robotham, J.L.; Yoon, Y. Mitochondrial Fission Mediates High Glucose-Induced Cell Death through Elevated Production of Reactive Oxygen Species. Cardiovasc Res. 2008, 79, 341–351. [Google Scholar] [CrossRef]
- Balteau, M.; Tajeddine, N.; de Meester, C.; Ginion, A.; Des Rosiers, C.; Brady, N.R.; Sommereyns, C.; Horman, S.; Vanoverschelde, J.-L.; Gailly, P.; et al. NADPH Oxidase Activation by Hyperglycaemia in Cardiomyocytes Is Independent of Glucose Metabolism but Requires SGLT1. Cardiovasc Res 2011, 92, 237–246. [Google Scholar] [CrossRef]
- Banerjee, D.; Sharma, P. Dual Effect of Glucose on Macrophage NADPH Oxidase Activity: A Possible Link between Diabetes and Tuberculosis. Oxid. Antioxid. Med. Sci. 2012, 1, 91–96. [Google Scholar] [CrossRef]
- Jansen, F.; Yang, X.; Franklin, B.S.; Hoelscher, M.; Schmitz, T.; Bedorf, J.; Nickenig, G.; Werner, N. High Glucose Condition Increases NADPH Oxidase Activity in Endothelial Microparticles That Promote Vascular Inflammation. Cardiovasc. Res. 2013, 98, 94–106. [Google Scholar] [CrossRef] [PubMed]
- Nowotny, K.; Jung, T.; Höhn, A.; Weber, D.; Grune, T. Advanced Glycation End Products and Oxidative Stress in Type 2 Diabetes Mellitus. Biomolecules 2015, 5, 194–222. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J. Pharmacology of Methylglyoxal: Formation, Modification of Proteins and Nucleic Acids, and Enzymatic Detoxification—A Role in Pathogenesis and Antiproliferative Chemotherapy. Gen. Pharm. 1996, 27, 565–573. [Google Scholar] [CrossRef]
- Lorenzi, M. The Polyol Pathway as a Mechanism for Diabetic Retinopathy: Attractive, Elusive, and Resilient. Exp. Diabetes Res. 2007, 2007, 61038. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, M.A.; Drury, S.; Fu, C.; Qu, W.; Taguchi, A.; Lu, Y.; Avila, C.; Kambham, N.; Bierhaus, A.; Nawroth, P.; et al. RAGE Mediates a Novel Proinflammatory Axis: A Central Cell Surface Receptor for S100/Calgranulin Polypeptides. Cell 1999, 97, 889–901. [Google Scholar] [CrossRef]
- Wautier, M.-P.; Chappey, O.; Corda, S.; Stern, D.M.; Schmidt, A.M.; Wautier, J.-L. Activation of NADPH Oxidase by AGE Links Oxidant Stress to Altered Gene Expression via RAGE. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E685–E694. [Google Scholar] [CrossRef]
- Reznikov, L.L.; Waksman, J.; Azam, T.; Kim, S.H.; Bufler, P.; Niwa, T.; Werman, A.; Zhang, X.; Pischetsrieder, M.; Shaldon, S.; et al. Effect of Advanced Glycation End Products on Endotoxin-Induced TNF-Alpha, IL-1beta and IL-8 in Human Peripheral Blood Mononuclear Cells. Clin. Nephrol. 2004, 61, 324–336. [Google Scholar] [CrossRef]
- Mohammed, A.M.; Syeda, K.; Hadden, T.; Kowluru, A. Upregulation of Phagocyte-like NADPH Oxidase by Cytokines in Pancreatic Beta-Cells: Attenuation of Oxidative and Nitrosative Stress by 2-Bromopalmitate. Biochem. Pharm. 2013, 85, 109–114. [Google Scholar] [CrossRef]
- Prasad, K. C-Reactive Protein Increases Oxygen Radical Generation by Neutrophils. J. Cardiovasc. Pharm. Ther. 2004, 9, 203–209. [Google Scholar] [CrossRef]
- Goldstein, B.J.; Mahadev, K.; Wu, X.; Zhu, L.; Motoshima, H. Role of Insulin-Induced Reactive Oxygen Species in the Insulin Signaling Pathway. Antioxid. Redox Signal. 2005, 7, 1021–1031. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, A.; Campos, C.; Díaz-Vegas, A.; Galgani, J.E.; Juretic, N.; Osorio-Fuentealba, C.; Bucarey, J.L.; Tapia, G.; Valenzuela, R.; Contreras-Ferrat, A.; et al. Insulin-Dependent H2O2 Production Is Higher in Muscle Fibers of Mice Fed with a High-Fat Diet. Int. J. Mol. Sci 2013, 14, 15740–15754. [Google Scholar] [CrossRef] [PubMed]
- Alleva, R.; Borghi, B.; Santarelli, L.; Strafella, E.; Carbonari, D.; Bracci, M.; Tomasetti, M. In Vitro Effect of Aspartame in Angiogenesis Induction. Toxicol. Vitr. 2011, 25, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Ranney, R.E.; Oppermann, J.A.; Muldoon, E.; McMahon, F.G. Comparative Metabolism of Aspartame in Experimental Animals and Humans. J. Toxicol. Environ. Health 1976, 2, 441–451. [Google Scholar] [CrossRef]
- Ashok, I.; Sheeladevi, R. Biochemical Responses and Mitochondrial Mediated Activation of Apoptosis on Long-Term Effect of Aspartame in Rat Brain. Redox Biol. 2014, 2, 820–831. [Google Scholar] [CrossRef]
- Iyyaswamy, A.; Rathinasamy, S. Effect of Chronic Exposure to Aspartame on Oxidative Stress in the Brain of Albino Rats. J. Biosci. 2012, 37, 679–688. [Google Scholar] [CrossRef] [PubMed]
- Ashok, I.; Poornima, P.S.; Wankhar, D.; Ravindran, R.; Sheeladevi, R. Oxidative Stress Evoked Damages on Rat Sperm and Attenuated Antioxidant Status on Consumption of Aspartame. Int. J. Impot. Res. 2017, 29, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Iyaswamy, A.; Wankhar, D.; Loganathan, S.; Shanmugam, S.; Rajan, R.; Rathinasamy, S. Disruption of Redox Homeostasis in Liver Function and Activation of Apoptosis on Consumption of Aspartame in Folate Deficient Rat Model. J. Nutr. Intermed. Metab. 2017, 8, 41–50. [Google Scholar] [CrossRef]
- Abhilash, M.; Paul, M.V.S.; Varghese, M.V.; Nair, R.H. Effect of Long Term Intake of Aspartame on Antioxidant Defense Status in Liver. Food Chem. Toxicol. 2011, 49, 1203–1207. [Google Scholar] [CrossRef]
- Abhilash, M.; Sauganth Paul, M.V.; Varghese, M.V.; Nair, R.H. Long-Term Consumption of Aspartame and Brain Antioxidant Defense Status. Drug. Chem. Toxicol. 2013, 36, 135–140. [Google Scholar] [CrossRef]
- Iyaswamy, A.; Kammella, A.K.; Thavasimuthu, C.; Wankupar, W.; Dapkupar, W.; Shanmugam, S.; Rajan, R.; Rathinasamy, S. Oxidative Stress Evoked Damages Leading to Attenuated Memory and Inhibition of NMDAR–CaMKII–ERK/CREB Signalling on Consumption of Aspartame in Rat Model. J. Food Drug Anal. 2018, 26, 903–916. [Google Scholar] [CrossRef]
- Jîtcă, G.; Fogarasi, E.; Ősz, B.-E.; Vari, C.E.; Tero-Vescan, A.; Miklos, A.; Bătrînu, M.-G.; Rusz, C.M.; Croitoru, M.D.; Dogaru, M.T. A Simple HPLC/DAD Method Validation for the Quantification of Malondialdehyde in Rodent’s Brain. Molecules 2021, 26, 5066. [Google Scholar] [CrossRef]
- Jîtcă, G.; Fogarasi, E.; Ősz, B.-E.; Vari, C.E.; Fülöp, I.; Croitoru, M.D.; Rusz, C.M.; Dogaru, M.T. Profiling the Concentration of Reduced and Oxidized Glutathione in Rat Brain Using HPLC/DAD Chromatographic System. Molecules 2021, 26, 6590. [Google Scholar] [CrossRef]
- Scherer, N.M.; Deamer, D.W. Oxidation of Thiols in the Ca2+-ATPase of Sarcoplasmic Reticulum Microsomes. Biochim. Biophys. Acta 1986, 862, 309–317. [Google Scholar] [CrossRef]
- Benedikter, B.J.; Weseler, A.R.; Wouters, E.F.M.; Savelkoul, P.H.M.; Rohde, G.G.U.; Stassen, F.R.M. Redox-Dependent Thiol Modifications: Implications for the Release of Extracellular Vesicles. Cell Mol. Life Sci. 2018, 75, 2321–2337. [Google Scholar] [CrossRef]
- Alkafafy, M.E.-S.; Ibrahim, Z.S.; Ahmed, M.M.; El-Shazly, S.A. Impact of Aspartame and Saccharin on the Rat Liver: Biochemical, Molecular, and Histological Approach. Int. J. Immunopathol. Pharm. 2015, 28, 247–255. [Google Scholar] [CrossRef]
- Azeez, O.H.; Alkass, S.Y.; Persike, D.S. Long-Term Saccharin Consumption and Increased Risk of Obesity, Diabetes, Hepatic Dysfunction, and Renal Impairment in Rats. Medicina 2019, 55, 681. [Google Scholar] [CrossRef]
- Heredia-García, G.; Gómez-Oliván, L.M.; Orozco-Hernández, J.M.; Luja-Mondragón, M.; Islas-Flores, H.; SanJuan-Reyes, N.; Galar-Martínez, M.; García-Medina, S.; Dublán-García, O. Alterations to DNA, Apoptosis and Oxidative Damage Induced by Sucralose in Blood Cells of Cyprinus Carpio. Sci. Total Environ. 2019, 692, 411–421. [Google Scholar] [CrossRef]
- Kundu, N.; Domingues, C.C.; Patel, J.; Aljishi, M.; Ahmadi, N.; Fakhri, M.; Sylvetsky, A.C.; Sen, S. Sucralose Promotes Accumulation of Reactive Oxygen Species (ROS) and Adipogenesis in Mesenchymal Stromal Cells. Stem. Cell Res. Ther. 2020, 11, 250. [Google Scholar] [CrossRef]
- Sánchez-Aceves, L.M.; Dublán-García, O.; López-Martínez, L.-X.; Novoa-Luna, K.A.; Islas-Flores, H.; Galar-Martínez, M.; García-Medina, S.; Hernández-Navarro, M.D.; Gómez-Oliván, L.M. Reduction of the Oxidative Stress Status Using Steviol Glycosides in a Fish Model (Cyprinus Carpio). Biomed. Res. Int. 2017, 2017, 2352594. [Google Scholar] [CrossRef]
- Prata, C.; Zambonin, L.; Rizzo, B.; Maraldi, T.; Angeloni, C.; Vieceli Dalla Sega, F.; Fiorentini, D.; Hrelia, S. Glycosides from Stevia Rebaudiana Bertoni Possess Insulin-Mimetic and Antioxidant Activities in Rat Cardiac Fibroblasts. Oxid. Med. Cell Longev. 2017, 2017, 3724545. [Google Scholar] [CrossRef]
- Li, C.; Lin, L.-M.; Sui, F.; Wang, Z.-M.; Huo, H.-R.; Dai, L.; Jiang, T.-L. Chemistry and Pharmacology of Siraitia Grosvenorii: A Review. Chin. J. Nat. Med. 2014, 12, 89–102. [Google Scholar] [CrossRef]
- Gong, X.; Chen, N.; Ren, K.; Jia, J.; Wei, K.; Zhang, L.; Lv, Y.; Wang, J.; Li, M. The Fruits of Siraitia Grosvenorii: A Review of a Chinese Food-Medicine. Front. Pharm. 2019, 10, 1400. [Google Scholar] [CrossRef]
- Li, H.; Li, R.; Jiang, W.; Zhou, L. Research Progress of Pharmacological Effects of Siraitia Grosvenorii Extract. J. Pharm. Pharm. 2022, 74, 953–960. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).