

Cell Type-Specific Patterns in the Accumulation of DNA Damage Following Multifractional Radiation Exposure

 , , and
, , and
Abstract
1. Introduction
2. Results
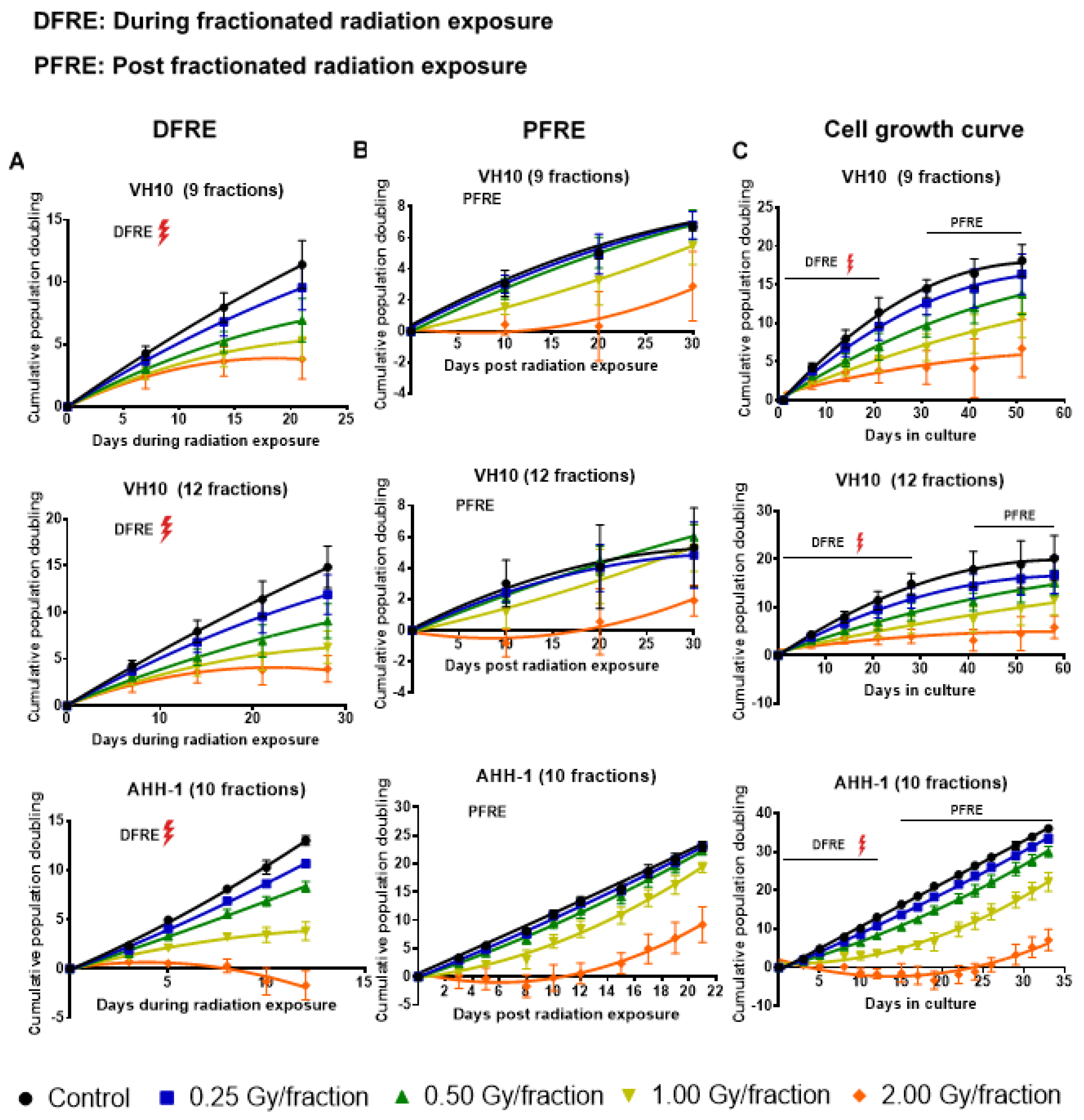
2.1. Delayed Growth of Cells Irradiated at High Doses-per-Fraction
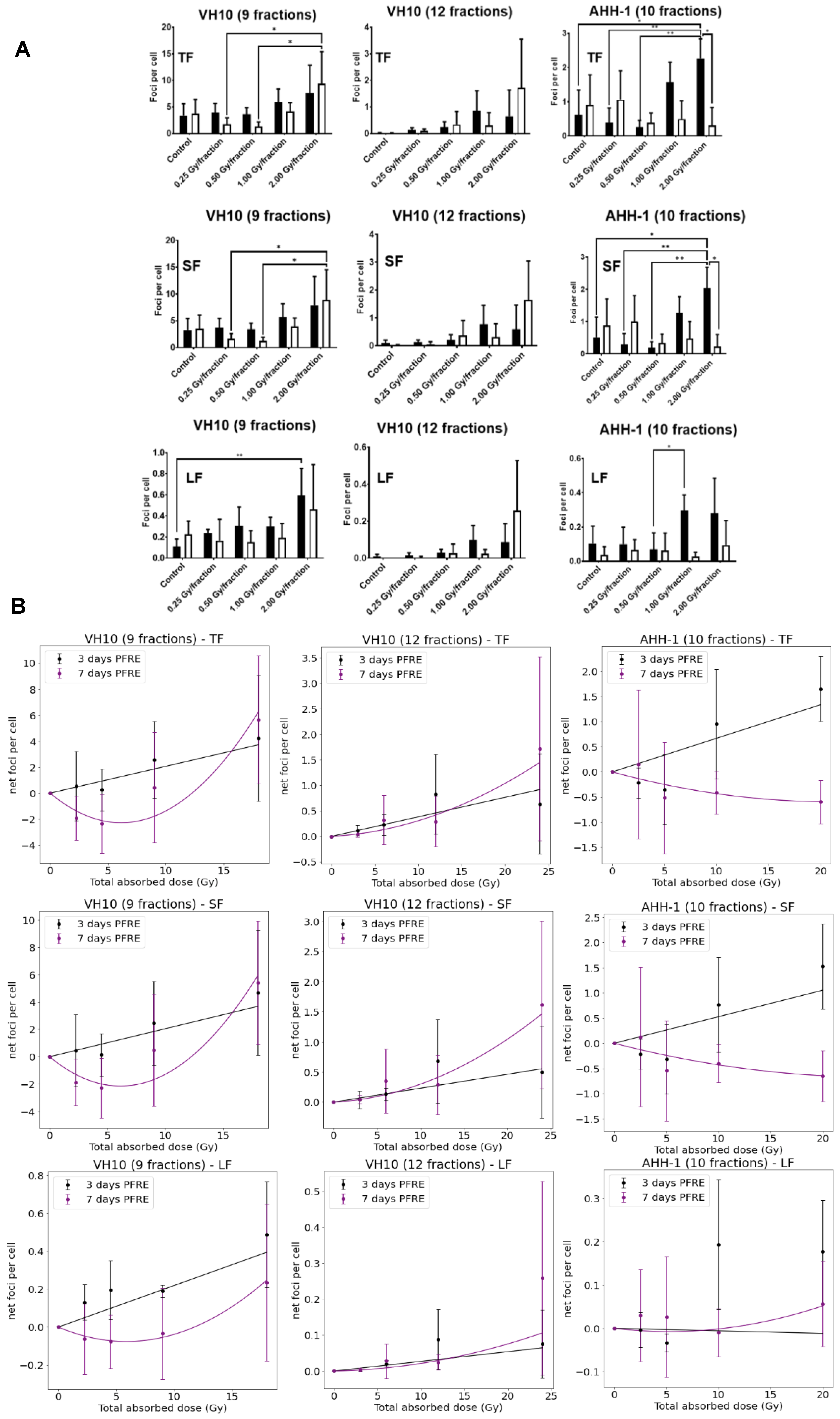
2.2. Excess DNA Damage at High Doses
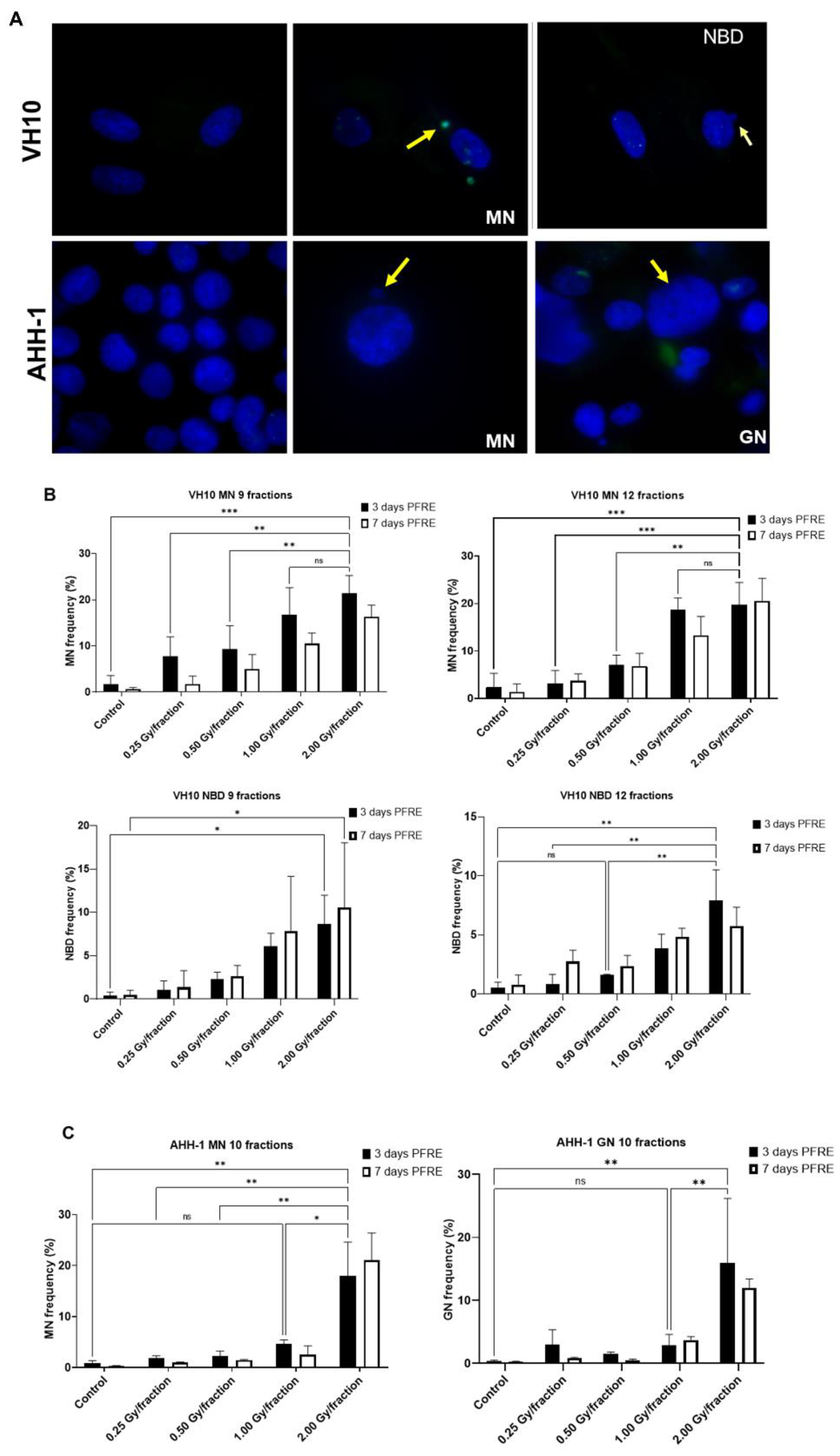
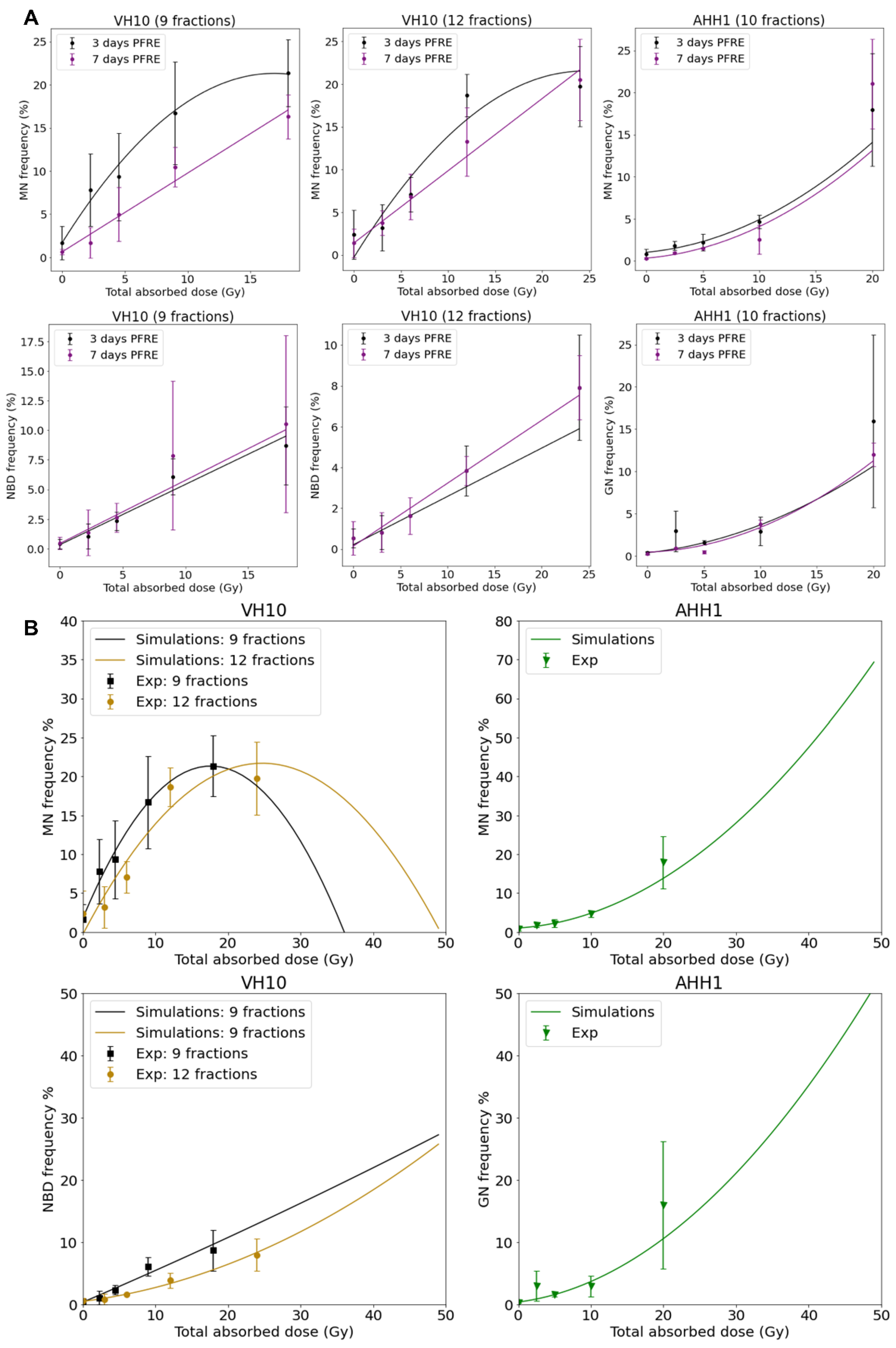
2.3. Cell Type-Specific Differences in the Accumulation of Genomic Instability
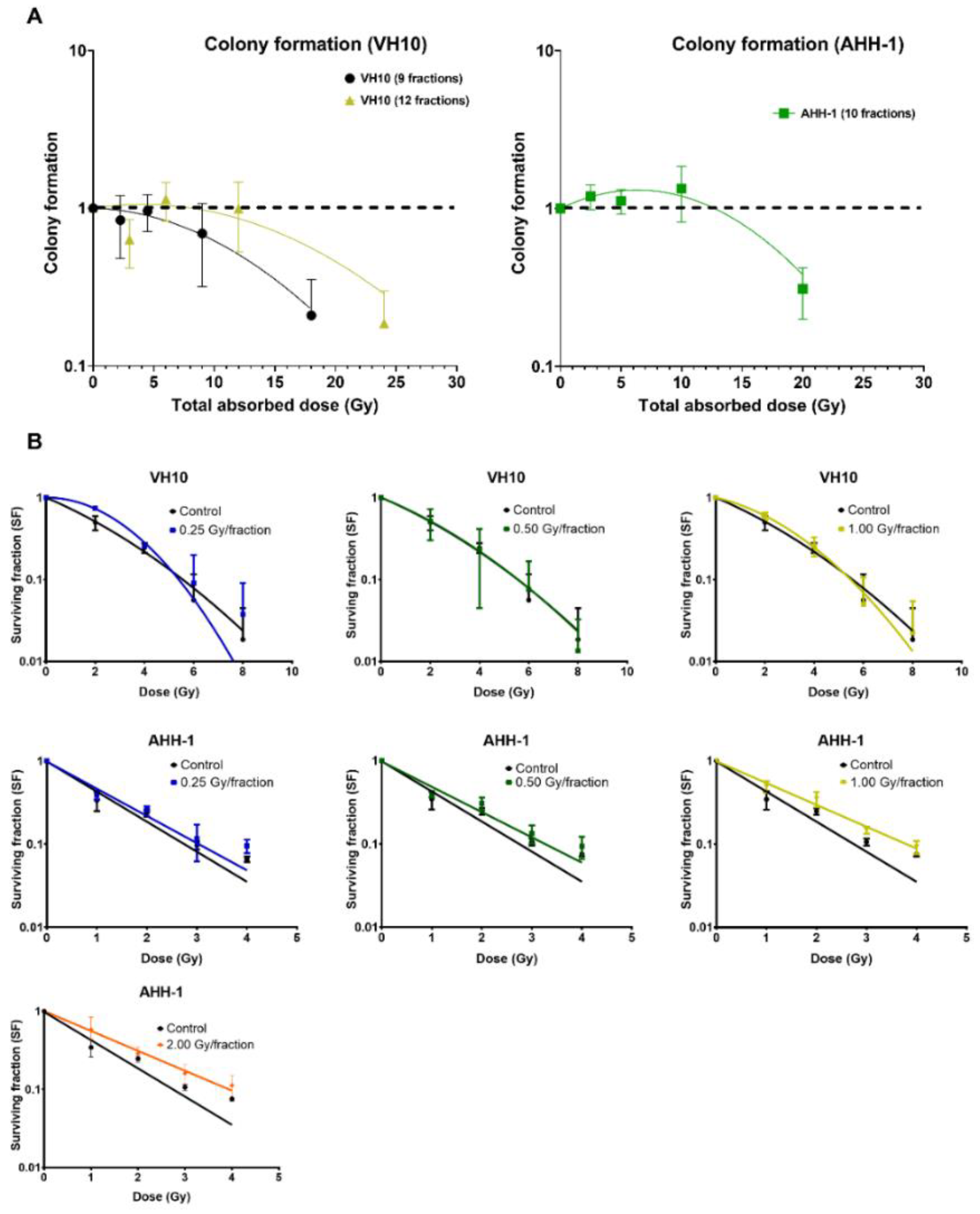
2.4. Differential Pattern in Cell Survival and Radiosensitivity
3. Discussion
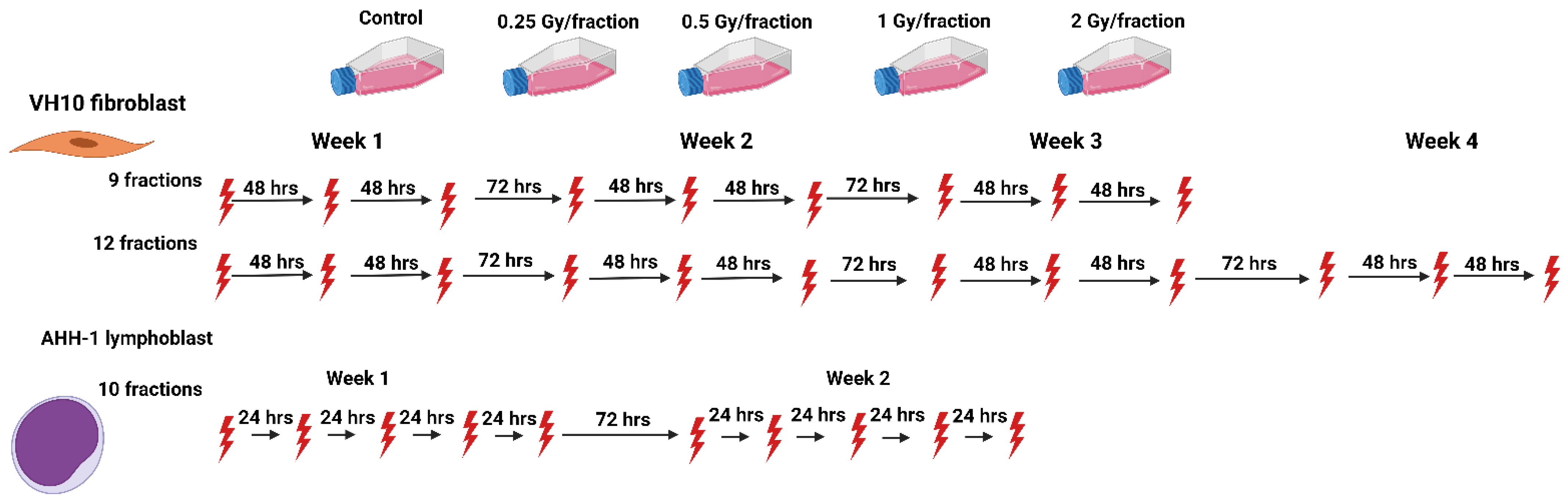
4. Materials and Methods
4.1. Cell Culture
4.2. Multi-Fraction Radiation Exposure
4.3. Cell Growth, Colony Formation Assay and Assessment of Radiosensitivity by Clonogenic Survival Assay
4.4. γH2AX Immunofluorescence Assay
4.5. Image Acquisition and Analysis of γH2AX Foci
4.6. Micronuclei, Nuclear Buds, and Giant Nuclei
4.7. Statistical Analysis and Data Fitting
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Kunos, C.A.; Coleman, C.N. Current and future initiatives for radiation oncology at the National Cancer Institute in the era of precision medicine. Int. J. Radiat. Oncol. Biol. Phys. 2018, 102, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Toma-Dasu, I.; Wojcik, A.; Lindblom, E.K. Risk of second cancer following radiotherapy. Phys. Med. Eur. J. Med. Phys. 2017, 42, 211–212. [Google Scholar] [CrossRef] [PubMed]
- Friedman, D.L.; Whitton, J.; Leisenring, W.; Mertens, A.C.; Hammond, S.; Stovall, M.; Donaldson, S.S.; Meadows, A.T.; Robison, L.L.; Neglia, J.P. Subsequent neoplasms in 5-year survivors of childhood cancer: The Childhood Cancer Survivor Study. JNCI J. Natl. Cancer Inst. 2010, 102, 1083–1095. [Google Scholar] [CrossRef] [PubMed]
- Berrington de Gonzalez, A.; Gilbert, E.; Curtis, R.; Inskip, P.; Kleinerman, R.; Morton, L.; Rajaraman, P.; Little, M.P. Second solid cancers after radiation therapy: A systematic review of the epidemiologic studies of the radiation dose-response relationship. Int. J. Radiat. Oncol. Biol. Phys. 2013, 86, 224–233. [Google Scholar] [CrossRef]
- Ng, A.K.; Bernardo, M.V.; Weller, E.; Backstrand, K.; Silver, B.; Marcus, K.C.; Tarbell, N.J.; Stevenson, M.A.; Friedberg, J.W.; Mauch, P.M. Second malignancy after Hodgkin disease treated with radiation therapy with or without chemotherapy: Long-term risks and risk factors. Blood 2002, 100, 1989–1996. [Google Scholar] [CrossRef]
- Nguyen, T.H.; Makena, M.R.; Yavvari, S.; Kaur, M.; Pham, T.; Urias, E.; Panapitiya, N.; Al-Rahawan, M.M. Sarcoma as Second Cancer in a Childhood Cancer Survivor: Case Report, Large Population Analysis and Literature Review. Medicina 2020, 56, 224. [Google Scholar] [CrossRef]
- Giulino-Roth, L.; Pei, Q.; Buxton, A.; Bush, R.; Wu, Y.; Wolden, S.L.; Constine, L.S.; Kelly, K.M.; Schwartz, C.L.; Friedman, D.L. Subsequent malignant neoplasms among children with Hodgkin lymphoma: A report from the Children’s Oncology Group. Blood 2021, 137, 1449–1456. [Google Scholar] [CrossRef]
- Turcotte, L.M.; Neglia, J.P.; Reulen, R.C.; Ronckers, C.M.; van Leeuwen, F.E.; Morton, L.M.; Hodgson, D.C.; Yasui, Y.; Oeffinger, K.C.; Henderson, T.O. Risk, Risk Factors, and Surveillance of Subsequent Malignant Neoplasms in Survivors of Childhood Cancer: A Review. J. Clin. Oncol. 2018, 36, 2145–2152. [Google Scholar] [CrossRef]
- Thariat, J.; Hannoun-Levi, J.M.; Sun Myint, A.; Vuong, T.; Gérard, J.P. Past, present, and future of radiotherapy for the benefit of patients. Nat. Rev. Clin. Oncol. 2013, 10, 52–60. [Google Scholar] [CrossRef]
- Lindsay, K.; Wheldon, E.; Deehan, C.; Wheldon, T. Radiation carcinogenesis modelling for risk of treatment-related second tumours following radiotherapy. Br. J. Radiol. 2001, 74, 529–536. [Google Scholar] [CrossRef]
- Schneider, U. Mechanistic model of radiation-induced cancer after fractionated radiotherapy using the linear-quadratic formula. Med. Phys. 2009, 36, 1138–1143. [Google Scholar] [CrossRef] [PubMed]
- Schneider, U. Modeling the risk of secondary malignancies after radiotherapy. Genes 2011, 2, 1033–1049. [Google Scholar] [CrossRef] [PubMed]
- Dörr, W.; Herrmann, T. Cancer induction by radiotherapy: Dose dependence and spatial relationship to irradiated volume. J. Radiol. Prot. 2002, 22, A117. [Google Scholar] [CrossRef]
- Diallo, I.; Haddy, N.; Adjadj, E.; Samand, A.; Quiniou, E.; Chavaudra, J.; Alziar, I.; Perret, N.; Guérin, S.; Lefkopoulos, D.; et al. Frequency distribution of second solid cancer locations in relation to the irradiated volume among 115 patients treated for childhood cancer. Int. J. Radiat. Oncol. Biol. Phys. 2009, 74, 876–883. [Google Scholar] [CrossRef] [PubMed]
- Hall, E.J.; Wuu, C.S. Radiation-induced second cancers: The impact of 3D-CRT and IMRT. Int. J. Radiat. Oncol. Biol. Phys. 2003, 56, 83–88. [Google Scholar] [CrossRef]
- Preston, D.L.; Shimizu, Y.; Pierce, D.A.; Suyama, A.; Mabuchi, K. Studies of mortality of atomic bomb survivors. Report 13: Solid cancer and noncancer disease mortality: 1950–1997. Radiat. Res. 2003, 160, 381–407. [Google Scholar] [CrossRef]
- Newhauser, W.D.; de Gonzalez, A.B.; Schulte, R.; Lee, C. A review of radiotherapy-induced late effects research after advanced technology treatments. Front. Oncol. 2016, 6, 13. [Google Scholar] [CrossRef]
- Turcotte, L.M.; Whitton, J.A.; Friedman, D.L.; Hammond, S.; Armstrong, G.T.; Leisenring, W.; Robison, L.L.; Neglia, J.P. Risk of Subsequent Neoplasms During the Fifth and Sixth Decades of Life in the Childhood Cancer Survivor Study Cohort. J. Clin. Oncol. 2015, 33, 3568–3575. [Google Scholar] [CrossRef]
- Daşu, A.; Toma-Daşu, I.; Olofsson, J.; Karlsson, M. The use of risk estimation models for the induction of secondary cancers following radiotherapy. Acta Oncol. 2005, 44, 339–347. [Google Scholar] [CrossRef]
- Han, A.; Elkind, M.M. Transformation of mouse C3H/10T1/2 cells by single and fractionated doses of X-rays and fission-spectrum neutrons. Cancer Res. 1979, 39, 123–130. [Google Scholar]
- Sachs, R.K.; Brenner, D.J. Solid tumor risks after high doses of ionizing radiation. Proc. Natl. Acad. Sci. USA 2005, 102, 13040–13045. [Google Scholar] [CrossRef] [PubMed]
- Newhauser, W.D.; Durante, M. Assessing the risk of second malignancies after modern radiotherapy. Nat. Rev. Cancer 2011, 11, 438–448. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H. DNA damage, aging, and cancer. New Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Fenech, M.; Kirsch-Volders, M.; Natarajan, A.T.; Surralles, J.; Crott, J.W.; Parry, J.; Norppa, H.; Eastmond, D.A.; Tucker, J.D.; Thomas, P. Molecular mechanisms of micronucleus, nucleoplasmic bridge and nuclear bud formation in mammalian and human cells. Mutagenesis 2011, 26, 125–132. [Google Scholar] [CrossRef]
- Mills, K.D.; Ferguson, D.O.; Alt, F.W. The role of DNA breaks in genomic instability and tumorigenesis. Immunol. Rev. 2003, 194, 77–95. [Google Scholar] [CrossRef]
- Gómez-Herreros, F. DNA Double Strand Breaks and Chromosomal Translocations Induced by DNA Topoisomerase II. Front. Mol. Biosci. 2019, 6, 141. [Google Scholar] [CrossRef]
- Shimizu, N.; Shingaki, K.; Kaneko-Sasaguri, Y.; Hashizume, T.; Kanda, T. When, where and how the bridge breaks: Anaphase bridge breakage plays a crucial role in gene amplification and HSR generation. Exp. Cell Res. 2005, 302, 233–243. [Google Scholar] [CrossRef]
- Shimizu, N.; Itoh, N.; Utiyama, H.; Wahl, G.M. Selective entrapment of extrachromosomally amplified DNA by nuclear budding and micronucleation during S phase. J. Cell Biol. 1998, 140, 1307–1320. [Google Scholar] [CrossRef]
- Song, Y.; Zhao, Y.; Deng, Z.; Zhao, R.; Huang, Q. Stress-induced polyploid giant cancer cells: Unique way of formation and non-negligible characteristics. Front. Oncol. 2021, 11, 3390. [Google Scholar] [CrossRef]
- Illidge, T.M.; Cragg, M.S.; Fringes, B.; Olive, P.; Erenpreisa, J.A. Polyploid giant cells provide a survival mechanism for p53 mutant cells after DNA damage. Cell Biol. Int. 2000, 24, 621–633. [Google Scholar] [CrossRef]
- Zhang, Z.; Feng, X.; Deng, Z.; Cheng, J.; Wang, Y.; Zhao, M.; Zhao, Y.; He, S.; Huang, Q. Irradiation-induced polyploid giant cancer cells are involved in tumor cell repopulation via neosis. Mol. Oncol. 2021, 15, 2219–2234. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.K. DNA Damage, Mutagenesis and Cancer. Int. J. Mol. Sci. 2018, 19, 970. [Google Scholar] [CrossRef] [PubMed]
- Loseva, O.; Shubbar, E.; Haghdoost, S.; Evers, B.; Helleday, T.; Harms-Ringdahl, M. Chronic Low Dose Rate Ionizing Radiation Exposure Induces Premature Senescence in Human Fibroblasts that Correlates with Up Regulation of Proteins Involved in Protection against Oxidative Stress. Proteomes 2014, 2, 341–362. [Google Scholar] [CrossRef] [PubMed]
- Vit, J.-P.; Rosselli, F. Role of the ceramide-signaling pathways in ionizing radiation-induced apoptosis. Oncogene 2003, 22, 8645–8652. [Google Scholar] [CrossRef]
- Bracalente, C.; Ibanez, I.; Molinari, B.; Palmieri, M.; Maglioco, A.; Policastro, L.; Kreiner, A.; Burlon, A.; Valda, A.; Davidson, J. Assessment of γH2AX nuclear foci number and size in normal and repair-deficient cells irradiated with low and high linear energy transfer radiation. Int. J. Low Radiat. 2010, 7, 393–408. [Google Scholar] [CrossRef]
- Elkind, M.M.; Sutton, H. Radiation response of mammalian cells grown in culture. 1. Repair of X-ray damage in surviving Chinese hamster cells. Radiat. Res. 1960, 13, 556–593. [Google Scholar] [CrossRef]
- Markova, E.; Schultz, N.; Belyaev, I. Kinetics and dose-response of residual 53BP1/γ-H2AX foci: Co-localization, relationship with DSB repair and clonogenic survival. Int. J. Radiat. Biol. 2007, 83, 319–329. [Google Scholar] [CrossRef]
- Dikomey, J.; Dahm-Daphi, I.; Brammer, R.; Martensen, B.; Kaina, E. Correlation between cellular radiosensitivity and non-repaired double-strand breaks studied in nine mammalian cell lines. Int. J. Radiat. Biol. 1998, 73, 269–278. [Google Scholar] [CrossRef]
- Rodier, F.; Coppé, J.-P.; Patil, C.K.; Hoeijmakers, W.A.M.; Muñoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef]
- Soto-Gamez, A.; Quax, W.J.; Demaria, M. Regulation of Survival Networks in Senescent Cells: From Mechanisms to Interventions. J. Mol. Biol. 2019, 431, 2629–2643. [Google Scholar] [CrossRef]
- Suzuki, M.; Boothman, D.A. Stress-induced premature senescence (SIPS)--influence of SIPS on radiotherapy. J. Radiat. Res. 2008, 49, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef] [PubMed]
- Ardenfors, O.; Josefsson, D.; Dasu, A. Are IMRT treatments in the head and neck region increasing the risk of secondary cancers? Acta Oncol. 2014, 53, 1041–1047. [Google Scholar] [CrossRef]
- Ivanov, A.; Cragg, M.S.; Erenpreisa, J.; Emzinsh, D.; Lukman, H.; Illidge, T.M. Endopolyploid cells produced after severe genotoxic damage have the potential to repair DNA double strand breaks. J. Cell Sci. 2003, 116, 4095–4106. [Google Scholar] [CrossRef]
- Olive, P.L.; Banáth, J.P.; Durand, R.E. Development of apoptosis and polyploidy in human lymphoblast cells as a function of position in the cell cycle at the time of irradiation. Radiat. Res. 1996, 146, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Sazonova, E.V.; Petrichuk, S.V.; Kopeina, G.S.; Zhivotovsky, B. A link between mitotic defects and mitotic catastrophe: Detection and cell fate. Biol. Direct 2021, 16, 25. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Cragg, M.S. Mitotic death: A mechanism of survival? A review. Cancer Cell Int. 2001, 1, 1. [Google Scholar] [CrossRef]
- Childs, B.G.; Baker, D.J.; Kirkland, J.L.; Campisi, J.; van Deursen, J.M. Senescence and apoptosis: Dueling or complementary cell fates? EMBO Rep. 2014, 15, 1139–1153. [Google Scholar] [CrossRef]
- Alamilla-Presuel, J.C.; Burgos-Molina, A.M.; González-Vidal, A.; Sendra-Portero, F.; Ruiz-Gómez, M.J. Factors and molecular mechanisms of radiation resistance in cancer cells. Int. J. Radiat. Biol. 2022, 98, 1301–1315. [Google Scholar] [CrossRef]
- Fernald, K.; Kurokawa, M. Evading apoptosis in cancer. Trends Cell Biol. 2013, 23, 620–633. [Google Scholar] [CrossRef]
- Ahmad, P.; Sana, J.; Slavik, M.; Slampa, P.; Smilek, P.; Slaby, O. MicroRNAs involvement in radioresistance of head and neck cancer. Dis. Mrk. 2017, 2017, 8245345. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Jin, G.; Cai, F.; Chen, X.; Cao, N.; Zhang, X.; Liu, J.; Chen, F.; Wang, F.; Dong, W. Extracellular signal-regulated kinase 5 increases radioresistance of lung cancer cells by enhancing the DNA damage response. Exp. Mol. Med. 2019, 51, 1–20. [Google Scholar] [CrossRef] [PubMed]
- McDonald, J.T.; Kim, K.; Norris, A.J.; Vlashi, E.; Phillips, T.M.; Lagadec, C.; Della Donna, L.; Ratikan, J.; Szelag, H.; Hlatky, L. Ionizing radiation activates the Nrf2 antioxidant response. Cancer Res. 2010, 70, 8886–8895. [Google Scholar] [CrossRef] [PubMed]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Morine, Y.; Yamada, S.; Saito, Y.; Ikemoto, T.; Tokuda, K.; Takasu, C.; Miyazaki, K.; Shimada, M. Nrf2 signaling promotes cancer stemness, migration, and expression of ABC transporter genes in sorafenib-resistant hepatocellular carcinoma cells. PLoS ONE 2021, 16, e0256755. [Google Scholar] [CrossRef]
- Kabakov, A.; Yakimova, A.; Matchuk, O. Molecular Chaperones in Cancer Stem Cells: Determinants of Stemness and Potential Targets for Antitumor Therapy. Cells 2020, 9, 892. [Google Scholar] [CrossRef]
- Gartel, A.L.; Tyner, A.L. The role of the cyclin-dependent kinase inhibitor p21 in apoptosis. Mol. Cancer Ther. 2002, 1, 639–649. [Google Scholar]
- Webley, K.; Bond, J.A.; Jones, C.J.; Blaydes, J.P.; Craig, A.; Hupp, T.; Wynford-Thomas, D. Posttranslational modifications of p53 in replicative senescence overlapping but distinct from those induced by DNA damage. Mol. Cell. Biol. 2000, 20, 2803–2808. [Google Scholar] [CrossRef]
- Strober, W. Trypan blue exclusion test of cell viability. Curr. Protoc. Immunol. 1997, 21, A.3B.1–A.3B.2. [Google Scholar]
- Chandna, S.; Dagur, R.S.; Mathur, A.; Natarajan, A.T.; Harms-Ringdahl, M.; Haghdoost, S. Agarose overlay selectively improves macrocolony formation and radiosensitivity assessment in primary fibroblasts. Int. J. Radiat. Biol. 2014, 90, 401–406. [Google Scholar] [CrossRef]
- Borowicz, S.; Van Scoyk, M.; Avasarala, S.; Karuppusamy Rathinam, M.K.; Tauler, J.; Bikkavilli, R.K.; Winn, R.A. The soft agar colony formation assay. J. Vis. Exp. JoVE 2014, 92, e51998. [Google Scholar] [CrossRef] [PubMed]
- Sollazzo, A.; Brzozowska, B.; Cheng, L.; Lundholm, L.; Haghdoost, S.; Scherthan, H.; Wojcik, A. Alpha particles and X rays interact in inducing DNA damage in U2OS cells. Radiat. Res. 2017, 188, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Fenech, M.; Chang, W.P.; Kirsch-Volders, M.; Holland, N.; Bonassi, S.; Zeiger, E. HUMN project: Detailed description of the scoring criteria for the cytokinesis-block micronucleus assay using isolated human lymphocyte cultures. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2003, 534, 65–75. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total Absorbed Doses | ||||
|---|---|---|---|---|
| 0.25 Gy/Fraction | 0.5 Gy/Fraction | 1.0 Gy/Fraction | 2.0 Gy/Fraction | |
| VH10 (9 fractions) | 2.25 Gy | 4.5 Gy | 9.0 Gy | 18.0 Gy |
| VH10 (12 fractions) | 3.0 Gy | 6.0 Gy | 12.0 Gy | 24.0 Gy |
| AHH-1 (10 fractions) | 2.5 Gy | 5.0 Gy | 10 Gy | 20.0 Gy |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akuwudike, P.; Tartas, A.; López-Riego, M.; Toma-Dasu, I.; Wojcik, A.; Lundholm, L. Cell Type-Specific Patterns in the Accumulation of DNA Damage Following Multifractional Radiation Exposure. Int. J. Mol. Sci. 2022, 23, 12861. https://doi.org/10.3390/ijms232112861
Akuwudike P, Tartas A, López-Riego M, Toma-Dasu I, Wojcik A, Lundholm L. Cell Type-Specific Patterns in the Accumulation of DNA Damage Following Multifractional Radiation Exposure. International Journal of Molecular Sciences. 2022; 23(21):12861. https://doi.org/10.3390/ijms232112861
Chicago/Turabian StyleAkuwudike, Pamela, Adrianna Tartas, Milagrosa López-Riego, Iuliana Toma-Dasu, Andrzej Wojcik, and Lovisa Lundholm. 2022. "Cell Type-Specific Patterns in the Accumulation of DNA Damage Following Multifractional Radiation Exposure" International Journal of Molecular Sciences 23, no. 21: 12861. https://doi.org/10.3390/ijms232112861
APA StyleAkuwudike, P., Tartas, A., López-Riego, M., Toma-Dasu, I., Wojcik, A., & Lundholm, L. (2022). Cell Type-Specific Patterns in the Accumulation of DNA Damage Following Multifractional Radiation Exposure. International Journal of Molecular Sciences, 23(21), 12861. https://doi.org/10.3390/ijms232112861