
Human Peripheral Blood Mononucleocyte Derived Myeloid Committed Progenitor Cells Mitigate H-ARS by Exosomal Paracrine Signal

 ,
,  , , ,
, , ,  and
and 
Abstract
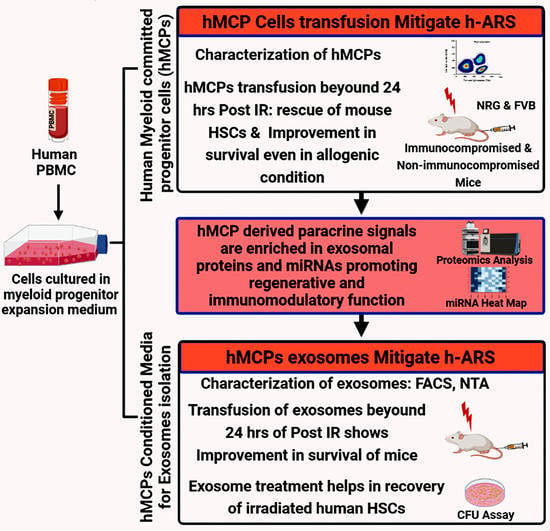
1. Introduction
2. Results
2.1. Transplantation of hMCP Cells Mitigates h-ARS and Improves Survival
2.2. hMCP Rescues HSCs and Induces Regenerative Potential against Radiation Injury
2.3. hMCPs Are Suitable for Allogenic Transplant
2.4. hMCP-Derived Paracrine Signals Are Enriched with Exosomal Proteins
2.5. hMCP-Derived sEVs Mitigates h-ARS
2.6. hMCP sEVs Promote Regenerative Response of HSCs
2.7. hMCP-Derived sEVs miRNAs-Associated Regenerative and Immune-Modulatory Process
3. Discussion
4. Materials and Methods
4.1. Human Myeloid-Committed Progenitor Cell (hMCP) Culture
4.2. Characterization of hMCP
4.3. Collection of hMCP-Conditioned Media
4.4. Isolation of Small Extracellular Vesicles (sEVs) from hMCP-Conditioned Media
4.5. Characterization of sEVs
4.6. sEVs Protein Estimation and Particle Count
4.7. Nanoparticle Tracking Analysis (NTA) of sEVs
4.8. Exosomal miRNA Analysis
4.9. T Cells Inhibition Assay
4.10. Mice
4.11. Irradiation
4.12. Assessment of Graft Vs Host Disease (GVHD)
4.13. Bone Marrow Cell Analysis
4.14. Histology of Bone Marrow
4.15. Ex Vivo Colony-Forming Unit (CFU) Assay
4.16. Proteomics Analysis of hMCP-Conditioned Media
4.17. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Clements, B.W.; Casani, J.A.P. 15-Nuclear and Radiological Disasters. In Disasters and Public Health, 2nd ed.; Clements, B.W., Casani, J.A.P., Eds.; Butterworth-Heinemann: Oxford, UK, 2016; pp. 357–383. [Google Scholar]
- Barabanova, A.V.; Bushmanov, A.J.; Kotenko, K.V. Acute Radiation Sickness From Chernobyl. In Reference Module in Earth Systems and Environmental Sciences; Elsevier: Amsterdam, The Netherlands, 2019. [Google Scholar]
- Iwai, K.; Hashimoto, K.; Nishizawa, K.; Sawada, K.; Honda, K. Evaluation of effective dose from a RANDO phantom in videofluorography diagnostic procedures for diagnosing dysphagia. Dentomaxillofacial Radiol. 2011, 40, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Chute, J.P. To survive radiation injury, remember your aPCs. Nat. Med. 2012, 18, 1013–1014. [Google Scholar] [CrossRef] [PubMed]
- Chinnapaka, S.; Yang, K.S.; Samadi, Y.; Epperly, M.W.; Hou, W.; Greenberger, J.S.; Ejaz, A.; Rubin, J.P. Allogeneic Adipose-Derived Stem Cells Mitigate Acute Radiation Syndrome by the Rescue of Damaged Bone Marrow Cells from Apoptosis. STEM CELLS Transl. Med. 2021, 10, 1095–1114. [Google Scholar] [CrossRef] [PubMed]
- Gluzman-Poltorak, Z.; Vainstein, V.; Basile, L.A. Recombinant interleukin-12, but not granulocyte-colony stimulating factor, improves survival in lethally irradiated nonhuman primates in the absence of supportive care: Evidence for the development of a frontline radiation medical countermeasure. Am. J. Hematol. 2014, 89, 868–873. [Google Scholar] [CrossRef]
- Mikkelsen, R.B.; Wardman, P. Biological chemistry of reactive oxygen and nitrogen and radiation-induced signal transduction mechanisms. Oncogene 2003, 22, 5734–5754. [Google Scholar] [CrossRef]
- Fliedner, T.M.; Graessle, D.; Meineke, V.; Dörr, H. Pathophysiological principles underlying the blood cell concentration responses used to assess the severity of effect after accidental whole-body radiation exposure: An essential basis for an evidence-based clinical triage. Exp. Hematol. 2007, 35, 8–16. [Google Scholar] [CrossRef]
- Macnaughton, W.K. Review article: New insights into the pathogenesis of radiation-induced intestinal dysfunction. Aliment. Pharmacol. Ther. 2000, 14, 523–528. [Google Scholar] [CrossRef]
- DiCarlo, A.L.; Maher, C.; Hick, J.L.; Hanfling, D.; Dainiak, N.; Chao, N.; Bader, J.L.; Coleman, C.N.; Weinstock, D.M. Radiation Injury After a Nuclear Detonation: Medical Consequences and the Need for Scarce Resources Allocation. Disaster Med. Public Health Prep. 2011, 5, S32–S44. [Google Scholar] [CrossRef]
- Green, D.E.; Rubin, C.T. Consequences of irradiation on bone and marrow phenotypes, and its relation to disruption of hematopoietic precursors. Bone 2014, 63, 87–94. [Google Scholar] [CrossRef]
- Shao, L.; Luo, Y.; Zhou, D. Hematopoietic Stem Cell Injury Induced by Ionizing Radiation. Antioxidants Redox Signal. 2014, 20, 1447–1462. [Google Scholar] [CrossRef]
- Stewart, F.M.; Zhong, S.; Lambert, J.-F.; Colvin, G.A.; Abedi, M.; Dooner, M.S.; McAuliffe, C.I.; Wang, H.; Hsieh, C.; Quesenberry, P.J. Host marrow stem cell potential and engraftability at varying times after low-dose whole-body irradiation. Blood 2001, 98, 1246–1251. [Google Scholar] [CrossRef] [PubMed]
- Hirouchi, T.; Ito, K.; Nakano, M.; Monzen, S.; Yoshino, H.; Chiba, M.; Hazawa, M.; Nakano, A.; Ishikawa, J.; Yamaguchi, M.; et al. Mitigative Effects of a Combination of Multiple Pharmaceutical Drugs on the Survival of Mice Exposed to Lethal Ionizing Radiation. Curr. Pharm. Biotechnol. 2015, 17, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Covic, A.; Abraham, I. State-of-the-art biosimilar erythropoietins in the management of renal anemia: Lessons learned from Europe and implications for US nephrologists. Int. Urol. Nephrol. 2015, 47, 1529–1539. [Google Scholar] [CrossRef] [PubMed]
- Gianoncelli, A.; Bonini, S.; Bertuzzi, M.; Guarienti, M.; Vezzoli, S.; Kumar, R.; Delbarba, A.; Mastinu, A.; Sigala, S.; Spano, P.; et al. An Integrated Approach for a Structural and Functional Evaluation of Biosimilars: Implications for Erythropoietin. BioDrugs 2015, 29, 285–300. [Google Scholar] [CrossRef]
- MacVittie, T.J.; Farese, A.M.; Smith, W.G.; Baum, C.M.; Burton, E.; Mckearn, J.P. Myelopoietin, an engineered chimeric IL-3 and G-CSF receptor agonist, stimulates multilineage hematopoietic recovery in a nonhuman primate model of radiation-induced myelosuppression. Blood 2000, 95, 837–845. [Google Scholar] [CrossRef]
- Satyamitra, M.M.; Cassatt, D.R.; Taliaferro, L.P. A Poly-Pharmacy Approach to Mitigate Acute Radiation Syndrome (ARS). Radiat. Res. 2021, 196, 423–428. [Google Scholar] [CrossRef]
- Lv, B.; Zhang, X.; Yuan, J.; Chen, Y.; Ding, H.; Cao, X.; Huang, A. Biomaterial-supported MSC transplantation enhances cell–cell communication for spinal cord injury. Stem Cell Res. Ther. 2021, 12, 36. [Google Scholar] [CrossRef]
- Aly, R.M. Current state of stem cell-based therapies: An overview. Stem Cell Investig. 2020, 7, 8. [Google Scholar] [CrossRef]
- Sémont, A.; Mouiseddine, M.; François, A.; Demarquay, C.; Mathieu, N.; Chapel, A.; Saché, A.; Thierry, D.; Laloi, P.; Gourmelon, P. Mesenchymal stem cells improve small intestinal integrity through regulation of endogenous epithelial cell homeostasis. Cell Death Differ. 2009, 17, 952–961. [Google Scholar] [CrossRef]
- Gong, W.; Guo, M.; Han, Z.; Wang, Y.; Yang, P.; Xu, C.; Wang, Q.; Du, L.; Li, Q.; Zhao, H.; et al. Mesenchymal stem cells stimulate intestinal stem cells to repair radiation-induced intestinal injury. Cell Death Dis. 2016, 7, e2387. [Google Scholar] [CrossRef]
- Sudres, M.; Norol, F.; Trenado, A.; Grégoire, S.; Charlotte, F.; Levacher, B.; Lataillade, J.-J.; Bourin, P.; Holy, X.; Vernant, J.-P.; et al. Bone Marrow Mesenchymal Stem Cells Suppress Lymphocyte Proliferation In Vitro but Fail to Prevent Graft-versus-Host Disease in Mice. J. Immunol. 2006, 176, 7761–7767. [Google Scholar] [CrossRef] [PubMed]
- Prigozhina, T.B.; Khitrin, S.; Elkin, G.; Eizik, O.; Morecki, S.; Slavin, S. Mesenchymal stromal cells lose their immunosuppressive potential after allotransplantation. Exp. Hematol. 2008, 36, 1370–1376. [Google Scholar] [CrossRef] [PubMed]
- Waselenko, J.K.; MacVittie, T.J.; Blakely, W.F.; Pesik, N.; Wiley, A.L.; Dickerson, W.E.; Tsu, H.; Confer, D.L.; Coleman, C.N.; Seed, T.; et al. Medical Management of the Acute Radiation Syndrome: Recommendations of the Strategic National Stockpile Radiation Working Group. Ann. Intern. Med. 2004, 140, 1037–1051. [Google Scholar] [CrossRef] [PubMed]
- Venkateswaran, K.; Shrivastava, A.; Agrawala, P.K.; Prasad, A.; Kalra, N.; Pandey, P.R.; Manda, K.; Raj, H.G.; Parmar, V.S.; Dwarakanath, B.S. Mitigation of radiation-induced hematopoietic injury by the polyphenolic acetate 7, 8-diacetoxy-4-methylthiocoumarin in mice. Sci. Rep. 2016, 6, 37305. [Google Scholar] [CrossRef]
- Singh, V.; Newman, V.L.; Berg, A.N.; MacVittie, T.J. Animal models for acute radiation syndrome drug discovery. Expert Opin. Drug Discov. 2015, 10, 497–517. [Google Scholar] [CrossRef]
- Matsukuma, K.E.; Wei, N.; Sun, K.; Ramsamooj, R.; Chen, M. Diagnosis and differential diagnosis of hepatic graft versus host disease (GVHD). J. Gastrointest. Oncol. 2016, 7, S21–S31. [Google Scholar] [CrossRef]
- De Serre, N.P.-M.; Reijasse, D.; Verkarre, V.; Canioni, D.; Colomb, V.; Haddad, E.; Brousse, N. Chronic intestinal graft-versus-host disease: Clinical, histological and immunohistochemical analysis of 17 children. Bone Marrow Transplant. 2002, 29, 223–230. [Google Scholar] [CrossRef][Green Version]
- Saha, S.; Bhanja, P.; Kabarriti, R.; Liu, L.; Alfieri, A.A.; Guha, C. Bone Marrow Stromal Cell Transplantation Mitigates Radiation-Induced Gastrointestinal Syndrome in Mice. PLoS ONE 2011, 6, e24072. [Google Scholar] [CrossRef]
- Fischer, U.M.; Harting, M.T.; Jimenez, F.; Monzon-Posadas, W.O.; Xue, H.; Savitz, S.I.; Laine, G.A.; Cox, C.S., Jr. Pulmonary Passage is a Major Obstacle for Intravenous Stem Cell Delivery: The Pulmonary First-Pass Effect. Stem Cells Dev. 2009, 18, 683–692. [Google Scholar] [CrossRef]
- Gorelik, M.; Orukari, I.; Wang, J.; Galpoththawela, S.; Kim, H.; Levy, M.; Gilad, A.A.; Bar-Shir, A.; Kerr, D.A.; Levchenko, A.; et al. Use of MR Cell Tracking to Evaluate Targeting of Glial Precursor Cells to Inflammatory Tissue by Exploiting the Very Late Antigen-4 Docking Receptor. Radiology 2012, 265, 175–185. [Google Scholar] [CrossRef]
- Whiteside, T.L. Tumor-Derived Exosomes and Their Role in Cancer Progression. Adv. Clin. Chem. 2016, 74, 103–141. [Google Scholar] [CrossRef] [PubMed]
- Gurung, S.; Perocheau, D.; Touramanidou, L.; Baruteau, J. The exosome journey: From biogenesis to uptake and intracellular signalling. Cell Commun. Signal. 2021, 19, 47. [Google Scholar] [CrossRef] [PubMed]
- Konoshenko, M.Y.; Lekchnov, E.A.; Vlassov, A.V.; Laktionov, P.P. Isolation of Extracellular Vesicles: General Methodologies and Latest Trends. BioMed Res. Int. 2018, 2018, 8545347. [Google Scholar] [CrossRef] [PubMed]
- Zhao, A.G.; Shah, K.; Cromer, B.; Sumer, H. Comparative analysis of extracellular vesicles isolated from human mesenchymal stem cells by different isolation methods and visualisation of their uptake. Exp. Cell Res. 2022, 414, 113097. [Google Scholar] [CrossRef]
- Campos-Silva, C.; Suárez, H.; Jara-Acevedo, R.; Linares-Espinós, E.; Martinez-Piñeiro, L.; Yáñez-Mó, M.; Valés-Gómez, M. High sensitivity detection of extracellular vesicles immune-captured from urine by conventional flow cytometry. Sci. Rep. 2019, 9, 2042. [Google Scholar] [CrossRef]
- Hu, G.; Drescher, K.M.; Chen, X.-M. Exosomal miRNAs: Biological Properties and Therapeutic Potential. Front. Genet. 2012, 3, 56. [Google Scholar] [CrossRef]
- Mittelbrunn, M.; Gutiérrez-Vázquez, C.; Villarroya-Beltri, C.; González, S.; Sánchez-Cabo, F.; González, M.A.; Bernad, A.; Sánchez-Madrid, F. Unidirectional transfer of microRNA-loaded exosomes from T cells to antigen-presenting cells. Nat. Commun. 2011, 2, 282. [Google Scholar] [CrossRef]
- Hunter, M.P.; Ismail, N.; Zhang, X.; Aguda, B.D.; Lee, E.J.; Yu, L.; Xiao, T.; Schafer, J.; Lee, M.-L.T.; Schmittgen, T.D.; et al. Detection of microRNA Expression in Human Peripheral Blood Microvesicles. PLoS ONE 2008, 3, e3694. [Google Scholar] [CrossRef]
- Vaidya, A.; Kale, V.P. TGF-β signaling and its role in the regulation of hematopoietic stem cells. Syst. Synth. Biol. 2015, 9, 1–10. [Google Scholar] [CrossRef]
- Juntilla, M.; Patil, V.D.; Calamito, M.; Joshi, R.P.; Birnbaum, M.; Koretzky, G.A. AKT1 and AKT2 maintain hematopoietic stem cell function by regulating reactive oxygen species. Blood 2010, 115, 4030–4038. [Google Scholar] [CrossRef]
- Sinclair, A.; Park, L.; Shah, M.; Drotar, M.; Calaminus, S.; Hopcroft, L.E.M.; Kinstrie, R.; Guitart, A.; Dunn, K.; Abraham, S.; et al. CXCR2 and CXCL4 regulate survival and self-renewal of hematopoietic stem/progenitor cells. Blood 2016, 128, 371–383. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.; Matsui, W. Hedgehog Signaling in Hematopoiesis. Crit. Rev. Eukaryot. Gene Expr. 2010, 20, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Richter, J.; Traver, D.; Willert, K. The role of Wnt signaling in hematopoietic stem cell development. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Tothova, Z.; Gilliland, D.G. FoxO Transcription Factors and Stem Cell Homeostasis: Insights from the Hematopoietic System. Cell Stem Cell 2007, 1, 140–152. [Google Scholar] [CrossRef]
- Kangari, P.; Talaei-Khozani, T.; Razeghian-Jahromi, I.; Razmkhah, M. Mesenchymal stem cells: Amazing remedies for bone and cartilage defects. Stem Cell Res. Ther. 2020, 11, 492. [Google Scholar] [CrossRef]
- Nanduri, L.S.Y.; Duddempudi, P.K.; Yang, W.-L.; Tamarat, R.; Guha, C. Extracellular Vesicles for the Treatment of Radiation Injuries. Front. Pharmacol. 2021, 12, 1200. [Google Scholar] [CrossRef]
- Oppezzo, A.; Rosselli, F. The underestimated role of the microphthalmia-associated transcription factor (MiTF) in normal and pathological haematopoiesis. Cell Biosci. 2021, 11, 18. [Google Scholar] [CrossRef]
- Hu, R.; Sharma, S.M.; Bronisz, A.; Srinivasan, R.; Sankar, U.; Ostrowski, M.C. Eos, MITF, and PU.1 Recruit Corepressors to Osteoclast-Specific Genes in Committed Myeloid Progenitors. Mol. Cell. Biol. 2007, 27, 4018–4027. [Google Scholar] [CrossRef]
- Taussig, D.C. Hematopoietic stem cells express multiple myeloid markers: Implications for the origin and targeted therapy of acute myeloid leukemia. Blood 2005, 106, 4086–4092. [Google Scholar] [CrossRef]
- Cimato, T.R.; Furlage, R.L.; Conway, A.; Wallace, P. Simultaneous measurement of human hematopoietic stem and progenitor cells in blood using multicolor flow cytometry. Cytom. Part. B Clin. Cytom. 2015, 90, 415–423. [Google Scholar] [CrossRef]
- Manz, M.G.; Miyamoto, T.; Akashi, K.; Weissman, I.L. Prospective isolation of human clonogenic common myeloid progenitors. Proc. Natl. Acad. Sci. USA 2002, 99, 11872–11877. [Google Scholar] [CrossRef] [PubMed]
- Bakhtyar, N.; Jeschke, M.G.; Herer, E.; Sheikholeslam, M.; Amini-Nik, S. Exosomes from acellular Wharton’s jelly of the human umbilical cord promotes skin wound healing. Stem Cell Res. Ther. 2018, 9, 193. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Jing, Y.; Zhang, S.; Liu, Y.; Shi, Y.; Wei, L. The role of immunosuppression of mesenchymal stem cells in tissue repair and tumor growth. Cell Biosci. 2012, 2, 8. [Google Scholar] [CrossRef] [PubMed]
- Nag, D.; Bhanja, P.; Riha, R.; Sanchez-Guerrero, G.; Kimler, B.F.; Tsue, T.T.; Lominska, C.; Saha, S. Auranofin Protects Intestine against Radiation Injury by Modulating p53/p21 Pathway and Radiosensitizes Human Colon Tumor. Clin. Cancer Res. 2019, 25, 4791–4807. [Google Scholar] [CrossRef]
- Sadeghi, B.; Aghdami, N.; Hassan, Z.; Forouzanfar, M.; Rozell, B.; Abedi-Valugerdi, M.; Hassan, M. GVHD after chemotherapy conditioning in allogeneic transplanted mice. Bone Marrow Transplant. 2008, 42, 807–818. [Google Scholar] [CrossRef]
- Sawai, C.M.; Babovic, S.; Upadhaya, S.; Knapp, D.J.; Lavin, Y.; Lau, C.M.; Goloborodko, A.; Feng, J.; Fujisaki, J.; Ding, L.; et al. Hematopoietic Stem Cells Are the Major Source of Multilineage Hematopoiesis in Adult Animals. Immunity 2016, 45, 597–609. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Identified miRNA | p-Value | Counts | Target Genes |
|---|---|---|---|
| Hematopoietic Stem Cell Proliferation | |||
| hsa-miR-22-3p | 0.007 | 52 | CTC1, MECOM, WNT1 |
| hsa-miR-148a-3p | 0.017 | 11 | WNT1, WNT10B, WNT2B |
| hsa-miR-1246 | 0.029 | 2038 | CTC1, PIM1 |
| hsa-miR-19b-3p | 0.012 | 69 | ARIH2, ATXN1L, CD34, EIF2AK2, MECOM, N4BP2L2, SFRP2, THPO, WNT1, WNT2B |
| hsa-miR-495-3p | 0.033 | 11 | ACE, ARIH2, ATXN1L, CTC1, EIF2AK2, MECOM, N4BP2L2, NKAP, RUNX1, SFRP2, WNT2B |
| hsa-miR-765 | 0.040 | 83 | ACE, ARIH2, ATXN1L, CD34, EIF2AK2, ETV6, NKAP, PIM1, RUNX1, WNT1, WNT10B, WNT2B |
| hsa-miR-582-5p | 0.004 | 12 | ACE, ARIH2, ATXN1L, CTC1, EIF2AK2, MECOM, N4BP2L2, NKAP, PDCD2, PIM1, RUNX1, SFRP2, THPO, WNT1, WNT10B, WNT2B |
| hsa-miR-1915-3p | 0.018 | 613 | ACE, ARIH2, ATXN1L, CD34, CTC1, EIF2AK2, ETV6, N4BP2L2, NKAP, PDCD2, PIM1, RUNX1, SFRP2, THPO, WNT1, WNT10B, WNT2B |
| hsa-miR-570-3p | 0.031 | 11 | ARIH2, ATXN1L, CD34, CTC1, EIF2AK2, ETV6, MECOM, N4BP2L2, NKAP, PDCD2, PIM1, RUNX1, SFRP2, WNT1, WNT10B, WNT2B |
| hsa-miR-148b-3p | 0.034 | 18 | ACE, ARIH2, ATXN1L, CTC1, EIF2AK2, ETV6, MECOM, N4BP2L2, PDCD2, RUNX1, THPO, WNT1, WNT10B, WNT2B |
| hsa-miR-4516 | 0.046 | 2637 | ACE, ARIH2, ATXN1L, CD34, CTC1, EIF2AK2, ETV6, N4BP2L2, NKAP, PDCD2, PIM1, RUNX1, THPO, WNT1, WNT10B, WNT2B |
| Hematopoietic Stem Cell Homeostasis | |||
| hsa-miR-130a-3p | 0.044 | 13 | CCN3, NLE1, TCIRG1, UBAP2L |
| Hematopoietic Cell Lineage | |||
| hsa-miR-451a | 0.010 | 10 | IL6, IL6R |
| hsa-miR-34a-5p | 0.013 | 16 | CD24, CD44, CSF1R, IL6R, ITGA6, ITGB3, KIT, TFRC, TNF |
| hsa-miR-148b-3p | 0.022 | 18 | CSF1, ITGA5 |
| hsa-miR-34a-5p | 0.031 | 16 | CD24, CD44, CSF1R, IL6R, KIT |
| hsa-miR-451a | 0.035 | 10 | IL6, IL6R |
| Hematopoietic Stem Cell Differentiation | |||
| hsa-miR-223-3p | 0.007 | 46 | IL6, LMO2, STAT5A |
| hsa-miR-146a-5p | 0.008 | 66 | CXCR4, FOS, IL6, NOTCH1 |
| hsa-miR-184 | 0.020 | 24 | CSF1, NFATC2 |
| hsa-miR-34a-5p | 0.028 | 16 | FOS, KCNH2, LEF1, MYB, NOTCH1 |
| hsa-miR-223-3p | 0.041 | 46 | IL6, LMO2, STAT5A |
| hsa-miR-155-5p | 0.044 | 268 | FLI1, FOS, IL6, MXI1, MYB, SPI1, THRB |
| hsa-miR-34a-5p | 0.047 | 16 | FOS, ITGB3, KCNH2, LEF1, MYB, NOTCH1 |
| Regulation of Hematopoietic Stem Cell Differentiation | |||
| hsa-miR-615-3p | 0.048 | 13 | CBFB, CDK6, HSPA9, PSMB7, PSMD13, PSMD2, PSMD8, PSMF1, TCF3, YTHDF2 |
| hsa-miR-105-5p | 0.018 | 17 | CDK6, MYB |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chugh, R.M.; Bhanja, P.; Olea, X.D.; Tao, F.; Schroeder, K.; Zitter, R.; Arora, T.; Pathak, H.; Kimler, B.F.; Godwin, A.K.; et al. Human Peripheral Blood Mononucleocyte Derived Myeloid Committed Progenitor Cells Mitigate H-ARS by Exosomal Paracrine Signal. Int. J. Mol. Sci. 2022, 23, 5498. https://doi.org/10.3390/ijms23105498
Chugh RM, Bhanja P, Olea XD, Tao F, Schroeder K, Zitter R, Arora T, Pathak H, Kimler BF, Godwin AK, et al. Human Peripheral Blood Mononucleocyte Derived Myeloid Committed Progenitor Cells Mitigate H-ARS by Exosomal Paracrine Signal. International Journal of Molecular Sciences. 2022; 23(10):5498. https://doi.org/10.3390/ijms23105498
Chicago/Turabian StyleChugh, Rishi Man, Payel Bhanja, Ximena Diaz Olea, Fang Tao, Kealan Schroeder, Ryan Zitter, Tanu Arora, Harsh Pathak, Bruce F. Kimler, Andrew K. Godwin, and et al. 2022. "Human Peripheral Blood Mononucleocyte Derived Myeloid Committed Progenitor Cells Mitigate H-ARS by Exosomal Paracrine Signal" International Journal of Molecular Sciences 23, no. 10: 5498. https://doi.org/10.3390/ijms23105498
APA StyleChugh, R. M., Bhanja, P., Olea, X. D., Tao, F., Schroeder, K., Zitter, R., Arora, T., Pathak, H., Kimler, B. F., Godwin, A. K., Perry, J. M., & Saha, S. (2022). Human Peripheral Blood Mononucleocyte Derived Myeloid Committed Progenitor Cells Mitigate H-ARS by Exosomal Paracrine Signal. International Journal of Molecular Sciences, 23(10), 5498. https://doi.org/10.3390/ijms23105498