
Autoimmune Pancreatitis: From Pathogenesis to Treatment


 , ,
, , 

Abstract
1. Materials and Methods
2. Introduction
2.1. Definitions

2.2. Epidemiology
2.3. Diagnosis
Laboratory Findings in Aip
- Electrophoresis changes: Jacobs et al. suggested that a characteristic focal band bridging the β- and γ-fractions in serum protein electrophoresis may be an initial serologic clue to IgG4–RD research [25].
- Hypocomplementemia was found in 36% with AIP by Muraki et al. [26].
- ANA and RF positivity [27].
- Other autoantibodies: efforts have focused on finding organ-specific autoantibodies because they may have pathogenetic significance in triggering the disease, but the truth is that most of them are not very common in AIP or are seen only in low titers. The diverse antibodies include anti-lactoferrin (Anti-LF), anti-carbonic anhydrase II (Anti-CA-IIAb), anti-carbonic anhydrase IV (Anti-CA-IVAb), anti-pancreas secretory trypsin inhibitor (anti-PSTI), anti-trypsinogen, anti-amylase alpha, anti-heat shock protein 10 (anti-HSP10), and anti-plasminogen-binding protein peptide (anti-PBP); none of these autoantibodies can be considered disease-specific [27].
- A quantitative polymerase chain reaction (qPCR) for the analysis of IgG4+ RNA molecules was developed by Tabibian et al. in 2016 and used in a prospective case-control study of patients at two European medical centers. It was found that IgG4+ RNA could be a more accurate diagnostic marker than IgG4 protein. However, further studies are needed to validate this method [28].
- Circulating plasmablasts and CC-chemokine ligand 18 (CCL18) measured by flow cytometry and enzyme-linked immunosorbent assay (ELISA) are also cited as valuable biomarkers, not only for diagnosis, but also for disease monitoring, as indicated in the evidence-based UEG and SGF recommendations [23].
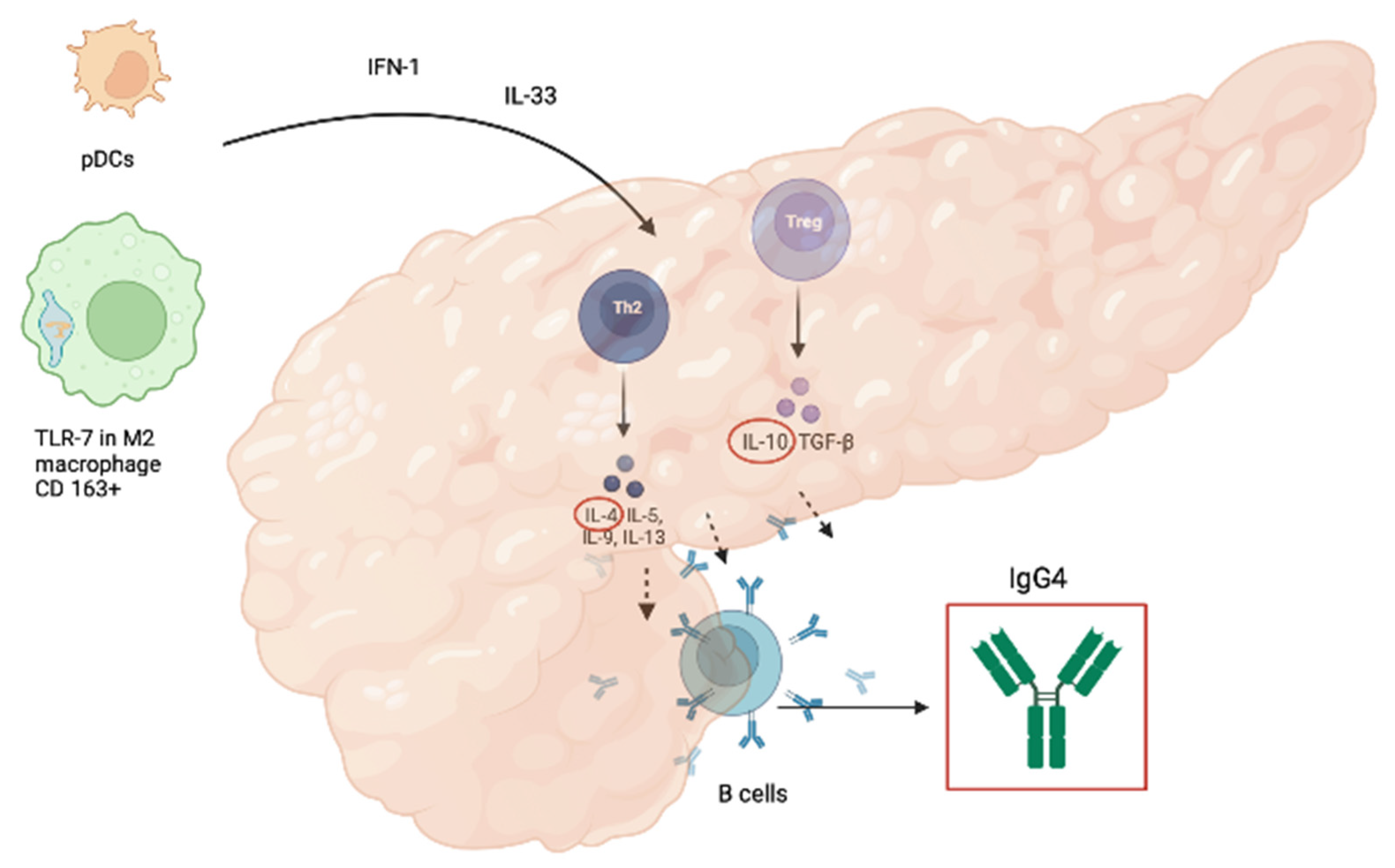
- IFN-alpha and IL-33, which are involved in the development of IgG4-RD, have diagnostic value as markers of type 1 AIP and/or IgG4-RD, comparable to that of serum IgG4 concentration, as calculated by Minaga et al.; they also showed markedly decreasing levels after induction of remission by prednisolone [29].
3. Pathogenesis
3.1. Pathogenesis of AIP-1
3.2. Pathogenesis of AIP-2
4. Therapy
4.1. Steroids
4.2. Steroid-Sparing Agents
- (1)
- The extensive metabolizers with wild-type genotype
- (2)
- The intermediate metabolizers with heterozygous variant genotype
- (3)
- The poor metabolizers with homozygous variant genotype, who should therefore receive alternative treatment [81].
4.3. Rituximab
4.4. Other Biological Therapies
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Shimosegawa, T.; Chari, S.T.; Frulloni, L.; Kamisawa, T.; Kawa, S.; Mino-Kenudson, M.; Kim, M.H.; Klöppel, G.; Lerch, M.M.; Löhr, M.; et al. International Consensus Diagnostic Criteria for Autoimmune Pancreatitis: Guidelines of the International Association of Pancreatology. Pancreas 2011, 40, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Sah, R.P.; Chari, S.T.; Pannala, R.; Sugumar, A.; Clain, J.E.; Levy, M.J.; Pearson, R.K.; Smyrk, T.C.; Petersen, B.T.; Topazian, M.D.; et al. Differences in Clinical Profile and Relapse Rate of Type 1 Versus Type 2 Autoimmune Pancreatitis. Gastroenterology 2010, 139, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Khandelwal, A.; Inoue, D.; Takahashi, N. Autoimmune Pancreatitis: An Update. Abdom. Radiol. 2020, 45, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Della-Torre, E.; Lanzillotta, M.; Doglioni, C. Immunology of IgG4-Related Disease. Clin. Exp. Immunol. 2015, 181, 191. [Google Scholar] [CrossRef]
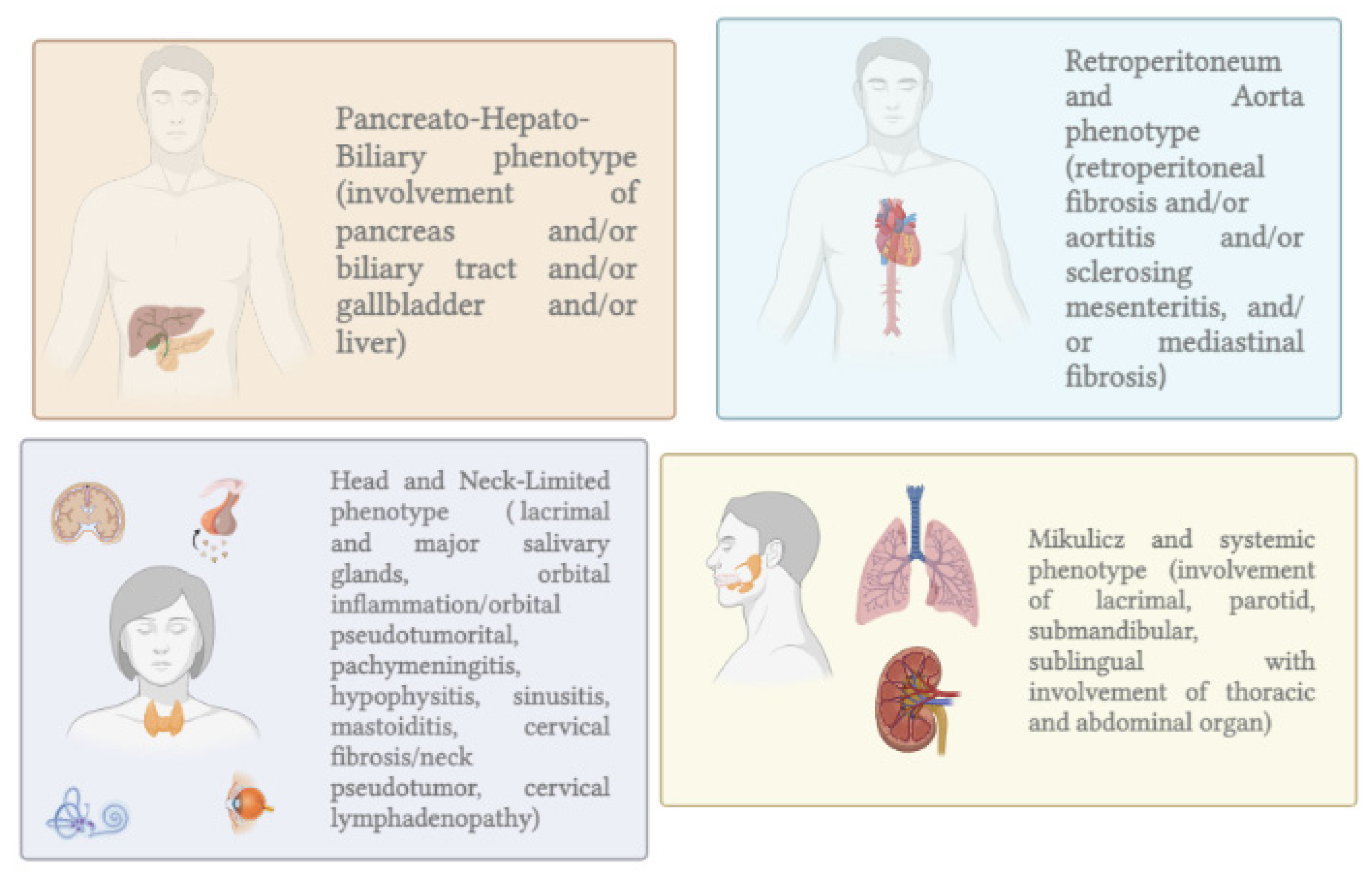
- Wallace, Z.S.; Zhang, Y.; Perugino, C.A.; Naden, R.; Choi, H.K.; Stone, J.H.; Takashi Akamizu, C.; Akiyama, M.; Bateman, A.; Blockmans, D.; et al. Clinical Phenotypes of IgG4-Related Disease: An Analysis of Two International Cross-Sectional Cohorts HHS Public Access. Eur. Leag. Against Rheum. 2018, 77, 70. [Google Scholar] [CrossRef]
- De Pretis, N.; Frulloni, L. Autoimmune Pancreatitis Type 2. Curr. Opin. Gastroenterol. 2020, 36, 417–420. [Google Scholar] [CrossRef]
- Zamboni, G.; Lüttges, J.; Capelli, P.; Frulloni, L.; Cavallini, G.; Pederzoli, P.; Leins, A.; Longnecker, D.; Klöppel, G. Histopathological Features of Diagnostic and Clinical Relevance in Autoimmune Pancreatitis: A Study on 53 Resection Specimens and 9 Biopsy Specimens. Virchows Arch. 2004, 445, 552–563. [Google Scholar] [CrossRef]
- de Pretis, N.; Vieceli, F.; Brandolese, A.; Brozzi, L.; Amodio, A.; Frulloni, L. Autoimmune Pancreatitis Not Otherwise Specified (NOS): Clinical Features and Outcomes of the Forgotten Type. Hepatobiliary Pancreat. Dis. Int. 2019, 18, 576–579. [Google Scholar] [CrossRef]
- Ikeura, T.; Manfredi, R.; Zamboni, G.; Negrelli, R.; Capelli, P.; Amodio, A.; Calió, A.; Colletta, G.; Gabbrielli, A.; Benini, L.; et al. Application of International Consensus Diagnostic Criteria to an Italian Series of Autoimmune Pancreatitis. United Eur. Gastroenterol. J. 2013, 1, 276. [Google Scholar] [CrossRef]
- Behzadi, F.; Suh, C.H.; Jo, V.Y.; Shanmugam, V.; Morgan, E.A.; Guenette, J.P. Imaging of IgG4-Related Disease in the Head and Neck: A Systematic Review, Case Series, and Pathophysiology Update. J. Neuroradiol. 2021, 48, 369–378. [Google Scholar] [CrossRef]
- Masamune, A.; Kikuta, K.; Hamada, S.; Tsuji, I.; Takeyama, Y.; Shimosegawa, T.; Okazaki, K. Nationwide Epidemiological Survey of Autoimmune Pancreatitis in Japan in 2016. J. Gastroenterol. 2016, 55, 462–470. [Google Scholar] [CrossRef]
- Kanno, A.; Masamune, A.; Okazaki, K.; Kamisawa, T.; Kawa, S.; Nishimori, I.; Tsuji, I.; Shimosegawa, T. Nationwide Epidemiological Survey of Autoimmune Pancreatitis in Japan in 2011. Pancreas 2015, 44, 535–539. [Google Scholar] [CrossRef]
- Kamisawa, T.; Chari, S.T.; Giday, S.A.; Kim, M.H.; Chung, J.B.; Lee, K.T.; Werner, J.; Bergmann, F.; Lerch, M.M.; Mayerle, J.; et al. Clinical Profile of Autoimmune Pancreatitis and Its Histological Subtypes: An International Multicenter Survey. Pancreas 2011, 40, 809–814. [Google Scholar] [CrossRef]
- Hart, P.A.; Kamisawa, T.; Brugge, W.R.; Chung, J.B.; Culver, E.L.; Czakó, L.; Frulloni, L.; Go, V.L.W.; Gress, T.M.; Kim, M.H.; et al. Original Article: Long-Term Outcomes of Autoimmune Pancreatitis: A Multicentre, International Analysis. Gut 2013, 62, 1771. [Google Scholar] [CrossRef]
- Barresi, L.; Tacelli, M.; Crinò, S.F.; Attili, F.; Chiara Petrone, M.; De Nucci, G.; Carrara, S.; Manfredi, G.; Capurso, G.; De Angelis, C.G.; et al. Multicentric Italian Survey on Daily Practice for Autoimmune Pancreatitis: Clinical Data, Diagnosis, Treatment, and Evolution toward Pancreatic Insufficiency. United Eur. Gastroenterol. J. 2020, 8, 705–715. [Google Scholar] [CrossRef]
- Okazaki, K.; Kawa, S.; Kamisawa, T.; Naruse, S.; Tanaka, S.; Nishimori, I.; Ohara, H.; Ito, T.; Kiriyama, S.; Inui, K.; et al. Clinical Diagnostic Criteria of Autoimmune Pancreatitis: Revised Proposal. J. Gastroenterol. 2006, 41, 626–631. [Google Scholar] [CrossRef]
- Chari, S.T.; Smyrk, T.C.; Levy, M.J.; Topazian, M.D.; Takahashi, N.; Zhang, L.; Clain, J.E.; Pearson, R.K.; Petersen, B.T.; Vege, S.S.; et al. Diagnosis of Autoimmune Pancreatitis: The Mayo Clinic Experience. Clin. Gastroenterol. Hepatol. 2006, 4, 1010–1016. [Google Scholar] [CrossRef]
- Chari, S.T. Diagnosis of Autoimmune Pancreatitis Using Its Fi ve Cardinal Features: Introducing the Mayo Clinic’s HISORt Criteria. J. Gastroenterol. 2007, 42, 39–41. [Google Scholar] [CrossRef]
- Maruyama, M.; Watanabe, T.; Kanai, K.; Oguchi, T.; Muraki, T.; Hamano, H.; Arakura, N.; Kawa, S. Clinical Study International Consensus Diagnostic Criteria for Autoimmune Pancreatitis and Its Japanese Amendment Have Improved Diagnostic Ability over Existing Criteria. Gastroenterol. Res. Pract. 2013, 2013, 456965. [Google Scholar] [CrossRef]
- Schneider, A.; Michaely, H.; Rückert, F.; Weiss, C.; Ströbel, P.; Belle, S.; Hirth, M.; Wilhelm, T.J.; Haas, S.L.; Jesenofsky, R.; et al. Diagnosing Autoimmune Pancreatitis with the Unifying-Autoimmune-Pancreatitis-Criteria. Pancreatology 2017, 17, 381–394. [Google Scholar] [CrossRef]
- Zen, Y. Type 2 Autoimmune Pancreatitis: Consensus and Controversies. Gut Liver 2022, 16, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Chari, S.T.; Takahashi, N.; Levy, M.J.; Smyrk, T.C.; Clain, J.E.; Pearson, R.K.; Petersen, B.T.; Topazian, M.A.; Vege, S.S. A Diagnostic Strategy to Distinguish Autoimmune Pancreatitis From Pancreatic Cancer. Clin. Gastroenterol. Hepatol. 2009, 7, 1097–1103. [Google Scholar] [CrossRef] [PubMed]
- Löhr, J.M.; Beuers, U.; Vujasinovic, M.; Alvaro, D.; Frøkjær, J.B.; Buttgereit, F.; Capurso, G.; Culver, E.L.; de-Madaria, E.; Della-Torre, E.; et al. European Guideline on IgG4-Related Digestive Disease–UEG and SGF Evidence-Based Recommendations. UEG J. 2020, 8, 637–666. [Google Scholar] [CrossRef] [PubMed]
- Lian, M.J.; Liu, S.; Wu, G.Y.; Liu, S.Y. Serum IgG4 and IgG for the Diagnosis of Autoimmune Pancreatitis: A Systematic Review with Meta-Analysis. Clin. Res. Hepatol. Gastroenterol. 2016, 40, 99–109. [Google Scholar] [CrossRef]
- Jacobs, J.F.M.; Van Der Molen, R.G.; Keren, D.F. Relatively Restricted Migration of Polyclonal IgG4 May Mimic a Monoclonal Gammopathy in IgG4-Related Disease. Am. J. Clin. Pathol. 2014, 142, 76–81. [Google Scholar] [CrossRef]
- Muraki, T.; Hamano, H.; Ochi, Y.; Komatsu, K.; Komiyama, Y.; Arakura, N.; Yoshizawa, K.; Ota, M.; Kawa, S.; Kiyosawa, K. Autoimmune Pancreatitis and Complement Activation System. Pancreas 2006, 32, 16–21. [Google Scholar] [CrossRef]
- Smyk, D.S.; Rigopoulou, E.I.; Koutsoumpas, A.L.; Kriese, S.; Burroughs, A.K.; Bogdanos, D.P. Review Article Autoantibodies in Autoimmune Pancreatitis. Int. J. Rheumatol. 2012, 2012, 940831. [Google Scholar] [CrossRef]
- Tabibian, J.H.; Lindor, K.D. Distinguishing Immunoglobulin G4-Related Disease from Its Pancreatobiliary Mimics: Are We There Now? Hepatology 2016, 64, 340–343. [Google Scholar] [CrossRef]
- Minaga, K.; Watanabe, T.; Hara, A.; Kamata, K.; Omoto, S.; Nakai, A.; Otsuka, Y.; Sekai, I.; Yoshikawa, T.; Yamao, K.; et al. Identification of Serum IFN-α and IL-33 as Novel Biomarkers for Type 1 Autoimmune Pancreatitis and IgG4-Related Disease. Sci. Rep. 2020, 10, 14879. [Google Scholar] [CrossRef]
- Kamisawa, T.; Anjiki, H.; Egawa, N.; Kubota, N. Allergic Manifestations in Autoimmune Pancreatitis. Eur. J. Gastroenterol. Hepatol. 2009, 21, 1136–1139. [Google Scholar] [CrossRef]
- Hirano, K.; Tada, M.; Isayama, H.; Kawakubo, K.; Sasaki, T.; Kogure, H.; Na-kai, Y.; Sasahira, N.; Tsujino, T.; Koike, K.; et al. Clinical Analysis of High Serum IgE in Autoimmune Pancreati-Tis. World J. Gastroenterol. 2010, 16, 5241–5246. [Google Scholar] [CrossRef]
- Watanabe, T.; Yamashita, K.; Fujikawa, S.; Sakurai, T.; Kudo, M.; Shiokawa, M.; Kodama, Y.; Uchida, K.; Okazaki, K.; Chiba, T. Involvement of Activation of Toll-like Receptors and Nucleotide-Binding Oligomerization Domain-like Receptors in Enhanced IgG4 Responses in Autoimmune Pancreatitis. Arthritis Rheum. 2012, 64, 914–924. [Google Scholar] [CrossRef]
- Ishiguro, N.; Moriyama, M.; Furusho, K.; Furukawa, S.; Shibata, T.; Murakami, Y.; Chinju, A.; Haque, A.S.M.R.; Gion, Y.; Ohta, M.; et al. Activated M2 Macrophages Contribute to the Pathogenesis of IgG4-Related Disease via Toll-like Receptor 7/Interleukin-33 Signaling. Arthritis Rheumatol. 2020, 72, 166–178. [Google Scholar] [CrossRef]
- Furukawa, S.; Moriyama, M.; Miyake, K.; Nakashima, H.; Tanaka, A.; Maehara, T.; Iizuka-Koga, M.; Tsuboi, H.; Hayashida, J.-N.; Ishiguro, N.; et al. Interleukin-33 Produced by M2 Macrophages and Other Immune Cells Contributes to Th2 Immune Reaction of IgG4-Related Disease. Sci. Rep. 2017, 7, srep42413. [Google Scholar] [CrossRef]
- Liew, F.Y.; Girard, J.-P.; Turnquist, H.R. IL-1 Family Interleukin-33 in Health and Disease. Nat. Rev. Immunol. 2016, 16, 676–689. [Google Scholar] [CrossRef]
- Cayrol, C.; Girard, J.P. IL-33: An Alarmin Cytokine with Crucial Roles in Innate Immunity, Inflammation and Allergy. Curr. Opin. Immunol. 2014, 31, 31–37. [Google Scholar] [CrossRef]
- Perugino, C.A.; Stone, J.H. IgG4-Related Disease: An Update on Pathophysiology and Implications for Clinical Care. Nat. Rev. Rheumatol. 2020, 16, 16–702. [Google Scholar] [CrossRef]
- Reizis, B. Plasmacytoid Dendritic Cells: Development, Regulation, and Function. Immunity 2019, 50, 37–50. [Google Scholar] [CrossRef]
- Swiecki, M.; Colonna, M. The Multifaceted Biology of Plasmacytoid Dendritic Cells. Nat. Rev. Immunol. 2015, 15, 471–485. [Google Scholar] [CrossRef]
- Psarras, A.; Emery, P.; Vital, E.M. Type I Interferon-Mediated Autoimmune Diseases: Pathogenesis, Diagnosis and Targeted Therapy. Rheumatology 2017, 56, 1662–1675. [Google Scholar] [CrossRef]
- Rodero, M.P.; Decalf, J.; Bondet, V.; Hunt, D.; Rice, G.I.; Werneke, S.; McGlasson, S.L.; Alyanakian, M.A.; Bader-Meunier, B.; Barnerias, C.; et al. Detection of Interferon Alpha Protein Reveals Differential Levels and Cellular Sources in Disease. J. Exp. Med. 2017, 214, 1547. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Yamashita, K.; Arai, Y.; Minaga, K.; Kamata, K.; Nagai, T.; Komeda, Y.; Takenaka, M.; Hagiwara, S.; Ida, H.; et al. Chronic Fibro-Inflammatory Responses in Autoimmune Pancreatitis Depend on IFN-α and IL-33 Produced by Plasmacytoid Dendritic Cells. J. Immunol. 2017, 198, 3886–3896. [Google Scholar] [CrossRef] [PubMed]
- Arai, Y.; Yamashita, K.; Kuriyama, K.; Shiokawa, M.; Kodama, Y.; Sakurai, T.; Mizugishi, K.; Uchida, K.; Kadowaki, N.; Takaori-Kondo, A.; et al. Plasmacytoid Dendritic Cell Activation and IFN-α Production Are Prominent Features of Murine Autoimmune Pancreatitis and Human IgG4-Related Autoimmune Pancreatitis. J. Immunol. 2015, 195, 3033–3044. [Google Scholar] [CrossRef] [PubMed]
- Kurimoto, M.; Watanabe, T.; Kamata, K.; Minaga, K.; Kudo, M. IL-33 as a Critical Cytokine for Inflammation and Fibrosis in Inflammatory Bowel Diseases and Pancreatitis. Front. Physiol. 2021, 12, 781012. [Google Scholar] [CrossRef]
- Fazekas, B.; Groth, D.S.; Cook, M.; Oliver, P.M.; Grados, A.; Ebbo, M.; Piperoglou, C.; Groh, M.; Regent, A.; Bonotte, B.; et al. T Cell Polarization toward T h 2/T Fh 2 and T h 17/T Fh 17 in Patients with Igg4-Related Disease. Amandine For. 2017, 8, 235. [Google Scholar] [CrossRef]
- Zen, Y.; Fujii, T.; Harada, K.; Kawano, M.; Yamada, K.; Takahira, M.; Nakanuma, Y. Th2 and Regulatory Immune Reactions Are Increased in Immunoglobin G4-Related Sclerosing Pancreatitis and Cholangitis. Hepatology 2007, 45, 1538–1546. [Google Scholar] [CrossRef]
- Romagnani, S. Regulation of the Development of Type 2 T-Helper Cells in Allergy. Curr. Opin. Immunol. 1994, 6, 838–846. [Google Scholar] [CrossRef]
- Nirula, A.; Glaser, S.M.; Kalled, S.L.; Taylora, F.R. What Is IgG4? A Review of the Biology of a Unique Immunoglobulin Subtype. Curr. Opin. Rheumatol. 2011, 23, 119–124. [Google Scholar] [CrossRef]
- Robinson, D.S.; Larché, M.; Durham, S.R. Tregs and Allergic Disease. J. Clin. Investig. 2004, 114, 1389–1397. [Google Scholar] [CrossRef]
- Zhang, L.; Guo, L.M.; Huang, Y.H.; Wang, T.L.; Shi, X.Y.; Chang, H.; Yao, W.; Huang, X.B. Allergic Diseases, Immunoglobulin E, and Autoimmune Pancreatitis: A Retrospective Study of 22 Patients. Chin. Med. J. 2014, 127, 4104–4109. [Google Scholar] [CrossRef]
- Della Torre, E.; Mattoo, H.; Mahajan, V.S.; Carruthers, M.; Pillai, S.; Stone, J.H.; John Stone, C.H. Prevalence of Atopy, Eosinophilia, and IgE Elevation in IgG4-Related Disease. Allergy 2014, 69, 269–272. [Google Scholar] [CrossRef]
- Kolfschoten, M.V.D.N.; Schuurman, J.; Losen, M.; Bleeker, W.K.; Martínez-Martínez, P.; Vermeulen, E.; Den Bleker, T.H.; Wiegman, L.; Vink, T.; Aarden, L.A.; et al. Anti-Inflammatory Activity of Human IgG4 Antibodies by Dynamic Fab Arm Exchange. Science 2007, 317, 1554–1557. [Google Scholar] [CrossRef]
- Kawa, S. The Immunobiology of Immunoglobulin G4 and Complement Activation Pathways in IgG4-Related Disease. Curr. Top. Microbiol. Immunol. 2017, 401, 61–73. [Google Scholar] [CrossRef]
- Sugimoto, M.; Watanabe, H.; Asano, T.; Sato, S.; Takagi, T.; Kobayashi, H.; Ohira, H. Possible Participation of IgG4 in the Activation of Complement in IgG4-Related Disease with Hypocomplementemia. Mod. Rheumatol. 2015, 26, 251–258. [Google Scholar] [CrossRef]
- Schepis, T.; De Lucia, S.S.; Nista, E.C.; Manilla, V.; Pignataro, G.; Ojetti, V.; Piccioni, A.; Gasbarrini, A.; Franceschi, F.; Candelli, M. Clinical Medicine Microbiota in Pancreatic Diseases: A Review of the Literature. J. Clin. Med. 2021, 10, 5920. [Google Scholar] [CrossRef]
- Ji, J.; Shu, D.; Zheng, M.; Wang, J.; Luo, C.; Wang, Y.; Guo, F.; Zou, X.; Lv, X.; Li, Y.; et al. Microbial Metabolite Butyrate Facilitates M2 Macrophage Polarization and Function. Sci. Rep. 2016, 6, 24838. [Google Scholar] [CrossRef]
- Kamata, K.; Watanabe, T.; Minaga, K.; Hara, A.; Yoshikawa, T.; Okamoto, A.; Yamao, K.; Takenaka, M.; Park, A.M.; Kudo, M. Intestinal Dysbiosis Mediates Experimental Autoimmune Pancreatitis via Activation of Plasmacytoid Dendritic Cells. Int. Immunol. 2019, 31, 795–809. [Google Scholar] [CrossRef]
- Kamata, K.; Watanabe, T.; Minaga, K.; Hara, A.; Sekai, I.; Otsuka, Y.; Yoshikawa, T.; Park, A.-M.; Kudo, M. Gut Microbiome Alterations in Type 1 Autoimmune Pancreatitis after Induction of Remission by Prednisolone. Clin. Exp. Immunol. 2020, 202, 308–320. [Google Scholar] [CrossRef]
- Yamaki, K.; Ohta, M.; Nakashima, I.; Noda, A.; Asai, J.; Kato, N. Microbial Adjuvant and Autoimmunity. Microbiol. Immunol. 1980, 24, 945–956. [Google Scholar] [CrossRef]
- Hsieh, S.C.; Shen, C.Y.; Liao, H.T.; Chen, M.H.; Wu, C.H.; Li, K.J.; Lu, C.S.; Kuo, Y.M.; Tsai, H.C.; Tsai, C.Y.; et al. The Cellular and Molecular Bases of Allergy, Inflammation and Tissue Fibrosis in Patients with IgG4-Related Disease. Int. J. Mol. Sci. 2020, 21, 1–24. [Google Scholar] [CrossRef]
- Frulloni, L.; Lunardi, C.; Simone, R.; Dolcino, M.; Scattolini, C.; Falconi, M.; Benini, L.; Vantini, I.; Corrocher, R.; Puccetti, A. Identification of a Novel Antibody Associated with Autoimmune Pancreatitis. N. Engl. J. Med. 2009, 361, 2135–2142. [Google Scholar] [CrossRef]
- Youssefi, M.; Tafaghodi, M.; Farsiani, H.; Ghazvini, K.; Keikha, M. Helicobacter Pylori Infection and Autoimmune Diseases; Is There an Association with Systemic Lupus Erythematosus, Rheumatoid Arthritis, Autoimmune Atrophy Gastritis and Autoimmune Pancreatitis? A Systematic Review and Meta-Analysis Study. J. Microbiol. Immunol. Infect. 2021, 54, 359–369. [Google Scholar] [CrossRef]
- Jun, S.Y.; Chun, J.; Kim, S.J.; Oh, D.; Kim, J.H.; Kim, M.H.; Hong, S.M. Granulocytic Epithelial Lesion (GEL) in Heterotopic Pancreas. Pancreatology 2022, 22, 435–442. [Google Scholar] [CrossRef]
- Loos, M.; Lauffer, F.; Schlitter, A.M.; Kleeff, J.; Friess, H.; Klöppel, G.; Esposito, I. Potential Role of Th17 Cells in the Pathogenesis of Type 2 Autoimmune Pancreatitis. Virchows Arch. 2015, 467, 641–648. [Google Scholar] [CrossRef]
- Dong, F.; Chen, Q.Q.; Zhuang, Z.H.; He, Q.L.; Wang, F.Q.; Liu, Q.C.; Liu, H.K.; Wang, Y. Multiple Gene Mutations in Patients with Type 2 Autoimmune Pancreatitis and Its Clinical Features. Cent. J. Immunol. 2014, 39, 77. [Google Scholar] [CrossRef]
- Ku, Y.; Hong, S.M.; Fujikura, K.; Kim, S.J.; Akita, M.; Abe-Suzuki, S.; Shiomi, H.; Masuda, A.; Itoh, T.; Azuma, T.; et al. IL-8 Expression in Granulocytic Epithelial Lesions of Idiopathic Duct-Centric Pancreatitis (Type 2 Autoimmune Pancreatitis). Am. J. Surg. Pathol. 2017, 41, 1129–1138. [Google Scholar] [CrossRef]
- Pearl, D.S.; Shah, K.; Whittaker, M.A.; Nitch-Smith, H.; Brown, J.F.; Shute, J.K.; Trebble, T.M. Cytokine Mucosal Expression in Ulcerative Colitis, the Relationship between Cytokine Release and Disease Activity. J. Crohn’s Colitis 2013, 7, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Hart, P.A.; Levy, M.J.; Smyrk, T.C.; Takahashi, N.; Dayyeh, B.K.A.; Clain, J.E.; Gleeson, F.C.; Pearson, R.K.; Petersen, B.T.; Topazian, M.D.; et al. Clinical Profiles and Outcomes in Idiopathic Duct-Centric Chronic Pancreatitis (Type 2 Autoimmune Pancreatitis): The Mayo Clinic Experience. Gut 2016, 65, 1702–1709. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Neyaz, A.; Chougule, A.; Akita, M.; Zen, Y.; Forcione, D.; Del Castillo, C.F.; Ferrone, C.R.; Deshpande, V. Autoimmune Pancreatitis Type 2: Diagnostic Utility of PD-L1 Immunohistochemistry. Am. J. Surg. Pathol. 2019, 43, 898–906. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, K.; Chari, S.T.; Frulloni, L.; Lerch, M.M.; Kamisawa, T.; Kawa, S.; Kim, M.H.; Lévy, P.; Masamune, A.; Webster, G.; et al. International Consensus for the Treatment of Autoimmune Pancreatitis. Pancreatology 2017, 17, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.M.; Chung, M.J.; Chung, J.B. Remission and Relapse of Autoimmune Pancreatitis: Focusing on Corticosteroid Treatment. Pancreas 2010, 39, 555–560. [Google Scholar] [CrossRef]
- Okazaki, K.; Kawa, S.; Kamisawa, T.; Ikeura, T.; Itoi, T.; Ito, T.; Inui, K.; Irisawa, A.; Uchida, K.; Ohara, H.; et al. Amendment of the Japanese Consensus Guidelines for Autoimmune Pancreatitis, 2020. J. Gastroenterol. 2022, 57, 225–245. [Google Scholar] [CrossRef]
- Kamisawa, T.; Okazaki, K.; Kawa, S.; Ito, T.; Inui, K.; Irie, H.; Nishino, T.; Notohara, K.; Nishimori, I.; Tanaka, S.; et al. Amendment of the Japanese Consensus Guidelines for Autoimmune Pancreatitis, 2013 III. Treatment and Prognosis of Autoimmune Pancreatitis. J. Gastroenterol. 2014, 49, 961–970. [Google Scholar] [CrossRef]
- Cain, D.W.; Cidlowski, J.A. Immune Regulation by Glucocorticoids. J. Gastroenterol. 2014, 49, 961–970. [Google Scholar] [CrossRef]
- Tacelli, M.; Celsa, C.; Magro, B.; Barresi, L.; Guastella, S.; Capurso, G.; Frulloni, L.; Cabibbo, G.; Cammà, C. Risk Factors for Rate of Relapse and Effects of Steroid Maintenance Therapy in Patients With Autoimmune Pancreatitis: Systematic Review and Meta-Analysis. Clin. Gastroenterol. Hepatol. 2019, 17, 1061–1072.e8. [Google Scholar] [CrossRef]
- Yoon, S.B.; Moon, S.H.; Kim, J.H.; Park, J.W.; Kim, S.E.; Kim, M.H. Determination of the Duration of Glucocorticoid Therapy in Type 1 Autoimmune Pancreatitis: A Systematic Review and Meta-Analysis. Pancreatology 2021, 21, 1199–1207. [Google Scholar] [CrossRef]
- Brito-Zerón, P.; Kostov, B.; Bosch, X.; Acar-Denizli, N.; Ramos-Casals, M.; Stone, J.H. Therapeutic Approach to IgG4-Related Disease: A Systematic Review. Medicine 2016, 95, e4002. [Google Scholar] [CrossRef]
- De Pretis, N.; Amodio, A.; Bernardoni, L.; Campagnola, P.; Capuano, F.; Chari, S.T.; Crinò, S.; Gabbrielli, A.; Massella, A.; Topazian, M.; et al. Azathioprine Maintenance Therapy to Prevent Relapses in Autoimmune. Clin. Transl. Gastroenterol. 2017, 8, e90. [Google Scholar] [CrossRef]
- Hart, P.A.; Topazian, M.D.; Witzig, T.E.; Clain, J.E.; Gleeson, F.C.; Klebig, R.R.; Levy, M.J.; Pearson, R.K.; Petersen, B.T.; Smyrk, T.C.; et al. Treatment of Relapsing Autoimmune Pancreatitis with Immunomodulators and Rituximab: The Mayo Clinic Experience. Gut 2013, 62, 1607–1615. [Google Scholar] [CrossRef]
- Masaki, Y.; Nakase, H.; Tsuji, Y.; Nojima, M.; Shimizu, K.; Mizuno, N.; Ikeura, T.; Uchida, K.; Ido, A.; Kodama, Y.; et al. The Clinical Efficacy of Azathioprine as Maintenance Treatment for Autoimmune Pancreatitis: A Systematic Review and Meta-Analysis. J. Gastroenterol. 2021, 56, 869–880. [Google Scholar] [CrossRef]
- Wilson, A.; Jansen, L.E.; Rose, R.V.; Gregor, J.C.; Ponich, T.; Chande, N.; Khanna, R.; Yan, B.; Jairath, V.; Khanna, N.; et al. HLA-DQA1-HLA-DRB1 Polymorphism Is a Major Predictor of Azathioprine-Induced Pancreatitis in Patients with Inflammatory Bowel Disease. Aliment. Pharmacol. Ther. 2018, 47, 615–620. [Google Scholar] [CrossRef]
- Leandro, M.; Isenberg, D.A. Rituximab–The First Twenty Years. Lupus 2021, 30, 371–377. [Google Scholar] [CrossRef]
- Nikolic, S.; Panic, N.; Hintikka, E.S.; Dani, L.; Rutkowski, W.; Hedström, A.; Steiner, C.; Löhr, J.M.; Vujasinovic, M. Efficacy and Safety of Rituximab in Autoimmune Pancreatitis Type 1: Our Experiences and Systematic Review of the Literature. Scand. J. Gastroenterol. 2021, 56, 1355–1362. [Google Scholar] [CrossRef]
- Carruthers, M.N.; Stone, J.H.; Deshpande, V.; Khosroshahi, A. Development of an IgG4-RD Responder Index. Int. J. Rheumatol. 2012, 2012, 259408. [Google Scholar] [CrossRef]
- Majumder, S.; Mohapatra, S.; Lennon, R.J.; Piovezani Ramos, G.; Postier, N.; Gleeson, F.C.; Levy, M.J.; Pearson, R.K.; Petersen, B.T.; Vege, S.S.; et al. Rituximab Maintenance Therapy Reduces Rate of Relapse of Pancreaticobiliary Immunoglobulin G4-Related Disease. Clin. Gastroenterol. Hepatol. 2018, 16, 1947–1953. [Google Scholar] [CrossRef]
- Carruthers, M.N.; Topazian, M.D.; Khosroshahi, A.; Witzig, T.E.; Wallace, Z.S.; Hart, P.A.; Deshpande, V.; Smyrk, T.C.; Chari, S.; Stone, J.H.; et al. Rituximab for IgG4-Related Disease: A Prospective, Open-Label Trial. Ann. Rheum. Dis. 2015, 74, 1171–1177. [Google Scholar] [CrossRef]
- Backhus, J.; Neumann, C.; Perkhofer, L.; Schulte, L.A.; Mayer, B.; Seufferlein, T.; Müller, M.; Kleger, A. A Follow-Up Study of a European IgG4-Related Disease Cohort Treated with Rituximab. J. Clin. Med. 2021, 10, 1329. [Google Scholar] [CrossRef]
- Ebbo, M.; lie Grados, A.; Samson, M.; Groh, M.; Loundou, A.; Rigolet, A.; Terrier, B.; Guillaud, C.; Carra-Dallière, C.; déric Renou, F.; et al. Long-Term Efficacy and Safety of Rituximab in IgG4-Related Disease: Data from a French Nationwide Study of Thirty-Three Patients. PLoS ONE 2017, 12, e0183844. [Google Scholar] [CrossRef] [PubMed]
- De Marchi, G.; de Pretis, N.; Gabrieletto, E.M.; Amodio, A.; Davì, V.; Crinò, S.F.; Gabbrielli, A.; Ciccocioppo, R.; Frulloni, L. Rituximab as Maintenance Therapy in Type 1 Autoimmune Pancreatitis: An Italian Experience. Pancreas 2021, 50, 1363–1367. [Google Scholar] [CrossRef] [PubMed]
- Soliman, H.; Vullierme, M.-P.; Maire, F.; Hentic, O.; Ruszniewski, P.; Lévy, P.; Rebours, V. Risk Factors and Treatment of Relapses in Autoimmune Pancreatitis: Rituximab Is Safe and Effective. United Eur. Gastroenterol. J. 2019, 7, 1073–1083. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Hao, M. Unique Properties of IgG4 Antibody and Its Clinical Application in Autoimmune Pancreatitis. Scand. J. Gastroenterol. 2018, 53, 1121–1131. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.H. Managing Premedications and the Risk for Reactions to Infusional Monoclonal Antibody Therapy. Oncologist 2008, 13, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, D.; Vullierme, M.P.; Rebours, V. Antitumor Necrosis Factor Therapy Is Effective for Autoimmune Pancreatitis Type 2. Am. J. Gastroenterol. 2020, 115, 1133–1134. [Google Scholar] [CrossRef]
- Naghibi, M.; Ahmed, A.; al Badri, A.M.; Bateman, A.C.; Shepherd, H.A.; Gordon, J.N. The Successful Treatment of IgG4-Positive Colitis with Adalimumab in a Patient with IgG4-Related Sclerosing Disease—A New Subtype of Aggressive Colitis? J. Crohns. Colitis 2013, 7, e81–e84. [Google Scholar] [CrossRef]
- Deeks, E.D. Anifrolumab: First Approval. Drugs 2021, 81, 1795–1802. [Google Scholar] [CrossRef]
- Takeuchi, T.; Tanaka, Y.; Matsumura, R.; Saito, K.; Yoshimura, M.; Amano, K.; Atsumi, T.; Suematsu, E.; Hayashi, N.; Wang, L.; et al. Safety and Tolerability of Sifalimumab, an Anti-Interferon-α Monoclonal Antibody, in Japanese Patients with Systemic Lupus Erythematosus: A Multicenter, Phase 2, Open-Label Study. Mod. Rheumatol. 2019, 30, 93–100. [Google Scholar] [CrossRef]
- Minaga, K.; Watanabe, T.; Hara, A.; Yoshikawa, T.; Kamata, K.; Kudo, M.; Mireille Rogers, N.; Bertsias, G.; Galgani, M. Plasmacytoid Dendritic Cells as a New Therapeutic Target for Autoimmune Pancreatitis and IgG4-Related Disease. Front. Immunol. 2021, 12, 713779. [Google Scholar] [CrossRef]
- Okazaki, K.; Ikeura, T.; Uchida, K. Recent Progress on the Treatment of Type 1 Autoimmune Pancreatitis and IgG4-Related Disease. Mod. Rheumatol. 2022, roac054. [Google Scholar] [CrossRef]
- Frampton, J.E. Inebilizumab: First Approval. Drugs 2020, 80, 1259–1264. [Google Scholar] [CrossRef]
- Oh, D.; Song, T.J.; Moon, S.H.; Kim, J.H.; Lee, J.N.; Hong, S.M.; Lee, J.S.; Jo, S.J.; Cho, D.H.; Park, D.H.; et al. Type 2 Autoimmune Pancreatitis (Idiopathic Duct-Centric Pancreatitis) Highlighting Patients Presenting as Clinical Acute Pancreatitis: A Single-Center Experience. Gut Liver 2019, 13, 461–470. [Google Scholar] [CrossRef]
- Poddighe, D. Autoimmune Pancreatitis and Pancreatic Cancer: Epidemiological Aspects and Immunological Considerations. World J. Gastroenterol. 2021, 27, 3825–3836. [Google Scholar] [CrossRef]
- Blaho, M.; Dítě, P.; Kunovský, L.; Kianička, B. Autoimmune PancreatitisAn Ongoing Challenge. Adv. Med. Sci. 2020, 65, 403–408. [Google Scholar] [CrossRef]
- Lanzillotta, M.; Della-Torre, E.; Wallace, Z.S.; Stone, J.H.; Karadag, O.; Fernández-Codina, A.; Arcidiacono, P.G.; Falconi, M.; Dagna, L.; Capurso, G. Efficacy and Safety of Rituximab for IgG4-Related Pancreato-Biliary Disease: A Systematic Review and Meta-Analysis. Pancreatology 2021, 21, 1395–1401. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
|
| For diagnosis of autoimmune pancreatitis, criterion 1 must be present, together with criterion 2, and/or 3. However, it is necessary to exclude malignant diseases such as pancreatic or biliary cancers. IgG: Immunoglobulin G; IgG4: Immunoglobulin G4 |
| Criterion | Level 1 | Level 2 |
|---|---|---|
| Parenchimal imaging Ductal imaging | Typical: diffuse enlargement with delayed enhancement (sometimes associated with rim-like enhancement) Long (>1/3 length of the main pancreatic duct) or multiple strictures without marked upstream dilatation | Indeterminate: segmental/focal enlargement with delayed enhancement (it also includes atypical findings, which include low-density mass, pancreatic ductal dilatation, and distal atrophy) Segmental or focal narrowing without marked upstream dilatation (duct size <5 mm) |
| Serology | IgG4 > 2x upper limit of normal value | IgG4 1–2 x upper limit of normal value |
| Other organ involvement | a or b a. Histology of extrapancreatic organs. Any of three of the following: -marked lynphoplasmacytic infiltration with fibrosis and without granulocytic infiltration -storiform fibrosis -obliterative phlebitis -abundant (>10 cells/HPF) IgG4-positive cells b. Typical radiology evidence At least one of the following: -segmental/multiple proximal or proximal and distal bile duct stricture -retroperitoneal fibrosis | a or b a. Histology of extrapancreatic organ including endoscopic biopsies of bile duct with both of the following: -marked lymphoplasmacytic infiltration without granulocytic infiltration -abundant (>10 cells/HPF) IgG4 positive cells b. Physical or radiological evidence of at least one of the following: -symmetrically enlarged salivary/lachrymal glands -radiological evidence of renal involvement in association with AIP |
| Histology of the pancreas | At least 3 of the following: -periductal lymphoplasmacitic infiltrate without granulocytic infiltration -obliterative phlebitis -storiform fibrosis -abundant (>10 cells/HPF) IgG4 positive cells | Any 2 of the following: -periductal lymphoplasmacitic infiltrate without granulocytic infiltration -obliterative phlebitis -storiform fibrosis -abundant (>10 cells/HPF) IgG4 positive cells |
| Response to steroid Rapid (<2 weeks) radiologically demonstrable resolution or marked improvement in pancreatic/extrapancreatic manifestations | ||
| Criterion | Level 1 | Level 2 |
|---|---|---|
| Parenchimal imaging Ductal imaging | Typical: diffuse enlargement with delayed enhancement (sometimes associated with rim-like enhancement) Long (>1/3 length of the main pancreatic duct) or multiple strictures without marked upstream dilatation | Indeterminate: segmental/focal enlargement with delayed enhancement (it also includes atypical findings, which include low-density mass, pancreatic ductal dilatation, and distal atrophy) Segmental or focal narrowing without marked upstream dilatation (duct size <5 mm) |
| Other organ involvement | / | Clinically diagnosed inflammatory bowel disease |
| Histology of the pancreas | IDCP Both of the following: -Granulocytic infiltration of duct wall (GEL) with or without granulocytic acinar inflammation -Absent or scant (0–10 cells/HPF) IgG4 positive cells | IDCP Both of the following: -granulocytic and lymphoplasmacytic acinar infiltrate -Absent or scant (0–10 cells/HPF) IgG4 positive cells |
| Response to steroid Rapid (<2 weeks) radiologically demonstrable resolution or marked improvement in pancreatic/extrapancreatic manifestations | ||
|
|
| 2.1 Histology: typical histological features (AIP-1; AIP-2) |
| 2.2 Imaging and serology: typical imaging findings and elevation of serum IgG4 other autoantibodies |
|
| Author, Year, [Ref.] | Patient’s Features | Type AIP | IBD | Treatment at AIP Recurrence | Anti TNF Therapy |
|---|---|---|---|---|---|
| Lorenzo D et al., 2020 [93] | Woman, 28 years old | AIP type 2 | Negative for IBD | Rituximab | Adalimumab |
| Lorenzo D et al., 2020 [93] | Woman, 16 years old | AIP type 2 | Ulcerative colitis | / | Adalimumab |
| Lorenzo D et al., 2020 [93] | Woman, 38 years old | AIP type 2 | Crohn disease | Azathioprine and then methotrexate | Infliximab |
| Lorenzo D et al., 2020 [93] | Woman, 18 years old | AIP type 2 | Crohn disease | Azathioprine | Adalimumab |
| Naghibi M et al., 2013 [94] | Woman, 16 years old | AIP type 1 | IgG4 positive colitis | Azathioprine and then infliximab | Adalimumab |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nista, E.C.; De Lucia, S.S.; Manilla, V.; Schepis, T.; Pellegrino, A.; Ojetti, V.; Pignataro, G.; Zileri dal Verme, L.; Franceschi, F.; Gasbarrini, A.; et al. Autoimmune Pancreatitis: From Pathogenesis to Treatment. Int. J. Mol. Sci. 2022, 23, 12667. https://doi.org/10.3390/ijms232012667
Nista EC, De Lucia SS, Manilla V, Schepis T, Pellegrino A, Ojetti V, Pignataro G, Zileri dal Verme L, Franceschi F, Gasbarrini A, et al. Autoimmune Pancreatitis: From Pathogenesis to Treatment. International Journal of Molecular Sciences. 2022; 23(20):12667. https://doi.org/10.3390/ijms232012667
Chicago/Turabian StyleNista, Enrico Celestino, Sara Sofia De Lucia, Vittoria Manilla, Tommaso Schepis, Antonio Pellegrino, Veronica Ojetti, Giulia Pignataro, Lorenzo Zileri dal Verme, Francesco Franceschi, Antonio Gasbarrini, and et al. 2022. "Autoimmune Pancreatitis: From Pathogenesis to Treatment" International Journal of Molecular Sciences 23, no. 20: 12667. https://doi.org/10.3390/ijms232012667
APA StyleNista, E. C., De Lucia, S. S., Manilla, V., Schepis, T., Pellegrino, A., Ojetti, V., Pignataro, G., Zileri dal Verme, L., Franceschi, F., Gasbarrini, A., & Candelli, M. (2022). Autoimmune Pancreatitis: From Pathogenesis to Treatment. International Journal of Molecular Sciences, 23(20), 12667. https://doi.org/10.3390/ijms232012667