Abstract
Amyloidoses is a group of diseases characterized by the accumulation of abnormal proteins (called amyloids) in different organs and tissues. For systemic amyloidoses, the disease is related to increased levels and/or abnormal synthesis of certain proteins in the organism due to pathological processes, e.g., monoclonal gammopathy and chronic inflammation in rheumatic arthritis. Treatment of amyloidoses is focused on reducing amyloidogenic protein production and inhibition of its aggregation. Therapeutic approaches critically depend on the type of amyloidosis, which underlines the importance of early differential diagnostics. In fact, the most accurate diagnostics of amyloidosis and its type requires analysis of a biopsy specimen from the disease-affected organ. However, absence of specific symptoms of amyloidosis and the invasive nature of biomaterial sampling causes the late diagnostics of these diseases, which leads to a delayed treatment, and significantly reduces its efficacy and patient survival. The establishment of noninvasive diagnostic methods and discovery of specific amyloidosis markers are essential for disease detection and identification of its type at earlier stages, which enables timely and targeted treatment. This review focuses on current approaches to the diagnostics of amyloidoses, primarily with renal involvement, and research perspectives in order to design new specific tests for early diagnosis.
1. Introduction
Amyloidoses is a heterogeneous group of diseases, which results from proteins misfolding with the formation of amyloid aggregates accumulated in tissues with subsequent progressive organ dysfunction and failure [1,2]. Amyloids are fibrils of orderly complexed proteins that are connected by hydrogen bonds to form an intermolecular cross-β-structure [3,4]. Amyloid fibrils are typically composed of two or more protofilaments or, in some cases, of a single protofilament. Protofilaments are connected to each other in a parallel fashion via their side chains [5]. Each protofilament has a cross-β structure, where β-strands are stacked perpendicular to the fibril axis [6]. Amyloids usually bind specific dyes, such as Thioflavin T (ThT) and Congo red [7,8,9], and exhibit resistance against the actions of proteases and various detergents [10,11]. The amyloidogenic properties may be a consequence of variations in the amino acid sequence and are manifested in different pathologies associated with increased levels of amyloidogenic protein [1,12,13,14]. Current classification of amyloidoses is based on the type of protein that predominantly forms fibrils in the deposits [5]. More than 30 amyloidogenic proteins and peptides have been found that accumulate in organs and tissues and cause approximately 70 different forms of amyloidoses in humans, with the majority being extremely rare [5,15].
The triggers of amyloidogenesis are not yet fully discovered. In the case of localized amyloidoses, individual organs may be affected by local production of amyloid protein in skin, soft tissues, urinary bladder, digestive tract, respiratory tract, and others [16].
The circulation of amyloidogenic proteins in systemic disease leads to amyloid deposition in multiple tissues throughout the body [17] (Figure 1). Systemic amyloidoses have a worse prognosis than the localized form of disease [18]. The most common types of systemic amyloidosis such as immunoglobulin (Ig) light chain (AL) amyloidosis, hereditary and wild-type transthyretin (ATTR) amyloidosis, serum amyloid A (AA) amyloidosis and leukocyte chemotactic factor 2 (ALECT2) amyloidosis cause progressive organ dysfunction and end-stage kidney disease [19,20,21]. The incidence of systemic amyloidosis exceeds 0.8/100,000 of the population [22].
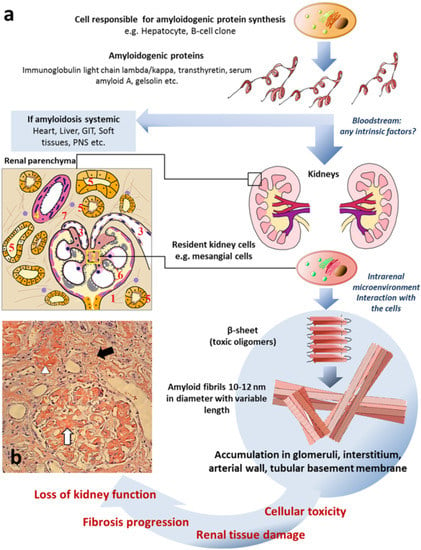
Figure 1.
Common mechanisms of renal amyloidosis. (a) Schematic representation of process of renal amyloidosis formation. GIT—gastrointestinal tract; PNS—peripheral nervous system; scheme of renal parenchyma: 1—glomerulus; 2—mesangium; 3—arterioles; 4—arterial wall; 5—tubules; 6—glomerular basement membrane; 7—interstitium. (b) The microphotograph demonstrating amyloid deposition in mesangium, capillary basement membranes and arterioles of glomerulus (white arrow), interstitium (black arrow) and arterial wall (arrowhead) presented as homogenous Congo-positive masses. Congo red stain, original magnification ×200 (the microphotograph was obtained by V.G. Sipovsky.
In the context of pathological processes, several diseases of the central nervous system (CNS), such as Alzheimer’s diseases and Parkinson’s diseases, form a specific distinct category in the amyloidosis entity. These disorders could result from the toxic effect of soluble oligomeric amyloid aggregates, rather than structural tissue alterations due to amyloid deposits, as it shown in cell culture and animal models [23,24]. Cellular toxicity of non-fibrillar (oligomeric) protein forms are also suggested for AL and ATTR amyloidoses [12,19,25].
The diagnosis of the amyloidosis type is critical for assigning therapies, reducing the specific protein level in the body, and inhibiting the pathological processes associated with this protein [26,27]. The effectiveness of the treatment largely depends on the stage at which the disease is diagnosed [28,29,30]. According to the current clinical criteria, the diagnosis of amyloidosis can be verified only by a morphological analysis of impaired organ biopsy samples, e.g., kidney biopsy in the case of renal amyloidosis [31]. The gold standard for diagnosis of amyloidosis is the positive staining of tissue specimens to amyloid-specific Congo red dye with apple green birefringence under polarized light microscopy [32,33].
Currently, there are different approaches based on immunodetection and mass spectrometric technologies that enable the type of amyloidosis to be determined by analysis of Congo red positive biopsy specimen (for review, see [34]). Current clinical practice based on clinical and histological presentation leaves many patients with systemic amyloidosis undiagnosed, especially those with earlier stages of the disease. Therefore, new approaches for amyloidosis confirmation and determination of its type are warranted. The only noninvasive method with 100% specificity and positive predictive value is based on infusion of 99mTc-labeled bone scintigraphy tracers and their accumulation in the myocardial amyloid deposits. This imaging technique, in fact, has been designed exclusively for the detection of cardiac ATTR amyloidosis in the absence of plasma cell dyscrasia [35]. Radioactively-labeled serum amyloid P component (SAP) can be applied for amyloid assessment, but only at a rather late stage, where massive tissue deposits are already apparent [36]. This method can hardly be used for differentiation of amyloidosis type (AA or AL) due to considerable overlap between patterns of organ involvement [36]. Hence, improvement in noninvasive diagnostics of amyloidosis and its types represents a significant clinical problem. The identification of new diagnostic markers for different amyloidosis types has clinical relevance, while being important for understanding the pathogenesis of the disease as well.
There have been previously published reviews considering methods of early noninvasive diagnosis of CNS amyloidosis and their significance for the effective monitoring and management of the disease [37,38,39]. This review focuses on the current advances and new approaches in noninvasive diagnostic modalities for systemic amyloidosis, primarily with renal involvement.
2. Different Types of Renal Amyloidosis
In systemic amyloidoses, kidney involvement is known to be very frequent and usually manifests with nephrotic syndrome and chronic kidney disease [19]. Nowadays, about 15 amyloid proteins are known to impair the kidneys (Table 1) with the pooled prevalence of AL, ALECT2, or AA types exceeding 88% [21]. The less common variants of renal amyloidosis are characterized by an accumulation of deposits of fibrinogen α chain, Ig heavy chains, apolipoproteins, and other precursor proteins (Table 1), as well as co-aggregation of Ig heavy and light chains [21,31]. Additionally, cases of two types of amyloidosis co-existing have also been described, either co-localizing in the same organ or affecting different organs [31,40]. Each type of amyloidosis has specific features depending on the affected renal structures (e.g., glomeruli, tubulointerstitium, and arteries) and the distribution of the renal deposits (diffuse or focal) [41]. Conversely, the wide range of morphological alterations and corresponding clinical manifestations may be observed at the same amyloidosis type [42,43]. In addition to kidney abnormalities, patients often have extrarenal disease manifestations (Table 1) [44,45]. Amyloid deposition in myocardium is associated with inferior patient survival [46], meanwhile kidney amyloid deposition results in end-stage kidney disease that requires renal replacement therapy [47,48].

Table 1.
Characteristics of renal amyloidoses [21,31,49,50,51,52].
AL-amyloidosis is the most common type of systemic amyloidosis in developed countries [22] and mainly relates to organ deposition of aberrant Ig kappa (Igκ) or Ig lambda (Igλ) light chains produced by the malignant clone of B-cell lineage (plasmacytic, lymphoplasmacytic, or lymphocytic). Due to numerous genetic changes, this clone co-produces light chain molecules with a destabilized structure of Ig variable domain [53]. The aggregation-prone state of the Ig variable domain of monoclonal protein is considered to be responsible for the progression of AL-amyloidosis [1].
AA-amyloidosis is another common type of renal amyloidosis [12], characterized by the tissue accumulation of serum amyloid A protein (SAA) aggregates. Increased SAA production is a response to chronic inflammation (rheumatoid arthritis, Crohn’s disease, chronic osteomyelitis, bronchiectasis, etc.). Other common types are hereditary and senile forms of ATTR amyloidoses, ATTRv and ATTRwt, respectively [54,55], caused by the accumulation of transthyretin protein (TTR). TTR accumulation primarily affects the heart, and only in rare cases, is ATTR associated with renal abnormalities [31]. ALECT2 and AFib amyloidoses are associated with deposition of leukocyte chemotactic factor 2 and fibrinogen α chain precursor proteins, respectively. New data indicate that ALECT2 amyloidosis may have a substantially higher prevalence than previously suspected. According to [21], ALECT2 accounts for 19.1% of the renal amyloidosis cases and yields only to AL amyloidosis. ALECT2 amyloidosis is most common in the Hispanic population [56] and primarily targets the kidneys among the elderly. AFib amyloidosis leads especially to end-stage kidney disease in the elderly, and the deposit formation is associated with a mutation in the FGA gene [57]. Amyloidoses associated with apolipoproteins and other proteins such as gelsolin and lysozyme do not involve kidneys.
3. Tissue- and Cell-Based Diagnostics of Amyloidosis
Early diagnostics of systemic amyloidosis is crucial as the disease has unfavorable prognosis if left untreated [22]. The median survival has been found to be about 6–11 years in AA amyloidosis [26,58] and about 1–3 years in AL amyloidosis [59,60] with the tendency toward declining mortality in the era of new treatment modalities [61]. Current available therapeutic options depend on the pathogenesis of particular amyloidosis type. The limitation of amyloid protein production by targeting specific pathological processes is the basic treatment approach. Such an approach, in particular, is used to reduce SAA levels by suppressing inflammation in rheumatoid arthritis, tuberculosis, periodontal disease, etc. [62]. A decrease in production of aberrant Ig light chains in AL-amyloidosis can be achieved by clone-directed chemotherapy, with or without autologous stem cell transplantation [63]. The treatment of hereditary ATTR amyloidosis is accomplished with liver transplantation, as the liver is a primary source of systemic TTR pool [64], and by prescribing functional tetramer TTR stabilizers [65,66]. The use of antisense oligonucleotides or silencing TTR mRNA has also found to be effective in phase 3 clinical trials [67,68]. Further efforts are in progress for AL amyloidosis in order to create stabilizers of the native dimeric structure of full-length Ig light chains or siRNA to reduce pathological λ-light-chain production [69,70]. Moreover, a number of amyloid-clearing immunotherapeutic agents are being clinically tested. Antibody targeting of amyloid proteins have not been proven as yet to be effective in systemic amyloidosis while studies on doxycycline treatment provided promising results (for review, see [27]).
The therapeutic efficacy is critically dependent on the stage of disease. The high mortality in AL amyloidosis [71] to a great extent refers to the difficulties in early diagnostics [72]. Disease recognition is complicated because clinical presentation of amyloidosis is largely non-specific and similar to that in other disease [73]. As a result, patients with amyloidosis often remain misdiagnosed and subjected to prolonged evaluation and unwarranted treatment. The assumption of amyloidosis is based on systemic organ damage, including nephrotic syndrome, unexplained cardiac dysfunction, autonomic or sensory neuropathies, periorbital purpura, weight loss, and other signs unrelated to the specific pathological processes [74].
In order to assess the degree of organs involvement and to clarify the type of amyloidosis, a number of noninvasive imaging tools could be used, such as echocardiography, magnetic resonance imaging, and imaging of radioactive tags injected into the circulation [75,76]. SAP protein with a radioactive tag, for instance, is trapped in amyloid deposits, thus making it possible to detect the localization of deposits by the radioactive signal. This method provides a high level of sensitivity and specificity (>90%) for the diagnosing of amyloidosis and determining the localization of the deposits in the body [36], however, without specifying the protein composition of the deposits. Important sensitive, although not specific, biomarkers of AL amyloidosis, are N-terminal pro-B-type natriuretic peptide (NT-proBNP) for the heart, proteinuria for the kidney, and alkaline phosphatase (ALP) for the liver. AL amyloidosis may occur in the presence of multiple myeloma (MM) most often being associated with monoclonal Igλ or λ free light chains (FLC) in the serum [63].
Suspected diagnosis of systemic amyloidosis with renal involvement should be proven by a morphological analysis of a kidney specimen. Alternatively, in a case of contraindications for kidney biopsy, one may consider an analysis of extrarenal tissue such as bone marrow, liver, and abdominal fat (for review, see [34]). However, the diagnostic value of non-renal samples is lower in comparison to renal ones [47,77].
Kidney biopsy specimens are studied using light microscopy, immunoassays, and electron microscopy. Special Congo red staining shows detected amyloid material as a red color under light microscopy and as apple green birefringence under polarized light as a result of dye binding with amyloids [32,33]. Immunofluorescence microscopy of frozen sections and immunogold labeling or immunohistochemistry (IHC) of formalin-fixed paraffin-embedded (FFPE) sections using antibodies against Igκ and Igλ, SAA, TTR and other amyloidogenic proteins make it possible to determine the type of amyloidosis [34]. The diagnostic effectiveness of IHC using antibodies against amyloid precursor proteins depends significantly on the quality of reagents, pathologist experience [78], and often requires antigen retrieval for antibodies in pretreatment with protease digestion, microwave heating, pressure cooking, and others [79]. In the case of AL amyloidosis, there is a possibility of false-negative results due to the presence of variable domains in Ig light chains [31], which can probably affect the folding of the proteins in aggregates and the epitope availability. Amyloids can include both full-length proteins and single fragments of Ig light chains, which also influences the efficiency of immunodetection [80,81] and the ability to specify the type of light chains, Igκ or Igλ [82]. In contrast, false-positive results are problematic for the diagnosis of AA amyloidosis [83]. At the same time, automatic assay systems with 100% sensitivity and specificity in diagnosing amyloidosis using an optimized antibody panel have been recently developed [84]. Immunoelectron microscopy (IEM) employing gold-labeled secondary antibodies provides more specificity in comparison to other antibody-based techniques due to the ability to detect nanostructures (amyloid fibrils and non-amyloid complexes) to which antibodies bind [85,86] and thereby exclude background staining.
The most effective approach for diagnosing amyloidosis and its type is laser microdissection followed by tandem mass spectrometry (LMD–MS) of biopsy tissue specimens [87]. Proteins isolated from Congo red-stained FFPE sections are tryptically digested into peptides that are sequenced by mass spectrometry (MS). This method also provides the identification of common amyloidosis marker proteins such as SAP and apolipoprotein E (amyloid signatures), for which its presence in samples serves as an internal control of procedure and confirms amyloid deposits [49,88]. LMD–MS tissue analysis provides the opportunity to correctly identify the amyloidosis type with sensitivity and specificity up to 100% [89,90,91]. It should be noted that the LMD–MS-approach has demonstrated greater efficiency compared to immunoassays for the identification of amyloidosis type on specimens from the same batch. According to [91], LMD–MS of FFPE affected organ biopsy samples enabled identification of amyloidosis type in 92% of cases, whereas the use of antibodies provided detection in only 45% of cases on specimens from the identical sampling. Gilbertson et al. demonstrated 100% concordance between positive IHC and LMD–MS in 142 sequential biopsy samples from 38 different tissue types; however, the diagnostic accuracy was 76% and 94%, respectively [90]. Gonzalez Suarez reported that immunofluorescence staining for Igκ or Igλ has inferior sensitivity and specificity compared with LMD–MS in the typing of 170 cases of renal immunoglobulin-derived amyloidosis [92]. Identification of 12.3% of cases failed in the immunofluorescence assay. LMD–MS is also an essential method for identifying extremely rare forms of amyloidosis [21]. Along with LMD–MS, there are other MS-based proteomics methods demonstrating their advantage over antibody-based techniques. The study by [93] reported that 2D-PAGE-based comparative proteomics allowed the identification of amyloidosis type in two cases in which the IEM assay was unsuccessful. However, immunoassays are still the most common diagnostic technique in clinical laboratories because the technique is more reproducible and lower in cost than LMD–MS.
Mutation studies for various hereditary amyloidosis are performed to verify the amyloidosis type identified by biopsy analysis and also to refine the disease predicting [94]. Specifically, ATTRv amyloidosis are related to the presence of point mutations in the TTR gene including Val30Met, Val122Ile, Thr60Ala, and AFib amyloidosis is caused by point mutations (most common Glu526Val) or frameshift mutations in the FGA gene [95,96]. When AA amyloidosis due to systemic autoinflammatory disease is suspected, the diagnostics can be complemented with genetic testing on mutations in MEFV, NLRP3, MVK, and other genes [97]. A current list of mutations could be viewed at http://amyloidosismutations.com/ (accessed on 19 September 2022) [98]. Genetic analysis is essential, as hereditary amyloidosis may mimic AL in immunoassays [99]. Notably, genetic analysis requires caution because the penetrance of mutations can be highly variable [57,100]. A more reliable method to verify the hereditary amyloidosis type (sensitivity 92% and specificity 100%) is mass spectrometric detection of variant proteins isolated from biopsy specimens [101].
Thus, the analysis of biopsy-derived material is an effective method of diagnosing the amyloidosis type. However, kidney biopsy is generally performed by indication in the case of pronounced organ dysfunctions and higher risks of treatment failure. Another major disadvantage of this approach is the invasive nature of the procedure and risk of complications that limit its use in certain cases.
4. Noninvasive Evaluation of Amyloidosis and Its Types
A major area of noninvasive diagnostics is the development of scanning research methods (radiography, computed tomography, ultrasound, magnetic resonance imaging, bone scintigraphy, and others). Currently, the application of those methods is limited to the late stages of amyloidosis in the presence of the significant deposition of amyloid aggregates in the organs [102,103,104]. Therefore, imaging is mainly applied to verify the amyloidosis diagnosis and clarify the type of amyloidosis. For example, 123I-labeled SAP scintigraphy provides detection of either AA- or AL- amyloidosis types with 90% sensitivity. However, this approach has limited value because of overlapping patterns of organ involvement for both types of amyloidosis [36]. Injection of 99mTc-labeled bone scintigraphy tracers into circulation followed by the visualization of their accumulation in myocardial amyloid deposits provides specificity and a positive predictive value of 100% for the diagnosis of ATTR cardiac amyloidosis, but only in the absence of plasma cell dyscrasia [35]. At the same time, plasma cell dyscrasia is a common competing disease, especially in the elderly, and was detected in 39% of ATTRwt cases and in 49% of patients with ATTRv amyloidoses [105]. Positron emission tomography and computed tomography with thioflavin-analogue tracers [106,107] enable the detection of Ig light chain deposits in organs not previously identified by clinical manifestations or biopsy. While being effective for the detection of asymptomatic amyloid in some extra-renal organs, PET identified renal involvement in fewer subjects than the international consensus diagnostic approach was able to [108]. Research applying diffusion-weighted magnetic resonance imaging, which is sensitive to local water motion in the tissue, becomes more promising for the detection of amyloid nephropathy [102]. However, the sensitivity and specificity of that method in the detection of amyloidosis are still low (79% and 60%, respectively). Collectively, imaging methods continue to be only a supporting tool in the diagnosis of renal amyloidosis.
Another area of noninvasive diagnosis is the analysis of body fluids (saliva, blood, urine, mucous membrane epithelial scrapes), which are collected without damaging the internal organs affected by amyloidosis. This approach provides opportunities for detecting factors and disease markers before the occurrence of severe systemic abnormalities that are recalcitrant to therapy. Fluid analysis, firstly, simplifies the condition monitoring of patients with already diagnosed amyloidosis and, secondly, enables the screening of the early stages of disease until progressive systemic abnormalities become apparent.
Since the 1970s, there have been several attempts to detect amyloid aggregates in the urine. Several studies have demonstrated the presence of different types of fibrillar structures in the urine in amyloidosis [109,110]. This approach, however, has been called into question by the fact that fibrils were not found in all specimens from patients with amyloidosis, and were also observed in control specimen groups [111,112,113].
Nowadays, there are a variety of methods for detecting the precursor protein of amyloidosis in serum and urine. Palladini et al. demonstrated that immunofixation electrophoresis of serum and urine (IFE) performed with anti-IgG, -IgA, -IgM, -Igκ, and -Igλ antibodies on gels could identify the amyloidogenic Ig light chains in all 115 patients with a monoclonal gammopathy [114]. The serum FLC immunoassay used for calculating the Igκ/Igλ ratio had less sensitivity (76%). However, a combination of these two methods had a 100% sensitivity [114]. Igκ/Igλ ratio outside of the physiological range (0.26–1.65) with high urine albumin levels implicates renal AL amyloidosis [115], and increased serum NT-proBNP values indicate cardiac AL amyloidosis in the absence of echographic features of heart involvement [116]. Despite the methods reaching 100% sensitivity in the detection of amyloidosis, they are nonspecific and require further verification of the diagnosis. Ig light chain is found in 100% of cases with other types of monoclonal gammopathies, including light chain monoclonal gammopathy of undetermined significance (MGUS) [117], which is significantly more prevalent than AL amyloidosis [118]. As noted above, monoclonal light chains could be found in the serum in patients with ATTRwt, AA, and other amyloidosis types [105,119]. On the contrary, increased levels of precursor protein SAA (>10 mg/mL) are associated with the progression of established AA amyloidosis in most cases [120]. However, SAA concentrations have no value for the prediction of this amyloid type incidence [121] and, hence, cannot substitute standard histological diagnostics.
The possibility of detection of precursor proteins in biological fluids by MS is currently being studied (for review, see [122]), despite the nonspecificity of the approaches to the identification of precursor proteins in fluids. These methods frequently involve the use of antibodies to enrich samples with putative precursor proteins because of the high content of non-target proteins in plasma and urine specimens (in the case of proteinuria) [123,124,125]. Using the MS provides an opportunity to detect the monoclonal protein in the serum and urine at concentrations below the threshold values for the IFE and FLC assay [124,126]. The MS of serum samples for ATTR amyloidosis enables the diagnosis of inherited forms of that disease [127]. The detection of peptides in serum and plasma samples with specific post-translational modifications at Cys-10 by liquid chromatography–MS is considered to be a promising approach for the diagnosis of ATTRv amyloidosis [123].
New perspectives in the analysis of precursor proteins of AL amyloidosis include the assessment of the Ig light chain functional properties detected in body fluids. The glycosylation of precursor proteins, their toxicity, and the genes encoding the monoclonal protein provide a high-level confidence in predicting of amyloidosis in a group of patients with MGUS, smoldering myeloma, or MM [13,128,129,130,131]. Proteomic analysis of urine exosome can be used for the differential diagnosis of renal disorders in monoclonal gammopathies [132]. In particular, in renal AL amyloidosis out of remission, immunoreactive proteins are found in urine exosomes corresponding to λ light chains, in contrast to non-amyloid renal injury in MM [132]. The use of the nematode Caenorhabditis elegans as an object for testing the toxicity of a monoclonal protein (“biosensor”) has demonstrated the possibility of diagnosing the cardiac AL amyloidosis in MM patients by the level of pathogenic effects on the pharynx of nematode [13]. Amyloidogenic Ig light chains isolated from the urine and serum of patients with cardiac AL amyloidosis caused cell death in the pharynx and reduced pumping rate. In contrast, non-cardiotoxic Ig light chains from patients with renal amyloidosis and MM had no effect on organ function [17]. Kumar et al. [131] found the presence of N-glycosylation of Ig light chains in 33 of 189 samples obtained by immunoprecipitation from the serum of patients with AL amyloidosis. Glycosylation of Igκ was detected for 32.8% of AL patients, and glycosylation of Igλ–for 10.2% of cases. The glycosylation rate in patients who had a detected monoclonal protein but without of AL amyloidosis was only 4.1% (5 of 122 cases) [131]. As a result, the risk group screening for Ig light chains glycosylation has a positive predictive value of 86.8%, which is extremely high for a noninvasive approach. Subsequent study has indicated that glycosylation in the MGUS group makes it possible to assess the risks of AL amyloidosis. Rates of progression at 20 years were 21% and 3% for AL patients with and without glycosylated light chains, respectively [129]. Kumar et al. also reported that glycosylation more frequently affects sites of polypeptides encoded by genes of the KV1 and LV3 families [131]. It has been previously demonstrated that proteomics-determined Ig germline gene usage provides a risk assessment of organ damage in AL amyloidosis [130]. Light chain variable region (IGVL) genes LV6-57, LV3-01, and LV3-21 are associated with renal injury, LV1-44 is more frequently identified in patients with cardiac AL amyloidosis, LV2-14 and KV1-33 usage are more probable if peripheral nerve and liver, respectively, are involved [130]. At the same time, in another recent study of Ig germline gene usage by cDNA analyses IGLV1-44 was the most dominant IGLV-subfamily for patients with dominant kidney involvement and IGLV3-21 with dominant heart involvement [128]. These and other inconsistencies indicate that the clinical significance of the identification of IGVL genes encoding the monoclonal protein in patients with AL amyloidosis requires further evaluation.
Thus, the available range of noninvasive methods in the amyloidosis evaluation seems to be useful in clarifying the organ involvement, assessment prognosis, and disease monitoring (Figure 2). However, these methods do not allow us to make an ultimate diagnosis of amyloidosis per se and to ascertain its type.
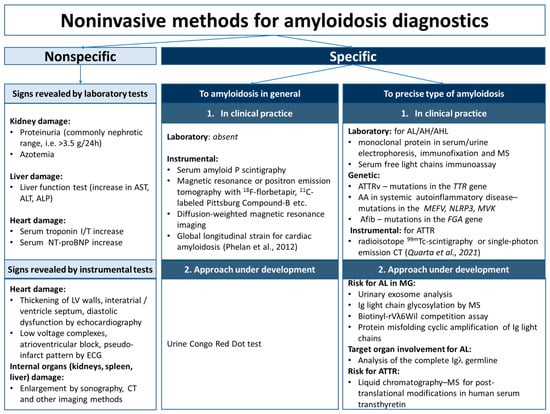
Figure 2.
Noninvasive methods for amyloidosis diagnostics. AA—serum amyloid A amyloidosis; AFib—fibrinogen α chain amyloidosis; AH—immunoglobulin heavy chains amyloidosis; AHL—immunoglobulin heavy and light chains amyloidosis; AL—immunoglobulin light chains amyloidosis; ALP—alkaline phosphatase; ALT—alanine aminotransferase; AST—aspartate aminotransferase; ATTRv—hereditary type of transthyretin amyloidosis; CT—computed tomography; ECG—electrocardiography; I/T—troponin I/troponin T; LV, left ventricle; MG—monoclonal gammopathy; MS—mass spectrometry; NT-proBNP—N-terminal pro-B-type natriuretic peptide [133,134].
5. Promising Trends in Noninvasive Diagnostics
In the absence of noninvasive methods of early diagnosis for the majority of amyloidosis, it is necessary to carry out research with new approaches. One of the trends is to use specific properties of amyloid aggregates and precursor proteins. These properties include the seeding of amyloidogenic protein monomers by preexisting aggregates [135], amyloid resistance to detergents and proteases [10,136], and the recruitment of amyloidogenic monomer protein by synthetic amyloid fibrils [137] (see Figure 3).
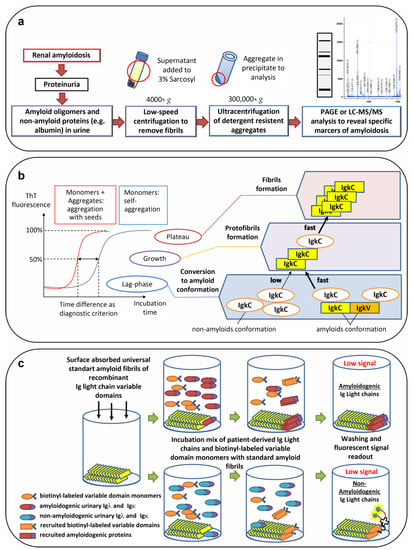
Figure 3.
Perspective approaches for noninvasive diagnostics of amyloidoses. (a) Methods based on the amyloid resistance to detergents. (b) Seeding of amyloidogenic protein monomers by preexisting aggregates. (c) Recruitment of amyloidogenic monomer proteins by synthetic amyloid fibrils. PAGE—polyacrylamide gel electrophoresis; LC-MS/MS—liquid chromatography with tandem mass spectrometry; ThT—thioflavin; IgκC—Igκ constant domain; IgκV—Igκ variable domain.
Amyloid aggregates, characterized by the presence of an array of β-strands, are known to induce and promote the formation of new aggregates by monomers of these proteins (nucleation and secondary seeding processes, for review, see [138]). The addition of aggregates to the monomer solution and periodic fragmentation of nascent fibrils leads to exponential growth of aggregated protein forms, making the detection of amyloids in biological fluids possible even at extremely low concentrations [139,140]. Both of these and related amyloid amplification methods are abbreviated as PMCA, from Protein Misfolding Cyclic Amplification, or QuIC, from Quaking-Induced Conversion. New-forming fibrils, typically, can be detected by Thioflavin T (ThT), the fluorescent dye. Previously, use of urine samples as seed in PMCA demonstrated high sensitivity for diagnosing diseases associated with amyloid deposition of prion proteins in the brain [141]. It is known that the Ig light chains isolated from the urine of patients with amyloidosis are capable of aggregation in vitro, as are prion proteins [142]. Alternatively, [143] reported that the ex vivo fibrils extracted from the heart of patient with cardiac AL amyloidosis accelerated fibril formation of homologous and non-homologous monomers of Igλ variable domain. The acceleration of time-resolved ThT fibrillation kinetics with ex vivo fibrils as seeds has also been demonstrated for TTR and SAA proteins [136,144]. Until now, no one has reported the possibility of detecting minor amounts of these protein aggregates in the urine or blood by PMCA or QuIC. In the future, since PMCA-based detection frequently demonstrates high sensitivity, that approach could provide detection of fibrils and specific oligomeric forms of pathological Ig light chains variants scarcely represented in the biological fluids of patients with amyloidosis.
One of the challenges in applying the protein amplification approach to AL amyloidosis is the unique nature of each Ig light chain due to the complementarity-determining regions of the variant domain. This problem could be solved by selection of the monomeric protein required to be induced by the aggregates from the biological fluids. The amino acid sequence of the selected protein will differ from the sequence of Ig light chains and their fragments in aggregates obtained from different patients. Therefore, the amplification reaction must proceed through cross-seeding between heterologous proteins [145]. Blancas-Mejía et al. show that the time of Igκ variable domain aggregation upon the addition of aggregates obtained in vitro from domains with a different sequence (sequence identity with the IGKV 1–33 germline gene 91–96%) varies significantly in various combinations [146]. Cross-seeding most often caused the acceleration of monomer aggregation, but the lag phase reduction could vary considerably for different monomeric proteins. The lag-phase reduction in cross-seeding was occasionally shorter compared to homologous seeding. Moreover, for one variant of the aggregates, an inhibition of the amplification rate during the cross-seeding of a monomeric protein variant was observed [146]. The authors generally conclude that the efficiency of seeding is determined mainly by the amyloidogenic properties of the monomers in solution rather than by the properties of the seed. Notably, the addition of aggregates formed by Igκ variable domains had no effect on the aggregation kinetics of monomeric variable domains from the λ1b subgroup (sequence identity with the germline gene IGKV 1-33 48%) [146].
Another interesting finding of [146] was the fact that aggregates of the germline gene IGKV 1-33 product can accelerate fibril formation in a solution of full-length Igκ monomers. That result raises the issue of considering the use of Igκ constant domain monomers for the detection of amyloidogenic monoclonal proteins in biological fluids. Since the constant domain is unchanged in all Igκ variants except for rare mutations, its use can provide a stable seeding with lower effects on aggregation kinetics varying between samples. It was demonstrated that monomers of the constant domain of Igκ light chains exhibit in vitro self-aggregation with the forming of amyloid fibrils [147]. In addition, amyloid aggregates formed by Igκ constant domain were described [148]. The successful use of protein fragments for amplification of aggregates in biological sample has been shown previously for the detection of human Creutzfeldt–Jakob disease using cerebrospinal fluid [149]. Amyloidogenic Ig light chains as monomers, oligomers, and as a part of aggregates can also possibly promote faster conversion of constant domain monomers to amyloid conformation both through direct interaction with constant domains in aggregates and through cross-seeding by amyloidogenic sites in the variable part of light chains [145].
A different approach for the optimization of monoclonal protein amyloidogenicity assessments is the development of universal standard aggregates of Ig light chains. For the testing amyloidogenicity of Ig light chains, [137] used synthetic amyloid fibrils composed of a λ6 variable domain isolated from an AL patient (rVλ6Wil). Four samples of AL-associated radiolabeled urinary Igλ and Igκ bound rVλ6Wil fibrils (recruitment) more efficiently than four samples of MM-associated proteins. Furthermore, in MM patients with abnormally high Ig light chain recruitment, AL amyloidosis was subsequently diagnosed, thus suggesting the prognostic potential of the proposed methodological approach. Notably, the AL-associated protein recruitment to Aβ(1–40) fibrils was more pronounced in comparison to MM proteins. The method was further improved by using biotinyl-λ6 variable domain monomers in solution together with patient-derived urinary proteins [150]. Competitive binding to rVλ6Wil fibrils between recombinant λ6 variable domain and urinary proteins permitted separation of MM and AL patient groups with 100% specificity and sensitivity by concentration-dependent inhibition of biotinyl-λ6 variable domain recruitment [150].
The assessment of amyloidogenicity of protein components and detection of amyloid aggregates in biological fluids are complicated by the presence of a large number of other proteins (not participating in amyloidogenesis). Specifically, albumin, the most abundant protein in serum and in urine samples from patients with nephrotic syndrome can inhibit amyloid formation [151]. Martin et al. isolated Ig light chains from urine samples [150] following the protocol proposed by [152] in several steps, including dialysis, zone electrophoresis, and gel filtration. In the mass spectrometric studies [124,127,153], the issue of target factor concentration was solved by preliminary immunoprecipitation of proteins suspected for amyloid formation.
Alternatively, one way to dispose of bulk protein and prepare a sample to assess the amyloidogenicity of the protein components is ultracentrifugation at speeds in the range of thousands g and treatment of the deposit with ionic detergents, such as sarcosyl and sodium dodecyl sulfate. It provides detergent-resistant aggregates from the sample and can be used for research and diagnostic purposes [11]. Ultracentrifugation eliminates the background of factors not involved in the aggregate formation, and treatment with detergents enables the avoidance of protein complexes of non-amyloid nature [154]. According to our preliminary data, the quantitative and qualitative composition of proteins in the fraction of aggregates resistant to the treatment with 3% sarcosyl significantly varies in urine samples with different etiologies of proteinuria and significantly differs in protein composition from the untreated samples (Figure 4).
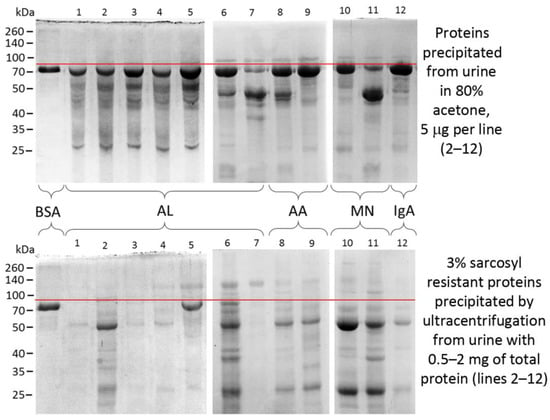
Figure 4.
Proteins of urine and detergent-resistant aggregates from patients with proteinuria. Samples are separated in polyacrylamide gel electrophoresis and stained by Coomassie Brilliant Blue. BSA—bovine serum albumin, AL—AL amyloidosis, AA—AA amyloidosis, MN—membranous nephropathy, IgA—immunoglobulin A nephropathy. Spectra Multicolor Broad Range Protein Ladder (no. 26634) (Thermo Fisher Scientific, Waltham, MA, USA) is shown to the left of the gels. Red line indicates the area just above the BSA band (2 µg) that runs at the level of human serum albumin in the samples on the right (lines 1–12). Images was obtained by S.A. Fedotov.
Generally, the obtained aggregates contain practically no albumin, and preliminary centrifugation at a speed of 4000× g enables the removal of large fibrils, the presence of which is not necessarily associated with amyloidogenic factors in the sample and can be found in healthy humans [112]. The analysis of the composition of detergent-resistant aggregates in specimens from patients with various diagnoses may have scientific and clinical relevance, not assessed by anyone to date. This is supported by the data on the detection in isolated urine aggregates the main factors for the progression of preeclampsia, a disease in pregnant women, which, as well as in renal amyloidosis, is associated with proteinuria and amyloid deposits in tissues ([155,156]; see review [157]). Of interest, the Congo red dot (CRD) test and the CRD paper test on urine samples have also demonstrated successful diagnostic applications for preeclampsia [155,158]. The design of those tests is based on the suggestion that Congo red binds amyloidogenic proteins in the urine of preeclampsia patients [155]. Performing CRD tests on the proteinuric specimens of urine in patients with various amyloidoses and non-amyloid diseases could clarify the diagnostic performance of such an approach.
6. Conclusions
The current arsenal of diagnostic approaches for renal amyloidosis is based primarily on invasive procedures applying typically at late stages. Since the effectiveness of renal amyloidoses treatment largely depends on the stage at which the disease is diagnosed the design of noninvasive screening techniques for renal amyloidosis might enable earlier detection of the disease, and significantly improve the renal and life prognosis in these patients. In this regard, further study of functional and pathological amyloid properties in humans is essential for the development of new diagnostic and treatment methods for amyloidosis.
Author Contributions
Conceptualization, S.A.F. and A.A.R.; validation, M.S.K., A.O.A., V.A.D. and A.A.R.; writing—original draft preparation, S.A.F.; writing—review and editing, S.A.F., M.S.K., A.O.A., V.A.D. and A.A.R.; figures and tables preparation, S.A.F.; supervision, and A.A.R.; project administration, S.A.F., V.A.D. and A.A.R.; funding acquisition, S.A.F. and A.A.R. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by grant 22-25-00315 from Russian Science Foundation.
Institutional Review Board Statement
The experimental part presented in this review was conducted in accordance with the Declaration of Helsinki, and approved by Ethics Committee of Pavlov University (No.: 21-250 date of approval 28 June 2021).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Not applicable.
Acknowledgments
The authors thank the large-scale research facilities no. 3076082 “Human Reproductive Health” D.O. Ott Research Institute of Obstetrics, Gynecology and Reproductology” for the samples from the biocollection. The authors acknowledge the support from the St. Petersburg State University (project 93025998) and the Center for Molecular and Cell Technologies (Research Park, St. Petersburg State University). The authors thank renal pathologist of Research Institute of Nephrology, V.G. Sipovsky, for kindly providing the microphotograph to Figure 1b. The authors also thank Maria Rubel for English editing.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AA | serum amyloid A amyloidosis |
| AFib | fibrinogen α chain amyloidosis |
| AH | immunoglobulin heavy chains amyloidosis |
| AHL | immunoglobulin heavy and light chains amyloidosis |
| AL | immunoglobulin light chains amyloidosis |
| ALECT2 | leukocyte chemotactic factor 2 amyloidoses |
| ALP | alkaline phosphatase |
| ALT | alanine aminotransferase |
| AST | aspartate aminotransferase |
| ATTR | transthyretin amyloidosis |
| ATTRv | hereditary type of transthyretin amyloidosis |
| ATTRwt | wild-type type of transthyretin amyloidosis |
| BSA | bovine serum albumin |
| CNS | central nervous system |
| CRD | Congo red dot |
| CT | computed tomography |
| ECG | electrocardiography |
| FFPE | formalin-fixed paraffin-embedded |
| FLC | free light chains |
| GIT | gastrointestinal tract |
| I/T | troponin I/troponin T |
| IEM | immunoelectron microscopy |
| IFE | immunofixation electrophoresis |
| Ig | immunoglobulin |
| IgA | immunoglobulin A nephropathy |
| IGVL | light chain variable region |
| Igκ | immunoglobulin kappa light chain |
| IgκC | immunoglobulin kappa constant domain |
| IgκV | immunoglobulin kappa variable domain. |
| Igλ | immunoglobulin lambda light chain |
| IHC | immunohistochemistry |
| LMD–MS | laser microdissection followed by tandem mass spectrometry |
| LV | left ventricle |
| MG | monoclonal gammopathy |
| MGUS | gammopathy of undetermined significance |
| MM | multiple myeloma |
| MN | membranous nephropathy |
| MS | mass spectrometry |
| NT-proBNP | N-terminal pro-B-type natriuretic peptide |
| PAGE | polyacrylamide gel electrophoresis |
| PMCA | protein misfolding cyclic amplification |
| PNS | peripheral nervous system |
| QuIC | quaking-induced conversion |
| SAA | serum amyloid A protein |
| SAP | serum amyloid P component |
| ThT | thioflavin T |
| TTR | transthyretin protein |
References
- Merlini, G.; Bellotti, V. Molecular Mechanisms of Amyloidosis. N. Engl. J. Med. 2003, 349, 583–596. [Google Scholar] [CrossRef] [PubMed]
- Pepys, M.B. Amyloidosis. Annu. Rev. Med. 2006, 57, 223–241. [Google Scholar] [CrossRef] [PubMed]
- Glenner, G.G. Amyloid Deposits and Amyloidosis. The Beta-Fibrilloses (First of Two Parts). N. Engl. J. Med. 1980, 302, 1283–1292. [Google Scholar] [CrossRef] [PubMed]
- Sunde, M.; Serpell, L.C.; Bartlam, M.; Fraser, P.E.; Pepys, M.B.; Blake, C.C.F. Common Core Structure of Amyloid Fibrils by Synchrotron X-ray Diffraction. J. Mol. Biol. 1997, 273, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Benson, M.D.; Buxbaum, J.N.; Eisenberg, D.S.; Merlini, G.; Saraiva, M.J.M.; Sekijima, Y.; Sipe, J.D.; Westermark, P. Amyloid Nomenclature 2020: Update and Recommendations by the International Society of Amyloidosis (ISA) Nomenclature Committee. Amyloid 2020, 27, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Chatani, E.; Yuzu, K.; Ohhashi, Y.; Goto, Y. Current Understanding of the Structure, Stability and Dynamic Properties of Amyloid Fibrils. Int. J. Mol. Sci. 2021, 22, 4349. [Google Scholar] [CrossRef] [PubMed]
- Maskevich, A.A.; Stsiapura, V.I.; Kuzmitsky, V.A.; Kuznetsova, I.M.; Povarova, O.I.; Uversky, V.N.; Turoverov, K.K. Spectral Properties of Thioflavin T in Solvents with Different Dielectric Properties and in a Fibril-Incorporated Form. J. Proteome Res. 2007, 6, 1392–1401. [Google Scholar] [CrossRef] [PubMed]
- Frieg, B.; Gremer, L.; Heise, H.; Willbold, D.; Gohlke, H. Binding Modes of Thioflavin T and Congo Red to the Fibril Structure of Amyloid-β(1–42). Chem. Commun. 2020, 56, 7589–7592. [Google Scholar] [CrossRef] [PubMed]
- Kachkin, D.V.; Volkov, K.V.; Sopova, J.V.; Bobylev, A.G.; Fedotov, S.A.; Inge-Vechtomov, S.G.; Galzitskaya, O.V.; Chernoff, Y.O.; Rubel, A.A.; Aksenova, A.Y. Human RAD51 Protein Forms Amyloid-like Aggregates In Vitro. Int. J. Mol. Sci. 2022, 23, 11657. [Google Scholar] [CrossRef] [PubMed]
- Riesner, D. Biochemistry and Structure of PrPC and PrPSc. Br. Med. Bull. 2003, 66, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Rubel, M.S.; Fedotov, S.A.; Grizel, A.V.; Sopova, J.V.; Malikova, O.A.; Chernoff, Y.O. Functional Mammalian Amyloids and Amyloid-Like Proteins. Life 2020, 10, 156. [Google Scholar] [CrossRef] [PubMed]
- Bunker, D.; Gorevic, P. AA Amyloidosis: Mount Sinai Experience, 1997-2012. Mt. Sinai J. Med. 2012, 79, 749–756. [Google Scholar] [CrossRef]
- Diomede, L.; Rognoni, P.; Lavatelli, F.; Romeo, M.; del Favero, E.; Cantù, L.; Ghibaudi, E.; di Fonzo, A.; Corbelli, A.; Fiordaliso, F.; et al. A Caenorhabditis Elegans–Based Assay Recognizes Immunoglobulin Light Chains Causing Heart Amyloidosis. Blood 2014, 123, 3543–3552. [Google Scholar] [CrossRef]
- Misra, P.; Blancas-Mejia, L.M.; Ramirez-Alvarado, M. Mechanistic Insights into the Early Events in the Aggregation of Immunoglobulin Light Chains. Biochemistry 2019, 58, 3155–3168. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Dobson, C.M. Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef] [PubMed]
- Biewend, M.L.; Menke, D.M.; Calamia, K.T. The Spectrum of Localized Amyloidosis: A Case Series of 20 Patients and Review of the Literature. Amyloid 2006, 13, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Nienhuis, H.L.A.; Bijzet, J.; Hazenberg, B.P.C. The Prevalence and Management of Systemic Amyloidosis in Western Countries. Kidney Dis. 2016, 2, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Baumgart, J.-V.; Stuhlmann-Laeisz, C.; Hegenbart, U.; Nattenmüller, J.; Schönland, S.; Krüger, S.; Behrens, H.-M.; Röcken, C. Local vs. Systemic Pulmonary Amyloidosis—Impact on Diagnostics and Clinical Management. Virchows Arch. 2018, 473, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Dember, L.M. Amyloidosis-Associated Kidney Disease. J. Am. Soc. Nephrol. 2006, 17, 3458–3471. [Google Scholar] [CrossRef] [PubMed]
- Linke, R.P. On Typing Amyloidosis Using Immunohistochemistry. Detailled Illustrations, Review and a Note on Mass Spectrometry. Prog. Histochem. Cytochem. 2012, 47, 61–132. [Google Scholar] [CrossRef]
- Dasari, S.; Theis, J.D.; Vrana, J.A.; Rech, K.L.; Dao, L.N.; Howard, M.T.; Dispenzieri, A.; Gertz, M.A.; Hasadsri, L.; Highsmith, W.E.; et al. Amyloid Typing by Mass Spectrometry in Clinical Practice: A Comprehensive Review of 16,175 Samples. Mayo Clin. Proc. 2020, 95, 1852–1864. [Google Scholar] [CrossRef]
- Pinney, J.H.; Whelan, C.J.; Petrie, A.; Dungu, J.; Banypersad, S.M.; Sattianayagam, P.; Wechalekar, A.; Gibbs, S.D.J.; Venner, C.P.; Wassef, N.; et al. Senile Systemic Amyloidosis: Clinical Features at Presentation and Outcome. J. Am. Hear. Assoc. 2013, 2, e000098. [Google Scholar] [CrossRef] [PubMed]
- Sevcuka, A.; White, K.; Terry, C. Factors That Contribute to HIAPP Amyloidosis in Type 2 Diabetes Mellitus. Life 2022, 12, 583. [Google Scholar] [CrossRef] [PubMed]
- Cline, E.N.; Bicca, M.A.; Viola, K.L.; Klein, W.L. The Amyloid-β Oligomer Hypothesis: Beginning of the Third Decade. J. Alzheimer’s Dis. 2018, 64, S567–S610. [Google Scholar] [CrossRef] [PubMed]
- Koike, H.; Katsuno, M. The Ultrastructure of Tissue Damage by Amyloid Fibrils. Molecules 2021, 26, 4611. [Google Scholar] [CrossRef] [PubMed]
- Lachmann, H.J.; Gallimore, J.R.; Sabin, C.A.; Hawkins, P.N. Natural History and Outcome in Systemic AA Amyloidosis. N. Engl. J. Med. 2007, 356, 2361–2371. [Google Scholar] [CrossRef] [PubMed]
- Nevone, A.; Merlini, G.; Nuvolone, M. Treating Protein Misfolding Diseases: Therapeutic Successes Against Systemic Amyloidoses. Front. Pharmacol. 2020, 11, 1024. [Google Scholar] [CrossRef]
- Wechalekar, A.; Whelan, C.; Lachmann, H.; Fontana, M.; Mahmood, S.; Gillmore, J.D.; Hawkins, P.N. Oral Doxycycline Improves Outcomes of Stage III AL Amyloidosis-a Matched Case Control Study. Blood 2015, 126, 732. [Google Scholar] [CrossRef]
- Ozawa, M.; Komatsuda, A.; Ohtani, H.; Nara, M.; Sato, R.; Togashi, M.; Takahashi, N.; Wakui, H. Long-Term Prognosis of AL and AA Renal Amyloidosis: A Japanese Single-Center Experience. Clin. Exp. Nephrol. 2017, 21, 212–227. [Google Scholar] [CrossRef]
- Oerlemans, M.I.F.J.; Rutten, K.H.G.; Minnema, M.C.; Raymakers, R.A.P.; Asselbergs, F.W.; de Jonge, N. Cardiac Amyloidosis: The Need for Early Diagnosis. Neth. Heart J. 2019, 27, 525–536. [Google Scholar] [CrossRef]
- Said, S.M.; Sethi, S.; Valeri, A.M.; Leung, N.; Cornell, L.D.; Fidler, M.E.; Hernandez, L.H.; Vrana, J.A.; Theis, J.D.; Quint, P.S.; et al. Renal Amyloidosis: Origin and Clinicopathologic Correlations of 474 Recent Cases. Clin. J. Am. Soc. Nephrol. 2013, 8, 1515–1523. [Google Scholar] [CrossRef] [PubMed]
- Puchtler, H.; Sweat, F.; Levine, M. On the binding of congo red by amyloid. J. Histochem. Cytochem. 1962, 10, 355–364. [Google Scholar] [CrossRef]
- Howie, A.J.; Brewer, D.B.; Howell, D.; Jones, A.P. Physical Basis of Colors Seen in Congo Red-Stained Amyloid in Polarized Light. Lab. Investig. 2008, 88, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Wisniowski, B.; Wechalekar, A. Confirming the Diagnosis of Amyloidosis. Acta Haematol. 2020, 143, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Gillmore, J.D.; Maurer, M.S.; Falk, R.H.; Merlini, G.; Damy, T.; Dispenzieri, A.; Wechalekar, A.D.; Berk, J.L.; Quarta, C.C.; Grogan, M.; et al. Nonbiopsy Diagnosis of Cardiac Transthyretin Amyloidosis. Circulation 2016, 133, 2404–2412. [Google Scholar] [CrossRef]
- Hazenberg, B.P.C.; van Rijswijk, M.H.; Piers, D.A.; Lub-de Hooge, M.N.; Vellenga, E.; Haagsma, E.B.; Hawkins, P.N.; Jager, P.L. Diagnostic Performance of 123I-Labeled Serum Amyloid P Component Scintigraphy in Patients with Amyloidosis. Am. J. Med. 2006, 119, 355.e15–355.e24. [Google Scholar] [CrossRef]
- Bature, F.; Pappas, Y.; Pang, D.; Guinn, B. Can Non-Invasive Biomarkers Lead to an Earlier Diagnosis of Alzheimer’s Disease? Curr. Alzheimer Res. 2021, 18, 908–913. [Google Scholar] [CrossRef]
- Kulichikhin, K.Y.; Fedotov, S.A.; Rubel, M.S.; Zalutskaya, N.M.; Zobnina, A.E.; Malikova, O.A.; Neznanov, N.G.; Chernoff, Y.O.; Rubel, A.A. Development of Molecular Tools for Diagnosis of Alzheimer’s Disease That Are Based on Detection of Amyloidogenic Proteins. Prion 2021, 15, 56–69. [Google Scholar] [CrossRef]
- Sharma, L.; Sharma, A.; Kumar, D.; Asthana, M.K.; Lalhlenmawia, H.; Kumar, A.; Bhattacharyya, S.; Kumar, D. Promising Protein Biomarkers in the Early Diagnosis of Alzheimer’s Disease. Metab. Brain Dis. 2022, 37, 1727–1744. [Google Scholar] [CrossRef]
- Sidiqi, M.H.; McPhail, E.D.; Theis, J.D.; Dasari, S.; Vrana, J.A.; Drosou, M.E.; Leung, N.; Hayman, S.; Rajkumar, S.V.; Warsame, R.; et al. Two Types of Amyloidosis Presenting in a Single Patient: A Case Series. Blood Cancer J. 2019, 9, 30. [Google Scholar] [CrossRef]
- Sethi, S.; Theis, J.D. Pathology and Diagnosis of Renal Non-AL Amyloidosis. J. Nephrol. 2018, 31, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Fenoglio, R.; Baldovino, S.; Barreca, A.; Bottasso, E.; Sciascia, S.; Sbaiz, L.; Papotti, M.; Roccatello, D. Renal Involvement in Transthyretin Amyloidosis: The Double Presentation of Transthyretin Amyloidosis Deposition Disease. Nephron 2022, 146, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Wang, S.-X.; Zhang, Y.-K.; Qu, Z.; Liu, G.; Zou, W.-Z. A Clinicopathological Analysis in a Large Cohort of Chinese Patients with Renal Amyloid Light-Chain Amyloidosis. Nephrol. Dial. Transplant. 2013, 28, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, M.; Gono, T.; Morita, H.; Katoh, N.; Kodaira, M.; Ikeda, S. Peripheral Nerve Involvement in Primary Systemic AL Amyloidosis: A Clinical and Electrophysiological Study: Polyneuropathy in AL Amyloidosis. Eur. J. Neurol. 2011, 18, 604–610. [Google Scholar] [CrossRef] [PubMed]
- Merlini, G.; Palladini, G. Light Chain Amyloidosis: The Heart of the Problem. Haematologica 2013, 98, 1492–1495. [Google Scholar] [CrossRef]
- Kapoor, P.; Thenappan, T.; Singh, E.; Kumar, S.; Greipp, P.R. Cardiac Amyloidosis: A Practical Approach to Diagnosis and Management. Am. J. Med. 2011, 124, 1006–1015. [Google Scholar] [CrossRef]
- Bergesio, F.; Ciciani, A.M.; Santostefano, M.; Brugnano, R.; Manganaro, M.; Palladini, G.; Palma, A.M.D.; Gallo, M.; Tosi, P.L.; Salvadori, M.; et al. Renal Involvement in Systemic Amyloidosis--an Italian Retrospective Study on Epidemiological and Clinical Data at Diagnosis. Nephrol. Dial. Transplant. 2007, 22, 1608–1618. [Google Scholar] [CrossRef]
- Bollée, G.; Guery, B.; Joly, M.; Snanoudj, R.; Terrier, B.; Allouache, M.; Mercadal, L.; Peraldi, M.-N.; Viron, B.; Fumeron, C.; et al. Presentation and Outcome of Patients with Systemic Amyloidosis Undergoing Dialysis. Clin. J. Am. Soc. Nephrol. 2008, 3, 375–381. [Google Scholar] [CrossRef]
- Sethi, S.; Vrana, J.A.; Theis, J.D.; Leung, N.; Sethi, A.; Nasr, S.H.; Fervenza, F.C.; Cornell, L.D.; Fidler, M.E.; Dogan, A. Laser Microdissection and Mass Spectrometry–Based Proteomics Aids the Diagnosis and Typing of Renal Amyloidosis. Kidney Int. 2012, 82, 226–234. [Google Scholar] [CrossRef]
- Larsen, C.P.; Walker, P.D.; Weiss, D.T.; Solomon, A. Prevalence and Morphology of Leukocyte Chemotactic Factor 2-Associated Amyloid in Renal Biopsies. Kidney Int. 2010, 77, 816–819. [Google Scholar] [CrossRef]
- von Hutten, H.; Mihatsch, M.; Lobeck, H.; Rudolph, B.; Eriksson, M.; Röcken, C. Prevalence and Origin of Amyloid in Kidney Biopsies. Am. J. Surg. Pathol. 2009, 33, 1198–1205. [Google Scholar] [CrossRef] [PubMed]
- Nasr, S.H.; Said, S.M.; Valeri, A.M.; Sethi, S.; Fidler, M.E.; Cornell, L.D.; Gertz, M.A.; Dispenzieri, A.; Buadi, F.K.; Vrana, J.A.; et al. The Diagnosis and Characteristics of Renal Heavy-Chain and Heavy/Light-Chain Amyloidosis and Their Comparison with Renal Light-Chain Amyloidosis. Kidney Int. 2013, 83, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Leung, N.; Bridoux, F.; Batuman, V.; Chaidos, A.; Cockwell, P.; D’Agati, V.D.; Dispenzieri, A.; Fervenza, F.C.; Fermand, J.-P.; Gibbs, S.; et al. The Evaluation of Monoclonal Gammopathy of Renal Significance: A Consensus Report of the International Kidney and Monoclonal Gammopathy Research Group. Nat. Rev. Nephrol. 2019, 15, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Benson, M.D. The Hereditary Amyloidoses. Best Pract. Res. Clin. Rheumatol. 2003, 17, 909–927. [Google Scholar] [CrossRef]
- Sekijima, Y. Transthyretin (ATTR) Amyloidosis: Clinical Spectrum, Molecular Pathogenesis and Disease-Modifying Treatments. J. Neurol. Neurosurg. Psychiatry 2015, 86, 1036–1043. [Google Scholar] [CrossRef]
- Said, S.M.; Sethi, S.; Valeri, A.M.; Chang, A.; Nast, C.C.; Krahl, L.; Molloy, P.; Barry, M.; Fidler, M.E.; Cornell, L.D.; et al. Characterization and Outcomes of Renal Leukocyte Chemotactic Factor 2-Associated Amyloidosis. Kidney Int. 2014, 86, 370–377. [Google Scholar] [CrossRef]
- Gillmore, J.D.; Lachmann, H.J.; Rowczenio, D.; Gilbertson, J.A.; Zeng, C.-H.; Liu, Z.-H.; Li, L.-S.; Wechalekar, A.; Hawkins, P.N. Diagnosis, Pathogenesis, Treatment, and Prognosis of Hereditary Fibrinogen Aα-Chain Amyloidosis. J. Am. Soc. Nephrol. 2009, 20, 444–451. [Google Scholar] [CrossRef]
- Bergesio, F.; Ciciani, A.M.; Manganaro, M.; Palladini, G.; Santostefano, M.; Brugnano, R.; Di Palma, A.M.; Gallo, M.; Rosati, A.; Tosi, P.L.; et al. Renal Involvement in Systemic Amyloidosis: An Italian Collaborative Study on Survival and Renal Outcome. Nephrol. Dial. Transplant. 2007, 23, 941–951. [Google Scholar] [CrossRef]
- Kyle, R.A.; Bayrd, E.D. Amyloidosis: Review of 236 Cases. Medicine 1975, 54, 271–299. [Google Scholar] [CrossRef]
- Hemminki, K.; Li, X.; Försti, A.; Sundquist, J.; Sundquist, K. Incidence and Survival in Non-Hereditary Amyloidosis in Sweden. BMC Public Health 2012, 12, 974. [Google Scholar] [CrossRef]
- Staron, A.; Zheng, L.; Doros, G.; Connors, L.H.; Mendelson, L.M.; Joshi, T.; Sanchorawala, V. Marked Progress in AL Amyloidosis Survival: A 40-Year Longitudinal Natural History Study. Blood Cancer J. 2021, 11, 139. [Google Scholar] [CrossRef] [PubMed]
- Papa, R.; Lachmann, H.J. Secondary, AA, Amyloidosis. Rheum. Dis. Clin. N. Am. 2018, 44, 585–603. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.A. Immunoglobulin Light Chain Amyloidosis: 2020 Update on Diagnosis, Prognosis, and Treatment. Am. J. Hematol. 2020, 95, 848–860. [Google Scholar] [CrossRef]
- Stangou, A.J.; Hawkins, P.N. Liver Transplantation in Transthyretin-Related Familial Amyloid Polyneuropathy. Curr. Opin. Neurol. 2004, 17, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Berk, J.L.; Suhr, O.B.; Obici, L.; Sekijima, Y.; Zeldenrust, S.R.; Yamashita, T.; Heneghan, M.A.; Gorevic, P.D.; Litchy, W.J.; Wiesman, J.F.; et al. Repurposing Diflunisal for Familial Amyloid Polyneuropathy: A Randomized Clinical Trial. JAMA 2013, 310, 2658. [Google Scholar] [CrossRef] [PubMed]
- Maurer, M.S.; Schwartz, J.H.; Gundapaneni, B.; Elliott, P.M.; Merlini, G.; Waddington-Cruz, M.; Kristen, A.V.; Grogan, M.; Witteles, R.; Damy, T.; et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N. Engl. J. Med. 2018, 379, 1007–1016. [Google Scholar] [CrossRef]
- Benson, M.D.; Buxbaum, J.N.; Eisenberg, D.S.; Merlini, G.; Saraiva, M.J.M.; Sekijima, Y.; Sipe, J.D.; Westermark, P. Amyloid Nomenclature 2018: Recommendations by the International Society of Amyloidosis (ISA) Nomenclature Committee. Amyloid 2018, 25, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.-C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Ma, X.; Iyer, L.; Chaulagain, C.; Comenzo, R.L. One SiRNA Pool Targeting the λ Constant Region Stops λ Light-Chain Production and Causes Terminal Endoplasmic Reticulum Stress. Blood 2014, 123, 3440–3451. [Google Scholar] [CrossRef] [PubMed]
- Morgan, G.J.; Yan, N.L.; Mortenson, D.E.; Rennella, E.; Blundon, J.M.; Gwin, R.M.; Lin, C.-Y.; Stanfield, R.L.; Brown, S.J.; Rosen, H.; et al. Stabilization of Amyloidogenic Immunoglobulin Light Chains by Small Molecules. Proc. Natl. Acad. Sci. USA 2019, 116, 8360–8369. [Google Scholar] [CrossRef]
- Kumar, S.K.; Gertz, M.A.; Lacy, M.Q.; Dingli, D.; Hayman, S.R.; Buadi, F.K.; Short-Detweiler, K.; Zeldenrust, S.R.; Leung, N.; Greipp, P.R.; et al. Recent Improvements in Survival in Primary Systemic Amyloidosis and the Importance of an Early Mortality Risk Score. Mayo Clin. Proc. 2011, 86, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Dispenzieri, A.; Merlini, G. Future Perspectives. Hematol./Oncol. Clin. N. Am. 2020, 34, 1205–1214. [Google Scholar] [CrossRef] [PubMed]
- The Inaugural Amyloidosis Forum Panelists; Blank, M.; Campbell, M.; Clarke, J.O.; Comenzo, R.; Dember, L.M.; Dispenzieri, A.; Dorbala, S.; Dunnmon, P.; Faller, D.V.; et al. The Amyloidosis Forum: A Public Private Partnership to Advance Drug Development in AL Amyloidosis. Orphanet J. Rare Dis. 2020, 15, 268. [Google Scholar] [CrossRef]
- Juneja, R.; Pati, H.P. Approach to the Diagnosis of Amyloidosis. Indian J. Hematol. Blood Transfus. 2020, 36, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Falk, R.H. Diagnosis and Management of the Cardiac Amyloidoses. Circulation 2005, 112, 2047–2060. [Google Scholar] [CrossRef] [PubMed]
- Banypersad, S.M.; Sado, D.M.; Flett, A.S.; Gibbs, S.D.J.; Pinney, J.H.; Maestrini, V.; Cox, A.T.; Fontana, M.; Whelan, C.J.; Wechalekar, A.D.; et al. Quantification of Myocardial Extracellular Volume Fraction in Systemic AL Amyloidosis: An Equilibrium Contrast Cardiovascular Magnetic Resonance Study. Circ. Cardiovasc. Imaging 2013, 6, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Swan, N.; Skinner, M.; O’Hara, C.J. Bone Marrow Core Biopsy Specimens in AL (Primary) Amyloidosis: A Morphologic and Immunohistochemical Study of 100 Cases. Am. J. Clin. Pathol. 2003, 120, 610–616. [Google Scholar] [CrossRef] [PubMed]
- Schönland, S.O.; Hegenbart, U.; Bochtler, T.; Mangatter, A.; Hansberg, M.; Ho, A.D.; Lohse, P.; Röcken, C. Immunohistochemistry in the Classification of Systemic Forms of Amyloidosis: A Systematic Investigation of 117 Patients. Blood 2012, 119, 488–493. [Google Scholar] [CrossRef]
- Owen, A.M.; Coleman, M.R.; Boly, M.; Davis, M.H.; Laureys, S.; Pickard, J.D. Detecting Awareness in the Vegetative State. Science 2006, 313, 1402. [Google Scholar] [CrossRef]
- Enqvist, S.; Sletten, K.; Westermark, P. Fibril Protein Fragmentation Pattern in Systemic AL-Amyloidosis. J. Pathol. 2009, 219, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Picken, M.M.; Herrera, G.A. The Burden of “Sticky” Amyloid: Typing Challenges. Arch. Pathol. Lab. Med. 2007, 131, 850–851. [Google Scholar] [CrossRef]
- Hoshii, Y.; Kiyama, M.; Cui, D.; Kawano, H.; Ishihara, T. Immunohistochemical Study of Immunoglobulin Light Chain Amyloidosis with Antibodies to the Immunoglobulin Light Chain Variable Region. Pathol. Int. 2006, 56, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Owen-Casey, M.P.; Sim, R.; Cook, H.T.; Roufosse, C.A.; Gillmore, J.D.; Gilbertson, J.A.; Hutchison, C.A.; Howie, A.J. Value of Antibodies to Free Light Chains in Immunoperoxidase Studies of Renal Biopsies. J. Clin. Pathol. 2014, 67, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Barreca, A.; Bottasso, E.; Veneziano, F.; Giarin, M.; Nocifora, A.; Martinetti, N.; Attanasio, A.; Biancone, L.; Benevolo, G.; Roccatello, D.; et al. Immunohistochemical Typing of Amyloid in Fixed Paraffin-Embedded Samples by an Automatic Procedure: Comparison with Immunofluorescence Data on Fresh-Frozen Tissue. PLoS ONE 2021, 16, e0256306. [Google Scholar] [CrossRef] [PubMed]
- de Larrea, C.F.; Verga, L.; Morbini, P.; Klersy, C.; Lavatelli, F.; Foli, A.; Obici, L.; Milani, P.; Capello, G.L.; Paulli, M.; et al. A Practical Approach to the Diagnosis of Systemic Amyloidoses. Blood 2015, 125, 2239–2244. [Google Scholar] [CrossRef] [PubMed]
- Abildgaard, N.; Rojek, A.M.; Møller, H.E.; Palstrøm, N.B.; Nyvold, C.G.; Rasmussen, L.M.; Hansen, C.T.; Beck, H.C.; Marcussen, N. Immunoelectron Microscopy and Mass Spectrometry for Classification of Amyloid Deposits. Amyloid 2020, 27, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Picken, M.M. Proteomics and Mass Spectrometry in the Diagnosis of Renal Amyloidosis. Clin. Kidney J. 2015, 8, 665–672. [Google Scholar] [CrossRef]
- Casadonte, R.; Kriegsmann, M.; Deininger, S.-O.; Amann, K.; Paape, R.; Belau, E.; Suckau, D.; Fuchser, J.; Beckmann, J.; Becker, M.; et al. Imaging Mass Spectrometry Analysis of Renal Amyloidosis Biopsies Reveals Protein Co-Localization with Amyloid Deposits. Anal. Bioanal. Chem. 2015, 407, 5323–5331. [Google Scholar] [CrossRef]
- Vrana, J.A.; Gamez, J.D.; Madden, B.J.; Theis, J.D.; Bergen, H.R.; Dogan, A. Classification of Amyloidosis by Laser Microdissection and Mass Spectrometry–Based Proteomic Analysis in Clinical Biopsy Specimens. Blood 2009, 114, 4957–4959. [Google Scholar] [CrossRef] [PubMed]
- Gilbertson, J.A.; Theis, J.D.; Vrana, J.A.; Lachmann, H.; Wechalekar, A.; Whelan, C.; Hawkins, P.N.; Dogan, A.; Gillmore, J.D. A Comparison of Immunohistochemistry and Mass Spectrometry for Determining the Amyloid Fibril Protein from Formalin-Fixed Biopsy Tissue. J. Clin. Pathol. 2015, 68, 314–317. [Google Scholar] [CrossRef]
- Mollee, P.; Boros, S.; Loo, D.; Ruelcke, J.E.; Lakis, V.A.; Cao, K.L.; Renaut, P.; Hill, M.M. Implementation and Evaluation of Amyloidosis Subtyping by Laser-Capture Microdissection and Tandem Mass Spectrometry. Clin. Proteom. 2016, 13, 30. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez Suarez, M.L.; Zhang, P.; Nasr, S.H.; Sathick, I.J.; Kittanamongkolchai, W.; Kurtin, P.J.; Alexander, M.P.; Cornell, L.D.; Fidler, M.E.; Grande, J.P.; et al. The Sensitivity and Specificity of the Routine Kidney Biopsy Immunofluorescence Panel Are Inferior to Diagnosing Renal Immunoglobulin-Derived Amyloidosis by Mass Spectrometry. Kidney Int. 2019, 96, 1005–1009. [Google Scholar] [CrossRef] [PubMed]
- Lavatelli, F.; Valentini, V.; Palladini, G.; Verga, L.; Russo, P.; Foli, A.; Obici, L.; Sarais, G.; Perfetti, V.; Casarini, S.; et al. Mass Spectrometry-Based Proteomics as a Diagnostic Tool When Immunoelectron Microscopy Fails in Typing Amyloid Deposits. Amyloid 2011, 18, 64–66. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.A.; Mauermann, M.L.; Grogan, M.; Coelho, T. Advances in the Treatment of Hereditary Transthyretin Amyloidosis: A Review. Brain Behav. 2019, 9, e01371. [Google Scholar] [CrossRef] [PubMed]
- Yazaki, M.; Yoshinaga, T.; Sekijima, Y.; Kametani, F.; Okumura, N. Hereditary Fibrinogen Aα-Chain Amyloidosis in Asia: Clinical and Molecular Characteristics. Int. J. Mol. Sci. 2018, 19, 320. [Google Scholar] [CrossRef] [PubMed]
- Maurer, M.S.; Bokhari, S.; Damy, T.; Dorbala, S.; Drachman, B.M.; Fontana, M.; Grogan, M.; Kristen, A.V.; Lousada, I.; Nativi-Nicolau, J.; et al. Expert Consensus Recommendations for the Suspicion and Diagnosis of Transthyretin Cardiac Amyloidosis. Circ. Heart Fail. 2019, 12, e006075. [Google Scholar] [CrossRef] [PubMed]
- Krainer, J.; Siebenhandl, S.; Weinhäusel, A. Systemic Autoinflammatory Diseases. J. Autoimmun. 2020, 109, 102421. [Google Scholar] [CrossRef] [PubMed]
- Rowczenio, D.M.; Noor, I.; Gillmore, J.D.; Lachmann, H.J.; Whelan, C.; Hawkins, P.N.; Obici, L.; Westermark, P.; Grateau, G.; Wechalekar, A.D. Online Registry for Mutations in Hereditary Amyloidosis Including Nomenclature Recommendations. Hum. Mutat. 2014, 35, E2403–E2412. [Google Scholar] [CrossRef] [PubMed]
- Lachmann, H.J.; Gilbertson, J.A.; Gillmore, J.D.; Hawkins, P.N. Misdiagnosis of Hereditary Amyloidosis as AL (Primary) Amyloidosis. N. Engl. J. Med. 2002, 346, 1786–1791. [Google Scholar] [CrossRef]
- Hellman, U.; Alarcon, F.; Lundgren, H.-E.; Suhr, O.B.; Bonaiti-PelliÉ, C.; Planté-Bordeneuve, V. Heterogeneity of Penetrance in Familial Amyloid Polyneuropathy, ATTR Val30Met, in the Swedish Population. Amyloid 2008, 15, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Dasari, S.; Theis, J.D.; Vrana, J.A.; Zenka, R.M.; Zimmermann, M.T.; Kocher, J.-P.A.; Highsmith, W.E.; Kurtin, P.J.; Dogan, A. Clinical Proteome Informatics Workbench Detects Pathogenic Mutations in Hereditary Amyloidoses. J. Proteome Res. 2014, 13, 2352–2358. [Google Scholar] [CrossRef]
- Rona, G.; Pasaoglu, L.; Ozkayar, N.; Ciliz, D.; Toprak, U.; Abat, G. Functional Evaluation of Secondary Renal Amyloidosis with Diffusion-Weighted MR Imaging. Ren. Fail. 2016, 38, 249–255. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Paeng, J.C.; Choi, J.Y. Nuclear Imaging for Cardiac Amyloidosis: Bone Scan, SPECT/CT, and Amyloid-Targeting PET. Nucl. Med. Mol. Imaging 2021, 55, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Razvi, Y.; Patel, R.K.; Fontana, M.; Gillmore, J.D. Cardiac Amyloidosis: A Review of Current Imaging Techniques. Front. Cardiovasc. Med. 2021, 8, 751293. [Google Scholar] [CrossRef] [PubMed]
- Phull, P.; Sanchorawala, V.; Connors, L.H.; Doros, G.; Ruberg, F.L.; Berk, J.L.; Sarosiek, S. Monoclonal Gammopathy of Undetermined Significance in Systemic Transthyretin Amyloidosis (ATTR). Amyloid 2018, 25, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Ezawa, N.; Katoh, N.; Oguchi, K.; Yoshinaga, T.; Yazaki, M.; Sekijima, Y. Visualization of Multiple Organ Amyloid Involvement in Systemic Amyloidosis Using 11C-PiB PET Imaging. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 452–461. [Google Scholar] [CrossRef]
- Gallegos, C.; Miller, E.J. Advances in PET-Based Cardiac Amyloid Radiotracers. Curr. Cardiol. Rep. 2020, 22, 40. [Google Scholar] [CrossRef]
- Ehman, E.C.; El-Sady, M.S.; Kijewski, M.F.; Khor, Y.M.; Jacob, S.; Ruberg, F.L.; Sanchorawala, V.; Landau, H.; Yee, A.J.; Bianchi, G.; et al. Early Detection of Multiorgan Light-Chain Amyloidosis by Whole-Body 18F-Florbetapir PET/CT. J. Nucl. Med. 2019, 60, 1234–1239. [Google Scholar] [CrossRef]
- Derosena, R.; Koss, M.N.; Pirani, C.L. Demonstration of Amyloid Fibrils in Urinary Sediment. N. Engl. J. Med. 1975, 293, 1131–1133. [Google Scholar] [CrossRef] [PubMed]
- Nimoityn, P.; Lasker, N.; Soriano, R.Z. Detection of Urinary Amyloid in Familial Mediterranean Fever. BMJ 1976, 2, 284. [Google Scholar] [CrossRef] [PubMed]
- Orfila, C.; Graeve, P.; Guilhem, A.; Suc, J.M. Study of Light-, Electron- and Immunofluorescence Microscopy of Urinary Sediment in Amyloidosis. Virchows Arch. A Path. Anat. Histol. 1978, 379, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Winer, R.L.; Wuerker, R.B.; Erickson, J.O.; Cooper, W.L. Ultrastructural Examination of Urinary Sediment: Value in Renal Amyloidosis. Am. J. Clin. Pathol. 1979, 71, 36–39. [Google Scholar] [CrossRef]
- Lindner, L.E.; Haber, M.H. Unsuitability of Electron Microscopic Examination of Urinary Sediment for the Diagnosis of Amyloidosis: Universal Presence of Fibrillar Proteins in Urine Containing Casts. Am. J. Clin. Pathol. 1979, 71, 40–42. [Google Scholar] [CrossRef] [PubMed]
- Palladini, G.; Russo, P.; Bosoni, T.; Verga, L.; Sarais, G.; Lavatelli, F.; Nuvolone, M.; Obici, L.; Casarini, S.; Donadei, S.; et al. Identification of Amyloidogenic Light Chains Requires the Combination of Serum-Free Light Chain Assay with Immunofixation of Serum and Urine. Clin. Chem. 2009, 55, 499–504. [Google Scholar] [CrossRef]
- Visram, A.; Al Saleh, A.S.; Parmar, H.; McDonald, J.S.; Lieske, J.C.; Vaxman, I.; Muchtar, E.; Hobbs, M.; Fonder, A.; Hwa, Y.L.; et al. Correlation between Urine ACR and 24-h Proteinuria in a Real-World Cohort of Systemic AL Amyloidosis Patients. Blood Cancer J. 2020, 10, 124. [Google Scholar] [CrossRef] [PubMed]
- Wechalekar, A.D.; Gillmore, J.D.; Wassef, N.; Lachmann, H.J.; Whelan, C.; Hawkins, P.N. Abnormal N-Terminal Fragment of Brain Natriuretic Peptide in Patients with Light Chain Amyloidosis without Cardiac Involvement at Presentation Is a Risk Factor for Development of Cardiac Amyloidosis. Haematologica 2011, 96, 1079–1080. [Google Scholar] [CrossRef]
- Katzmann, J.A.; Dispenzieri, A.; Kyle, R.A.; Snyder, M.R.; Plevak, M.F.; Larson, D.R.; Abraham, R.S.; Lust, J.A.; Melton, L.J.; Rajkumar, S.V. Elimination of the Need for Urine Studies in the Screening Algorithm for Monoclonal Gammopathies by Using Serum Immunofixation and Free Light Chain Assays. Mayo Clin. Proc. 2006, 81, 1575–1578. [Google Scholar] [CrossRef] [PubMed]
- Dispenzieri, A.; Katzmann, J.A.; Kyle, R.A.; Larson, D.R.; Melton, L.J.; Colby, C.L.; Therneau, T.M.; Clark, R.; Kumar, S.K.; Bradwell, A.; et al. Prevalence and Risk of Progression of Light-Chain Monoclonal Gammopathy of Undetermined Significance: A Retrospective Population-Based Cohort Study. Lancet 2010, 375, 1721–1728. [Google Scholar] [CrossRef]
- Terré, A.; Colombat, M.; Cez, A.; Martin, C.; Diet, C.; Brechignac, S.; Oghina, S.; Bodez, D.; Faguer, S.; Savey, L.; et al. AA Amyloidosis Complicating Monoclonal Gammopathies, an Unusual Feature Validating the Concept of “Monoclonal Gammopathy of Inflammatory Significance”? Int. J. Clin. Pract. 2021, 75, e14817. [Google Scholar] [CrossRef] [PubMed]
- Gillmore, J.D.; Lovat, L.B.; Persey, M.R.; Pepys, M.B.; Hawkins, P.N. Amyloid Load and Clinical Outcome in AA Amyloidosis in Relation to Circulating Concentration of Serum Amyloid A Protein. Lancet 2001, 358, 24–29. [Google Scholar] [CrossRef]
- Hosman, I.S.; Kos, I.; Lamot, L. Serum Amyloid a in Inflammatory Rheumatic Diseases: A Compendious Review of a Renowned Biomarker. Front. Immunol. 2021, 11, 631299. [Google Scholar] [CrossRef] [PubMed]
- Lavatelli, F.; di Fonzo, A.; Palladini, G.; Merlini, G. Systemic Amyloidoses and Proteomics: The State of the Art. EuPA Open Proteom. 2016, 11, 4–10. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vilà-Rico, M.; Colomé-Calls, N.; Martín-Castel, L.; Gay, M.; Azorín, S.; Vilaseca, M.; Planas, A.; Canals, F. Quantitative Analysis of Post-Translational Modifications in Human Serum Transthyretin Associated with Familial Amyloidotic Polyneuropathy by Targeted LC-MS and Intact Protein MS. J. Proteom. 2015, 127, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Mills, J.R.; Barnidge, D.R.; Murray, D.L. Detecting Monoclonal Immunoglobulins in Human Serum Using Mass Spectrometry. Methods 2015, 81, 56–65. [Google Scholar] [CrossRef] [PubMed]
- VanDuijn, M.M.; Jacobs, J.F.M.; Wevers, R.A.; Engelke, U.F.; Joosten, I.; Luider, T.M. Quantitative Measurement of Immunoglobulins and Free Light Chains Using Mass Spectrometry. Anal. Chem. 2015, 87, 8268–8274. [Google Scholar] [CrossRef] [PubMed]
- Barnidge, D.R.; Dasari, S.; Botz, C.M.; Murray, D.H.; Snyder, M.R.; Katzmann, J.A.; Dispenzieri, A.; Murray, D.L. Using Mass Spectrometry to Monitor Monoclonal Immunoglobulins in Patients with a Monoclonal Gammopathy. J. Proteome Res. 2014, 13, 1419–1427. [Google Scholar] [CrossRef]
- Tasaki, M.; Ueda, M.; Obayashi, K.; Motokawa, H.; Kinoshita, Y.; Suenaga, G.; Yanagisawa, A.; Toyoshima, R.; Misumi, Y.; Masuda, T.; et al. Rapid Detection of Wild-Type and Mutated Transthyretins. Ann. Clin. Biochem. 2016, 53, 508–510. [Google Scholar] [CrossRef]
- Berghaus, N.; Schreiner, S.; Granzow, M.; Müller-Tidow, C.; Hegenbart, U.; Schönland, S.O.; Huhn, S. Analysis of the Complete Lambda Light Chain Germline Usage in Patients with AL Amyloidosis and Dominant Heart or Kidney Involvement. PLoS ONE 2022, 17, e0264407. [Google Scholar] [CrossRef]
- Dispenzieri, A.; Larson, D.R.; Rajkumar, S.V.; Kyle, R.A.; Kumar, S.K.; Kourelis, T.; Arendt, B.; Willrcih, M.; Dasari, S.; Murray, D. N-Glycosylation of Monoclonal Light Chains on Routine MASS-FIX Testing Is a Risk Factor for MGUS Progression. Leukemia 2020, 34, 2749–2753. [Google Scholar] [CrossRef]
- Kourelis, T.V.; Dasari, S.; Theis, J.D.; Ramirez-Alvarado, M.; Kurtin, P.J.; Gertz, M.A.; Zeldenrust, S.R.; Zenka, R.M.; Dogan, A.; Dispenzieri, A. Clarifying Immunoglobulin Gene Usage in Systemic and Localized Immunoglobulin Light-Chain Amyloidosis by Mass Spectrometry. Blood 2017, 129, 299–306. [Google Scholar] [CrossRef]
- Kumar, S.; Murray, D.; Dasari, S.; Milani, P.; Barnidge, D.; Madden, B.; Kourelis, T.; Arendt, B.; Merlini, G.; Ramirez-Alvarado, M.; et al. Assay to rapidly screen for immunoglobulin light chain glycosylation: A potential path to earlier AL diagnosis for a subset of patients. Leukemia 2019, 33, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Alvarado, M.; Ward, C.J.; Huang, B.Q.; Gong, X.; Hogan, M.C.; Madden, B.J.; Charlesworth, M.C.; Leung, N. Differences in Immunoglobulin Light Chain Species Found in Urinary Exosomes in Light Chain Amyloidosis (AL). PLoS ONE 2012, 7, e38061. [Google Scholar] [CrossRef] [PubMed]
- Phelan, D.; Collier, P.; Thavendiranathan, P.; Popović, Z.B.; Hanna, M.; Plana, J.C.; Marwick, T.H.; Thomas, J.D. Relative Apical Sparing of Longitudinal Strain Using Two-Dimensional Speckle-Tracking Echocardiography Is Both Sensitive and Specific for the Diagnosis of Cardiac Amyloidosis. Heart 2012, 98, 1442–1448. [Google Scholar] [CrossRef] [PubMed]
- Quarta, C.C.; Zheng, J.; Hutt, D.; Grigore, S.F.; Manwani, R.; Sachchithanantham, S.; Mahmood, S.A.; Whelan, C.J.; Fontana, M.; Martinez-Naharro, A.; et al. 99mTc-DPD Scintigraphy in Immunoglobulin Light Chain (AL) Cardiac Amyloidosis. Eur. Heart J. Cardiovasc. Imaging 2021, 22, 1304–1311. [Google Scholar] [CrossRef]
- Colby, D.W.; Zhang, Q.; Wang, S.; Groth, D.; Legname, G.; Riesner, D.; Prusiner, S.B. Prion Detection by an Amyloid Seeding Assay. Proc. Natl. Acad. Sci. USA 2007, 104, 20914–20919. [Google Scholar] [CrossRef]
- Heerde, T.; Rennegarbe, M.; Biedermann, A.; Savran, D.; Pfeiffer, P.B.; Hitzenberger, M.; Baur, J.; Puscalau-Girtu, I.; Zacharias, M.; Schwierz, N.; et al. Cryo-EM Demonstrates the in Vitro Proliferation of an Ex Vivo Amyloid Fibril Morphology by Seeding. Nat. Commun. 2022, 13, 85. [Google Scholar] [CrossRef]
- Martin, E.; Williams, A.; Wooliver, C.; Heidel, R.E.; Adams, S.; Dunlap, J.; Ramirez-Alvarado, M.; Blancas-Mejia, L.M.; Lands, R.H.; Kennel, S.J.; et al. Differential Recruitment Efficacy of Patient-Derived Amyloidogenic and Myeloma Light Chain Proteins by Synthetic Fibrils-A Metric for Predicting Amyloid Propensity. PLoS ONE 2017, 12, E0174152. [Google Scholar] [CrossRef]
- Arosio, P.; Knowles, T.P.J.; Linse, S. On the Lag Phase in Amyloid Fibril Formation. Phys. Chem. Chem. Phys. 2015, 17, 7606–7618. [Google Scholar] [CrossRef]
- Saborio, G.P.; Permanne, B.; Soto, C. Sensitive Detection of Pathological Prion Protein by Cyclic Amplification of Protein Misfolding. Nature 2001, 411, 810–813. [Google Scholar] [CrossRef]
- Atarashi, R.; Satoh, K.; Sano, K.; Fuse, T.; Yamaguchi, N.; Ishibashi, D.; Matsubara, T.; Nakagaki, T.; Yamanaka, H.; Shirabe, S.; et al. Ultrasensitive Human Prion Detection in Cerebrospinal Fluid by Real-Time Quaking-Induced Conversion. Nat. Med. 2011, 17, 175–178. [Google Scholar] [CrossRef]
- Moda, F.; Gambetti, P.; Notari, S.; Concha-Marambio, L.; Catania, M.; Park, K.-W.; Maderna, E.; Suardi, S.; Haïk, S.; Brandel, J.-P.; et al. Prions in the Urine of Patients with Variant Creutzfeldt–Jakob Disease. N. Engl. J. Med. 2014, 371, 530–539. [Google Scholar] [CrossRef] [PubMed]
- Andrich, K.; Hegenbart, U.; Kimmich, C.; Kedia, N.; Bergen, H.R.; Schönland, S.; Wanker, E.; Bieschke, J. Aggregation of Full-Length Immunoglobulin Light Chains from Systemic Light Chain Amyloidosis (AL) Patients Is Remodeled by Epigallocatechin-3-Gallate. J. Biol. Chem. 2017, 292, 2328–2344. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, T.; Annamalai, K.; Sarkar, R.; Huhn, S.; Hegenbart, U.; Schönland, S.; Fändrich, M.; Reif, B. Seeded Fibrils of the Germline Variant of Human λ-III Immunoglobulin Light Chain FOR005 Have a Similar Core as Patient Fibrils with Reduced Stability. J. Biol. Chem. 2020, 295, 18474–18484. [Google Scholar] [CrossRef] [PubMed]
- Saelices, L.; Chung, K.; Lee, J.H.; Cohn, W.; Whitelegge, J.P.; Benson, M.D.; Eisenberg, D.S. Amyloid Seeding of Transthyretin by Ex Vivo Cardiac Fibrils and Its Inhibition. Proc. Natl. Acad. Sci. USA 2018, 115, E6741–E6750. [Google Scholar] [CrossRef] [PubMed]
- Morales, R.; Moreno-Gonzalez, I.; Soto, C. Cross-Seeding of Misfolded Proteins: Implications for Etiology and Pathogenesis of Protein Misfolding Diseases. PLoS Pathog. 2013, 9, e1003537. [Google Scholar] [CrossRef]
- Blancas-Mejía, L.M.; Ramirez-Alvarado, M. Recruitment of Light Chains by Homologous and Heterologous Fibrils Shows Distinctive Kinetic and Conformational Specificity. Biochemistry 2016, 55, 2967–2978. [Google Scholar] [CrossRef]
- Yamamoto, K.; Yagi, H.; Lee, Y.-H.; Kardos, J.; Hagihara, Y.; Naiki, H.; Goto, Y. The Amyloid Fibrils of the Constant Domain of Immunoglobulin Light Chain. FEBS Lett. 2010, 584, 3348–3353. [Google Scholar] [CrossRef]
- Solomon, A.; Weiss, D.T.; Murphy, C.L.; Hrncic, R.; Wall, J.S.; Schell, M. Light chain-associated amyloid deposits comprised of a novel kappa constant domain. Proc. Natl. Acad. Sci. USA 1998, 95, 9547–9551. [Google Scholar] [CrossRef]
- Orrú, C.D.; Groveman, B.R.; Hughson, A.G.; Zanusso, G.; Coulthart, M.B.; Caughey, B. Rapid and Sensitive RT-QuIC Detection of Human Creutzfeldt-Jakob Disease Using Cerebrospinal Fluid. mBio 2015, 6, e02451-14. [Google Scholar] [CrossRef]
- Martin, E.B.; Williams, A.D.; Heidel, R.E.; Foster, J.S.; Lands, R.H.; Kennel, S.J.; Wall, J.S. A Functional Assay to Identify Amyloidogenic Light Chains. Amyloid 2018, 25, 93–100. [Google Scholar] [CrossRef]
- Finn, T.E.; Nunez, A.C.; Sunde, M.; Easterbrook-Smith, S.B. Serum Albumin Prevents Protein Aggregation and Amyloid Formation and Retains Chaperone-like Activity in the Presence of Physiological Ligands. J. Biol. Chem. 2012, 287, 21530–21540. [Google Scholar] [CrossRef] [PubMed]
- Solomon, A. [6] Light Chains of Human Immunoglobulins. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1985; Volume 116, pp. 101–121. ISBN 978-0-12-182016-9. [Google Scholar]
- Corlin, D.B.; Sen, J.W.; Ladefoged, S.; Lund, G.B.; Nissen, M.H.; Heegaard, N.H. Quantification of Cleaved Β2-Microglobulin in Serum from Patients Undergoing Chronic Hemodialysis. Clin. Chem. 2005, 51, 1177–1184. [Google Scholar] [CrossRef] [PubMed]
- Nizhnikov, A.A.; Alexandrov, A.I.; Ryzhova, T.A.; Mitkevich, O.V.; Dergalev, A.A.; Ter-Avanesyan, M.D.; Galkin, A.P. Proteomic Screening for Amyloid Proteins. PLoS ONE 2014, 9, e116003. [Google Scholar] [CrossRef]
- Buhimschi, I.A.; Nayeri, U.A.; Zhao, G.; Shook, L.L.; Pensalfini, A.; Funai, E.F.; Bernstein, I.M.; Glabe, C.G.; Buhimschi, C.S. Protein Misfolding, Congophilia, Oligomerization, and Defective Amyloid Processing in Preeclampsia. Sci. Transl. Med. 2014, 6, 245ra92. [Google Scholar] [CrossRef]
- Fedotov, S.A.; Gerasimova, E.M.; Vashukova, E.S.; Pakin, V.S.; Kapustin, R.V.; Pachulia, O.V.; Glotov, A.S.; Kulichikhin, K.Y.; Chernoff, Y.O.; Rubel, A.A. Evaluation of the effectiveness of modified CRD tests in the diagnosis of preeclampsia. J. Obstet. Women’s Dis. 2022, 71, 65–74. [Google Scholar] [CrossRef]
- Gerasimova, E.M.; Fedotov, S.A.; Kachkin, D.V.; Vashukova, E.S.; Glotov, A.S.; Chernoff, Y.O.; Rubel, A.A. Protein Misfolding during Pregnancy: New Approaches to Preeclampsia Diagnostics. Int. J. Mol. Sci. 2019, 20, 6183. [Google Scholar] [CrossRef]
- Rood, K.M.; Buhimschi, C.S.; Dible, T.; Webster, S.; Zhao, G.; Samuels, P.; Buhimschi, I.A. Congo Red Dot Paper Test for Antenatal Triage and Rapid Identification of Preeclampsia. eClinicalMedicine 2019, 8, 47–56. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).