
Pathophysiology of Mild Hypercortisolism: From the Bench to the Bedside


 ,
,  ,
, 
 , and
, and 
Abstract
1. Introduction
2. Material and Methods
3. Pathophysiology of Pituitary and Adrenal Mild Hypercortisolism
3.1. Physiology of Cortisol Secretion and Metabolism
3.2. Pathophysiology of Pituitary Mild Hypercortisolism
3.3. Pathophysiology of Adrenal Mild Hypercortisolism
4. Peripheral Glucocorticoid Sensitivity and Activation: From Eucortisolism to Mild Hypercortisolism
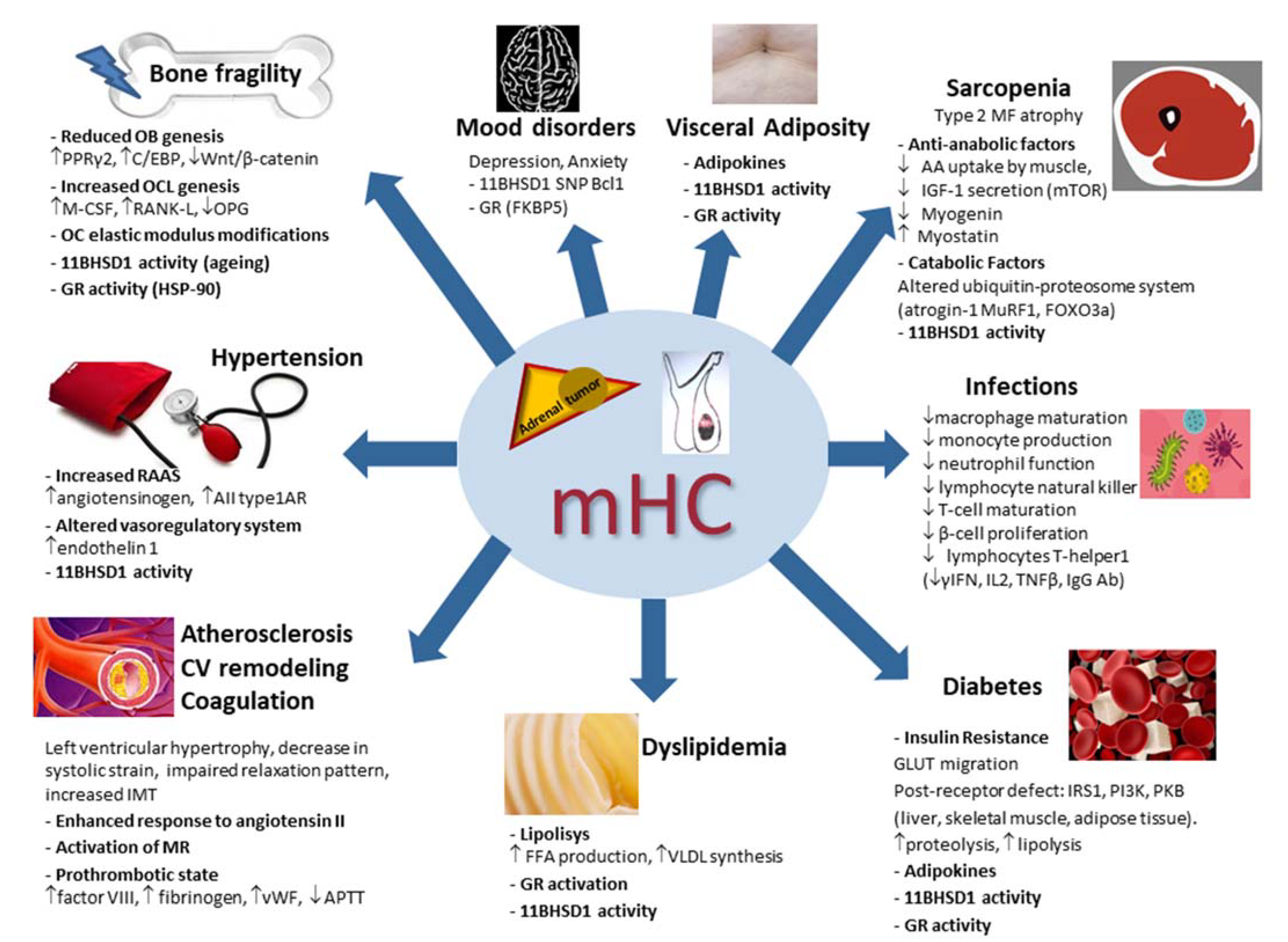
5. Pathophysiology of the Systemic Consequences of Mild Hypercortisolism
5.1. Bone Fragility in Mild Hypercortisolism
5.2. Hypertension in Mild Hypercortisolism
5.3. Subclinical Atherosclerosis, Cardiovascular Remodeling and Coagulation in Mild Hypercortisolism
5.4. Lipid and Glucose Metabolism in Mild Hypercortisolism
5.5. Skeletal Muscle in Mild Hypercortisolism
5.6. Mood Disorders in Mild Hypercortisolism
5.7. Infections in Mild Hypercortisolism
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Araujo-Castro, M.; Sampedro Núñez, M.A.; Marazuela, M. Autonomous cortisol secretion in adrenal incidentalomas. Endocrine 2019, 64, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Nieman, L.K. Update on subclinical Cushing’s syndrome. Curr. Opin. Endocrinol. Diabetes Obes. 2015, 22, 180–184. [Google Scholar] [CrossRef]
- Delivanis, D.A.; Athimulam, S.; Bancos, I. Modern Management of Mild Autonomous Cortisol Secretion. Clin. Pharmacol. Ther. 2019, 106, 1209–1221. [Google Scholar] [CrossRef]
- Chiodini, I.; Albani, A.; Ambrogio, A.G.; Campo, M.; De Martino, M.C.; Marcelli, G.; Morelli, V.; Zampetti, B.; Colao, A.; Pivonello, R. Six controversial issues on subclinical Cushing’s syndrome. Endocrine 2017, 56, 262–266. [Google Scholar] [CrossRef]
- Chiodini, I. Diagnosis and treatment of subclinical hypercortisolism. J. Clin. Endocrinol. Metab. 2011, 96, 1223–1236. [Google Scholar] [CrossRef] [PubMed]
- Giovanelli, L.; Aresta, C.; Favero, V.; Bonomi, M.; Cangiano, B.; Eller-Vainicher, C.; Grassi, G.; Morelli, V.; Pugliese, F.; Falchetti, A.; et al. Hidden hypercortisolism: A too frequently neglected clinical condition. J. Endocrinol. Investig. 2021, 44, 1581–1596. [Google Scholar] [CrossRef] [PubMed]
- Aresta, C.; Soranna, D.; Giovanelli, L.; Favero, V.; Parazzoli, C.; Gennari, L.; Persani, L.; Scillitani, A.; Blevins, L.S.; Brown, D.; et al. Endocrine Practice When to suspect hidden hypercortisolism in type 2 diabetes: A meta-analysis. Endocr. Pract. 2021, 27, 1216–1224. [Google Scholar] [CrossRef]
- Fassnacht, M.; Arlt, W.; Bancos, I.; Dralle, H.; Newell-Price, J.; Sahdev, A.; Tabarin, A.; Terzolo, M.; Tsagarakis, S.; Dekkers, O.M. Management of adrenal incidentalomas: European Society of Endocrinology Clinical Practice Guideline in collaboration with the European Network for the Study of Adrenal Tumors. Eur. J. Endocrinol. 2016, 175, G1–G34. [Google Scholar] [CrossRef] [PubMed]
- Vassilatou, E.; Vryonidou, A.; Ioannidis, D.; Paschou, S.A.; Panagou, M.; Tzavara, I. Bilateral adrenal incidentalomas differ from unilateral adrenal incidentalomas in subclinical cortisol hypersecretion but not in potential clinical implications. Eur. J. Endocrinol. 2014, 171, 37–45. [Google Scholar] [CrossRef]
- Zavatta, G.; Di Dalmazi, G. Recent Advances on Subclinical Hypercortisolism. Endocrinol. Metab. Clin. N. Am. 2018, 47, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Toini, A.; Dolci, A.; Ferrante, E.; Verrua, E.; Malchiodi, E.; Sala, E.; Lania, A.G.; Chiodini, I.; Beck-Peccoz, P.; Arosio, M.; et al. Screening for ACTH-dependent hypercortisolism in patients affected with pituitary incidentaloma. Eur. J. Endocrinol. 2015, 172, 363–369. [Google Scholar] [CrossRef]
- Favero, V.; Cremaschi, A.; Falchetti, A.; Gaudio, A.; Gennari, L.; Scillitani, A.; Vescini, F.; Morelli, V.; Aresta, C.; Chiodini, I. Management and Medical Therapy of Mild Hypercortisolism. Int. J. Mol. Sci. 2021, 22, 11521. [Google Scholar] [CrossRef] [PubMed]
- Loerz, C.; Maser, E. The cortisol-activating enzyme 11β-hydroxysteroid dehydrogenase type 1 in skeletal muscle in the pathogenesis of the metabolic syndrome. J. Steroid. Biochem. Mol. Biol. 2017, 174, 65–71. [Google Scholar] [CrossRef]
- Hassan-Smith, Z.K.; Morgan, S.A.; Sherlock, M.; Hughes, B.; Taylor, A.E.; Lavery, G.G.; Tomlinson, J.W.; Stewart, P.M. Gender-specific differences in skeletal muscle 11β- HSD1 expression across healthy aging. J. Clin. Endocrinol. Metab. 2015, 100, 2673–2681. [Google Scholar] [CrossRef]
- Morgan, S.A.; Hassan-Smith, Z.K.; Lavery, G.G. Mechanisms in endocrinology: Tissue-specific activation of cortisol in Cushing’s syndrome. Eur. J. Endocrinol. 2016, 175, R81–R87. [Google Scholar] [CrossRef]
- Motavalli, R.; Majidi, T.; Pourlak, T.; Abediazar, S.; Shoja, M.M.; Zununi Vahed, S.; Etemadi, J. The clinical significance of the glucocorticoid receptors: Genetics and epigenetics. J. Steroid. Biochem. Mol. Biol. 2021, 213, 105952. [Google Scholar] [CrossRef]
- Morelli, V.; Aresta, C.; Gaudio, A.; Eller-Vainicher, C.; Zhukouskaya, V.V.; Merlotti, D.; Orsi, E.; Maria Barbieri, A.; Fustinoni, S.; Polledri, E.; et al. Prediction of hypertension, diabetes and fractures in eucortisolemic women by measuring parameters of cortisol milieu. Endocrine 2020, 68, 411–419. [Google Scholar] [CrossRef]
- Chiodini, I.; Gaudio, A.; Eller-Vainicher, C.; Morelli, V.; Aresta, C.; Zhukouskaya, V.V.; Merlotti, D.; Orsi, E.; Barbieri, A.M.; Fustinoni, S.; et al. Cortisol Secretion, Sensitivity, and Activity Are Associated with Hypertension in Postmenopausal Eucortisolemic Women. J. Clin. Endocrinol. Metab. 2019, 104, 4441–4448. [Google Scholar] [CrossRef] [PubMed]
- Journal, E.; Findling, J.W.; Raff, H. Differentiation of pathologic/neoplastic hypercortisolism (Cushing’s syndrome) from physiologic/non-neoplastic hypercortisolism (formerly known as pseudo-Cushing’s syndrome). Eur. J. Endocrinol. 2017, 176, 205–216. [Google Scholar] [CrossRef]
- Tirabassi, G.; Boscaro, M.; Arnaldi, G. Harmful effects of functional hypercortisolism: A working hypothesis. Endocrine 2014, 46, 370–386. [Google Scholar] [CrossRef] [PubMed]
- Peverelli, E.; Catalano, R.; Giardino, E.; Treppiedi, D.; Morelli, V.; Ronchi, C.L.; Vaczlavik, A.; Fusco, N.; Ferrero, S.; Bertherat, J.; et al. Cofilin is a cAMP effector in mediating actin cytoskeleton reorganization and steroidogenesis in mouse and human adrenocortical tumor cells. Cancer Lett. 2017, 406, 54–63. [Google Scholar] [CrossRef]
- Raff, H.; Carroll, T. The Journal of Physiology Cushing’s syndrome: From physiological principles to diagnosis and clinical care. J. Physiol. 2015, 593, 493–506. [Google Scholar] [CrossRef]
- Zennaro, M.C.; Boulkroun, S.; Fernandes-Rosa, F. Genetic causes of functional adrenocortical adenomas. Endocr. Rev. 2017, 38, 516–537. [Google Scholar] [CrossRef]
- Morgan, S.A.; McCabe, E.L.; Gathercole, L.L.; Hassan-Smith, Z.K.; Larner, D.P.; Bujalska, I.J.; Stewart, P.M.; Tomlinson, J.W.; Lavery, G.G. 11β-HSD1 is the major regulator of the tissue-specific effects of circulating glucocorticoid excess. Proc. Natl. Acad. Sci. USA 2014, 111, E2482–E2491. [Google Scholar] [CrossRef]
- Vitellius, G.; Trabado, S.; Bouligand, J.; Delemer, B.; Lombès, M. Pathophysiology of Glucocorticoid Signaling. Ann. Endocrinol. 2018, 79, 98–106. [Google Scholar] [CrossRef] [PubMed]
- van Rossum, E.F.C.; Lamberts, S.W.J. Polymorphisms in the glucocorticoid receptor gene and their associations with metabolic parameters and body composition. Recent Prog. Horm. Res. 2004, 59, 333–357. [Google Scholar] [CrossRef]
- Kageyama, K.; Oki, Y.; Nigawara, T.; Suda, T.; Daimon, M. Pathophysiology and treatment of subclinical Cushing’s disease and pituitary silent corticotroph adenomas. Endocr. J. 2014, 61, 941–948. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Nakao, T.; Ogawa, W.; Fukuoka, H. Aggressive Cushing’s Disease: Molecular Pathology and Its Therapeutic Approach. Front. Endocrinol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Ebisawa, T.; Tojo, K.; Tajima, N.; Kamio, M.; Oki, Y.; Ono, K.; Sasano, H. Immunohistochemical Analysis of 11-β-Hydroxysteroid Dehydrogenase Type 2 and Glucocorticoid Receptor in Subclinical Cushing’s Disease due to Pituitary Macroadenoma. Endocr. Pathol. 2008, 19, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Fukuoka, H.; Shichi, H.; Yamamoto, M.; Takahashi, Y. Molecular Sciences The Mechanisms Underlying Autonomous Adrenocorticotropic Hormone Secretion in Cushing’s Disease. Int. J. Mol. Sci. 2020, 21, 9132. [Google Scholar] [CrossRef]
- Sonino, N.; Fava, G.; Boscaro, M. A role for life events in the pathogenesis of Cushing’s disease. Clin. Endocrinol. 1993, 38, 261–264. [Google Scholar] [CrossRef] [PubMed]
- Sonino, N.; Fava, M.; Grandi, S.; Mantero, F.; Boscaro, M. Stressful life events in the pathogenesis of Cushing’s syndrome. Clin. Endocrinol. 1988, 29, 617–623. [Google Scholar] [CrossRef]
- Campana, G.; Loizzo, S.; Fortuna, A.; Rimondini, R.; Maroccia, Z.; Scillitani, A.; Falchetti, A.; Spampinato, S.M.; Persani, L.; Chiodini, I. Early post-natal life stress induces permanent adrenocorticotropin-dependent hypercortisolism in male mice. Endocrine 2021, 73, 186–195. [Google Scholar] [CrossRef]
- Kokkinopoulou, I.; Diakoumi, A.; Moutsatsou, P. Glucocorticoid Receptor Signaling in Diabetes. Int. J. Mol. Sci. 2021, 22, 11173. [Google Scholar] [CrossRef] [PubMed]
- Sherlock, M.; Scarsbrook, A.; Abbas, A.; Fraser, S.; Limumpornpetch, P.; Dineen, R.; Stewart, P.M. Adrenal Incidentaloma. Endocr. Rev. 2020, 41, 775–820. [Google Scholar] [CrossRef]
- Bonnet-Serrano, F.; Bertherat, J. Genetics of tumors of the adrenal cortex. Endocr.-Relat. Cancer 2018, 25, R131–R152. [Google Scholar] [CrossRef] [PubMed]
- Kamilaris, C.D.C.; Hannah-Shmouni, F.; Stratakis, C.A. Adrenocortical tumorigenesis: Lessons from genetics. Best Pract. Res. Clin. Endocrinol. Metab. 2020, 34, 101428. [Google Scholar] [CrossRef]
- Maillet, M.; Bourdeau, I.; Lacroix, A. Update on primary micronodular bilateral adrenocortical diseases. Curr. Opin. Endocrinol. Diabetes Obes. 2020, 27, 132–139. [Google Scholar] [CrossRef]
- Tirosh, A.; Valdés, N.; Stratakis, C.A. Genetics of micronodular adrenal hyperplasia and Carney complex. Presse Med. 2018, 47 Pt 2, e127–e137. [Google Scholar] [CrossRef]
- Vassiliadi, D.A.; Tsagarakis, S. Diagnosis and management of primary bilateral macronodular adrenal hyperplasia. Endocr.-Relat. Cancer 2019, 26, R567–R581. [Google Scholar] [CrossRef]
- Alencar, G.A.; Lerario, A.M.; Nishi, M.Y.; Mariani, B.M.; Almeida, M.Q.; Tremblay, J.; Hamet, P.; Bourdeau, I.; Zerbini, M.C.; Pereira, M.A.; et al. ARMC5 mutations are a frequent cause of primary macronodular adrenal Hyperplasia. J. Clin. Endocrinol. Metab. 2014, 99, E1501–E1509. [Google Scholar] [CrossRef]
- Reznik, Y.; Lefebvre, H.; Rohmer, V.; Charbonnel, B.; Tabarin, A.; Rodien, P.; Lecomte, P.; Bardet, S.; Coffin, C.; Mahoudeau, J. Aberrant adrenal sensitivity to multiple ligands in unilateral incidentaloma with subclinical autonomous cortisol hypersecretion: A prospective clinical study. Clin. Endocrinol. 2004, 61, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; He, M.; Gao, Z.; Peng, Y.; Li, Y.; Li, L.; Zhou, W.; Li, X.; Zhong, X.; Lei, Y.; et al. Activating hotspot L205R mutation in PRKACA and adrenal Cushing’s syndrome. Science 2014, 344, 913–917. [Google Scholar] [CrossRef]
- Beuschlein, F.; Fassnacht, M.; Assié, G.; Calebiro, D.; Stratakis, C.A.; Osswald, A.; Ronchi, C.L.; Wieland, T.; Sbiera, S.; Faucz, F.R.; et al. Constitutive activation of PKA catalytic subunit in adrenal Cushing’s syndrome. N. Engl. J. Med. 2014, 370, 1019–1028. [Google Scholar] [CrossRef] [PubMed]
- Goh, G.; Scholl, U.I.; Healy, J.M.; Choi, M.; Prasad, M.L.; Nelson-Williams, C.; Kunstman, J.W.; Korah, R.; Suttorp, A.C.; Dietrich, D.; et al. Recurrent activating mutation in PRKACA in cortisol-producing adrenal tumors. Nat. Genet. 2014, 46, 613–617. [Google Scholar] [CrossRef]
- Di Dalmazi, G.; Kisker, C.; Calebiro, D.; Mannelli, M.; Canu, L.; Arnaldi, G.; Quinkler, M.; Rayes, N.; Tabarin, A.; Laure Jullié, M.; et al. Novel somatic mutations in the catalytic subunit of the protein kinase A as a cause of adrenal Cushing’s syndrome: A European multicentric study. J. Clin. Endocrinol. Metab. 2014, 99, E2093–E2100. [Google Scholar] [CrossRef]
- Sato, Y.; Maekawa, S.; Ishii, R.; Sanada, M.; Morikawa, T.; Shiraishi, Y.; Yoshida, K.; Nagata, Y.; Sato-Otsubo, A.; Yoshizato, T.; et al. Recurrent somatic mutations underlie corticotropin-independent Cushing’s syndrome. Science 2014, 344, 917–920. [Google Scholar] [CrossRef] [PubMed]
- Thiel, A.; Reis, A.C.; Haase, M.; Goh, G.; Schott, M.; Willenberg, H.S.; Scholl, U.I. PRKACA mutations in cortisol-producing adenomas and adrenal hyperplasia: A single-center study of 60 cases. Eur. J. Endocrinol. 2015, 172, 677–685. [Google Scholar] [CrossRef]
- Ronchi, C.L.; Di Dalmazi, G.; Faillot, S.; Sbiera, S.; Assié, G.; Weigand, I.; Calebiro, D.; Schwarzmayr, T.; Appenzeller, S.; Rubin, B.; et al. Genetic landscape of sporadic unilateral adrenocortical adenomas without PRKACA p.Leu206Arg mutation. J. Clin. Endocrinol. Metab. 2016, 101, 3526–3538. [Google Scholar] [CrossRef]
- Lyraki, R.; Schedl, A. Adrenal cortex renewal in health and disease. Nat. Rev. Endocrinol. 2021, 17, 421–434. [Google Scholar] [CrossRef]
- Wilmot Roussel, H.; Vezzosi, D.; Rizk-Rabin, M.; Barreau, O.; Ragazzon, B.; René-Corail, F.; de Reynies, A.; Bertherat, J.; Assié, G. Identification of gene expression profiles associated with cortisol secretion in adrenocortical adenomas. J. Clin. Endocrinol. Metab. 2013, 98, E1109–E1121. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, S.; Serysheva, I. Ryanodine receptor structure: Progress and challenges. J. Biol. Chem. 2009, 284, 4047–4051. [Google Scholar] [CrossRef] [PubMed]
- Enyeart, J.; Enyeart, J. Adrenal fasciculata cells express T-type and rapidly and slowly activating L-type Ca2+ channels that regulate cortisol secretion. Am. J. Physiol. Cell Physiol. 2015, 308, C899–C918. [Google Scholar] [CrossRef] [PubMed]
- Stewart, T.A.; Yapa, K.T.; Monteith, G.R. Altered calcium signaling in cancer cells. Biochim. Biophys. Acta 2015, 1848, 2502–2511. [Google Scholar] [CrossRef]
- Di Dalmazi, G.; Altieri, B.; Scholz, C.; Sbiera, S.; Luconi, M.; Waldman, J.; Kastelan, D.; Ceccato, F.; Chiodini, I.; Arnaldi, G.; et al. RNA Sequencing and Somatic Mutation Status of Adrenocortical Tumors: Novel Pathogenetic Insights. J. Clin. Endocrinol. Metab. 2020, 105, e4459–e4473. [Google Scholar] [CrossRef]
- Kometani, M.; Yoneda, T.; Demura, M.; Koide, H.; Nishimoto, K.; Mukai, K.; Gomez-Sanchez, C.E.; Akagi, T.; Yokota, T.; Horike, S.I.; et al. Cortisol overproduction results from DNA methylation of CYP11B1 in hypercortisolemia. Sci. Rep. 2017, 7, 11205. [Google Scholar] [CrossRef] [PubMed]
- Di Dalmazi, G.; Morandi, L.; Rubin, B.; Pilon, C.; Asioli, S.; Vicennati, V.; de Leo, A.; Ambrosi, F.; Santini, D.; Pagotto, U.; et al. DNA Methylation of Steroidogenic Enzymes in Benign Adrenocortical Tumors: New Insights in Aldosterone-Producing Adenomas. J. Clin. Endocrinol. Metab. 2020, 105, e4605–e4615. [Google Scholar] [CrossRef]
- Wang, C.; Sun, Y.; Feng, R.; Feng, R.; Xu, M.; Yin, X.; Liang, K.; Zhao, R.; Gu, G.; Jiang, X.; et al. Alterations of DNA Methylation Were Associated with the Rapid Growth of Cortisol-Producing Adrenocortical Adenoma During Pregnancy. SSRN Electron. J. 2021, 13, 1–14. [Google Scholar] [CrossRef]
- Rossi, R.; Tauchmanova, L.; Luciano, A.; Di Martino, C.; Battista, C.; Del Viscovo, L.; Nuzzo, V.; Lombardi, G. Subclinical Cushing’s syndrome in patients with adrenal incidentaloma: Clinical and biochemical features. J. Clin. Endocrinol. Metab. 2000, 85, 1440–1448. [Google Scholar] [CrossRef] [PubMed]
- Masjkur, J.; Gruber, M.; Peitzsch, M.; Kaden, D.; Di Dalmazi, G.; Bidlingmaier, M.; Zopp, S.; Langton, K.; Fazel, J.; Beuschlein, F.; et al. Plasma Steroid. Profiles in Subclinical Compared With Overt Adrenal Cushing Syndrome. J. Clin. Endocrinol. Metab. 2019, 104, 4331–4340. [Google Scholar] [CrossRef]
- Di Dalmazi, G.; Fanelli, F.; Mezzullo, M.; Casadio, E.; Rinaldi, E.; Garelli, S.; Giampalma, E.; Mosconi, C.; Golfieri, R.; Vicennati, V.; et al. Steroid. Profiling by LC-MS/MS in Nonsecreting and Subclinical Cortisol-Secreting Adrenocortical Adenomas. J. Clin. Endocrinol. Metab. 2015, 100, 3529–3538. [Google Scholar] [CrossRef]
- Damjanovic, S.S.; Antic, J.A.; Ilic, B.B.; Cokic, B.B.; Ivovic, M.; Ognjanovic, S.I.; Isailovic, T.V.; Popovic, B.M.; Bozic, I.B.; Tatic, S.; et al. Glucocorticoid Receptor and Molecular Chaperones in the Pathogenesis of Adrenal Incidentalomas: Potential Role of Reduced Sensitivity to Glucocorticoids. Mol. Med. 2012, 18, 1456–1465. [Google Scholar] [CrossRef]
- Tzanela, M.; Mantzou, E.; Saltiki, K.; Tampourlou, M.; Kalogeris, N.; Hadjidakis, D.; Tsagarakis, S.; Alevizaki, M. Clinical and biochemical impact of BCL1 polymorphic genotype of the glucocorticoid receptor gene in patients with adrenal incidentalomas. J. Endocrinol. Investig. 2012, 35, 395–400. [Google Scholar] [CrossRef]
- Majnik, J.; Patocs, A.; Balogh, K.; Toth, M.; Gergics, P.; Szappanos, A.; Mondok, A.; Borgulya, G.; Panczel, P.; Prohaszka, Z.; et al. Overrepresentation of the N363S variant of the glucocorticoid receptor gene in patients with bilateral adrenal incidentalomas. J. Clin. Endocrinol. Metab. 2006, 91, 2796–2799. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Trementino, L.; Appolloni, G.; Concettoni, C.; Cardinaletti, M.; Boscaro, M.; Arnaldi, G. Association of glucocorticoid receptor polymorphism A3669G with decreased risk of developing diabetes in patients with Cushing’s syndrome. Eur. J. Endocrinol. 2012, 166, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Szappanos, Á.; Patócs, A.; Tõke, J.; Boyle, B.; Sereg, M.; Majnik, J.; Borgulya, G.; Varga, I.; Likó, I.; Rácz, K.; et al. BclI polymorphism of the glucocorticoid receptor gene is associated with decreased bone mineral density in patients with endogenous hypercortisolism. Clin. Endocrinol. 2009, 71, 636–643. [Google Scholar] [CrossRef] [PubMed]
- Reimondo, G.; Chiodini, I.; Puglisi, S.; Pia, A.; Morelli, V.; Kastelan, D.; Cannavo, S.; Berchialla, P.; Giachino, D.; Perotti, P.; et al. Analysis of BCLI, N363S and ER22/23EK polymorphisms of the glucocorticoid receptor gene in adrenal incidentalomas. PLoS ONE 2016, 11, e0162437. [Google Scholar] [CrossRef]
- Tomlinson, J.; Draper, N.; Mackie, J.; Johnson, A.; Holder, G.; Wood, P.; Stewart, P. Absence of Cushingoid phenotype in a patient with Cushing’s disease due to defective cortisone to cortisol conversion. J. Clin. Endocrinol. Metab. 2002, 87, 57–62. [Google Scholar] [CrossRef]
- Masuzaki, H.; Paterson, J.; Shinyama, H.; Morton, N.M.; Mullins, J.J.; Seckl, J.R.; Flier, J.S. A transgenic model of visceral obesity and the metabolic syndrome. Science 2001, 294, 2166–2170. [Google Scholar] [CrossRef]
- Iwasaki, Y.; Takayasu, S.; Nishiyama, M.; Tsugita, M.; Taguchi, T.; Asai, M.; Yoshida, M.; Kambayashi, M.; Hashimoto, K. Is the metabolic syndrome an intracellular Cushing state? Effects of multiple humoral factors on the transcriptional activity of the hepatic glucocorticoid-activating enzyme (11β-hydroxysteroid dehydrogenase type 1) gene. Mol. Cell. Endocrinol. 2008, 285, 10–18. [Google Scholar] [CrossRef]
- Morelli, V.; Donadio, F.; Eller-Vainicher, C.; Cirello, V.; Olgiati, L.; Savoca, C.; Cairoli, E.; Salcuni, A.S.; Beck-Peccoz, P.; Chiodini, I. Role of glucocorticoid receptor polymorphism in adrenal incidentalomas. Eur. J. Clin. Investig. 2010, 40, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Canalis, E.; Mazziotti, G.; Giustina, A.; Bilezikian, J. Glucocorticoid-induced osteoporosis: Pathophysiology and therapy. Osteoporos. Int. 2007, 18, 1319–1328. [Google Scholar] [CrossRef] [PubMed]
- Chotiyarnwong, P.; McCloskey, E.V. Pathogenesis of glucocorticoid-induced osteoporosis and options for treatment. Nat. Rev Endocrinol. 2020, 16, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Chiodini, I.; Falchetti, A.; Merlotti, D.; Eller Vainicher, C.; Gennari, L. Updates in epidemiology, pathophysiology and management strategies of glucocorticoid-induced osteoporosis. Expert Rev. Endocrinol. Metab. 2020, 15, 283–298. [Google Scholar] [CrossRef]
- Cooper, M.S.; Walker, E.A.; Bland, R.; Fraser, W.D.; Hewison, M.; Stewart, P.M. Expression and Functional Consequences of 11-Hydroxysteroid Dehydrogenase Activity in Human Bone. Bone 2000, 27, 375–381. [Google Scholar] [CrossRef]
- Park, J.S.; Bae, S.J.; Choi, S.W.; Son, Y.H.; Park, S.B.; Rhee, S.D.; Kim, H.Y.; Jung, W.H.; Kang, S.K.; Ahn, J.H.; et al. A novel 11β-HSD1 inhibitor improves diabesity and osteoblast differentiation. J. Mol. Endocrinol. 2014, 52, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhou, F.; Pan, Z.; Bu, X.; Wang, Y.; Chen, F. 11β-hydroxysteroid dehydrogenases-2 decreases the apoptosis of MC3T3/MLO-Y4 cells induced by glucocorticoids. Biochem. Biophys. Res. Commun. 2017, 490, 1399–1406. [Google Scholar] [CrossRef]
- Fenton, C.G.; Doig, C.L.; Fareed, S.; Naylor, A.; Morrell, A.P.; Addison, O.; Wehmeyer, C.; Buckley, C.D.; Cooper, M.S.; Lavery, G.G.; et al. 11β-HSD1 plays a critical role in trabecular bone loss associated with systemic glucocorticoid therapy. Arthritis. Res. Ther. 2019, 21, 1–10. [Google Scholar] [CrossRef]
- Cooper, M.S.; Syddall, H.E.; Fall, C.H.D.; Wood, P.J.; Stewart, P.M.; Cooper, C.; Dennison, E.M. Circulating cortisone levels are associated with biochemical markers of bone formation and lumbar spine BMD: The Hertfordshire Cohort Study. Clin. Endocrinol. 2005, 62, 692–697. [Google Scholar] [CrossRef]
- Hwang, J.Y.; Lee, S.H.; Kim, G.S.; Koh, J.M.; Go, M.J.; Kim, Y.J.; Kim, H.C.; Kim, T.H.; Hong, J.M.; Park, E.K.; et al. HSD11B1 polymorphisms predicted bone mineral density and fracture risk in postmenopausal women without a clinically apparent hypercortisolemia. Bone 2009, 45, 1098–1103. [Google Scholar] [CrossRef] [PubMed]
- Siggelkow, H.; Etmanski, M.; Bozkurt, S.; Groß, P.; Koepp, R.; Brockmöller, J.; Tzvetkov, M.V. Genetic polymorphisms in 11β-hydroxysteroid dehydrogenase type 1 correlate with the postdexamethasone cortisol levels and bone mineral density in patients evaluated for osteoporosis. J. Clin. Endocrinol. Metab. 2014, 99, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.S.; Cooper, M.S.; Hardy, R.S. Endogenous Glucocorticoid Metabolism in Bone: Friend or Foe. Front. Endocrinol. 2021, 12, 1–13. [Google Scholar] [CrossRef]
- Aresta, C.; Favero, V.; Morelli, V.; Giovanelli, L.; Parazzoli, C.; Falchetti, A.; Pugliese, F.; Gennari, L.; Vescini, F.; Salcuni, A.; et al. Cardiovascular complications of mild autonomous cortisol secretion. Best Pract. Res. Clin. Endocrinol. Metab. 2021, 35, 101494. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; El Kawkgi, O.; Henriquez, A.; Bancos, I. Cardiovascular risk and mortality in patients with active and treated hypercortisolism. Gland Surg. 2020, 9, 43–58. [Google Scholar] [CrossRef]
- Shibata, H.; Suzuki, H.; Maruyama, T.; Saruta, T. Gene expression of angiotensin II receptor in blood cells of Cushing’s syndrome. Hypertension 1995, 26, 1003–1010. [Google Scholar] [CrossRef]
- Kirilov, G.; Tomova, A.; Dakovska, L.; Kumanov, P.; Shinkov, A.; Alexandrov, A. Elevated plasma endothelin as an additional cardiovascular risk factor in patients with Cushing’s syndrome. Eur. J. Endocrinol. 2003, 149, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Vitellius, G.; Delemer, B.; Caron, P.; Chabre, O.; Bouligand, J.; Pussard, E.; Trabado, S.; Lombes, M. Impaired 11β-Hydroxysteroid Dehydrogenase Type 2 in Glucocorticoid-Resistant Patients. J. Clin. Endocrinol. Metab. 2019, 104, 5205–5216. [Google Scholar] [CrossRef]
- St-Jean, M.; Lim, D.S.T.; Langlois, F. Hypercoagulability in Cushing’s syndrome: From arterial to venous disease. Best Pract. Res. Clin. Endocrinol. Metab. 2021, 35, 101496. [Google Scholar] [CrossRef]
- Di Dalmazi, G.; Vicennati, V.; Garelli, S.; Casadio, E.; Rinaldi, E.; Giampalma, E.; Mosconi, C.; Golfieri, R.; Paccapelo, A.; Pagotto, U.; et al. Cardiovascular events and mortality in patients with adrenal incidentalomas that are either non-secreting or associated with intermediate phenotype or subclinical Cushing’s syndrome: A 15-year retrospective study. Lancet Diabetes Endocrinol. 2014, 2, 396–405. [Google Scholar] [CrossRef]
- Morelli, V.; Reimondo, G.; Giordano, R.; Della Casa, S.; Policola, C.; Palmieri, S.; Salcuni, A.S.; Dolci, A.; Mendola, M.; Arosio, M.; et al. Long-term follow-up in adrenal incidentalomas: An Italian multicenter study. J. Clin. Endocrinol. Metab. 2014, 99, 827–834. [Google Scholar] [CrossRef]
- Patrova, J.; Kjellman, M.; Wahrenberg, H.; Falhammar, H. Increased mortality in patients with adrenal incidentalomas and autonomous cortisol secretion: A 13-year retrospective study from one center. Endocrine 2017, 58, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Debono, M.; Bradburn, M.; Bull, M.; Harrison, B.; Ross, R.J.; Newell-Price, J. Cortisol as a marker for increased mortality in patients with incidental adrenocortical adenomas. J. Clin. Endocrinol. Metab. 2014, 99, 4462–4470. [Google Scholar] [CrossRef] [PubMed]
- Androulakis, I.I.; Kaltsas, G.A.; Kollias, G.E.; Markou, A.C.; Gouli, A.K.; Thomas, D.A.; Alexandraki, K.I.; Papamichael, C.M.; Hadjidakis, D.J.; Piaditis, G.P. Patients With Apparently Nonfunctioning Adrenal Incidentalomas May Be at Increased Cardiovascular Risk Due to Excessive Cortisol Secretion. J. Clin. Endocrinol. Metab. 2014, 99, 2754–2762. [Google Scholar] [CrossRef]
- Ermetici, F.; Dall’Asta, C.; Malavazos, A.; Coman, C.; Morricone, L.; Montericcio, V.; Ambrosi, B. Echocardiographic alterations in patients with non-functioning adrenal incidentaloma. J. Endocrinol. Investig. 2008, 31, 573–577. [Google Scholar] [CrossRef]
- Yener, S.; Genc, S.; Akinci, B.; Secil, M.; Demir, T.; Comlekci, A.; Ertilav, S.; Yesil, S. Carotid intima media thickness is increased and associated with morning cortisol in subjects with non-functioning adrenal incidentaloma. Endocrine 2009, 35, 365–370. [Google Scholar] [CrossRef]
- Sereg, M.; Szappanos, A.; Toke, J.; Karlinger, K.; Feldman, K.; Kaszper, E.; Varga, I.; Gláz, E.; Rácz, K.; Tóth, M. Atherosclerotic risk factors and complications in patients with non-functioning adrenal adenomas treated with or without adrenalectomy: A long-term follow-up study. Eur. J. Endocrinol. 2009, 160, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Tuna, M.M.; Imga, N.N.; Doğan, B.A.; Yılmaz, F.M.; Topçuoğlu, C.; Akbaba, G.; Berker, D.; Güler, S. Non-functioning adrenal incidentalomas are associated with higher hypertension prevalence and higher risk of atherosclerosis. J. Endocrinol. Investig. 2014, 37, 765–768. [Google Scholar] [CrossRef]
- Pivonello, R.; Isidori, A.M.; De Martino, M.C.; Newell-Price, J.; Biller, B.M.K.; Colao, A. Complications of Cushing’s syndrome: State of the art. Lancet Diabetes Endocrinol. 2016, 4, 611–629. [Google Scholar] [CrossRef]
- Światkowska-Stodulska, R.; Sworczak, K. Disorders of hemostasis in overt and subclinical hypercortisolism. Exp. Clin. Endocrinol. Diabetes 2013, 121, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Swiatkowska-Stodulska, R.; Skibowska-Bielinska, A.; Wisniewski, P.; Sworczak, K. Activity of selected coagulation factors in overt and subclinical hypercortisolism. Endocr. J. 2015, 62, 687–694. [Google Scholar] [CrossRef]
- Świątkowska-Stodulska, R.; Kaniuka-Jakubowska, S.; Wiśniewski, P.; Skibowska-Bielińska, A.; Sworczak, K. The estimation of selected endogenous anticoagulation system parameters in patients with subclinical Cushing’s syndrome. Eur. J. Endocrinol. 2011, 165, 865–871. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Masserini, B.; Morelli, V.; Palmieri, S.; Eller-Vainicher, C.; Zhukouskaya, V.; Cairoli, E.; Orsi, E.; Beck-Peccoz, P.; Spada, A.; Chiodini, I. Lipid abnormalities in patients with adrenal incidentalomas: Role of subclinical hypercortisolism and impaired glucose metabolism. J. Endocrinol. Investig. 2015, 38, 623–628. [Google Scholar] [CrossRef]
- Arnaldi, G.; Scandali, V.; Trementino, L.; Cardinaletti, M.; Appolloni, G.; Boscaro, M. Pathophysiology of dyslipidemia in Cushing’s syndrome. Neuroendocrinology 2010, 92 (Suppl. 1), 86–90. [Google Scholar] [CrossRef] [PubMed]
- Mazziotti, G.; Formenti, A.; Frara, S.; Maffezzoni, F.; Doga, M.; Giustina, A. Diabetes in Cushing Disease. Curr. Diab. Rep. 2017, 17, 32. [Google Scholar] [CrossRef]
- Sharma, A.; Vella, A. Glucose Metabolism in Cushing Syndrome. Curr. Opin. Endocrinol. Diabetes. Obes. 2020, 27, 140. [Google Scholar] [CrossRef]
- Ermetici, F.; Malavazos, A.; Corbetta, S.; Morricone, L.; Dall’Asta, C.; Corsi, M.; Ambrosi, B. Adipokine levels and cardiovascular risk in patients with adrenal incidentaloma. Metabolism 2007, 56, 686–692. [Google Scholar] [CrossRef]
- Oakley, R.H.; Sar, M.; Cidlowski, J.A. The Human Glucocorticoid Receptor β Isoform: Expression, biochemical properties, and putative function (∗). J. Biol. Chem. 1996, 271, 9550–9559. [Google Scholar] [CrossRef]
- Reincke, M. Cushing Syndrome Associated Myopathy: It Is Time for a Change. Endocrinol. Metab. 2021, 36, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Karsenty, G.; Olson, E. Bone and Muscle Endocrine Functions: Unexpected Paradigms of Inter-organ Communication. Cell 2016, 164, 1248–1256. [Google Scholar] [CrossRef]
- Schakman, O.; Kalista, S.; Barbé, C.; Loumaye, A.; Thissen, J. Glucocorticoid-induced skeletal muscle atrophy. Int. J. Biochem. Cell Biol. 2013, 45, 2163–2172. [Google Scholar] [CrossRef] [PubMed]
- Canepari, M.; Agoni, V.; Brocca, L.; Ghigo, E.; Gnesi, M.; Minetto, M.; Bottinelli, R. Structural and molecular adaptations to dexamethasone and unacylated ghrelin administration in skeletal muscle of the mice. J. Physiol. Pharmacol. 2018, 69. [Google Scholar] [CrossRef]
- Morgan, S.; Hassan-Smith, Z.; Doig, C.; Sherlock, M.; Stewart, P.; Lavery, G. Glucocorticoids and 11β-HSD1 are major regulators of intramyocellular protein metabolism. J. Endocrinol. 2016, 229, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Oda, S.; Ashida, K.; Uchiyama, M.; Sakamoto, S.; Hasuzawa, N.; Nagayama, A.; Wang, L.; Nagata, H.; Sakamoto, R.; Kishimoto, J.; et al. An Open-label Phase I/IIa Clinical Trial of 11β-HSD1 Inhibitor for Cushing’s Syndrome and Autonomous Cortisol Secretion. J. Clin. Endocrinol. Metab. 2021, 106, e3865–e3880. [Google Scholar] [CrossRef] [PubMed]
- Berr, C.M.; Stieg, M.R.; Deutschbein, T.; Quinkler, M.; Schmidmaier, R.; Osswald, A.; Reisch, N.; Ritzel, K.; Dimopoulou, C.; Fazel, J.; et al. Persistence of myopathy in Cushing’s syndrome: Evaluation of the German Cushing’s Registry. Eur. J. Endocrinol. 2017, 176, 737–746. [Google Scholar] [CrossRef]
- Vogel, F.; Braun, L.; Rubinstein, G.; Zopp, S.; Benedix, S.; Schneider, H.; Ritzel, K.; Schilbach, K.; Schmidmaier, R.; Beuschlein, F.; et al. Patients with low IGF-I after curative surgery for Cushing’s syndrome have an adverse long-term outcome of hypercortisolism-induced myopathy. Eur. J. Endocrinol. 2021, 184, 813–821. [Google Scholar] [CrossRef] [PubMed]
- Geer, E.; Shen, W.; Strohmayer, E.; Post, K.; Freda, P. Body composition and cardiovascular risk markers after remission of Cushing’s disease: A prospective study using whole-body MRI. J. Clin. Endocrinol. Metab. 2012, 97, 1702–1711. [Google Scholar] [CrossRef]
- Delivanis, D.A.; Andrade, M.D.H.; Cortes, T.; Athimulam, S.; Khanna, A.; Atkinson, E.; McKenzie, T.; Takahashi, N.; Moynagh, M.R.; Bancos, I. Abnormal body composition in patients with adrenal adenomas. Eur. J. Endocrinol. 2021, 185, 653–662. [Google Scholar] [CrossRef]
- Hong, N.; Lee, J.; Ku, C.R.; Han, K.; Lee, C.R.; Kang, S.W.; Rhee, Y. Changes of computed tomography-based body composition after adrenalectomy in patients with endogenous hypercortisolism. Clin. Endocrinol. 2019, 90, 267–276. [Google Scholar] [CrossRef]
- Pivonello, R.; Simeoli, C.; De Martino, M.; Cozzolino, A.; De Leo, M.; Iacuaniello, D.; Pivonello, C.; Negri, M.; Pellecchia, M.; Iasevoli, F.; et al. Neuropsychiatric disorders in Cushing’s syndrome. Front. Neurosci. 2015, 9. [Google Scholar] [CrossRef]
- Lin, T.Y.; Hanna, J.; Ishak, W.W. Psychiatric Symptoms in Cushing’s Syndrome: A Systematic Review. Innov. Clin. Neurosci. 2020, 17, 30. [Google Scholar]
- Piasecka, M.; Papakokkinou, E.; Valassi, E.; Santos, A.; Webb, S.M.; de Vries, F.; Pereira, A.M.; Ragnarsson, O. Psychiatric and neurocognitive consequences of endogenous hypercortisolism. J. Intern. Med. 2020, 288, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.; Resmini, E.; Pascual, J.C.; Crespo, I.; Webb, S.M. Psychiatric Symptoms in Patients with Cushing’s Syndrome: Prevalence, Diagnosis and Management. Drugs 2017, 77, 829–842. [Google Scholar] [CrossRef]
- Ragnarsson, O.; Glad, C.; Berglund, P.; Bergthorsdottir, R.; Eder, D.; Johannsson, G. Common genetic variants in the glucocorticoid receptor and the 11β-hydroxysteroid dehydrogenase type 1 genes influence long-term cognitive impairments in patients with Cushing’s syndrome in remission. J. Clin. Endocrinol. Metab. 2014, 99, E1803–E1807. [Google Scholar] [CrossRef]
- Resmini, E.; Santos, A.; Aulinas, A.; Webb, S.; Vives-Gilabert, Y.; Cox, O.; Wand, G.; Lee, R. Reduced DNA methylation of FKBP5 in Cushing’s syndrome. Endocrine 2016, 54, 768–777. [Google Scholar] [CrossRef]
- Morelli, V.; Ghielmetti, A.; Caldiroli, A.; Grassi, S.; Siri, F.M.; Caletti, E.; Mucci, F.; Aresta, C.; Passeri, E.; Pugliese, F.; et al. Mental Health in Patients With Adrenal Incidentalomas: Is There a Relation With Different Degrees of Cortisol Secretion? J. Clin. Endocrinol. Metab. 2020, 106, e130–e139. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, O.; Jørgensen, J.; Cannegieter, S.; Ehrenstein, V.; Vandenbroucke, J.; Pereira, A.; Sørensen, H. Multisystem morbidity and mortality in Cushing’s syndrome: A cohort study. J. Clin. Endocrinol. Metab. 2013, 98, 2277–2284. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, Y.; Kameda, H.; Omori, K.; Tani, M.; Cho, K.Y.; Nakamura, A.; Miyoshi, H.; Tanaka, S.; Atsumi, T. Severe infection including disseminated herpes zoster triggered by subclinical Cushing’s disease: A case report. BMC Endocr. Disord. 2021, 21, 84. [Google Scholar] [CrossRef]
- Chiodini, I.; Gennari, L. Grand Challenge in Adrenal Endocrinology: Is the Legacy of the Past a Challenge for the Future of Precision Medicine? Front. Endocrinol. 2021, 12, 1–7. [Google Scholar] [CrossRef]
- Eller-Vainicher, C.; Morelli, V.; Aresta, C.; Salcuni, A.S.; Falchetti, A.; Carnevale, V.; Persani, L.; Scillitani, A.; Chiodini, I. Defining Nonfunctioning Adrenal Adenomas on the Basis of the Occurrence of Hypocortisolism after Adrenalectomy. J. Endocr. Soc. 2020. [Google Scholar] [CrossRef]
- Crawford, A.A.; Soderberg, S.; Kirschbaum, C.; Murphy, L.; Eliasson, M.; Ebrahim, S.; Smith, G.D.; Olsson, T.; Sattar, N.; Lawlor, D.A.; et al. Morning plasma cortisol as a cardiovascular risk factor: Findings from prospective cohort and Mendelian randomization studies. Eur. J. Endocrinol. 2019, 181, 429–438. [Google Scholar] [CrossRef]
- Mourtzi, N.; Sertedaki, A.; Charmandari, E. Glucocorticoid Signaling and Epigenetic Alterations in Stress-Related Disorders. Int. J. Mol. Sci. 2021, 22, 5964. [Google Scholar] [CrossRef] [PubMed]
- Tafet, G.; Nemeroff, C. Pharmacological Treatment of Anxiety Disorders: The Role of the HPA Axis. Front. Psychiatry 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Rosenstock, J.; Banarer, S.; Fonseca, V.A.; Inzucchi, S.E.; William, S.; Wenqing, Y.; Hollis, G.; Flores, R.; Levy, R.; Williams, W.V.; et al. The 11-β-hydroxysteroid dehydrogenase type 1 inhibitor INCB13739 improves hyperglycemia in patients with type 2 diabetes inadequately controlled by metformin monotherapy. Diabetes Care 2010, 33, 1516–1522. [Google Scholar] [CrossRef]
- Haas, A.V.; Hopkins, P.N.; Brown, N.J.; Pojoga, L.H.; Williams, J.S.; Adler, G.K.; Williams, G.H. Higher urinary cortisol levels associate with increased cardiovascular risk. Endocr. Connect. 2019, 8, 634–640. [Google Scholar] [CrossRef] [PubMed]
- Chamarthi, B.; Kolatkar, N.S.; Hunt, S.C.; Williams, J.S.; Seely, E.W.; Brown, N.J.; Murphey, L.J.; Jeunemaitre, X.; Williams, G.H. Urinary free cortisol: An intermediate phenotype and a potential genetic marker for a salt-resistant subset of essential hypertension. J. Clin. Endocrinol. Metab. 2007, 92, 1340–1346. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cushing’s Syndrome | Mild Hypercortisolism | |
|---|---|---|
| Definition | Large group of signs and symptoms that reflect prolonged and inappropriately high exposure of tissue to glucocorticoids | Alteration of hypothalamic–pituitary–adrenal axis secretion in the absence of signs or symptoms of cortisol excess |
| Prevalence | 1/500,000 | 0.8–2/1000 |
| Origin | ACTH-dependent: 80% | ACTH-dependent: 10–15% |
| ACTH-independent: 20% | ACTH-independent: 85–90% | |
| Specific signs and symptoms | Easy bruising | None |
| Facial plethora | ||
| Proximal myopathy (or proximal muscle weakness) | ||
| Striae (especially if reddish purple and >1 cm wide) | ||
| Associated conditions | Bone fragility | Bone fragility |
| Diabetes | Diabetes | |
| Hypertension | Hypertension | |
| Obesity | Obesity | |
| Dyslipidemia | Dyslipidemia | |
| Mood disorders | Mood disorders | |
| Diagnostic delay | 3–5 years | Not known |
| Macronodular Bilateral Adrenocortical Hyperplasia | Micronodular Bilateral Adrenocortical Hyperplasia | |
|---|---|---|
| Size | Bilateral benign adrenal macronodules >1 cm. | Bilateral adrenal nodules <1 cm in diameter. Primary pigmented nodular adrenal disease is the most frequent form. |
| Cortisol secretion degree/clinical presentation | Associated with variable degree of cortisol excess. Responsible for less than 2% of all cases of Cushing’s syndrome. | Clinically overt cortisol excess (Cushing’s syndrome). |
| Age of onset | Late median age in sporadic cases (around 55 years). | Diagnosis is often made before the age of 30 years, with 50% of patients being less than 15 years. |
| Syndromic cases | Rarely may be part of hereditary familial tumor syndromes including multiple endocrine neoplasia type 1 (MEN1), familial adenomatous polyposis (APC) and hereditary leiomyomatosis and renal cell cancer syndrome (fumarate hydrogenase, FH). | Primary pigmented nodular adrenal disease can be the presenting manifestation of Carney complex (PRKAR1A mutations). |
| Familial cases | Related to germline mutations of the Armadillo Repeat Containing 5 (ARMC5). A mutation of ARMC5 gene is found in around 20–25% of all bilateral macronodular adrenocortical hyperplasia cases (40% in patients with overt Cushing’s syndrome and 11% in patients with mHC). | Isolated micronodular bilateral adrenocortical hyperplasia can be related to germline PRKA1RA mutation (c.709-7del6 is the most frequent form). |
| Sporadic cases | Cortisol secretion is in part regulated by the expression of multiple aberrant G protein-coupled receptors (GPCRs). Many of these aberrant receptors stimulate the cAMP/PKA pathway, as does ACTH in normal adrenals. | Micronodular bilateral adrenocortical hyperplasia can be sporadic or familial. |
| Adrenal Cushing’s Syndrome | Adrenal mHC | |||
|---|---|---|---|---|
| PRKACA (n. Mutated Patients/ n. Studied Patients) | CTNNB1 (n. Mutated Patients/ n. Studied Patients) | PRKACA (n. Mutated Patients/ n. Studied Patients) | CTNNB1 (n. Mutated Patients/ n. Studied Patients) | |
| Cao et al. [43] | 57/87 | 1/87 | - | - |
| Beuschlein et al. [44] | 22/59 | 0/59 | 0/40 | 0/40 |
| Goh et al. [45] | 13/36 | 3/36 | 3/27 | 6/27 |
| Di Dalmazi et al. [46] | 22/64 | - | 0/36 | - |
| Sato et al. [47] | 33/55 | 0/55 | 1/9 | 0/9 |
| Thiel et al. [48] | 11/36 | 5/36 | 1/22 | 8/22 |
| Ronchi et al. [49] | - | 7/39 | - | 19/35 |
| Overall (%) | 158/337 (46%) | 16/312 (5%) | 5/109 (4.6%) | 33/133 (25%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Favero, V.; Cremaschi, A.; Parazzoli, C.; Falchetti, A.; Gaudio, A.; Gennari, L.; Scillitani, A.; Vescini, F.; Morelli, V.; Aresta, C.; et al. Pathophysiology of Mild Hypercortisolism: From the Bench to the Bedside. Int. J. Mol. Sci. 2022, 23, 673. https://doi.org/10.3390/ijms23020673
Favero V, Cremaschi A, Parazzoli C, Falchetti A, Gaudio A, Gennari L, Scillitani A, Vescini F, Morelli V, Aresta C, et al. Pathophysiology of Mild Hypercortisolism: From the Bench to the Bedside. International Journal of Molecular Sciences. 2022; 23(2):673. https://doi.org/10.3390/ijms23020673
Chicago/Turabian StyleFavero, Vittoria, Arianna Cremaschi, Chiara Parazzoli, Alberto Falchetti, Agostino Gaudio, Luigi Gennari, Alfredo Scillitani, Fabio Vescini, Valentina Morelli, Carmen Aresta, and et al. 2022. "Pathophysiology of Mild Hypercortisolism: From the Bench to the Bedside" International Journal of Molecular Sciences 23, no. 2: 673. https://doi.org/10.3390/ijms23020673
APA StyleFavero, V., Cremaschi, A., Parazzoli, C., Falchetti, A., Gaudio, A., Gennari, L., Scillitani, A., Vescini, F., Morelli, V., Aresta, C., & Chiodini, I. (2022). Pathophysiology of Mild Hypercortisolism: From the Bench to the Bedside. International Journal of Molecular Sciences, 23(2), 673. https://doi.org/10.3390/ijms23020673