
Pathophysiological Link between Insulin Resistance and Adrenal Incidentalomas

 ,
, 
Abstract
1. Introduction
2. Adrenal Incidentaloma

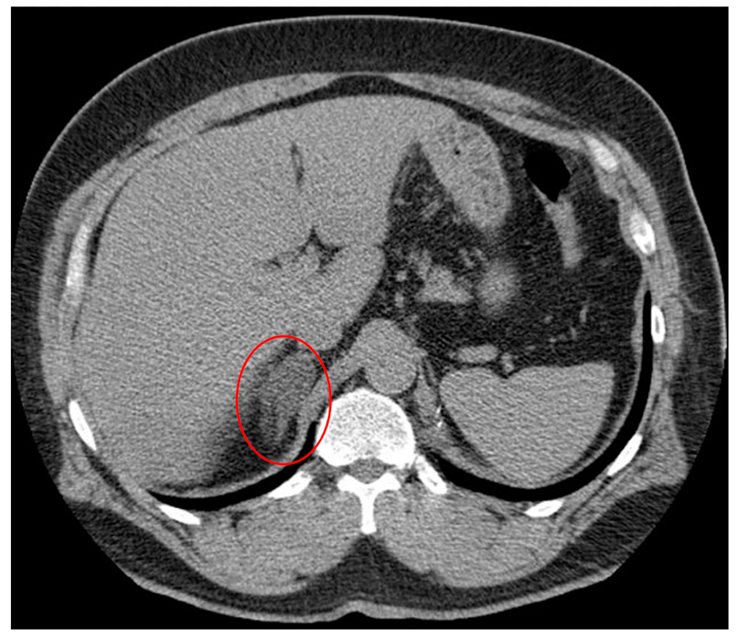{kind=link}
{kind=link}
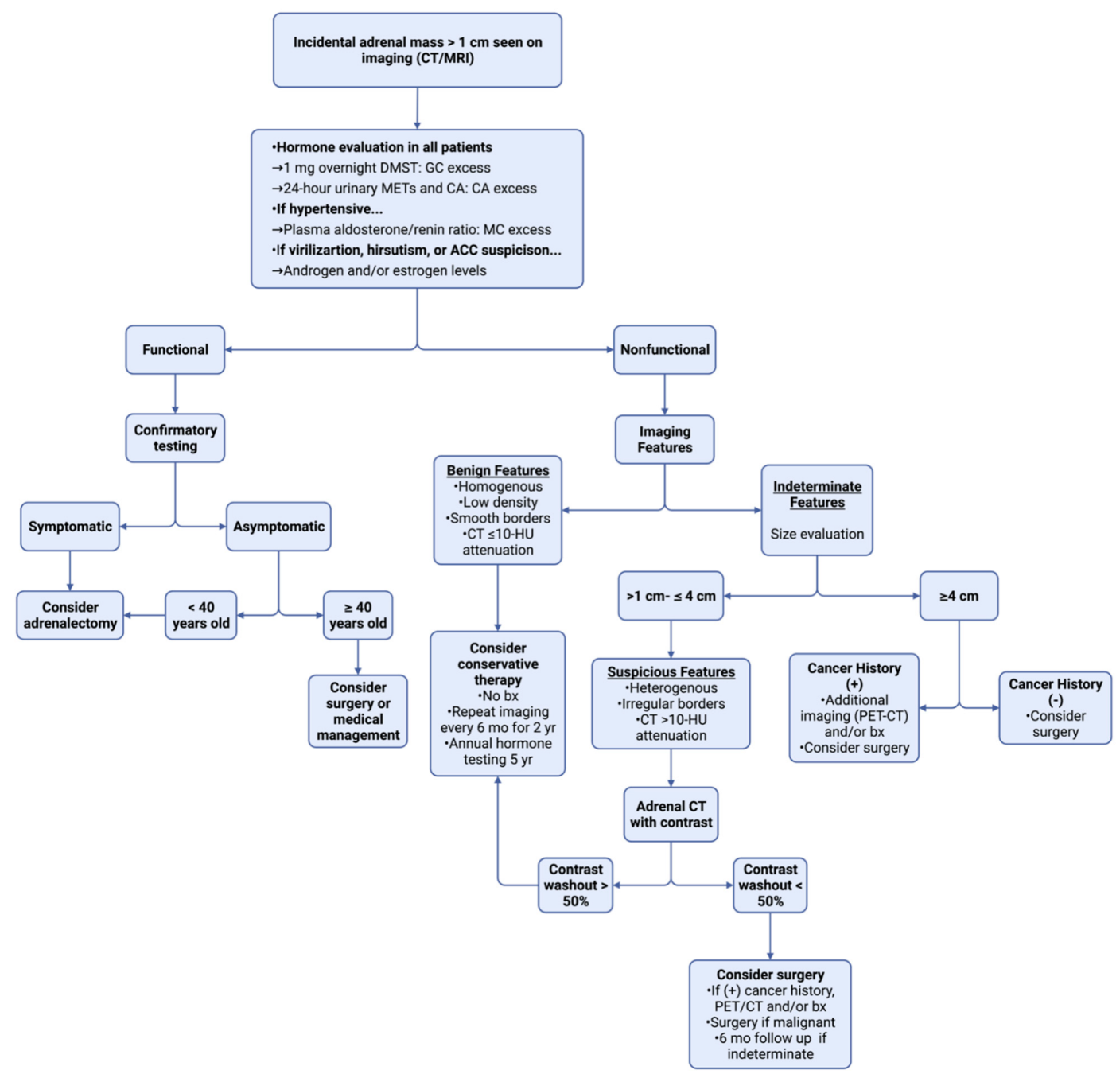{kind=link}
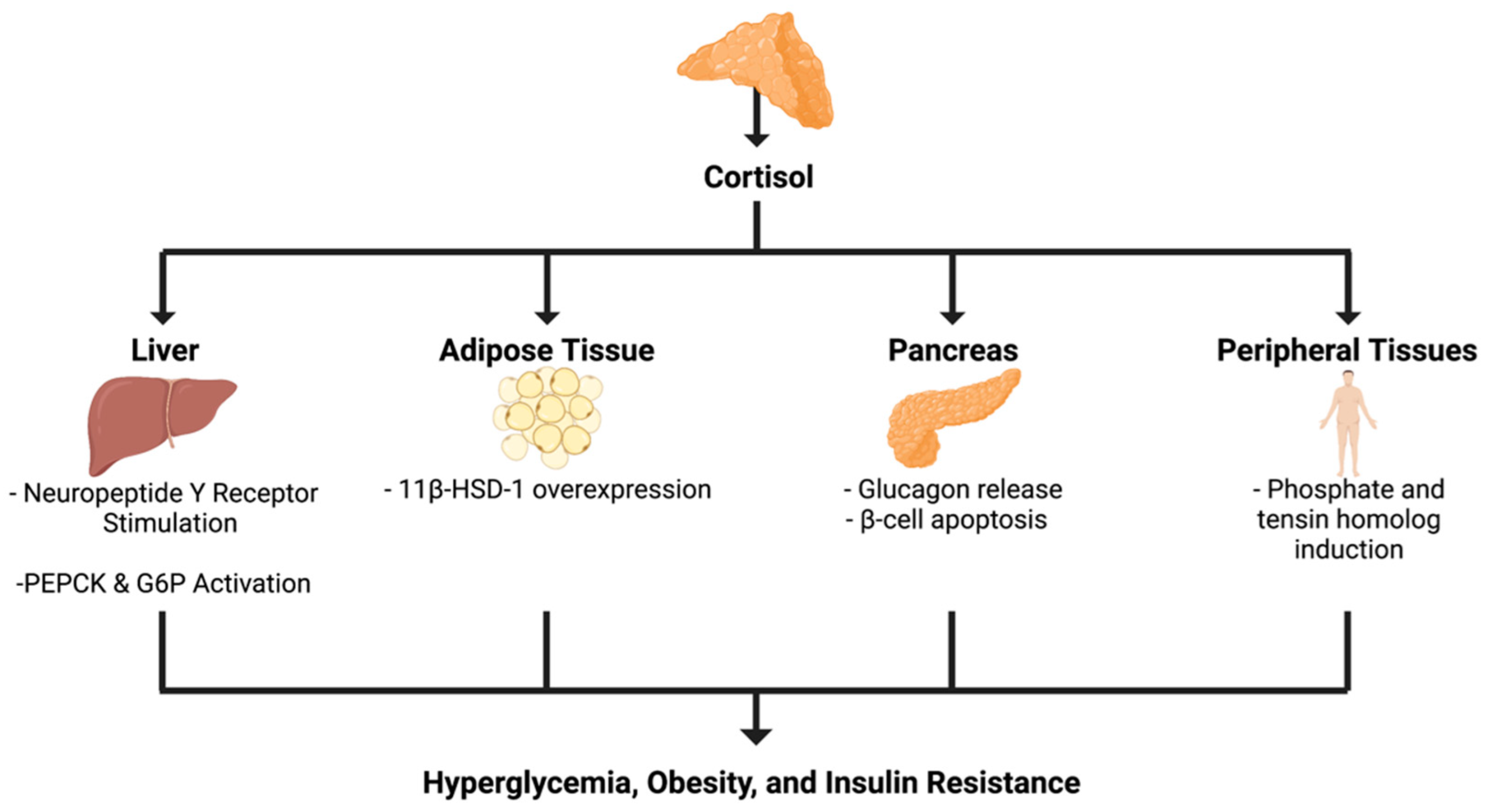{kind=link}
{kind=link}
| Type of Benign Mass | Possible Clinical Presentation | References |
|---|---|---|
| Adrenocortical Adenoma (~80%) | [1,2,44] | |
| Non-functional (~75%) | Asymptomatic, discovered on imaging | [1,2,44] |
| Cortisol-Producing (~12%) | Muscle weakness, easy bleeding/bruising, obesity, flushing, CV events, osteoporosis; overt Cushing’s syndrome | [1,2,44] |
| Aldosterone-Producing (~2.5%) | Muscle cramping/weakness, hypertension, headache, fatigue, polydipsia, polyuria, osteoporosis | [1,2,44] |
| Androgen-Producing (~2.5%) | Feminization, virilization (i.e., excessive facial hair, acne, clitoromegaly, male pattern baldness, deepened voice), hirsutism | [1,2,44] |
| Estrogen-Producing (rare) | Men: decreased libido, testicular atrophy, gynecomastiaWomen: IUB 1, breast tenderness | [1,44] |
| Pheochromocytoma (~7%) | Paroxysmal headaches, hypertension, weight loss, sweating, palpitations, anxiety, hot flashes (50%) | [1,2,44] |
| Myelolipoma (rare) | Possible flank/abdominal pain, shock due to rupture/hemorrhage | [44,49] |
| Adrenal Cyst (rare) | Acute abdominal pain | [44,63] |
| Schwannoma (rare) | Compressive symptoms/abdominal discomfort with increased size | [44,64] |
| Ganglioneuroma (rare) | Primarily asymptomatic, even if large | [44,65] |
| Hematoma/Hemorrhage (rare) | Asymptomatic—history of trauma, stress, sepsis, surgery, pregnancySymptomatic—nausea, abdominal pain, fever, hypotension, vomiting | [44,66] |
| Malignancy | ||
| Adrenocortical Carcinoma (~8%) | Compressive symptoms (abdominal and/or flank pain) in 30%, symptoms of GC 2, MC 3, or androgen excess, if functional—40–60% | [1,2,44] |
| Metastatic Cancer (~5%) | Weight loss, vomiting, history of smoking or cancer (primarily lung, then GI, kidney, breast); symptoms of adrenal insufficiency if bilateral (i.e., postural hypotension, hyponatremia, hyperkalemia) | [1,44] |
| Adrenal Lymphoma | Abdominal pain, B symptoms (fever, night sweats, weight loss) | [1,67] |
3. Insulin Resistance
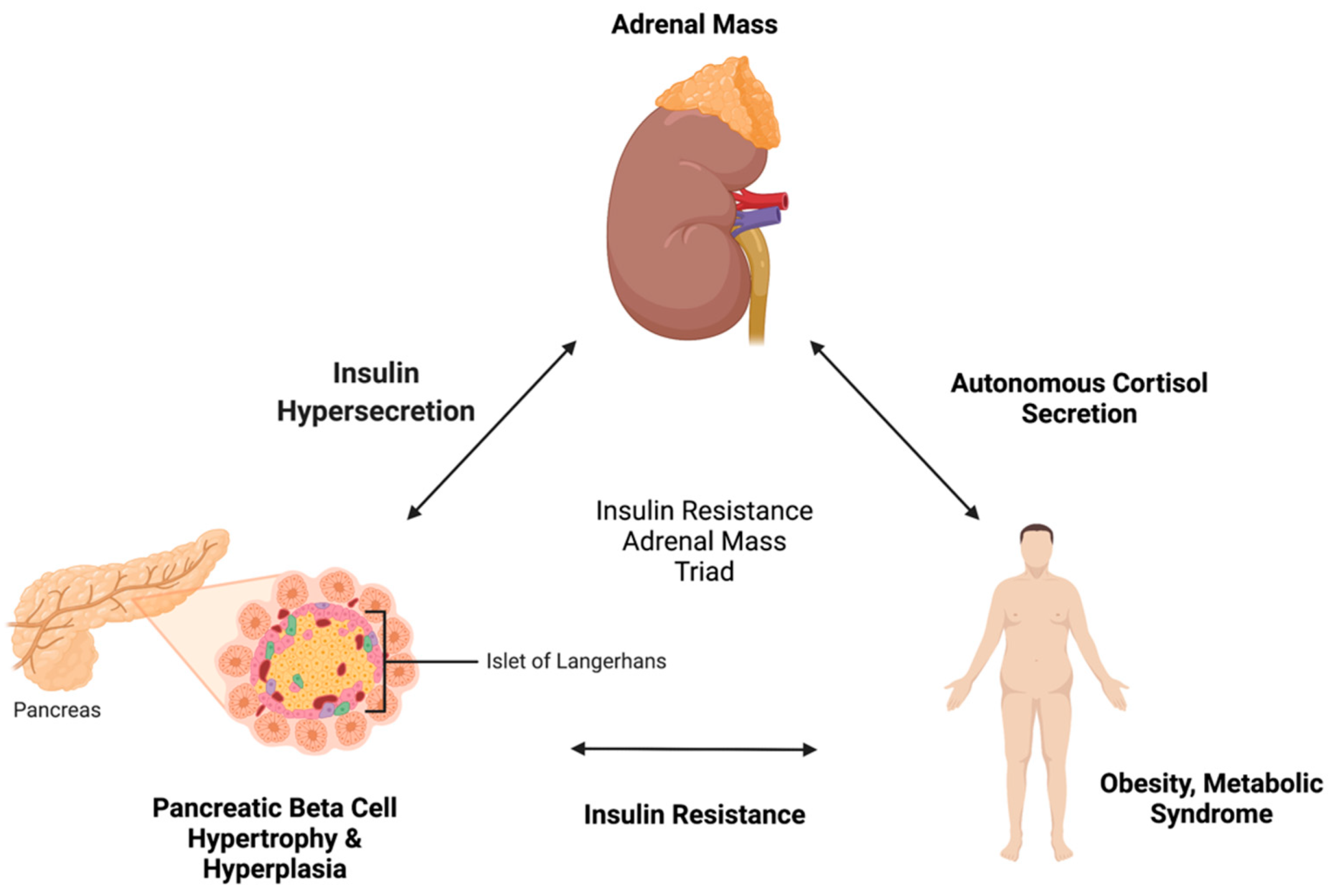
4. Pathophysiological Link between Insulin Resistance and Adrenal Incidentaloma
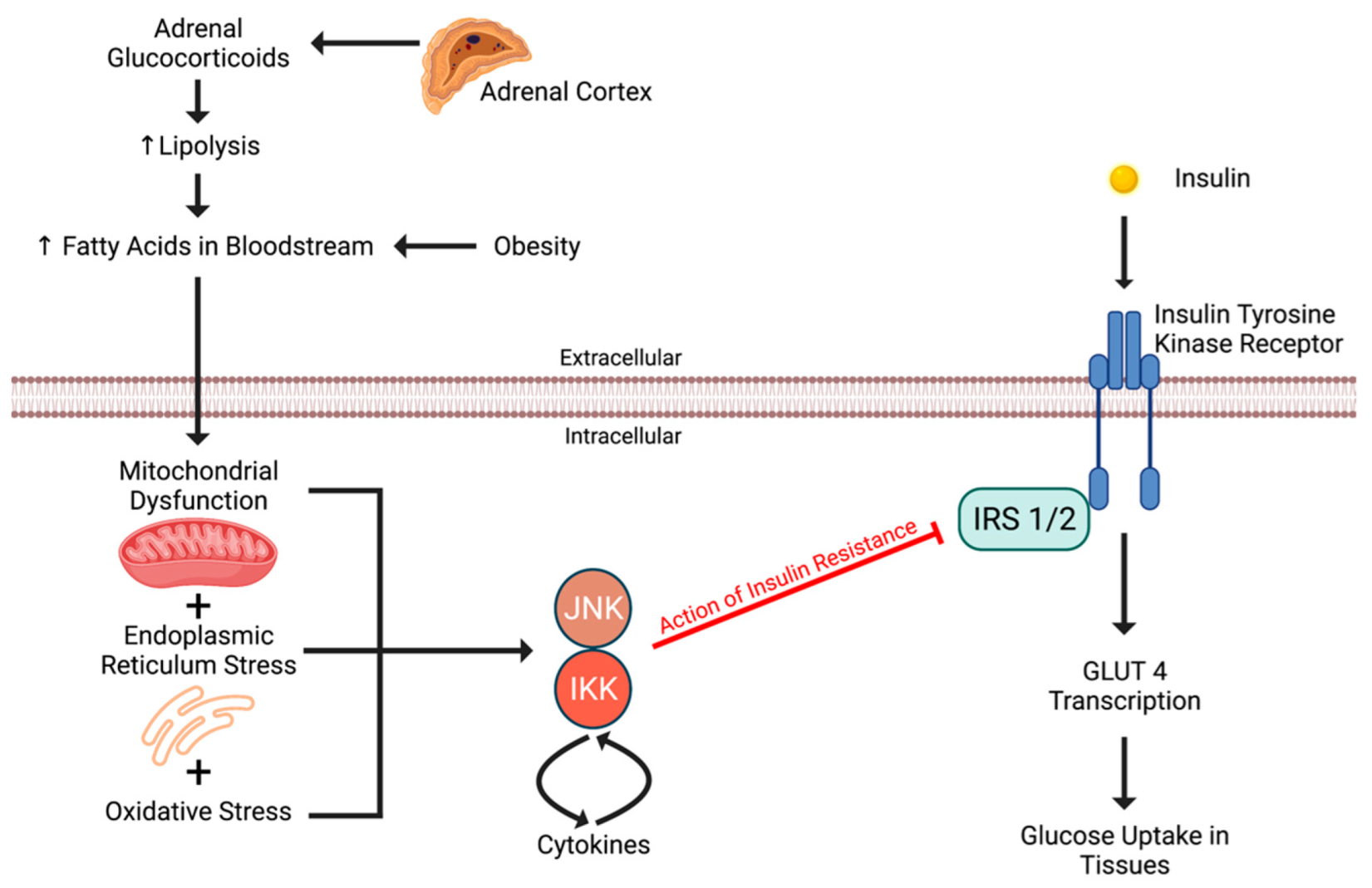
4.1. Insulin Resistance to Adrenal Incidentaloma
4.2. Adrenal Incidentaloma to Insulin Resistance
5. Areas of Future Research
6. Synthesis and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Sherlock, M.; Scarsbrook, A.; Abbas, A.; Fraser, S.; Limumpornpetch, P.; Dineen, R.; Stewart, P.M. Adrenal Incidentaloma. Endocr. Rev. 2020, 41, bnaa008. [Google Scholar] [CrossRef] [PubMed]
- Jason, D.S.; Oltmann, S.C. Evaluation of an Adrenal Incidentaloma. Surg. Clin. N. Am. 2019, 99, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Bovio, S.; Cataldi, A.; Reimondo, G.; Sperone, P.; Novello, S.; Berruti, A.; Borasio, P.; Fava, C.; Dogliotti, L.; Scagliotti, G.V.; et al. Prevalence of Adrenal Incidentaloma in a Contemporary Computerized Tomography Series. J. Endocrinol. Investig. 2006, 29, 298–302. [Google Scholar] [CrossRef]
- Crimì, F.; Quaia, E.; Cabrelle, G.; Zanon, C.; Pepe, A.; Regazzo, D.; Tizianel, I.; Scaroni, C.; Ceccato, F. Diagnostic Accuracy of CT Texture Analysis in Adrenal Masses: A Systematic Review. Int. J. Mol. Sci. 2022, 23, 637. [Google Scholar] [CrossRef] [PubMed]
- Muscogiuri, G.; De Martino, M.C.; Negri, M.; Pivonello, C.; Simeoli, C.; Orio, F.; Pivonello, R.; Colao, A. Adrenal Mass: Insight into Pathogenesis and a Common Link with Insulin Resistance. Endocrinology 2017, 158, 1527–1532. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lee, J.M.; Kim, M.K.; Ko, S.H.; Koh, J.M.; Kim, B.Y.; Kim, S.W.; Kim, S.K.; Kim, H.J.; Ryu, O.H.; Park, J.; et al. Clinical Guidelines for the Management of Adrenal Incidentaloma. Endocrinol. Metab. Seoul Korea 2017, 32, 200–218. [Google Scholar] [CrossRef]
- Robinson, D.Y. Adrenal Mass Causing Secondary Hypertension. J. Emerg. Med. 2015, 49, 638–640. [Google Scholar] [CrossRef]
- Martínez Steele, E.; Juul, F.; Neri, D.; Rauber, F.; Monteiro, C.A. Dietary Share of Ultra-Processed Foods and Metabolic Syndrome in the US Adult Population. Prev. Med. 2019, 125, 40–48. [Google Scholar] [CrossRef]
- Gaillard, F. Incidentaloma|Radiology Reference Article|Radiopaedia.Org. Available online: https://radiopaedia.org/articles/incidentaloma?lang=us (accessed on 25 February 2022).
- Zavatta, G.; Di Dalmazi, G. Recent Advances on Subclinical Hypercortisolism. Endocrinol. Metab. Clin. N. Am. 2018, 47, 375–383. [Google Scholar] [CrossRef]
- Kamilaris, C.D.C.; Stratakis, C.A.; Hannah-Shmouni, F. Molecular Genetic and Genomic Alterations in Cushing’s Syndrome and Primary Aldosteronism. Front. Endocrinol. 2021, 12, 632543. [Google Scholar] [CrossRef]
- Di Dalmazi, G. Hyperandrogenism and Adrenocortical Tumors. Front. Horm. Res. 2019, 53, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Calissendorff, J.; Juhlin, C.C.; Sundin, A.; Bancos, I.; Falhammar, H. Adrenal Myelolipomas. Lancet Diabetes Endocrinol. 2021, 9, 767–775. [Google Scholar] [CrossRef]
- Addeo, P.; Mazzucotelli, J.-P.; Bachellier, P. A Two-Stage Surgical Approach for Malignant Adrenocortical Carcinoma with Intracardiac Extension. Ann. Thorac. Surg. 2022, in press. [CrossRef] [PubMed]
- Geiker, N.R.W.; Astrup, A.; Hjorth, M.F.; Sjödin, A.; Pijls, L.; Markus, C.R. Does Stress Influence Sleep Patterns, Food Intake, Weight Gain, Abdominal Obesity and Weight Loss Interventions and Vice Versa? Obes. Rev. 2018, 19, 81–97. [Google Scholar] [CrossRef]
- Miller, A.H.; Yeung, S.-C.J. Hypercortisolism Manifesting as Severe Weight Loss, Hypokalemia, and Hyperglycemia in the Emergency Department. J. Emerg. Med. 2016, 50, e187–e190. [Google Scholar] [CrossRef]
- Ferraù, F.; Korbonits, M. Metabolic Comorbidities in Cushing’s Syndrome. Eur. J. Endocrinol. 2015, 173, M133–M157. [Google Scholar] [CrossRef]
- Favero, V.; Cremaschi, A.; Parazzoli, C.; Falchetti, A.; Gaudio, A.; Gennari, L.; Scillitani, A.; Vescini, F.; Morelli, V.; Aresta, C.; et al. Pathophysiology of Mild Hypercortisolism: From the Bench to the Bedside. Int. J. Mol. Sci. 2022, 23, 673. [Google Scholar] [CrossRef]
- Gupta, A.; Gupta, Y. Glucocorticoid-Induced Myopathy: Pathophysiology, Diagnosis, and Treatment. Indian J. Endocrinol. Metab. 2013, 17, 913–916. [Google Scholar] [CrossRef]
- Glad, C.A.M.; Andersson-Assarsson, J.C.; Berglund, P.; Bergthorsdottir, R.; Ragnarsson, O.; Johannsson, G. Reduced DNA Methylation and Psychopathology Following Endogenous Hypercortisolism—A Genome-Wide Study. Sci. Rep. 2017, 7, 44445. [Google Scholar] [CrossRef]
- Oki, K.; Yamane, K.; Nakanishi, S.; Shiwa, T.; Kohno, N. Influence of Adrenal Subclinical Hypercortisolism on Hypertension in Patients with Adrenal Incidentaloma. Exp. Clin. Endocrinol. Diabetes 2012, 120, 244–247. [Google Scholar] [CrossRef]
- Dias, J.P.; Joseph, J.J.; Kluwe, B.; Zhao, S.; Shardell, M.; Seeman, T.; Needham, B.L.; Wand, G.S.; Kline, D.; Brock, G.; et al. The Longitudinal Association of Changes in Diurnal Cortisol Features with Fasting Glucose: MESA. Psychoneuroendocrinology 2020, 119, 104698. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.-H.; Chen, L.-R.; Chen, K.-H. Osteoporosis Due to Hormone Imbalance: An Overview of the Effects of Estrogen Deficiency and Glucocorticoid Overuse on Bone Turnover. Int. J. Mol. Sci. 2022, 23, 1376. [Google Scholar] [CrossRef] [PubMed]
- Saei Ghare Naz, M.; Rostami Dovom, M.; Ramezani Tehrani, F. The Menstrual Disturbances in Endocrine Disorders: A Narrative Review. Int. J. Endocrinol. Metab. 2020, 18, e106694. [Google Scholar] [CrossRef] [PubMed]
- Geer, E.B.; Islam, J.; Buettner, C. Mechanisms of Glucocorticoid-Induced Insulin Resistance: Focus on Adipose Tissue Function and Lipid Metabolism. Endocrinol. Metab. Clin. N. Am. 2014, 43, 75–102. [Google Scholar] [CrossRef]
- Da Silva, A.A.; do Carmo, J.M.; Li, X.; Wang, Z.; Mouton, A.J.; Hall, J.E. Role of Hyperinsulinemia and Insulin Resistance in Hypertension: Metabolic Syndrome Revisited. Can. J. Cardiol. 2020, 36, 671–682. [Google Scholar] [CrossRef]
- Sacerdote, A.; Dave, P.; Lokshin, V.; Bahtiyar, G. Type 2 Diabetes Mellitus, Insulin Resistance, and Vitamin D. Curr. Diabetes Rep. 2019, 19, 101. [Google Scholar] [CrossRef]
- Yaribeygi, H.; Farrokhi, F.R.; Butler, A.E.; Sahebkar, A. Insulin Resistance: Review of the Underlying Molecular Mechanisms. J. Cell. Physiol. 2019, 234, 8152–8161. [Google Scholar] [CrossRef]
- Azarpazhooh, M.R.; Najafi, F.; Darbandi, M.; Kiarasi, S.; Oduyemi, T.; Spence, J.D. Triglyceride/High-Density Lipoprotein Cholesterol Ratio: A Clue to Metabolic Syndrome, Insulin Resistance, and Severe Atherosclerosis. Lipids 2021, 56, 405–412. [Google Scholar] [CrossRef]
- Gluvic, Z.; Zaric, B.; Resanovic, I.; Obradovic, M.; Mitrovic, A.; Radak, D.; Isenovic, E.R. Link between Metabolic Syndrome and Insulin Resistance. Curr. Vasc. Pharmacol. 2017, 15, 30–39. [Google Scholar] [CrossRef]
- Hill, M.A.; Yang, Y.; Zhang, L.; Sun, Z.; Jia, G.; Parrish, A.R.; Sowers, J.R. Insulin Resistance, Cardiovascular Stiffening and Cardiovascular Disease. Metabolism 2021, 119, 154766. [Google Scholar] [CrossRef]
- Morigny, P.; Houssier, M.; Mouisel, E.; Langin, D. Adipocyte Lipolysis and Insulin Resistance. Biochimie 2016, 125, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Varewijck, A.J.; Janssen, J.A.M.J.L. Insulin and Its Analogues and Their Affinities for the IGF1 Receptor. Endocr. Relat. Cancer 2012, 19, F63–F75. [Google Scholar] [CrossRef] [PubMed]
- Laron, Z. Insulin–A Growth Hormone. Arch. Physiol. Biochem. 2008, 114, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Leung, K.C.; Doyle, N.; Ballesteros, M.; Waters, M.J.; Ho, K.K. Insulin Regulation of Human Hepatic Growth Hormone Receptors: Divergent Effects on Biosynthesis and Surface Translocation. J. Clin. Endocrinol. Metab. 2000, 85, 4712–4720. [Google Scholar] [CrossRef] [PubMed]
- Tzanavari, T.; Giannogonas, P.; Karalis, K.P. TNF-Alpha and Obesity. Curr. Dir. Autoimmun. 2010, 11, 145–156. [Google Scholar] [CrossRef]
- Shanik, M.H.; Xu, Y.; Skrha, J.; Dankner, R.; Zick, Y.; Roth, J. Insulin Resistance and Hyperinsulinemia: Is Hyperinsulinemia the Cart or the Horse? Diabetes Care 2008, 31 (Suppl. 2), S262–S268. [Google Scholar] [CrossRef]
- Kebebew, E. Adrenal Incidentaloma. N. Engl. J. Med. 2021, 384, 1542–1551. [Google Scholar] [CrossRef]
- Mantero, F.; Terzolo, M.; Arnaldi, G.; Osella, G.; Masini, A.M.; Alì, A.; Giovagnetti, M.; Opocher, G.; Angeli, A. A Survey on Adrenal Incidentaloma in Italy1. J. Clin. Endocrinol. Metab. 2000, 85, 637–644. [Google Scholar] [CrossRef]
- Kasperlik-Załuska, A.A.; Otto, M.; Cichocki, A.; Rosłonowska, E.; Słowińska-Srzednicka, J.; Jeske, W.; Papierska, L.; Zgliczyński, W. Incidentally Discovered Adrenal Tumors: A Lesson from Observation of 1444 Patients. Horm. Metab. Res. 2008, 40, 338–341. [Google Scholar] [CrossRef]
- Comlekci, A.; Yener, S.; Ertilav, S.; Secil, M.; Akinci, B.; Demir, T.; Kebapcilar, L.; Bayraktar, F.; Yesil, S.; Eraslan, S. Adrenal Incidentaloma, Clinical, Metabolic, Follow-up Aspects: Single Centre Experience. Endocrine 2010, 37, 40–46. [Google Scholar] [CrossRef]
- Ahn, S.H.; Kim, J.H.; Baek, S.H.; Kim, H.; Cho, Y.Y.; Suh, S.; Kim, B.J.; Hong, S.; Koh, J.M.; Lee, S.H.; et al. Characteristics of Adrenal Incidentalomas in a Large, Prospective Computed Tomography-Based Multicenter Study: The COAR Study in Korea. Yonsei Med. J. 2018, 59, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Hao, M.; Lopez, D.; Luque-Fernandez, M.A.; Cote, K.; Newfield, J.; Connors, M.; Vaidya, A. The Lateralizing Asymmetry of Adrenal Adenomas. J. Endocr. Soc. 2018, 2, 374–385. [Google Scholar] [CrossRef] [PubMed]
- Fassnacht, M.; Arlt, W.; Bancos, I.; Dralle, H.; Newell-Price, J.; Sahdev, A.; Tabarin, A.; Terzolo, M.; Tsagarakis, S.; Dekkers, O.M. Management of Adrenal Incidentalomas: European Society of Endocrinology Clinical Practice Guideline in Collaboration with the European Network for the Study of Adrenal Tumors. Eur. J. Endocrinol. 2016, 175, G1–G34. [Google Scholar] [CrossRef] [PubMed]
- Gendy, R.; Rashid, P. Incidental Adrenal Masses—A Primary Care Approach. Aust. Fam. Physician 2017, 46, 385–390. [Google Scholar] [PubMed]
- Mayo-Smith, W.W.; Song, J.H.; Boland, G.L.; Francis, I.R.; Israel, G.M.; Mazzaglia, P.J.; Berland, L.L.; Pandharipande, P.V. Management of Incidental Adrenal Masses: A White Paper of the ACR Incidental Findings Committee. J. Am. Coll. Radiol. 2017, 14, 1038–1044. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.Z.; Blute, M.L.; Seitz, C.; Habra, M.A.; Karam, J.A. Management of the Incidental Adrenal Mass. Eur. Urol. Focus 2016, 1, 223–230. [Google Scholar] [CrossRef]
- Lew, J.I. Clinical Management of Adrenal Tumors; IntechOpen: London, UK, 2017; ISBN 978-953-51-3266-0. [Google Scholar]
- Ramirez, M.; Misra, S. Adrenal Myelolipoma: To Operate or Not? A Case Report and Review of the Literature. Int. J. Surg. Case Rep. 2014, 5, 494–496. [Google Scholar] [CrossRef]
- Duh, Q.-Y. Adrenal Gland: New Guidelines for Adrenal Incidentalomas. Nat. Rev. Endocrinol. 2016, 12, 561–562. [Google Scholar] [CrossRef]
- Elijovich, F.; Laffer, C.L. Chapter 69-Cushing’s Syndrome and Human Glucocorticoid Hypertension. In Comprehensive Hypertension; Lip, G.Y.H., Hall, J.E., Eds.; Mosby: Philadelphia, PA, USA, 2007; pp. 835–864. ISBN 978-0-323-03961-1. [Google Scholar]
- Araujo-Castro, M.; Sampedro Núñez, M.A.; Marazuela, M. Autonomous Cortisol Secretion in Adrenal Incidentalomas. Endocrine 2019, 64, 1–13. [Google Scholar] [CrossRef]
- Debono, M.; Newell-Price, J. Subclinical Hypercortisolism in Adrenal Incidentaloma. Curr. Opin. Endocrinol. Diabetes Obes. 2015, 22, 185–192. [Google Scholar] [CrossRef]
- Sbardella, E.; Minnetti, M.; D’Aluisio, D.; Rizza, L.; Di Giorgio, M.R.; Vinci, F.; Pofi, R.; Giannetta, E.; Venneri, M.A.; Vestri, A.; et al. Cardiovascular Features of Possible Autonomous Cortisol Secretion in Patients with Adrenal Incidentalomas. Eur. J. Endocrinol. 2018, 178, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Morelli, V.; Palmieri, S.; Lania, A.; Tresoldi, A.; Corbetta, S.; Cairoli, E.; Eller-Vainicher, C.; Arosio, M.; Copetti, M.; Grossi, E.; et al. Cardiovascular Events in Patients with Mild Autonomous Cortisol Secretion: Analysis with Artificial Neural Networks. Eur. J. Endocrinol. 2017, 177, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Yener, S.; Baris, M.; Peker, A.; Demir, O.; Ozgen, B.; Secil, M. Autonomous Cortisol Secretion in Adrenal Incidentalomas and Increased Visceral Fat Accumulation during Follow-Up. Clin. Endocrinol. 2017, 87, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Belokovskaya, R.; Ravikumar, A.; Arumugam, D.; Izadmehr, S.; Goddard, G.M.; Geer, E.B.; Levine, A.C. Mifepristone treatment for mild autonomous cortisol secretion due to adrenal adenomas: A pilot study. Endocr. Pract. 2019, 25, 846–853. [Google Scholar] [CrossRef] [PubMed]
- Elhassan, Y.S.; Alahdab, F.; Prete, A.; Delivanis, D.A.; Khanna, A.; Prokop, L.; Murad, M.H.; O’Reilly, M.W.; Arlt, W.; Bancos, I. Natural History of Adrenal Incidentalomas with and without Mild Autonomous Cortisol Excess. Ann. Intern. Med. 2019, 171, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Patrova, J.; Kjellman, M.; Wahrenberg, H.; Falhammar, H. Increased Mortality in Patients with Adrenal Incidentalomas and Autonomous Cortisol Secretion: A 13-Year Retrospective Study from One Center. Endocrine 2017, 58, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Blake, M.A.; Holalkere, N.-S.; Boland, G.W. Imaging Techniques for Adrenal Lesion Characterization. Radiol. Clin. N. Am. 2008, 46, 65–78. [Google Scholar] [CrossRef]
- Petramala, L.; Olmati, F.; Concistrè, A.; Russo, R.; Mezzadri, M.; Soldini, M.; De Vincentis, G.; Iannucci, G.; De Toma, G.; Letizia, C. Cardiovascular and Metabolic Risk Factors in Patients with Subclinical Cushing. Endocrine 2020, 70, 150–163. [Google Scholar] [CrossRef]
- Androulakis, I.I.; Kaltsas, G.; Piaditis, G.; Grossman, A.B. The Clinical Significance of Adrenal Incidentalomas. Eur. J. Clin. Investig. 2011, 41, 552–560. [Google Scholar] [CrossRef]
- Nerli, R.B.; Guntaka, A.; Devaraju, S.; Patil, S.; Hiremath, M.B. Adrenal Cysts: Our Laparoscopic Experience. J. Minimal Access Surg. 2012, 8, 145–148. [Google Scholar] [CrossRef]
- Li, S.-Q.; Zhang, Y.-S.; Shi, J.; Li, H.-Z. Clinical Features and Retroperitoneal Laparoscopic Resection of Adrenal Schwannoma in 19 Patients. Endocr. Pract. 2015, 21, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Sarwal, A.; Khullar, R.; Sharma, A.; Soni, V.; Baijal, M.; Chowbey, P. Laparoscopic Adrenalectomy for Ganglioneuroma Presenting as an Adrenal Incidentaloma. J. Minimal Access Surg. 2019, 15, 259–261. [Google Scholar] [CrossRef] [PubMed]
- Kılınç, İ.; Dumlu, E.G.; Öztürk, V.; Çuhacı, N.; Balcı, S.; Yalçın, A.; Kılıç, M. Idiopathic Adrenal Hematoma Mimicking Neoplasia: A Case Report. Int. J. Surg. Case Rep. 2016, 28, 15–17. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ren, Y.; Ma, L.; Li, J.; Zhu, Y.; Zhao, L.; Tian, H.; Chen, T. Clinical Features of 50 Patients with Primary Adrenal Lymphoma. Front. Endocrinol. 2020, 11, 595. [Google Scholar] [CrossRef]
- E.Hormone|Endocrine System: Types of Hormones. Available online: http://e.hormone.tulane.edu/learning/types-of-hormones.html (accessed on 23 February 2022).
- Copps, K.D.; White, M.F. Regulation of Insulin Sensitivity by Serine/Threonine Phosphorylation of Insulin Receptor Substrate Proteins IRS1 and IRS2. Diabetologia 2012, 55, 2565–2582. [Google Scholar] [CrossRef]
- Leto, D.; Saltiel, A.R. Regulation of Glucose Transport by Insulin: Traffic Control of GLUT4. Nat. Rev. Mol. Cell Biol. 2012, 13, 383–396. [Google Scholar] [CrossRef]
- Bolli, G.B.; Porcellati, F.; Lucidi, P.; Fanelli, C.G. The Physiological Basis of Insulin Therapy in People with Diabetes Mellitus. Diabetes Res. Clin. Pract. 2021, 175, 108839. [Google Scholar] [CrossRef]
- Liu, K.; Jin, X.; Zhang, X.; Lian, H.; Ye, J. The Mechanisms of Nucleotide Actions in Insulin Resistance. J. Genet. Genom. Yi Chuan Xue Bao 2022, in press. [CrossRef]
- Zatterale, F.; Longo, M.; Naderi, J.; Raciti, G.A.; Desiderio, A.; Miele, C.; Beguinot, F. Chronic Adipose Tissue Inflammation Linking Obesity to Insulin Resistance and Type 2 Diabetes. Front. Physiol. 2020, 10, 1607. [Google Scholar] [CrossRef]
- Perseghin, G.; Calori, G.; Lattuada, G.; Ragogna, F.; Dugnani, E.; Garancini, M.P.; Crosignani, P.; Villa, M.; Bosi, E.; Ruotolo, G.; et al. Insulin Resistance/Hyperinsulinemia and Cancer Mortality: The Cremona Study at the 15th Year of Follow-Up. Acta Diabetol. 2012, 49, 421–428. [Google Scholar] [CrossRef]
- Jiralerspong, S.; Kim, E.S.; Dong, W.; Feng, L.; Hortobagyi, G.N.; Giordano, S.H. Obesity, Diabetes, and Survival Outcomes in a Large Cohort of Early-Stage Breast Cancer Patients. Ann. Oncol. 2013, 24, 2506–2514. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-H.; Park, S.-Y.; Choi, C.S. Insulin Resistance: From Mechanisms to Therapeutic Strategies. Diabetes Metab. J. 2022, 46, 15–37. [Google Scholar] [CrossRef] [PubMed]
- Coelho, M.; Oliveira, T.; Fernandes, R. Biochemistry of Adipose Tissue: An Endocrine Organ. Arch. Med. Sci. AMS 2013, 9, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Cypess, A.M. Reassessing Human Adipose Tissue. N. Engl. J. Med. 2022, 386, 768–779. [Google Scholar] [CrossRef] [PubMed]
- Odegaard, J.I.; Chawla, A. Pleiotropic Actions of Insulin Resistance and Inflammation in Metabolic Homeostasis. Science 2013, 339, 172–177. [Google Scholar] [CrossRef]
- Akash, M.S.H.; Rehman, K.; Liaqat, A. Tumor Necrosis Factor-Alpha: Role in Development of Insulin Resistance and Pathogenesis of Type 2 Diabetes Mellitus. J. Cell. Biochem. 2018, 119, 105–110. [Google Scholar] [CrossRef]
- Reddy, P.; Lent-Schochet, D.; Ramakrishnan, N.; McLaughlin, M.; Jialal, I. Metabolic Syndrome Is an Inflammatory Disorder: A Conspiracy between Adipose Tissue and Phagocytes. Clin. Chim. Acta 2019, 496, 35–44. [Google Scholar] [CrossRef]
- Calle, E.E.; Rodriguez, C.; Walker-Thurmond, K.; Thun, M.J. Overweight, Obesity, and Mortality from Cancer in a Prospectively Studied Cohort of U.S. Adults. N. Engl. J. Med. 2003, 348, 1625–1638. [Google Scholar] [CrossRef]
- Giovannucci, E.; Harlan, D.M.; Archer, M.C.; Bergenstal, R.M.; Gapstur, S.M.; Habel, L.A.; Pollak, M.; Regensteiner, J.G.; Yee, D. Diabetes and Cancer: A Consensus Report. Diabetes Care 2010, 33, 1674–1685. [Google Scholar] [CrossRef]
- Zhou, X.H.; Qiao, Q.; Zethelius, B.; Pyörälä, K.; Söderberg, S.; Pajak, A.; Stehouwer, C.D.A.; Heine, R.J.; Jousilahti, P.; Ruotolo, G.; et al. Diabetes, Prediabetes and Cancer Mortality. Diabetologia 2010, 53, 1867–1876. [Google Scholar] [CrossRef]
- Sydney, G.I.; Ioakim, K.J.; Paschou, S.A. Insulin Resistance and Adrenal Incidentalomas: A Bidirectional Relationship. Maturitas 2019, 121, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Altieri, B.; Tirabassi, G.; Della Casa, S.; Ronchi, C.L.; Balercia, G.; Orio, F.; Pontecorvi, A.; Colao, A.; Muscogiuri, G. Adrenocortical Tumors and Insulin Resistance: What Is the First Step? Int. J. Cancer 2016, 138, 2785–2794. [Google Scholar] [CrossRef] [PubMed]
- Angelousi, A.; Kyriakopoulos, G.; Nasiri-Ansari, N.; Karageorgou, M.; Kassi, E. The Role of Epithelial Growth Factors and Insulin Growth Factors in the Adrenal Neoplasms. Ann. Transl. Med. 2018, 6, 253. [Google Scholar] [CrossRef] [PubMed]
- Haase, M.; Thiel, A.; Scholl, U.I.; Ashmawy, H.; Schott, M.; Ehlers, M. Subcellular Localization of Fibroblast Growth Factor Receptor Type 2 and Correlation with CTNNB1 Genotype in Adrenocortical Carcinoma. BMC Res. Notes 2020, 13, 282. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, T.C.; Latronico, A.C. Insulin-like Growth Factor System on Adrenocortical Tumorigenesis. Mol. Cell. Endocrinol. 2012, 351, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.Q.; Fragoso, M.C.B.V.; Lotfi, C.F.P.; Santos, M.G.; Nishi, M.Y.; Costa, M.H.S.; Lerario, A.M.; Maciel, C.C.; Mattos, G.E.; Jorge, A.A.L.; et al. Expression of Insulin-Like Growth Factor-II and Its Receptor in Pediatric and Adult Adrenocortical Tumors. J. Clin. Endocrinol. Metab. 2008, 93, 3524–3531. [Google Scholar] [CrossRef]
- Drelon, C.; Berthon, A.; Ragazzon, B.; Tissier, F.; Bandiera, R.; Sahut-Barnola, I.; de Joussineau, C.; Batisse-Lignier, M.; Lefrançois-Martinez, A.-M.; Bertherat, J.; et al. Analysis of the Role of Igf2 in Adrenal Tumour Development in Transgenic Mouse Models. PLoS ONE 2012, 7, e44171. [Google Scholar] [CrossRef]
- Heaton, J.H.; Wood, M.A.; Kim, A.C.; Lima, L.O.; Barlaskar, F.M.; Almeida, M.Q.; Fragoso, M.C.B.V.; Kuick, R.; Lerario, A.M.; Simon, D.P.; et al. Progression to Adrenocortical Tumorigenesis in Mice and Humans through Insulin-Like Growth Factor 2 and β-Catenin. Am. J. Pathol. 2012, 181, 1017–1033. [Google Scholar] [CrossRef]
- Wang, S.; Wu, J.; Wang, N.; Zeng, L.; Wu, Y. The Role of Growth Hormone Receptor in β Cell Function. Growth Horm. IGF Res. 2017, 36, 30–35. [Google Scholar] [CrossRef]
- Blum, W.F.; Alherbish, A.; Alsagheir, A.; Awwa, A.E.; Kaplan, W.; Koledova, E.; Savage, M.O. The Growth Hormone–Insulin-like Growth Factor-I Axis in the Diagnosis and Treatment of Growth Disorders. Endocr. Connect. 2018, 7, R212–R222. [Google Scholar] [CrossRef]
- Rostoker, R.; Abelson, S.; Bitton-Worms, K.; Genkin, I.; Ben-Shmuel, S.; Dakwar, M.; Orr, Z.S.; Caspi, A.; Tzukerman, M.; LeRoith, D. Highly Specific Role of the Insulin Receptor in Breast Cancer Progression. Endocr. Relat. Cancer 2015, 22, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Roth, R.A.; Steele-Perkins, G.; Hari, J.; Stover, C.; Pierce, S.; Turner, J.; Edman, J.C.; Rutter, W.J. Insulin and Insulin-like Growth Factor Receptors and Responses. Cold Spring Harb. Symp. Quant. Biol. 1988, 53, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Bergman, D.; Halje, M.; Nordin, M.; Engström, W. Insulin-Like Growth Factor 2 in Development and Disease: A Mini-Review. Gerontology 2013, 59, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Belfiore, A.; Malaguarnera, R.; Vella, V.; Lawrence, M.C.; Sciacca, L.; Frasca, F.; Morrione, A.; Vigneri, R. Insulin Receptor Isoforms in Physiology and Disease: An Updated View. Endocr. Rev. 2017, 38, 379–431. [Google Scholar] [CrossRef]
- Frasca, F.; Pandini, G.; Scalia, P.; Sciacca, L.; Mineo, R.; Costantino, A.; Goldfine, I.D.; Belfiore, A.; Vigneri, R. Insulin Receptor Isoform A, a Newly Recognized, High-Affinity Insulin-like Growth Factor II Receptor in Fetal and Cancer Cells. Mol. Cell. Biol. 1999, 19, 3278–3288. [Google Scholar] [CrossRef]
- Jiang, L.; Zhu, W.; Streicher, K.; Morehouse, C.; Brohawn, P.; Ge, X.; Dong, Z.; Yin, X.; Zhu, G.; Gu, Y.; et al. Increased IR-A/IR-B Ratio in Non-Small Cell Lung Cancers Associates with Lower Epithelial-Mesenchymal Transition Signature and Longer Survival in Squamous Cell Lung Carcinoma. BMC Cancer 2014, 14, 131. [Google Scholar] [CrossRef]
- Denley, A.; Bonython, E.R.; Booker, G.W.; Cosgrove, L.J.; Forbes, B.E.; Ward, C.W.; Wallace, J.C. Structural Determinants for High-Affinity Binding of Insulin-like Growth Factor II to Insulin Receptor (IR)-A, the Exon 11 Minus Isoform of the IR. Mol. Endocrinol. 2004, 18, 2502–2512. [Google Scholar] [CrossRef]
- Harrington, S.C.; Weroha, S.J.; Reynolds, C.; Suman, V.J.; Lingle, W.L.; Haluska, P. Quantifying Insulin Receptor Isoform Expression in FFPE Breast Tumors. Growth Horm. IGF Res. 2012, 22, 108–115. [Google Scholar] [CrossRef]
- Yakar, S.; Pennisi, P.; Zhao, H.; Zhang, Y.; LeRoith, D. Circulating IGF-1 and Its Role in Cancer: Lessons from the IGF-1 Gene Deletion (LID) Mouse. Novartis Found. Symp. 2004, 262, 265–268. [Google Scholar]
- Brismar, K.; Fernqvist-Forbes, E.; Wahren, J.; Hall, K. Effect of Insulin on the Hepatic Production of Insulin-like Growth Factor-Binding Protein-1 (IGFBP-1), IGFBP-3, and IGF-I in Insulin-Dependent Diabetes. J. Clin. Endocrinol. Metab. 1994, 79, 872–878. [Google Scholar] [CrossRef]
- Vassilakos, G.; Lei, H.; Yang, Y.; Puglise, J.; Matheny, M.; Durzynska, J.; Ozery, M.; Bennett, K.; Spradlin, R.; Bonanno, H.; et al. Deletion of Muscle IGF-I Transiently Impairs Growth and Progressively Disrupts Glucose Homeostasis in Male Mice. FASEB J. 2019, 33, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, M.S.; Dunger, D.B.; Giovannucci, E.L. Insulin, Insulin-Like Growth Factor-I (IGF-I), IGF Binding Proteins, Their Biologic Interactions, and Colorectal Cancer. JNCI J. Natl. Cancer Inst. 2002, 94, 972–980. [Google Scholar] [CrossRef] [PubMed]
- Rajwani, A.; Ezzat, V.; Smith, J.; Yuldasheva, N.Y.; Duncan, E.R.; Gage, M.; Cubbon, R.M.; Kahn, M.B.; Imrie, H.; Abbas, A.; et al. Increasing Circulating IGFBP1 Levels Improves Insulin Sensitivity, Promotes Nitric Oxide Production, Lowers Blood Pressure, and Protects Against Atherosclerosis. Diabetes 2012, 61, 915–924. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Chanmee, T.; Itano, N. Hyaluronan: Metabolism and Function. Biomolecules 2020, 10, E1525. [Google Scholar] [CrossRef]
- Twarock, S.; Reichert, C.; Peters, U.; Gorski, D.J.; Röck, K.; Fischer, J.W. Hyperglycaemia and Aberrated Insulin Signalling Stimulate Tumour Progression via Induction of the Extracellular Matrix Component Hyaluronan. Int. J. Cancer 2017, 141, 791–804. [Google Scholar] [CrossRef]
- Mohd Azmi, N.A.S.; Juliana, N.; Azmani, S.; Mohd Effendy, N.; Abu, I.F.; Mohd Fahmi Teng, N.I.; Das, S. Cortisol on Circadian Rhythm and Its Effect on Cardiovascular System. Int. J. Environ. Res. Public Health 2021, 18, 676. [Google Scholar] [CrossRef]
- Leistner, C.; Menke, A. Hypothalamic-Pituitary-Adrenal Axis and Stress. Handb. Clin. Neurol. 2020, 175, 55–64. [Google Scholar] [CrossRef]
- Lopez, D.; Luque-Fernandez, M.A.; Steele, A.; Adler, G.K.; Turchin, A.; Vaidya, A. “Nonfunctional” Adrenal Tumors and the Risk for Incident Diabetes and Cardiovascular Outcomes: A Cohort Study. Ann. Intern. Med. 2016, 165, 533–542. [Google Scholar] [CrossRef]
- Debono, M.; Chadarevian, R.; Eastell, R.; Ross, R.J.; Newell-Price, J. Mifepristone Reduces Insulin Resistance in Patient Volunteers with Adrenal Incidentalomas That Secrete Low Levels of Cortisol: A Pilot Study. PLoS ONE 2013, 8, e60984. [Google Scholar] [CrossRef]
- Androulakis, I.I.; Kaltsas, G.A.; Kollias, G.E.; Markou, A.C.; Gouli, A.K.; Thomas, D.A.; Alexandraki, K.I.; Papamichael, C.M.; Hadjidakis, D.J.; Piaditis, G.P. Patients with Apparently Nonfunctioning Adrenal Incidentalomas May Be at Increased Cardiovascular Risk Due to Excessive Cortisol Secretion. J. Clin. Endocrinol. Metab. 2014, 99, 2754–2762. [Google Scholar] [CrossRef]
- Kuo, T.; McQueen, A.; Chen, T.-C.; Wang, J.-C. Regulation of Glucose Homeostasis by Glucocorticoids. Adv. Exp. Med. Biol. 2015, 872, 99–126. [Google Scholar] [CrossRef] [PubMed]
- Asensio, C.; Muzzin, P.; Rohner-Jeanrenaud, F. Role of Glucocorticoids in the Physiopathology of Excessive Fat Deposition and Insulin Resistance. Int. J. Obes. 2004, 28 (Suppl. 4), S45–S52. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Meng, S.; Xiang, M.; Ma, H. Phosphoenolpyruvate Carboxykinase in Cell Metabolism: Roles and Mechanisms beyond Gluconeogenesis. Mol. Metab. 2021, 53, 101257. [Google Scholar] [CrossRef] [PubMed]
- Lemke, U.; Krones-Herzig, A.; Berriel Diaz, M.; Narvekar, P.; Ziegler, A.; Vegiopoulos, A.; Cato, A.C.B.; Bohl, S.; Klingmüller, U.; Screaton, R.A.; et al. The Glucocorticoid Receptor Controls Hepatic Dyslipidemia through Hes1. Cell Metab. 2008, 8, 212–223. [Google Scholar] [CrossRef]
- Morton, N.M.; Paterson, J.M.; Masuzaki, H.; Holmes, M.C.; Staels, B.; Fievet, C.; Walker, B.R.; Flier, J.S.; Mullins, J.J.; Seckl, J.R. Novel Adipose Tissue-Mediated Resistance to Diet-Induced Visceral Obesity in 11 Beta-Hydroxysteroid Dehydrogenase Type 1-Deficient Mice. Diabetes 2004, 53, 931–938. [Google Scholar] [CrossRef]
- Qi, J.; Wang, W.; Zhu, Q.; He, Y.; Lu, Y.; Wang, Y.; Li, X.; Chen, Z.; Sun, Y. Local Cortisol Elevation Contributes to Endometrial Insulin Resistance in Polycystic Ovary Syndrome. J. Clin. Endocrinol. Metab. 2018, 103, 2457–2467. [Google Scholar] [CrossRef]
- Thau, L.; Gandhi, J.; Sharma, S. Physiology, Cortisol. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Finan, B.; Capozzi, M.E.; Campbell, J.E. Repositioning Glucagon Action in the Physiology and Pharmacology of Diabetes. Diabetes 2020, 69, 532–541. [Google Scholar] [CrossRef]
- Fichna, M.; Fichna, P. Glucocorticoids and Beta-Cell Function. Endokrynol. Pol. 2017, 68, 568–573. [Google Scholar] [CrossRef]
- Esguerra, J.L.S.; Ofori, J.K.; Nagao, M.; Shuto, Y.; Karagiannopoulos, A.; Fadista, J.; Sugihara, H.; Groop, L.; Eliasson, L. Glucocorticoid Induces Human Beta Cell Dysfunction by Involving Riborepressor GAS5 LincRNA. Mol. Metab. 2020, 32, 160–167. [Google Scholar] [CrossRef]
- Xu, W.; Cui, J.; Zhou, F.; Bai, M.; Deng, R.; Wang, W. Leonurine Protects against Dexamethasone-Induced Cytotoxicity in Pancreatic β-Cells via PI3K/Akt Signaling Pathway. Biochem. Biophys. Res. Commun. 2020, 529, 652–658. [Google Scholar] [CrossRef]
- Ferris, H.A.; Kahn, C.R. New Mechanisms of Glucocorticoid-Induced Insulin Resistance: Make No Bones about It. J. Clin. Investig. 2012, 122, 3854–3857. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Judd, R.L. Adiponectin Regulation and Function. Compr. Physiol. 2018, 8, 1031–1063. [Google Scholar] [CrossRef] [PubMed]
- Achari, A.E.; Jain, S.K. Adiponectin, a Therapeutic Target for Obesity, Diabetes, and Endothelial Dysfunction. Int. J. Mol. Sci. 2017, 18, 1321. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased Oxidative Stress in Obesity and Its Impact on Metabolic Syndrome. J. Clin. Investig. 2004, 114, 1752–1761. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, M.; Shimomura, I. Roles of Adiponectin and Oxidative Stress in Obesity-Associated Metabolic and Cardiovascular Diseases. Rev. Endocr. Metab. Disord. 2014, 15, 1–10. [Google Scholar] [CrossRef]
- Diez, J.J.; Iglesias, P. The Role of the Novel Adipocyte-Derived Hormone Adiponectin in Human Disease. Eur. J. Endocrinol. 2003, 148, 293–300. [Google Scholar] [CrossRef]
- Kadowaki, T.; Yamauchi, T.; Kubota, N.; Hara, K.; Ueki, K.; Tobe, K. Adiponectin and Adiponectin Receptors in Insulin Resistance, Diabetes, and the Metabolic Syndrome. J. Clin. Investig. 2006, 116, 1784–1792. [Google Scholar] [CrossRef]
- Rabin, D.; Gold, P.W.; Margioris, A.N.; Chrousos, G.P. Stress and Reproduction: Physiologic and Pathophysiologic Interactions between the Stress and Reproductive Axes. In Mechanisms of Physical and Emotional Stress; Chrousos, G.P., Loriaux, D.L., Gold, P.W., Eds.; Advances in Experimental Medicine and Biology; Springer: Boston, MA, USA, 1988; pp. 377–387. ISBN 978-1-4899-2064-5. [Google Scholar]
- Yoon, V.; Heyliger, A.; Maekawa, T.; Sasano, H.; Carrick, K.; Woodruff, S.; Rabaglia, J.; Auchus, R.J.; Ghayee, H.K. Benign Adrenal Adenomas Secreting Excess Mineralocorticoids and Glucocorticoids. Endocrinol. Diabetes Metab. Case Rep. 2013, 2013, 130042. [Google Scholar] [CrossRef]
- Saltiel, A.R. Insulin Signaling in Health and Disease. J. Clin. Investig. 2021, 131, e142241. [Google Scholar] [CrossRef]
- Perry, R.J.; Resch, J.M.; Douglass, A.M.; Madara, J.C.; Rabin-Court, A.; Kucukdereli, H.; Wu, C.; Song, J.D.; Lowell, B.B.; Shulman, G.I. Leptin’s Hunger-Suppressing Effects Are Mediated by the Hypothalamic-Pituitary-Adrenocortical Axis in Rodents. Proc. Natl. Acad. Sci. USA 2019, 116, 13670–13679. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Higgs, J.A.; Quinn, A.P.; Seely, K.D.; Richards, Z.; Mortensen, S.P.; Crandall, C.S.; Brooks, A.E. Pathophysiological Link between Insulin Resistance and Adrenal Incidentalomas. Int. J. Mol. Sci. 2022, 23, 4340. https://doi.org/10.3390/ijms23084340
Higgs JA, Quinn AP, Seely KD, Richards Z, Mortensen SP, Crandall CS, Brooks AE. Pathophysiological Link between Insulin Resistance and Adrenal Incidentalomas. International Journal of Molecular Sciences. 2022; 23(8):4340. https://doi.org/10.3390/ijms23084340
Chicago/Turabian StyleHiggs, Jordan A., Alyssa P. Quinn, Kevin D. Seely, Zeke Richards, Shad P. Mortensen, Cody S. Crandall, and Amanda E. Brooks. 2022. "Pathophysiological Link between Insulin Resistance and Adrenal Incidentalomas" International Journal of Molecular Sciences 23, no. 8: 4340. https://doi.org/10.3390/ijms23084340
APA StyleHiggs, J. A., Quinn, A. P., Seely, K. D., Richards, Z., Mortensen, S. P., Crandall, C. S., & Brooks, A. E. (2022). Pathophysiological Link between Insulin Resistance and Adrenal Incidentalomas. International Journal of Molecular Sciences, 23(8), 4340. https://doi.org/10.3390/ijms23084340