

Cellular Senescence in Immunity against Infections


Abstract
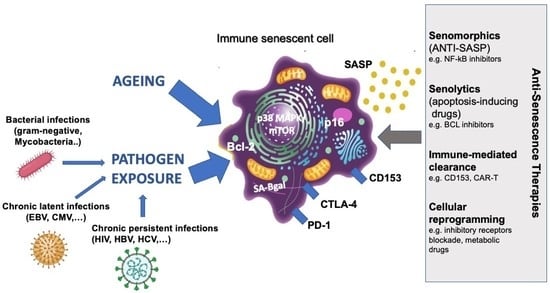
1. Introduction
2. Aging and Immunosenescence
2.1. Immunosenescence in the Adaptive Immune System
2.2. Immunosenescence in the Innate Immune System
3. Immune Response to Infection in Aging
4. Immune Senescence Driven by Pathogen Exposure
4.1. Immune Senescence during Chronic Persistent Viral Infections
4.2. Immune Senescence during Chronic Latent Viral Infections
4.3. Immune Senescence during Bacterial Infections
5. Immunometabolism and Cellular Senescence
6. Targeting of Senescent Immune Cells
6.1. Therapeutic Opportunities for Aging
6.2. Therapeutic Opportunities for Persistent Infections
7. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AKT | Protein kinase B |
| ATM | Ataxia Telangiectasia Mutated |
| BCL-xl | Bcl-2-Like Protein-1 |
| BCR | B-cell receptor |
| B-gal | B-galactosidase |
| BTLA | B- and T-Lymphocyte Attenuator |
| CAR-T | Chimeric Antigen Receptor-T |
| CD122 | Interleukin-2 receptor-b |
| CD127 | Interleukin-7 receptor |
| CDKN | Cyclin-Dependent Kinase inhibitor |
| CMV | Human cytomegalovirus |
| CR | Calorie restriction |
| CTLA-4 | Cytotoxic T-lymphocyte antigen-4 |
| CXCR | C-X-C Motif Chemokine Receptor |
| DDR | DNA damage response |
| DNMTs | DNA methyltransferases |
| EBV | Epstein–Barr virus |
| ERK | Extracellular signal-regulated kinase |
| FOXP3 | Forkhead box P3 |
| gH2AX | Gamma H2A histone family member X |
| HAT | Histone Acetyltransferases |
| HBV | Hepatitis B virus |
| HCV | Hepatitis C virus |
| HDAC | Histone Deacetylases |
| HIV | Human immunodeficiency virus |
| HHV-8 | Human herpesvirus-8 |
| HLA-DR | Human Leukocyte Antigen DR |
| HSV-1/2 | Herpes simplex-1/2 virus |
| HTLV-I | Human T-cell leukemia virus type-1 (HTLV-I) |
| JNK/AP-1 | c-Jun N-terminal kinase/activator protein-1 |
| KLRG1 | Killer Cell Lectin-Like Receptor G1 |
| LAG-3 | Lymphocyte activation gene 3 |
| LC3 | Microtubule-associated protein 1A/1B-light chain 3 |
| LCMV | Lymphocytic choriomeningitis virus |
| LPS | Lipopolysaccharide |
| MAP | Mitogen-activated protein |
| Mtb | Mycobacterium tuberculosis |
| mTOR | Mammalian target of rapamycin |
| NKCC | Natural Killer Cellular Cytotoxicity |
| NET | Neutrophil extracellular trap |
| NLRP3 | NLR family pyrin domain containing 3 |
| PD-1 | Programmed cell death protein 1 |
| PI3K | Phosphatidylinositol 3-kinase |
| PTEN | Phosphatase and Tensin Homolog |
| ROS | Reactive oxygen species |
| RSV | Respiratory syncytial virus |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| SASP | Senescence-Associated Secretory Phenotype |
| SIV | Simian immunodeficiency virus |
| STAT | Signal Transducer and Activator of Transcription |
| TCF1 | T Cell Factor 1 |
| TCR | T-cell receptor |
| TFH | T follicular helper |
| TIGIT | T-cell immunoglobulin and ITIM domain |
| TH | T helper |
| TIM-3 | T-cell immunoglobulin and mucin-domain containing-3 |
| TLR | Toll-Like Receptor |
| TNF | Tumor Necrosis Factor |
| VZV | Varicella-zoster virus |
References
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef] [PubMed]
- Rodier, F.; Coppé, J.P.; Patil, C.K.; Hoeijmakers, W.A.; Muñoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Prata, L.G.P.L.; Ovsyannikova, I.G.; Tchkonia, T.; Kirkland, J.L. Senescent cell clearance by the immune system: Emerging therapeutic opportunities. Semin. Immunol. 2018, 40, 101275. [Google Scholar] [CrossRef]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef]
- Frasca, D.; Blomberg, B.B. Inflammaging decreases adaptive and innate immune responses in mice and humans. Biogerontology 2016, 17, 7–19. [Google Scholar] [CrossRef]
- Camell, C.D.; Yousefzadeh, M.J.; Zhu, Y.; Prata, L.G.P.L.; Huggins, M.A.; Pierson, M.; Zhang, L.; O’Kelly, R.D.; Pirtskhalava, T.; Xun, P.; et al. Senolytics reduce coronavirus-related mortality in old mice. Science 2021, 373, eabe4832. [Google Scholar] [CrossRef]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; d’Adda di Fagagna, F. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 2021, 22, 75–95. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Fulop, T.; Larbi, A.; Dupuis, G.; Le Page, A.; Frost, E.H.; Cohen, A.A.; Witkowski, J.M.; Franceschi, C. Immunosenescence and Inflamm-Aging As Two Sides of the Same Coin: Friends or Foes? Front. Immunol. 2017, 8, 1960. [Google Scholar] [CrossRef]
- Pawelec, G. Hallmarks of human “immunosenescence”: Adaptation or dysregulation? Immun. Ageing 2012, 9, 15. [Google Scholar] [CrossRef]
- Vicente, R.; Mausset-Bonnefont, A.L.; Jorgensen, C.; Louis-Plence, P.; Brondello, J.M. Cellular senescence impact on immune cell fate and function. Aging Cell 2016, 15, 400–406. [Google Scholar] [CrossRef]
- Chambers, E.S.; Vukmanovic-Stejic, M.; Shih, B.B.; Trahair, H.; Subramanian, P.; Devine, O.P.; Glanville, J.; Gilroy, D.; Rustin, M.H.A.; Freeman, T.C.; et al. Recruitment of inflammatory monocytes by senescent fibroblasts inhibits antigen-specific tissue immunity during human aging. Nat. Aging 2021, 1, 101–113. [Google Scholar] [CrossRef]
- Aw, D.; Silva, A.B.; Palmer, D.B. Immunosenescence: Emerging challenges for an ageing population. Immunology 2007, 120, 435–446. [Google Scholar] [CrossRef]
- McElhaney, J.E.; Zhou, X.; Talbot, H.K.; Soethout, E.; Bleackley, R.C.; Granville, D.J.; Pawelec, G. The unmet need in the elderly: How immunosenescence, CMV infection, co-morbidities and frailty are a challenge for the development of more effective influenza vaccines. Vaccine 2012, 30, 2060–2067. [Google Scholar] [CrossRef]
- Yousefzadeh, M.J.; Flores, R.R.; Zhu, Y.; Schmiechen, Z.C.; Brooks, R.W.; Trussoni, C.E.; Cui, Y.; Angelini, L.; Lee, K.A.; McGowan, S.J.; et al. An aged immune system drives senescence and ageing of solid organs. Nature 2021, 594, 100–105. [Google Scholar] [CrossRef]
- Desdín-Micó, G.; Soto-Heredero, G.; Aranda, J.F.; Oller, J.; Carrasco, E.; Gabandé-Rodríguez, E.; Blanco, E.M.; Alfranca, A.; Cussó, L.; Desco, M.; et al. T cells with dysfunctional mitochondria induce multimorbidity and premature senescence. Science 2020, 368, 1371–1376. [Google Scholar] [CrossRef]
- Goronzy, J.J.; Fang, F.; Cavanagh, M.M.; Qi, Q.; Weyand, C.M. Naive T cell maintenance and function in human aging. J. Immunol. 2015, 194, 4073–4080. [Google Scholar] [CrossRef]
- Stulnig, T.; Maczek, C.; Böck, G.; Majdic, O.; Wick, G. Reference intervals for human peripheral blood lymphocyte subpopulations from ‘healthy’ young and aged subjects. Int. Arch. Allergy Immunol. 1995, 108, 205–210. [Google Scholar] [CrossRef]
- Garrido-Rodríguez, V.; Herrero-Fernández, I.; Castro, M.J.; Castillo, A.; Rosado-Sánchez, I.; Galvá, M.I.; Ramos, R.; Olivas-Martínez, I.; Bulnes-Ramos, Á.; Cañizares, J.; et al. Immunological features beyond CD4/CD8 ratio values in older individuals. Aging 2021, 13, 13443–13459. [Google Scholar] [CrossRef]
- Zanni, F.; Vescovini, R.; Biasini, C.; Fagnoni, F.; Zanlari, L.; Telera, A.; Di Pede, P.; Passeri, G.; Pedrazzoni, M.; Passeri, M.; et al. Marked increase with age of type 1 cytokines within memory and effector/cytotoxic CD8+ T cells in humans: A contribution to understand the relationship between inflammation and immunosenescence. Exp. Gerontol. 2003, 38, 981–987. [Google Scholar] [CrossRef]
- Weng, N.P.; Akbar, A.N.; Goronzy, J. CD28− T cells: Their role in the age-associated decline of immune function. Trends Immunol. 2009, 30, 306–312. [Google Scholar] [CrossRef]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef]
- Henson, S.M.; Akbar, A.N. KLRG1—More than a marker for T cell senescence. Age 2009, 31, 285–291. [Google Scholar] [CrossRef]
- Brenchley, J.M.; Karandikar, N.J.; Betts, M.R.; Ambrozak, D.R.; Hill, B.J.; Crotty, L.E.; Casazza, J.P.; Kuruppu, J.; Migueles, S.A.; Connors, M.; et al. Expression of CD57 defines replicative senescence and antigen-induced apoptotic death of CD8+ T cells. Blood 2003, 101, 2711–2720. [Google Scholar] [CrossRef]
- Xu, W.; Larbi, A. Markers of T Cell Senescence in Humans. Int. J. Mol. Sci. 2017, 18, 1742. [Google Scholar] [CrossRef]
- Tarazona, R.; DelaRosa, O.; Alonso, C.; Ostos, B.; Espejo, J.; Peña, J.; Solana, R. Increased expression of NK cell markers on T lymphocytes in aging and chronic activation of the immune system reflects the accumulation of effector/senescent T cells. Mech. Ageing Dev. 2000, 121, 77–88. [Google Scholar] [CrossRef]
- Lanna, A.; Gomes, D.C.; Muller-Durovic, B.; McDonnell, T.; Escors, D.; Gilroy, D.W.; Lee, J.H.; Karin, M.; Akbar, A.N. A sestrin-dependent Erk-Jnk-p38 MAPK activation complex inhibits immunity during aging. Nat. Immunol. 2017, 18, 354–363. [Google Scholar] [CrossRef]
- Pereira, B.I.; De Maeyer, R.P.H.; Covre, L.P.; Nehar-Belaid, D.; Lanna, A.; Ward, S.; Marches, R.; Chambers, E.S.; Gomes, D.C.O.; Riddell, N.E.; et al. Sestrins induce natural killer function in senescent-like CD8. Nat. Immunol. 2020, 21, 684–694. [Google Scholar] [CrossRef]
- Henson, S.M.; Lanna, A.; Riddell, N.E.; Franzese, O.; Macaulay, R.; Griffiths, S.J.; Puleston, D.J.; Watson, A.S.; Simon, A.K.; Tooze, S.A.; et al. p38 signaling inhibits mTORC1-independent autophagy in senescent human CD8⁺ T cells. J. Clin. Investig. 2014, 124, 4004–4016. [Google Scholar] [CrossRef] [PubMed]
- Shirakawa, K.; Yan, X.; Shinmura, K.; Endo, J.; Kataoka, M.; Katsumata, Y.; Yamamoto, T.; Anzai, A.; Isobe, S.; Yoshida, N.; et al. Obesity accelerates T cell senescence in murine visceral adipose tissue. J. Clin. Investig. 2016, 126, 4626–4639. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Yu, M.; Lee, W.W.; Tsang, M.; Krishnan, E.; Weyand, C.M.; Goronzy, J.J. Decline in miR-181a expression with age impairs T cell receptor sensitivity by increasing DUSP6 activity. Nat. Med. 2012, 18, 1518–1524. [Google Scholar] [CrossRef] [PubMed]
- Sansoni, P.; Cossarizza, A.; Brianti, V.; Fagnoni, F.; Snelli, G.; Monti, D.; Marcato, A.; Passeri, G.; Ortolani, C.; Forti, E. Lymphocyte subsets and natural killer cell activity in healthy old people and centenarians. Blood 1993, 82, 2767–2773. [Google Scholar] [CrossRef]
- Pangrazzi, L.; Meryk, A.; Naismith, E.; Koziel, R.; Lair, J.; Krismer, M.; Trieb, K.; Grubeck-Loebenstein, B. “Inflamm-aging” influences immune cell survival factors in human bone marrow. Eur. J. Immunol. 2017, 47, 481–492. [Google Scholar] [CrossRef]
- McLeod, J.D. Apoptotic capability in ageing T cells. Mech. Ageing Dev. 2000, 121, 151–159. [Google Scholar] [CrossRef]
- Callender, L.A.; Carroll, E.C.; Beal, R.W.J.; Chambers, E.S.; Nourshargh, S.; Akbar, A.N.; Henson, S.M. Human CD8. Aging Cell 2018, 17, e12675. [Google Scholar] [CrossRef]
- Martínez-Zamudio, R.I.; Dewald, H.K.; Vasilopoulos, T.; Gittens-Williams, L.; Fitzgerald-Bocarsly, P.; Herbig, U. Senescence-associated β-galactosidase reveals the abundance of senescent CD8+ T cells in aging humans. Aging Cell 2021, 20, e13344. [Google Scholar] [CrossRef]
- Pustavoitau, A.; Barodka, V.; Sharpless, N.E.; Torrice, C.; Nyhan, D.; Berkowitz, D.E.; Shah, A.S.; Bandeen Roche, K.J.; Walston, J.D. Role of senescence marker p16 INK4a measured in peripheral blood T-lymphocytes in predicting length of hospital stay after coronary artery bypass surgery in older adults. Exp. Gerontol. 2016, 74, 29–36. [Google Scholar] [CrossRef]
- Quinn, K.M.; Fox, A.; Harland, K.L.; Russ, B.E.; Li, J.; Nguyen, T.H.O.; Loh, L.; Olshanksy, M.; Naeem, H.; Tsyganov, K.; et al. Age-related decline in primary CD8+ T cell responses is associated with the development of senescence in virtual memory CD8+ T cells. Cell Rep. 2018, 23, 3512–3524. [Google Scholar] [CrossRef]
- Abbas, A.K.; Murphy, K.M.; Sher, A. Functional diversity of helper T lymphocytes. Nature 1996, 383, 787–793. [Google Scholar] [CrossRef]
- Rink, L.; Cakman, I.; Kirchner, H. Altered cytokine production in the elderly. Mech. Ageing Dev. 1998, 102, 199–209. [Google Scholar] [CrossRef]
- Goronzy, J.J.; Weyand, C.M. Mechanisms underlying T cell ageing. Nat. Rev. Immunol. 2019, 19, 573–583. [Google Scholar] [CrossRef]
- Utzschneider, D.T.; Delpoux, A.; Wieland, D.; Huang, X.; Lai, C.Y.; Hofmann, M.; Thimme, R.; Hedrick, S.M. Active Maintenance of T Cell Memory in Acute and Chronic Viral Infection Depends on Continuous Expression of FOXO1. Cell Rep. 2018, 22, 3454–3467. [Google Scholar] [CrossRef]
- Linterman, M.A. How T follicular helper cells and the germinal centre response change with age. Immunol. Cell Biol. 2014, 92, 72–79. [Google Scholar] [CrossRef]
- Schmitt, V.; Rink, L.; Uciechowski, P. The Th17/Treg balance is disturbed during aging. Exp. Gerontol. 2013, 48, 1379–1386. [Google Scholar] [CrossRef]
- Rocamora-Reverte, L.; Melzer, F.L.; Würzner, R.; Weinberger, B. The Complex Role of Regulatory T Cells in Immunity and Aging. Front. Immunol. 2020, 11, 616949. [Google Scholar] [CrossRef]
- Frasca, D.; Diaz, A.; Romero, M.; Garcia, D.; Blomberg, B.B. B Cell Immunosenescence. Annu. Rev. Cell Dev. Biol. 2020, 36, 551–574. [Google Scholar] [CrossRef]
- Ratliff, M.; Alter, S.; Frasca, D.; Blomberg, B.B.; Riley, R.L. In senescence, age-associated B cells secrete TNFα and inhibit survival of B-cell precursors. Aging Cell 2013, 12, 303–311. [Google Scholar] [CrossRef]
- Hao, Y.; O’Neill, P.; Naradikian, M.S.; Scholz, J.L.; Cancro, M.P. A B-cell subset uniquely responsive to innate stimuli accumulates in aged mice. Blood 2011, 118, 1294–1304. [Google Scholar] [CrossRef]
- Rubtsov, A.V.; Rubtsova, K.; Fischer, A.; Meehan, R.T.; Gillis, J.Z.; Kappler, J.W.; Marrack, P. Toll-like receptor 7 (TLR7)-driven accumulation of a novel CD11c⁺ B-cell population is important for the development of autoimmunity. Blood 2011, 118, 1305–1315. [Google Scholar] [CrossRef]
- Cancro, M.P. Age-Associated B Cells. Annu. Rev. Immunol. 2020, 38, 315–340. [Google Scholar] [CrossRef]
- Ademokun, A.; Wu, Y.C.; Dunn-Walters, D. The ageing B cell population: Composition and function. Biogerontology 2010, 11, 125–137. [Google Scholar] [CrossRef]
- Mauri, C.; Menon, M. The expanding family of regulatory B cells. Int Immunol 2015, 27, 479–486. [Google Scholar] [CrossRef]
- Duggal, N.A.; Upton, J.; Phillips, A.C.; Sapey, E.; Lord, J.M. An age-related numerical and functional deficit in CD19+ CD24hi CD38hi B cells is associated with an increase in systemic autoimmunity. Aging Cell 2013, 12, 873–881. [Google Scholar] [CrossRef]
- van der Geest, K.S.; Lorencetti, P.G.; Abdulahad, W.H.; Horst, G.; Huitema, M.; Roozendaal, C.; Kroesen, B.J.; Brouwer, E.; Boots, A.M. Aging-dependent decline of IL-10 producing B cells coincides with production of antinuclear antibodies but not rheumatoid factors. Exp. Gerontol. 2016, 75, 24–29. [Google Scholar] [CrossRef]
- Borgoni, S.; Kudryashova, K.S.; Burka, K.; de Magalhães, J.P. Targeting immune dysfunction in aging. Ageing Res. Rev. 2021, 70, 101410. [Google Scholar] [CrossRef]
- Bulati, M.; Buffa, S.; Candore, G.; Caruso, C.; Dunn-Walters, D.K.; Pellicanò, M.; Wu, Y.C.; Colonna Romano, G. B cells and immunosenescence: A focus on IgG+IgD−CD27− (DN) B cells in aged humans. Ageing Res. Rev. 2011, 10, 274–284. [Google Scholar] [CrossRef]
- Frasca, D.; Diaz, A.; Romero, M.; Blomberg, B.B. Human peripheral late/exhausted memory B cells express a senescent-associated secretory phenotype and preferentially utilize metabolic signaling pathways. Exp. Gerontol. 2017, 87, 113–120. [Google Scholar] [CrossRef]
- Buffa, S.; Bulati, M.; Pellicanò, M.; Dunn-Walters, D.K.; Wu, Y.C.; Candore, G.; Vitello, S.; Caruso, C.; Colonna-Romano, G. B cell immunosenescence: Different features of naive and memory B cells in elderly. Biogerontology 2011, 12, 473–483. [Google Scholar] [CrossRef]
- Shi, Y.; Yamazaki, T.; Okubo, Y.; Uehara, Y.; Sugane, K.; Agematsu, K. Regulation of aged humoral immune defense against pneumococcal bacteria by IgM memory B cell. J. Immunol. 2005, 175, 3262–3267. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.Y.; Li, Y.; Kaplan, D.E. Hepatitis C viraemia reversibly maintains subset of antigen-specific T-bet+ tissue-like memory B cells. J. Viral Hepat. 2017, 24, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Illingworth, J.; Butler, N.S.; Roetynck, S.; Mwacharo, J.; Pierce, S.K.; Bejon, P.; Crompton, P.D.; Marsh, K.; Ndungu, F.M. Chronic exposure to Plasmodium falciparum is associated with phenotypic evidence of B and T cell exhaustion. J. Immunol. 2013, 190, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Moir, S.; Ho, J.; Malaspina, A.; Wang, W.; DiPoto, A.C.; O’Shea, M.A.; Roby, G.; Kottilil, S.; Arthos, J.; Proschan, M.A.; et al. Evidence for HIV-associated B cell exhaustion in a dysfunctional memory B cell compartment in HIV-infected viremic individuals. J. Exp. Med. 2008, 205, 1797–1805. [Google Scholar] [CrossRef]
- Colonna-Romano, G.; Bulati, M.; Aquino, A.; Pellicanò, M.; Vitello, S.; Lio, D.; Candore, G.; Caruso, C. A double-negative (IgD−CD27−) B cell population is increased in the peripheral blood of elderly people. Mech. Ageing Dev. 2009, 130, 681–690. [Google Scholar] [CrossRef]
- Martorana, A.; Balistreri, C.R.; Bulati, M.; Buffa, S.; Azzarello, D.M.; Camarda, C.; Monastero, R.; Caruso, C.; Colonna-Romano, G. Double negative (CD19+IgG+IgD−CD27−) B lymphocytes: A new insight from telomerase in healthy elderly, in centenarian offspring and in Alzheimer’s disease patients. Immunol. Lett. 2014, 162, 303–309. [Google Scholar] [CrossRef]
- Simell, B.; Vuorela, A.; Ekström, N.; Palmu, A.; Reunanen, A.; Meri, S.; Käyhty, H.; Väkeväinen, M. Aging reduces the functionality of anti-pneumococcal antibodies and the killing of Streptococcus pneumoniae by neutrophil phagocytosis. Vaccine 2011, 29, 1929–1934. [Google Scholar] [CrossRef]
- MacGregor, R.R.; Shalit, M. Neutrophil function in healthy elderly subjects. J. Gerontol. 1990, 45, M55-60. [Google Scholar] [CrossRef]
- Butcher, S.; Chahel, H.; Lord, J.M. Review article: Ageing and the neutrophil: No appetite for killing? Immunology 2000, 100, 411–416. [Google Scholar] [CrossRef]
- Brubaker, A.L.; Rendon, J.L.; Ramirez, L.; Choudhry, M.A.; Kovacs, E.J. Reduced neutrophil chemotaxis and infiltration contributes to delayed resolution of cutaneous wound infection with advanced age. J. Immunol. 2013, 190, 1746–1757. [Google Scholar] [CrossRef]
- Butcher, S.K.; Chahal, H.; Nayak, L.; Sinclair, A.; Henriquez, N.V.; Sapey, E.; O’Mahony, D.; Lord, J.M. Senescence in innate immune responses: Reduced neutrophil phagocytic capacity and CD16 expression in elderly humans. J. Leukoc. Biol. 2001, 70, 881–886. [Google Scholar] [CrossRef]
- Fulop, T.; Larbi, A.; Douziech, N.; Fortin, C.; Guérard, K.P.; Lesur, O.; Khalil, A.; Dupuis, G. Signal transduction and functional changes in neutrophils with aging. Aging Cell 2004, 3, 217–226. [Google Scholar] [CrossRef]
- Sapey, E.; Greenwood, H.; Walton, G.; Mann, E.; Love, A.; Aaronson, N.; Insall, R.H.; Stockley, R.A.; Lord, J.M. Phosphoinositide 3-kinase inhibition restores neutrophil accuracy in the elderly: Toward targeted treatments for immunosenescence. Blood 2014, 123, 239–248. [Google Scholar] [CrossRef]
- Kolaczkowska, E. The older the faster: Aged neutrophils in inflammation. Blood 2016, 128, 2280–2282. [Google Scholar] [CrossRef]
- Wenisch, C.; Patruta, S.; Daxböck, F.; Krause, R.; Hörl, W. Effect of age on human neutrophil function. J. Leukoc. Biol. 2000, 67, 40–45. [Google Scholar] [CrossRef]
- Emanuelli, G.; Lanzio, M.; Anfossi, T.; Romano, S.; Anfossi, G.; Calcamuggi, G. Influence of age on polymorphonuclear leukocytes in vitro: Phagocytic activity in healthy human subjects. Gerontology 1986, 32, 308–316. [Google Scholar] [CrossRef]
- Tseng, C.W.; Kyme, P.A.; Arruda, A.; Ramanujan, V.K.; Tawackoli, W.; Liu, G.Y. Innate immune dysfunctions in aged mice facilitate the systemic dissemination of methicillin-resistant S. aureus. PLoS ONE 2012, 7, e41454. [Google Scholar] [CrossRef]
- Hazeldine, J.; Harris, P.; Chapple, I.L.; Grant, M.; Greenwood, H.; Livesey, A.; Sapey, E.; Lord, J.M. Impaired neutrophil extracellular trap formation: A novel defect in the innate immune system of aged individuals. Aging Cell 2014, 13, 690–698. [Google Scholar] [CrossRef]
- Gayoso, I.; Sanchez-Correa, B.; Campos, C.; Alonso, C.; Pera, A.; Casado, J.G.; Morgado, S.; Tarazona, R.; Solana, R. Immunosenescence of human natural killer cells. J. Innate Immun. 2011, 3, 337–343. [Google Scholar] [CrossRef]
- Muzzioli, M.; Stecconi, R.; Moresi, R.; Provinciali, M. Zinc improves the development of human CD34+ cell progenitors towards NK cells and increases the expression of GATA-3 transcription factor in young and old ages. Biogerontology 2009, 10, 593–604. [Google Scholar] [CrossRef]
- Hazeldine, J.; Hampson, P.; Lord, J.M. Reduced release and binding of perforin at the immunological synapse underlies the age-related decline in natural killer cell cytotoxicity. Aging Cell 2012, 11, 751–759. [Google Scholar] [CrossRef]
- Almeida-Oliveira, A.; Smith-Carvalho, M.; Porto, L.C.; Cardoso-Oliveira, J.; Ribeiro, A.o.S.; Falcão, R.R.; Abdelhay, E.; Bouzas, L.F.; Thuler, L.C.; Ornellas, M.H.; et al. Age-related changes in natural killer cell receptors from childhood through old age. Hum. Immunol. 2011, 72, 319–329. [Google Scholar] [CrossRef]
- Solana, R.; Campos, C.; Pera, A.; Tarazona, R. Shaping of NK cell subsets by aging. Curr. Opin. Immunol. 2014, 29, 56–61. [Google Scholar] [CrossRef]
- Ogata, K.; An, E.; Shioi, Y.; Nakamura, K.; Luo, S.; Yokose, N.; Minami, S.; Dan, K. Association between natural killer cell activity and infection in immunologically normal elderly people. Clin. Exp. Immunol. 2001, 124, 392–397. [Google Scholar] [CrossRef]
- Lutz, C.T.; Moore, M.B.; Bradley, S.; Shelton, B.J.; Lutgendorf, S.K. Reciprocal age related change in natural killer cell receptors for MHC class I. Mech. Ageing Dev. 2005, 126, 722–731. [Google Scholar] [CrossRef]
- Rajagopalan, S.; Lee, E.C.; DuPrie, M.L.; Long, E.O. TNFR-associated factor 6 and TGF-β-activated kinase 1 control signals for a senescence response by an endosomal NK cell receptor. J. Immunol. 2014, 192, 714–721. [Google Scholar] [CrossRef]
- Kohut, M.L.; Senchina, D.S.; Madden, K.S.; Martin, A.E.; Felten, D.L.; Moynihan, J.A. Age effects on macrophage function vary by tissue site, nature of stimulant, and exercise behavior. Exp. Gerontol. 2004, 39, 1347–1360. [Google Scholar] [CrossRef]
- Geiger, H.; Van Zant, G. The aging of lympho-hematopoietic stem cells. Nat. Immunol. 2002, 3, 329–333. [Google Scholar] [CrossRef] [PubMed]
- De Maeyer, R.P.H.; Chambers, E.S. The impact of ageing on monocytes and macrophages. Immunol. Lett. 2021, 230, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, T.; Kitagawa, M.; Hirokawa, K. Age-related changes of human bone marrow: A histometric estimation of proliferative cells, apoptotic cells, T cells, B cells and macrophages. Mech. Ageing Dev. 2000, 117, 57–68. [Google Scholar] [CrossRef]
- Herrero, C.; Marqués, L.; Lloberas, J.; Celada, A. IFN-gamma-dependent transcription of MHC class II IA is impaired in macrophages from aged mice. J. Clin. Investig. 2001, 107, 485–493. [Google Scholar] [CrossRef]
- Wong, C.O.; Gregory, S.; Hu, H.; Chao, Y.; Sepúlveda, V.E.; He, Y.; Li-Kroeger, D.; Goldman, W.E.; Bellen, H.J.; Venkatachalam, K. Lysosomal Degradation Is Required for Sustained Phagocytosis of Bacteria by Macrophages. Cell Host Microbe 2017, 21, 719–730. [Google Scholar] [CrossRef]
- Renshaw, M.; Rockwell, J.; Engleman, C.; Gewirtz, A.; Katz, J.; Sambhara, S. Cutting edge: Impaired Toll-like receptor expression and function in aging. J. Immunol. 2002, 169, 4697–4701. [Google Scholar] [CrossRef]
- Chelvarajan, R.L.; Liu, Y.; Popa, D.; Getchell, M.L.; Getchell, T.V.; Stromberg, A.J.; Bondada, S. Molecular basis of age-associated cytokine dysregulation in LPS-stimulated macrophages. J. Leukoc. Biol. 2006, 79, 1314–1327. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- van Beek, A.A.; Van den Bossche, J.; Mastroberardino, P.G.; de Winther, M.P.J.; Leenen, P.J.M. Metabolic Alterations in Aging Macrophages: Ingredients for Inflammaging? Trends Immunol. 2019, 40, 113–127. [Google Scholar] [CrossRef]
- Mahbub, S.; Deburghgraeve, C.R.; Kovacs, E.J. Advanced age impairs macrophage polarization. J. Interfer. Cytokine Res. 2012, 32, 18–26. [Google Scholar] [CrossRef]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef]
- Qian, F.; Wang, X.; Zhang, L.; Chen, S.; Piecychna, M.; Allore, H.; Bockenstedt, L.; Malawista, S.; Bucala, R.; Shaw, A.C.; et al. Age-associated elevation in TLR5 leads to increased inflammatory responses in the elderly. Aging Cell 2012, 11, 104–110. [Google Scholar] [CrossRef]
- Nyunoya, T.; Powers, L.S.; Yarovinsky, T.O.; Butler, N.S.; Monick, M.M.; Hunninghake, G.W. Hyperoxia induces macrophage cell cycle arrest by adhesion-dependent induction of p21Cip1 and activation of the retinoblastoma protein. J. Biol. Chem. 2003, 278, 36099–36106. [Google Scholar] [CrossRef]
- Wang, H.; Fu, H.; Zhu, R.; Wu, X.; Ji, X.; Li, X.; Jiang, H.; Lin, Z.; Tang, X.; Sun, S.; et al. BRD4 contributes to LPS-induced macrophage senescence and promotes progression of atherosclerosis-associated lipid uptake. Aging 2020, 12, 9240–9259. [Google Scholar] [CrossRef]
- Prattichizzo, F.; De Nigris, V.; Mancuso, E.; Spiga, R.; Giuliani, A.; Matacchione, G.; Lazzarini, R.; Marcheselli, F.; Recchioni, R.; Testa, R.; et al. Short-term sustained hyperglycaemia fosters an archetypal senescence-associated secretory phenotype in endothelial cells and macrophages. Redox Biol. 2018, 15, 170–181. [Google Scholar] [CrossRef]
- Kloc, M.; Uosef, A.; Subuddhi, A.; Kubiak, J.Z.; Piprek, R.P.; Ghobrial, R.M. Giant Multinucleated Cells in Aging and Senescence-An Abridgement. Biology 2022, 11, 1121. [Google Scholar] [CrossRef]
- Metcalf, T.U.; Wilkinson, P.A.; Cameron, M.J.; Ghneim, K.; Chiang, C.; Wertheimer, A.M.; Hiscott, J.B.; Nikolich-Zugich, J.; Haddad, E.K. Human Monocyte Subsets Are Transcriptionally and Functionally Altered in Aging in Response to Pattern Recognition Receptor Agonists. J. Immunol. 2017, 199, 1405–1417. [Google Scholar] [CrossRef]
- Molony, R.D.; Nguyen, J.T.; Kong, Y.; Montgomery, R.R.; Shaw, A.C.; Iwasaki, A. Aging impairs both primary and secondary RIG-I signaling for interferon induction in human monocytes. Sci. Signal. 2017, 10, eaan2392. [Google Scholar] [CrossRef]
- Wong, K.L.; Yeap, W.H.; Tai, J.J.; Ong, S.M.; Dang, T.M.; Wong, S.C. The three human monocyte subsets: Implications for health and disease. Immunol. Res. 2012, 53, 41–57. [Google Scholar] [CrossRef]
- Ong, S.M.; Hadadi, E.; Dang, T.M.; Yeap, W.H.; Tan, C.T.; Ng, T.P.; Larbi, A.; Wong, S.C. The pro-inflammatory phenotype of the human non-classical monocyte subset is attributed to senescence. Cell Death Dis. 2018, 9, 266. [Google Scholar] [CrossRef]
- Connors, J.; Taramangalam, B.; Cusimano, G.; Bell, M.R.; Matt, S.M.; Runner, K.; Gaskill, P.J.; DeFilippis, V.; Nikolich-Žugich, J.; Kutzler, M.A.; et al. Aging alters antiviral signaling pathways resulting in functional impairment in innate immunity in response to pattern recognition receptor agonists. Geroscience 2022, 1–18. [Google Scholar] [CrossRef]
- Bleve, A.; Motta, F.; Durante, B.; Pandolfo, C.; Selmi, C.; Sica, A. Immunosenescence, Inflammaging, and Frailty: Role of Myeloid Cells in Age-Related Diseases. Clin. Rev. Allergy Immunol. 2022, 1–22. [Google Scholar] [CrossRef]
- Consonni, F.M.; Porta, C.; Marino, A.; Pandolfo, C.; Mola, S.; Bleve, A.; Sica, A. Myeloid-Derived Suppressor Cells: Ductile Targets in Disease. Front. Immunol. 2019, 10, 949. [Google Scholar] [CrossRef]
- Jing, Y.; Shaheen, E.; Drake, R.R.; Chen, N.; Gravenstein, S.; Deng, Y. Aging is associated with a numerical and functional decline in plasmacytoid dendritic cells, whereas myeloid dendritic cells are relatively unaltered in human peripheral blood. Hum. Immunol. 2009, 70, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Granot, T.; Senda, T.; Carpenter, D.J.; Matsuoka, N.; Weiner, J.; Gordon, C.L.; Miron, M.; Kumar, B.V.; Griesemer, A.; Ho, S.H.; et al. Dendritic Cells Display Subset and Tissue-Specific Maturation Dynamics over Human Life. Immunity 2017, 46, 504–515. [Google Scholar] [CrossRef] [PubMed]
- Chambers, E.S.; Vukmanovic-Stejic, M. Skin barrier immunity and ageing. Immunology 2020, 160, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Panda, A.; Qian, F.; Mohanty, S.; van Duin, D.; Newman, F.K.; Zhang, L.; Chen, S.; Towle, V.; Belshe, R.B.; Fikrig, E.; et al. Age-associated decrease in TLR function in primary human dendritic cells predicts influenza vaccine response. J. Immunol. 2010, 184, 2518–2527. [Google Scholar] [CrossRef]
- Agrawal, A.; Gupta, S. Impact of aging on dendritic cell functions in humans. Ageing Res. Rev. 2011, 10, 336–345. [Google Scholar] [CrossRef]
- Gustafson, C.E.; Kim, C.; Weyand, C.M.; Goronzy, J.J. Influence of immune aging on vaccine responses. J. Allergy Clin. Immunol. 2020, 145, 1309–1321. [Google Scholar] [CrossRef]
- Kulkarni, D.H.; Gustafsson, J.K.; Knoop, K.A.; McDonald, K.G.; Bidani, S.S.; Davis, J.E.; Floyd, A.N.; Hogan, S.P.; Hsieh, C.S.; Newberry, R.D. Goblet cell associated antigen passages support the induction and maintenance of oral tolerance. Mucosal Immunol. 2020, 13, 271–282. [Google Scholar] [CrossRef]
- Josset, L.; Engelmann, F.; Haberthur, K.; Kelly, S.; Park, B.; Kawoaka, Y.; García-Sastre, A.; Katze, M.G.; Messaoudi, I. Increased viral loads and exacerbated innate host responses in aged macaques infected with the 2009 pandemic H1N1 influenza A virus. J. Virol. 2012, 86, 11115–11127. [Google Scholar] [CrossRef]
- Tang, B.M.; Shojaei, M.; Teoh, S.; Meyers, A.; Ho, J.; Ball, T.B.; Keynan, Y.; Pisipati, A.; Kumar, A.; Eisen, D.P.; et al. Neutrophils-related host factors associated with severe disease and fatality in patients with influenza infection. Nat. Commun. 2019, 10, 3422. [Google Scholar] [CrossRef]
- Dunning, J.; Blankley, S.; Hoang, L.T.; Cox, M.; Graham, C.M.; James, P.L.; Bloom, C.I.; Chaussabel, D.; Banchereau, J.; Brett, S.J.; et al. Author Correction: Progression of whole-blood transcriptional signatures from interferon-induced to neutrophil-associated patterns in severe influenza. Nat. Immunol. 2019, 20, 373. [Google Scholar] [CrossRef]
- Wong, C.K.; Smith, C.A.; Sakamoto, K.; Kaminski, N.; Koff, J.L.; Goldstein, D.R. Aging Impairs Alveolar Macrophage Phagocytosis and Increases Influenza-Induced Mortality in Mice. J. Immunol. 2017, 199, 1060–1068. [Google Scholar] [CrossRef]
- De Maeyer, R.P.H.; van de Merwe, R.C.; Louie, R.; Bracken, O.V.; Devine, O.P.; Goldstein, D.R.; Uddin, M.; Akbar, A.N.; Gilroy, D.W. Blocking elevated p38 MAPK restores efferocytosis and inflammatory resolution in the elderly. Nat. Immunol. 2020, 21, 615–625. [Google Scholar] [CrossRef]
- Aprahamian, T.; Takemura, Y.; Goukassian, D.; Walsh, K. Ageing is associated with diminished apoptotic cell clearance in vivo. Clin. Exp. Immunol. 2008, 152, 448–455. [Google Scholar] [CrossRef]
- Decman, V.; Laidlaw, B.J.; Dimenna, L.J.; Abdulla, S.; Mozdzanowska, K.; Erikson, J.; Ertl, H.C.; Wherry, E.J. Cell-intrinsic defects in the proliferative response of antiviral memory CD8 T cells in aged mice upon secondary infection. J. Immunol. 2010, 184, 5151–5159. [Google Scholar] [CrossRef]
- Yager, E.J.; Ahmed, M.; Lanzer, K.; Randall, T.D.; Woodland, D.L.; Blackman, M.A. Age-associated decline in T cell repertoire diversity leads to holes in the repertoire and impaired immunity to influenza virus. J. Exp. Med. 2008, 205, 711–723. [Google Scholar] [CrossRef]
- Lanzer, K.G.; Johnson, L.L.; Woodland, D.L.; Blackman, M.A. Impact of ageing on the response and repertoire of influenza virus-specific CD4 T cells. Immun. Ageing 2014, 11, 9. [Google Scholar] [CrossRef]
- Lanfermeijer, J.; Borghans, J.A.M.; van Baarle, D. How age and infection history shape the antigen-specific CD8. Aging Cell 2020, 19, e13262. [Google Scholar] [CrossRef]
- Po, J.L.; Gardner, E.M.; Anaraki, F.; Katsikis, P.D.; Murasko, D.M. Age-associated decrease in virus-specific CD8+ T lymphocytes during primary influenza infection. Mech. Ageing Dev. 2002, 123, 1167–1181. [Google Scholar] [CrossRef]
- Toapanta, F.R.; Ross, T.M. Impaired immune responses in the lungs of aged mice following influenza infection. Respir. Res. 2009, 10, 112. [Google Scholar] [CrossRef]
- Jiang, J.; Bennett, A.J.; Fisher, E.; Williams-Bey, Y.; Shen, H.; Murasko, D.M. Limited expansion of virus-specific CD8 T cells in the aged environment. Mech. Ageing Dev. 2009, 130, 713–721. [Google Scholar] [CrossRef]
- Lefebvre, J.S.; Masters, A.R.; Hopkins, J.W.; Haynes, L. Age-related impairment of humoral response to influenza is associated with changes in antigen specific T follicular helper cell responses. Sci. Rep. 2016, 6, 25051. [Google Scholar] [CrossRef]
- Zacca, E.R.; Crespo, M.I.; Acland, R.P.; Roselli, E.; Núñez, N.G.; Maccioni, M.; Maletto, B.A.; Pistoresi-Palencia, M.C.; Morón, G. Aging Impairs the Ability of Conventional Dendritic Cells to Cross-Prime CD8+ T Cells upon Stimulation with a TLR7 Ligand. PLoS ONE 2015, 10, e0140672. [Google Scholar] [CrossRef]
- Zhao, J.; Legge, K.; Perlman, S. Age-related increases in PGD(2) expression impair respiratory DC migration, resulting in diminished T cell responses upon respiratory virus infection in mice. J. Clin. Investig. 2011, 121, 4921–4930. [Google Scholar] [CrossRef]
- Agrawal, A.; Agrawal, S.; Cao, J.N.; Su, H.; Osann, K.; Gupta, S. Altered innate immune functioning of dendritic cells in elderly humans: A role of phosphoinositide 3-kinase-signaling pathway. J. Immunol. 2007, 178, 6912–6922. [Google Scholar] [CrossRef]
- Aksoy, E.; Saveanu, L.; Manoury, B. The Isoform Selective Roles of PI3Ks in Dendritic Cell Biology and Function. Front. Immunol. 2018, 9, 2574. [Google Scholar] [CrossRef]
- Frasca, D.; Diaz, A.; Romero, M.; Blomberg, B.B. The generation of memory B cells is maintained, but the antibody response is not, in the elderly after repeated influenza immunizations. Vaccine 2016, 34, 2834–2840. [Google Scholar] [CrossRef]
- Nipper, A.J.; Smithey, M.J.; Shah, R.C.; Canaday, D.H.; Landay, A.L. Diminished antibody response to influenza vaccination is characterized by expansion of an age-associated B-cell population with low PAX5. Clin Immunol 2018, 193, 80–87. [Google Scholar] [CrossRef]
- Goodwin, K.; Viboud, C.; Simonsen, L. Antibody response to influenza vaccination in the elderly: A quantitative review. Vaccine 2006, 24, 1159–1169. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Y.; Gilmore, X.; Xu, K.; Wyde, P.R.; Mbawuike, I.N. An aged mouse model for RSV infection and diminished CD8+ CTL responses. Exp. Biol. Med. 2002, 227, 133–140. [Google Scholar] [CrossRef]
- Pennings, J.L.A.; Mariman, R.; Hodemaekers, H.M.; Reemers, S.S.N.; Janssen, R.; Guichelaar, T. Transcriptomics in lung tissue upon respiratory syncytial virus infection reveals aging as important modulator of immune activation and matrix maintenance. Sci. Rep. 2018, 8, 16653. [Google Scholar] [CrossRef]
- Cherukuri, A.; Patton, K.; Gasser, R.A.; Zuo, F.; Woo, J.; Esser, M.T.; Tang, R.S. Adults 65 years old and older have reduced numbers of functional memory T cells to respiratory syncytial virus fusion protein. Clin. Vaccine Immunol. 2013, 20, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, J.; Parsons, R.; Botelho, F.; Millar, J.; McNeil, S.; Fulop, T.; McElhaney, J.; Andrew, M.K.; Walter, S.D.; Devereaux, P.J.; et al. Immune biomarkers predictive of respiratory viral infection in elderly nursing home residents. PLoS ONE 2014, 9, e108481. [Google Scholar] [CrossRef] [PubMed]
- Falsey, A.R.; Dallal, G.E.; Formica, M.A.; Andolina, G.G.; Hamer, D.H.; Leka, L.L.; Meydani, S.N. Long-term care facilities: A cornucopia of viral pathogens. J. Am. Geriatr. Soc. 2008, 56, 1281–1285. [Google Scholar] [CrossRef] [PubMed]
- Krone, C.L.; van de Groep, K.; Trzciński, K.; Sanders, E.A.; Bogaert, D. Immunosenescence and pneumococcal disease: An imbalance in host-pathogen interactions. Lancet Respir. Med. 2014, 2, 141–153. [Google Scholar] [CrossRef]
- Williams, A.E.; José, R.J.; Brown, J.S.; Chambers, R.C. Enhanced inflammation in aged mice following infection with Streptococcus pneumoniae is associated with decreased IL-10 and augmented chemokine production. Am. J. Physiol. Cell. Mol. Physiol. 2015, 308, L539–L549. [Google Scholar] [CrossRef]
- Menter, T.; Giefing-Kroell, C.; Grubeck-Loebenstein, B.; Tzankov, A. Characterization of the inflammatory infiltrate in Streptococcus pneumoniae pneumonia in young and elderly patients. Pathobiology 2014, 81, 160–167. [Google Scholar] [CrossRef]
- Bhalla, M.; Heinzinger, L.R.; Morenikeji, O.B.; Marzullo, B.; Thomas, B.N.; Bou Ghanem, E.N. Transcriptome Profiling Reveals CD73 and Age-Driven Changes in Neutrophil Responses against Streptococcus pneumoniae. Infect. Immun. 2021, 89, e00258-21. [Google Scholar] [CrossRef]
- Hinojosa, E.; Boyd, A.R.; Orihuela, C.J. Age-associated inflammation and toll-like receptor dysfunction prime the lungs for pneumococcal pneumonia. J. Infect. Dis. 2009, 200, 546–554. [Google Scholar] [CrossRef]
- Boyd, A.R.; Shivshankar, P.; Jiang, S.; Berton, M.T.; Orihuela, C.J. Age-related defects in TLR2 signaling diminish the cytokine response by alveolar macrophages during murine pneumococcal pneumonia. Exp. Gerontol. 2012, 47, 507–518. [Google Scholar] [CrossRef]
- Cho, S.J.; Rooney, K.; Choi, A.M.K.; Stout-Delgado, H.W. NLRP3 inflammasome activation in aged macrophages is diminished during Streptococcus pneumoniae infection. Am. J. Physiol. Cell. Mol. Physiol. 2018, 314, L372–L387. [Google Scholar] [CrossRef]
- Inomata, M.; Xu, S.; Chandra, P.; Meydani, S.N.; Takemura, G.; Philips, J.A.; Leong, J.M. Macrophage LC3-associated phagocytosis is an immune defense against. Proc. Natl. Acad. Sci. USA 2020, 117, 33561–33569. [Google Scholar] [CrossRef]
- Birjandi, S.Z.; Ippolito, J.A.; Ramadorai, A.K.; Witte, P.L. Alterations in marginal zone macrophages and marginal zone B cells in old mice. J. Immunol. 2011, 186, 3441–3451. [Google Scholar] [CrossRef]
- Bruunsgaard, H.; Skinhøj, P.; Qvist, J.; Pedersen, B.K. Elderly humans show prolonged in vivo inflammatory activity during pneumococcal infections. J. Infect. Dis. 1999, 180, 551–554. [Google Scholar] [CrossRef]
- Aiello, A.; Farzaneh, F.; Candore, G.; Caruso, C.; Davinelli, S.; Gambino, C.M.; Ligotti, M.E.; Zareian, N.; Accardi, G. Immunosenescence and Its Hallmarks: How to Oppose Aging Strategically? A Review of Potential Options for Therapeutic Intervention. Front. Immunol. 2019, 10, 2247. [Google Scholar] [CrossRef]
- Virgin, H.W.; Wherry, E.J.; Ahmed, R. Redefining chronic viral infection. Cell 2009, 138, 30–50. [Google Scholar] [CrossRef]
- Kahan, S.M.; Wherry, E.J.; Zajac, A.J. T cell exhaustion during persistent viral infections. Virology 2015, 479–480, 180–193. [Google Scholar] [CrossRef]
- Velu, V.; Titanji, K.; Zhu, B.; Husain, S.; Pladevega, A.; Lai, L.; Vanderford, T.H.; Chennareddi, L.; Silvestri, G.; Freeman, G.J.; et al. Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature 2009, 458, 206–210. [Google Scholar] [CrossRef]
- Wherry, E.J.; Ha, S.J.; Kaech, S.M.; Haining, W.N.; Sarkar, S.; Kalia, V.; Subramaniam, S.; Blattman, J.N.; Barber, D.L.; Ahmed, R. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 2007, 27, 670–684. [Google Scholar] [CrossRef]
- Shin, H.; Blackburn, S.D.; Blattman, J.N.; Wherry, E.J. Viral antigen and extensive division maintain virus-specific CD8 T cells during chronic infection. J. Exp. Med. 2007, 204, 941–949. [Google Scholar] [CrossRef]
- Moro-García, M.A.; Alonso-Arias, R.; López-Larrea, C. Molecular mechanisms involved in the aging of the T-cell immune response. Curr. Genom. 2012, 13, 589–602. [Google Scholar] [CrossRef]
- Voehringer, D.; Blaser, C.; Brawand, P.; Raulet, D.H.; Hanke, T.; Pircher, H. Viral infections induce abundant numbers of senescent CD8 T cells. J. Immunol. 2001, 167, 4838–4843. [Google Scholar] [CrossRef]
- Gillespie, G.M.; Wills, M.R.; Appay, V.; O’Callaghan, C.; Murphy, M.; Smith, N.; Sissons, P.; Rowland-Jones, S.; Bell, J.I.; Moss, P.A. Functional heterogeneity and high frequencies of cytomegalovirus-specific CD8+ T lymphocytes in healthy seropositive donors. J. Virol. 2000, 74, 8140–8150. [Google Scholar] [CrossRef]
- Park, H.J.; Park, J.S.; Jeong, Y.H.; Son, J.; Ban, Y.H.; Lee, B.H.; Chen, L.; Chang, J.; Chung, D.H.; Choi, I.; et al. Correction: PD-1 Upregulated on Regulatory T Cells during Chronic Virus Infection Enhances the Suppression of CD8+ T Cell Immune Response via the Interaction with PD-L1 Expressed on CD8+ T Cells. J. Immunol. 2015, 195, 5841–5842. [Google Scholar] [CrossRef]
- Catakovic, K.; Klieser, E.; Neureiter, D.; Geisberger, R. T cell exhaustion: From pathophysiological basics to tumor immunotherapy. Cell Commun. Signal. 2017, 15, 1. [Google Scholar] [CrossRef]
- Strauss, G.; Knape, I.; Melzner, I.; Debatin, K.M. Constitutive caspase activation and impaired death-inducing signaling complex formation in CD95-resistant, long-term activated, antigen-specific T cells. J. Immunol. 2003, 171, 1172–1182. [Google Scholar] [CrossRef]
- Collaboration, A.T.C. Life expectancy of individuals on combination antiretroviral therapy in high-income countries: A collaborative analysis of 14 cohort studies. Lancet 2008, 372, 293–299. [Google Scholar] [CrossRef]
- Trautmann, L.; Janbazian, L.; Chomont, N.; Said, E.A.; Gimmig, S.; Bessette, B.; Boulassel, M.R.; Delwart, E.; Sepulveda, H.; Balderas, R.S.; et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat. Med. 2006, 12, 1198–1202. [Google Scholar] [CrossRef]
- Cao, W.; Mehraj, V.; Kaufmann, D.E.; Li, T.; Routy, J.P. Elevation and persistence of CD8 T-cells in HIV infection: The Achilles heel in the ART era. J. Int. AIDS Soc. 2016, 19, 20697. [Google Scholar] [CrossRef]
- Palmer, L.D.; Weng, N.; Levine, B.L.; June, C.H.; Lane, H.C.; Hodes, R.J. Telomere length, telomerase activity, and replicative potential in HIV infection: Analysis of CD4+ and CD8+ T cells from HIV-discordant monozygotic twins. J. Exp. Med. 1997, 185, 1381–1386. [Google Scholar] [CrossRef]
- Dagarag, M.; Evazyan, T.; Rao, N.; Effros, R.B. Genetic manipulation of telomerase in HIV-specific CD8+ T cells: Enhanced antiviral functions accompany the increased proliferative potential and telomere length stabilization. J. Immunol. 2004, 173, 6303–6311. [Google Scholar] [CrossRef]
- Golden-Mason, L.; Palmer, B.E.; Kassam, N.; Townshend-Bulson, L.; Livingston, S.; McMahon, B.J.; Castelblanco, N.; Kuchroo, V.; Gretch, D.R.; Rosen, H.R. Negative immune regulator Tim-3 is overexpressed on T cells in hepatitis C virus infection and its blockade rescues dysfunctional CD4+ and CD8+ T cells. J. Virol. 2009, 83, 9122–9130. [Google Scholar] [CrossRef] [PubMed]
- Hoare, M.; Shankar, A.; Shah, M.; Rushbrook, S.; Gelson, W.; Davies, S.; Akbar, A.; Alexander, G.J. γ-H2AX + CD8+ T lymphocytes cannot respond to IFN-α, IL-2 or IL-6 in chronic hepatitis C virus infection. J. Hepatol. 2013, 58, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Rha, M.S.; Shin, E.C. Activation or exhaustion of CD8. Cell. Mol. Immunol. 2021, 18, 2325–2333. [Google Scholar] [CrossRef] [PubMed]
- James, S.F.; Traina-Dorge, V.; Deharo, E.; Wellish, M.; Palmer, B.E.; Gilden, D.; Mahalingam, R. T cells increase before zoster and PD-1 expression increases at the time of zoster in immunosuppressed nonhuman primates latently infected with simian varicella virus. J. Neurovirol. 2014, 20, 309–313. [Google Scholar] [CrossRef]
- Allen, S.J.; Mott, K.R.; Zandian, M.; Ghiasi, H. Immunization with different viral antigens alters the pattern of T cell exhaustion and latency in herpes simplex virus type 1-infected mice. J. Virol. 2010, 84, 12315–12324. [Google Scholar] [CrossRef]
- O’Bryan, J.M.; Woda, M.; Co, M.; Mathew, A.; Rothman, A.L. Telomere length dynamics in human memory T cells specific for viruses causing acute or latent infections. Immun. Ageing 2013, 10, 37. [Google Scholar] [CrossRef]
- Gonçalves, D.U.; Proietti, F.A.; Ribas, J.G.; Araújo, M.G.; Pinheiro, S.R.; Guedes, A.C.; Carneiro-Proietti, A.B. Epidemiology, treatment, and prevention of human T-cell leukemia virus type 1-associated diseases. Clin. Microbiol. Rev. 2010, 23, 577–589. [Google Scholar] [CrossRef]
- Kozako, T.; Yoshimitsu, M.; Akimoto, M.; White, Y.; Matsushita, K.; Soeda, S.; Shimeno, H.; Kubota, R.; Izumo, S.; Arima, N. Programmed death-1 (PD-1)/PD-1 ligand pathway-mediated immune responses against human T-lymphotropic virus type 1 (HTLV-1) in HTLV-1-associated myelopathy/tropical spastic paraparesis and carriers with autoimmune disorders. Hum. Immunol. 2011, 72, 1001–1006. [Google Scholar] [CrossRef]
- Ezinne, C.C.; Yoshimitsu, M.; White, Y.; Arima, N. HTLV-1 specific CD8+ T cell function augmented by blockade of 2B4/CD48 interaction in HTLV-1 infection. PLoS ONE 2014, 9, e87631. [Google Scholar] [CrossRef]
- Sabouri, A.H.; Usuku, K.; Hayashi, D.; Izumo, S.; Ohara, Y.; Osame, M.; Saito, M. Impaired function of human T-lymphotropic virus type 1 (HTLV-1)-specific CD8+ T cells in HTLV-1-associated neurologic disease. Blood 2008, 112, 2411–2420. [Google Scholar] [CrossRef]
- Datta, A.; Nicot, C. Telomere attrition induces a DNA double-strand break damage signal that reactivates p53 transcription in HTLV-I leukemic cells. Oncogene 2008, 27, 1135–1141. [Google Scholar] [CrossRef]
- Jones, R.B.; Ndhlovu, L.C.; Barbour, J.D.; Sheth, P.M.; Jha, A.R.; Long, B.R.; Wong, J.C.; Satkunarajah, M.; Schweneker, M.; Chapman, J.M.; et al. Tim-3 expression defines a novel population of dysfunctional T cells with highly elevated frequencies in progressive HIV-1 infection. J. Exp. Med. 2008, 205, 2763–2779. [Google Scholar] [CrossRef]
- Larsen, M.; Sauce, D.; Deback, C.; Arnaud, L.; Mathian, A.; Miyara, M.; Boutolleau, D.; Parizot, C.; Dorgham, K.; Papagno, L.; et al. Exhausted cytotoxic control of Epstein-Barr virus in human lupus. PLoS Pathog. 2011, 7, e1002328. [Google Scholar] [CrossRef]
- Pender, M.P.; Csurhes, P.A.; Burrows, J.M.; Burrows, S.R. Defective T-cell control of Epstein-Barr virus infection in multiple sclerosis. Clin. Transl. Immunol. 2017, 6, e126. [Google Scholar] [CrossRef]
- Annels, N.E.; Callan, M.F.; Tan, L.; Rickinson, A.B. Changing patterns of dominant TCR usage with maturation of an EBV-specific cytotoxic T cell response. J. Immunol. 2000, 165, 4831–4841. [Google Scholar] [CrossRef]
- van Baarle, D.; Hovenkamp, E.; Callan, M.F.; Wolthers, K.C.; Kostense, S.; Tan, L.C.; Niesters, H.G.; Osterhaus, A.D.; McMichael, A.J.; van Oers, M.H.; et al. Dysfunctional Epstein-Barr virus (EBV)-specific CD8+ T lymphocytes and increased EBV load in HIV-1 infected individuals progressing to AIDS-related non-Hodgkin lymphoma. Blood 2001, 98, 146–155. [Google Scholar] [CrossRef]
- Plunkett, F.J.; Franzese, O.; Belaramani, L.L.; Fletcher, J.M.; Gilmour, K.C.; Sharifi, R.; Khan, N.; Hislop, A.D.; Cara, A.; Salmon, M.; et al. The impact of telomere erosion on memory CD8+ T cells in patients with X-linked lymphoproliferative syndrome. Mech. Ageing Dev. 2005, 126, 855–865. [Google Scholar] [CrossRef]
- van de Berg, P.J.; van Stijn, A.; Ten Berge, I.J.; van Lier, R.A. A fingerprint left by cytomegalovirus infection in the human T cell compartment. J. Clin. Virol. 2008, 41, 213–217. [Google Scholar] [CrossRef]
- Pourgheysari, B.; Khan, N.; Best, D.; Bruton, R.; Nayak, L.; Moss, P.A. The cytomegalovirus-specific CD4+ T-cell response expands with age and markedly alters the CD4+ T-cell repertoire. J. Virol. 2007, 81, 7759–7765. [Google Scholar] [CrossRef]
- van Leeuwen, E.M.; van Buul, J.D.; Remmerswaal, E.B.; Hordijk, P.L.; ten Berge, I.J.; van Lier, R.A. Functional re-expression of CCR7 on CMV-specific CD8+ T cells upon antigenic stimulation. Int. Immunol. 2005, 17, 713–719. [Google Scholar] [CrossRef]
- Cicin-Sain, L.; Brien, J.D.; Uhrlaub, J.L.; Drabig, A.; Marandu, T.F.; Nikolich-Zugich, J. Cytomegalovirus infection impairs immune responses and accentuates T-cell pool changes observed in mice with aging. PLoS Pathog. 2012, 8, e1002849. [Google Scholar] [CrossRef]
- Chiu, Y.L.; Lin, C.H.; Sung, B.Y.; Chuang, Y.F.; Schneck, J.P.; Kern, F.; Pawelec, G.; Wang, G.C. Cytotoxic polyfunctionality maturation of cytomegalovirus-pp65-specific CD4 + and CD8 + T-cell responses in older adults positively correlates with response size. Sci. Rep. 2016, 6, 19227. [Google Scholar] [CrossRef]
- Unemori, P.; Leslie, K.S.; Hunt, P.W.; Sinclair, E.; Epling, L.; Mitsuyasu, R.; Effros, R.B.; Dock, J.; Dollard, S.G.; Deeks, S.G.; et al. Immunosenescence is associated with presence of Kaposi’s sarcoma in antiretroviral treated HIV infection. AIDS 2013, 27, 1735–1742. [Google Scholar] [CrossRef]
- Beldi-Ferchiou, A.; Lambert, M.; Dogniaux, S.; Vély, F.; Vivier, E.; Olive, D.; Dupuy, S.; Levasseur, F.; Zucman, D.; Lebbé, C.; et al. PD-1 mediates functional exhaustion of activated NK cells in patients with Kaposi sarcoma. Oncotarget 2016, 7, 72961–72977. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Song, Y.; Lu, Y.L.; Sun, J.Z.; Wang, H.W. Increased expression of programmed death (PD)-1 and its ligand PD-L1 correlates with impaired cell-mediated immunity in high-risk human papillomavirus-related cervical intraepithelial neoplasia. Immunology 2013, 139, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Woltman, A.M.; Janssen, H.L.; Boonstra, A. Modulation of dendritic cell function by persistent viruses. J. Leukoc. Biol. 2009, 85, 205–214. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mathiasen, S.L.; Gall-Mas, L.; Pateras, I.S.; Theodorou, S.D.P.; Namini, M.R.J.; Hansen, M.B.; Martin, O.C.B.; Vadivel, C.K.; Ntostoglou, K.; Butter, D.; et al. Bacterial genotoxins induce T cell senescence. Cell Rep. 2021, 35, 109220. [Google Scholar] [CrossRef]
- Ibler, A.E.M.; ElGhazaly, M.; Naylor, K.L.; Bulgakova, N.A.; F El-Khamisy, S.; Humphreys, D. Typhoid toxin exhausts the RPA response to DNA replication stress driving senescence and Salmonella infection. Nat. Commun. 2019, 10, 4040. [Google Scholar] [CrossRef]
- Sakai, S.; Mayer-Barber, K.D.; Barber, D.L. Defining features of protective CD4 T cell responses to Mycobacterium tuberculosis. Curr. Opin. Immunol. 2014, 29, 137–142. [Google Scholar] [CrossRef]
- McNab, F.W.; Berry, M.P.; Graham, C.M.; Bloch, S.A.; Oni, T.; Wilkinson, K.A.; Wilkinson, R.J.; Kon, O.M.; Banchereau, J.; Chaussabel, D.; et al. Programmed death ligand 1 is over-expressed by neutrophils in the blood of patients with active tuberculosis. Eur. J. Immunol. 2011, 41, 1941–1947. [Google Scholar] [CrossRef]
- Jurado, J.O.; Alvarez, I.B.; Pasquinelli, V.; Martínez, G.J.; Quiroga, M.F.; Abbate, E.; Musella, R.M.; Chuluyan, H.E.; García, V.E. Programmed death (PD)-1:PD-ligand 1/PD-ligand 2 pathway inhibits T cell effector functions during human tuberculosis. J. Immunol. 2008, 181, 116–125. [Google Scholar] [CrossRef]
- Singh, A.; Mohan, A.; Dey, A.B.; Mitra, D.K. Inhibiting the programmed death 1 pathway rescues Mycobacterium tuberculosis-specific interferon γ-producing T cells from apoptosis in patients with pulmonary tuberculosis. J. Infect. Dis. 2013, 208, 603–615. [Google Scholar] [CrossRef]
- Barber, D.L.; Mayer-Barber, K.D.; Feng, C.G.; Sharpe, A.H.; Sher, A. CD4 T cells promote rather than control tuberculosis in the absence of PD-1-mediated inhibition. J. Immunol. 2011, 186, 1598–1607. [Google Scholar] [CrossRef]
- Shankar, E.M.; Velu, V.; Kamarulzaman, A.; Larsson, M. Mechanistic insights on immunosenescence and chronic immune activation in HIV-tuberculosis co-infection. World J. Virol. 2015, 4, 17–24. [Google Scholar] [CrossRef]
- Covre, L.P.; Martins, R.F.; Devine, O.P.; Chambers, E.S.; Vukmanovic-Stejic, M.; Silva, J.A.; Dietze, R.; Rodrigues, R.R.; de Matos Guedes, H.L.; Falqueto, A.; et al. Circulating Senescent T Cells Are Linked to Systemic Inflammation and Lesion Size During Human Cutaneous Leishmaniasis. Front. Immunol. 2018, 9, 3001. [Google Scholar] [CrossRef]
- Conway, J.; Certo, M.; Lord, J.M.; Mauro, C.; Duggal, N.A. Understanding the role of host metabolites in the induction of immune senescence: Future strategies for keeping the ageing population healthy. Br. J. Pharmacol. 2022, 179, 1808–1824. [Google Scholar] [CrossRef]
- Lee, K.A.; Robbins, P.D.; Camell, C.D. Intersection of immunometabolism and immunosenescence during aging. Curr. Opin. Pharmacol. 2021, 57, 107–116. [Google Scholar] [CrossRef]
- Wiley, C.D.; Campisi, J. The metabolic roots of senescence: Mechanisms and opportunities for intervention. Nat. Metab. 2021, 3, 1290–1301. [Google Scholar] [CrossRef]
- Jeng, M.Y.; Hull, P.A.; Fei, M.; Kwon, H.S.; Tsou, C.L.; Kasler, H.; Ng, C.P.; Gordon, D.E.; Johnson, J.; Krogan, N.; et al. Metabolic reprogramming of human CD8. J. Exp. Med. 2018, 215, 51–62. [Google Scholar] [CrossRef]
- Li, L.; Liu, X.; Sanders, K.L.; Edwards, J.L.; Ye, J.; Si, F.; Gao, A.; Huang, L.; Hsueh, E.C.; Ford, D.A.; et al. TLR8-Mediated Metabolic Control of Human Treg Function: A Mechanistic Target for Cancer Immunotherapy. Cell Metab. 2019, 29, 103–123. [Google Scholar] [CrossRef]
- Kurupati, R.K.; Haut, L.H.; Schmader, K.E.; Ertl, H.C. Age-related changes in B cell metabolism. Aging 2019, 11, 4367–4381. [Google Scholar] [CrossRef]
- Frasca, D.; Diaz, A.; Romero, M.; Thaller, S.; Blomberg, B.B. Metabolic requirements of human pro-inflammatory B cells in aging and obesity. PLoS ONE 2019, 14, e0219545. [Google Scholar] [CrossRef]
- Chougnet, C.A.; Thacker, R.I.; Shehata, H.M.; Hennies, C.M.; Lehn, M.A.; Lages, C.S.; Janssen, E.M. Loss of Phagocytic and Antigen Cross-Presenting Capacity in Aging Dendritic Cells Is Associated with Mitochondrial Dysfunction. J. Immunol. 2015, 195, 2624–2632. [Google Scholar] [CrossRef]
- Ma, T.; Patel, H.; Babapoor-Farrokhran, S.; Franklin, R.; Semenza, G.L.; Sodhi, A.; Montaner, S. KSHV induces aerobic glycolysis and angiogenesis through HIF-1-dependent upregulation of pyruvate kinase 2 in Kaposi’s sarcoma. Angiogenesis 2015, 18, 477–488. [Google Scholar] [CrossRef]
- Palmer, C.S.; Duette, G.A.; Wagner, M.C.E.; Henstridge, D.C.; Saleh, S.; Pereira, C.; Zhou, J.; Simar, D.; Lewin, S.R.; Ostrowski, M.; et al. Metabolically active CD4+ T cells expressing Glut1 and OX40 preferentially harbor HIV during in vitro infection. FEBS Lett. 2017, 591, 3319–3332. [Google Scholar] [CrossRef]
- Palmer, C.S.; Anzinger, J.J.; Zhou, J.; Gouillou, M.; Landay, A.; Jaworowski, A.; McCune, J.M.; Crowe, S.M. Glucose transporter 1-expressing proinflammatory monocytes are elevated in combination antiretroviral therapy-treated and untreated HIV+ subjects. J. Immunol. 2014, 193, 5595–5603. [Google Scholar] [CrossRef]
- Wakisaka, N.; Kondo, S.; Yoshizaki, T.; Murono, S.; Furukawa, M.; Pagano, J.S. Epstein-Barr virus latent membrane protein 1 induces synthesis of hypoxia-inducible factor 1 alpha. Mol. Cell. Biol. 2004, 24, 5223–5234. [Google Scholar] [CrossRef]
- Teng, C.F.; Hsieh, W.C.; Wu, H.C.; Lin, Y.J.; Tsai, H.W.; Huang, W.; Su, I.J. Hepatitis B Virus Pre-S2 Mutant Induces Aerobic Glycolysis through Mammalian Target of Rapamycin Signal Cascade. PLoS ONE 2015, 10, e0122373. [Google Scholar] [CrossRef]
- Russell, S.L.; Lamprecht, D.A.; Mandizvo, T.; Jones, T.T.; Naidoo, V.; Addicott, K.W.; Moodley, C.; Ngcobo, B.; Crossman, D.K.; Wells, G.; et al. Compromised Metabolic Reprogramming Is an Early Indicator of CD8. Cell Rep. 2019, 29, 3564–3579. [Google Scholar] [CrossRef]
- Lagoumtzi, S.M.; Chondrogianni, N. Senolytics and senomorphics: Natural and synthetic therapeutics in the treatment of aging and chronic diseases. Free Radic. Biol. Med. 2021, 171, 169–190. [Google Scholar] [CrossRef]
- Tchkonia, T.; Zhu, Y.; van Deursen, J.; Campisi, J.; Kirkland, J.L. Cellular senescence and the senescent secretory phenotype: Therapeutic opportunities. J. Clin. Investig. 2013, 123, 966–972. [Google Scholar] [CrossRef] [PubMed]
- Ovadya, Y.; Krizhanovsky, V. Strategies targeting cellular senescence. J. Clin. Investig. 2018, 128, 1247–1254. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.M.; Balan, V.; Gleiberman, A.S.; Strom, E.; Krasnov, P.; Virtuoso, L.P.; Rydkina, E.; Vujcic, S.; Balan, K.; Gitlin, I.; et al. Aging of mice is associated with p16(Ink4a)- and β-galactosidase-positive macrophage accumulation that can be induced in young mice by senescent cells. Aging 2016, 8, 1294–1315. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Zhou, H.; Zhu, Y.; Sun, Q.; Ji, Y.; Xue, A.; Wang, Y.; Chen, W.; Yu, X.; Wang, L.; et al. Elimination of senescent cells by β-galactosidase-targeted prodrug attenuates inflammation and restores physical function in aged mice. Cell Res. 2020, 30, 574–589. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Rovira, M.; Galiana, I.; Giménez, C.; Lozano-Torres, B.; Paez-Ribes, M.; Llanos, S.; Chaib, S.; Muñoz-Martín, M.; Ucero, A.C.; et al. A versatile drug delivery system targeting senescent cells. EMBO Mol. Med. 2018, 10, e9355. [Google Scholar] [CrossRef]
- Yoshida, S.; Nakagami, H.; Hayashi, H.; Ikeda, Y.; Sun, J.; Tenma, A.; Tomioka, H.; Kawano, T.; Shimamura, M.; Morishita, R.; et al. The CD153 vaccine is a senotherapeutic option for preventing the accumulation of senescent T cells in mice. Nat. Commun. 2020, 11, 2482. [Google Scholar] [CrossRef]
- Sallin, M.A.; Kauffman, K.D.; Riou, C.; Du Bruyn, E.; Foreman, T.W.; Sakai, S.; Hoft, S.G.; Myers, T.G.; Gardina, P.J.; Sher, A.; et al. Host resistance to pulmonary Mycobacterium tuberculosis infection requires CD153 expression. Nat. Microbiol. 2018, 3, 1198–1205. [Google Scholar] [CrossRef]
- Liu, Y.; Sanoff, H.K.; Cho, H.; Burd, C.E.; Torrice, C.; Ibrahim, J.G.; Thomas, N.E.; Sharpless, N.E. Expression of p16(INK4a) in peripheral blood T-cells is a biomarker of human aging. Aging Cell 2009, 8, 439–448. [Google Scholar] [CrossRef]
- Liu, Y.; Johnson, S.M.; Fedoriw, Y.; Rogers, A.B.; Yuan, H.; Krishnamurthy, J.; Sharpless, N.E. Expression of p16(INK4a) prevents cancer and promotes aging in lymphocytes. Blood 2011, 117, 3257–3267. [Google Scholar] [CrossRef]
- Lucas, C.L.; Kuehn, H.S.; Zhao, F.; Niemela, J.E.; Deenick, E.K.; Palendira, U.; Avery, D.T.; Moens, L.; Cannons, J.L.; Biancalana, M.; et al. Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110δ result in T cell senescence and human immunodeficiency. Nat. Immunol. 2014, 15, 88–97. [Google Scholar] [CrossRef]
- Paez-Ribes, M.; González-Gualda, E.; Doherty, G.J.; Muñoz-Espín, D. Targeting senescent cells in translational medicine. EMBO Mol. Med. 2019, 11, e10234. [Google Scholar] [CrossRef]
- Rao, V.K.; Webster, S.; Dalm, V.A.S.H.; Šedivá, A.; van Hagen, P.M.; Holland, S.; Rosenzweig, S.D.; Christ, A.D.; Sloth, B.; Cabanski, M.; et al. Effective “activated PI3Kδ syndrome”-targeted therapy with the PI3Kδ inhibitor leniolisib. Blood 2017, 130, 2307–2316. [Google Scholar] [CrossRef]
- Dörr, J.R.; Yu, Y.; Milanovic, M.; Beuster, G.; Zasada, C.; Däbritz, J.H.; Lisec, J.; Lenze, D.; Gerhardt, A.; Schleicher, K.; et al. Synthetic lethal metabolic targeting of cellular senescence in cancer therapy. Nature 2013, 501, 421–425. [Google Scholar] [CrossRef]
- Heilbronn, L.K.; Ravussin, E. Calorie restriction and aging: Review of the literature and implications for studies in humans. Am. J. Clin. Nutr. 2003, 78, 361–369. [Google Scholar] [CrossRef]
- Cantó, C.; Auwerx, J. Calorie restriction: Is AMPK a key sensor and effector? Physiology 2011, 26, 214–224. [Google Scholar] [CrossRef]
- Barbosa, M.C.; Grosso, R.A.; Fader, C.M. Hallmarks of Aging: An Autophagic Perspective. Front. Endocrinol. 2018, 9, 790. [Google Scholar] [CrossRef]
- Messaoudi, I.; Warner, J.; Fischer, M.; Park, B.; Hill, B.; Mattison, J.; Lane, M.A.; Roth, G.S.; Ingram, D.K.; Picker, L.J.; et al. Delay of T cell senescence by caloric restriction in aged long-lived nonhuman primates. Proc. Natl. Acad. Sci. USA 2006, 103, 19448–19453. [Google Scholar] [CrossRef]
- White, M.J.; Beaver, C.M.; Goodier, M.R.; Bottomley, C.; Nielsen, C.M.; Wolf, A.F.; Boldrin, L.; Whitmore, C.; Morgan, J.; Pearce, D.J.; et al. Calorie Restriction Attenuates Terminal Differentiation of Immune Cells. Front. Immunol. 2016, 7, 667. [Google Scholar] [CrossRef]
- Smallwood, H.S.; Duan, S.; Morfouace, M.; Rezinciuc, S.; Shulkin, B.L.; Shelat, A.; Zink, E.E.; Milasta, S.; Bajracharya, R.; Oluwaseum, A.J.; et al. Targeting Metabolic Reprogramming by Influenza Infection for Therapeutic Intervention. Cell Rep. 2017, 19, 1640–1653. [Google Scholar] [CrossRef]
- Mannick, J.B.; Del Giudice, G.; Lattanzi, M.; Valiante, N.M.; Praestgaard, J.; Huang, B.; Lonetto, M.A.; Maecker, H.T.; Kovarik, J.; Carson, S.; et al. mTOR inhibition improves immune function in the elderly. Sci. Transl. Med. 2014, 6, 268ra179. [Google Scholar] [CrossRef]
- Mannick, J.B.; Morris, M.; Hockey, H.P.; Roma, G.; Beibel, M.; Kulmatycki, K.; Watkins, M.; Shavlakadze, T.; Zhou, W.; Quinn, D.; et al. TORC1 inhibition enhances immune function and reduces infections in the elderly. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef]
- Mannick, J.B.; Teo, G.; Bernardo, P.; Quinn, D.; Russell, K.; Klickstein, L.; Marshall, W.; Shergill, S. Targeting the biology of ageing with mTOR inhibitors to improve immune function in older adults: Phase 2b and phase 3 randomised trials. Lancet Healthy Longev 2021, 2, e250–e262. [Google Scholar] [CrossRef]
- Patocka, J.; Kuca, K.; Oleksak, P.; Nepovimova, E.; Valis, M.; Novotny, M.; Klimova, B. Rapamycin: Drug Repurposing in SARS-CoV-2 Infection. Pharmaceuticals 2021, 14, 217. [Google Scholar] [CrossRef]
- Bischof, E.; Siow, R.C.; Zhavoronkov, A.; Kaeberlein, M. The potential of rapalogs to enhance resilience against SARS-CoV-2 infection and reduce the severity of COVID-19. Lancet Healthy Longev. 2021, 2, e105–e111. [Google Scholar] [CrossRef]
- Omarjee, L.; Janin, A.; Perrot, F.; Laviolle, B.; Meilhac, O.; Mahe, G. Targeting T-cell senescence and cytokine storm with rapamycin to prevent severe progression in COVID-19. Clin. Immunol. 2020, 216, 108464. [Google Scholar] [CrossRef]
- Bharath, L.P.; Agrawal, M.; McCambridge, G.; Nicholas, D.A.; Hasturk, H.; Liu, J.; Jiang, K.; Liu, R.; Guo, Z.; Deeney, J.; et al. Metformin Enhances Autophagy and Normalizes Mitochondrial Function to Alleviate Aging-Associated Inflammation. Cell Metab. 2020, 32, 44–55. [Google Scholar] [CrossRef]
- Martin-Montalvo, A.; Mercken, E.M.; Mitchell, S.J.; Palacios, H.H.; Mote, P.L.; Scheibye-Knudsen, M.; Gomes, A.P.; Ward, T.M.; Minor, R.K.; Blouin, M.J.; et al. Metformin improves healthspan and lifespan in mice. Nat. Commun. 2013, 4, 2192. [Google Scholar] [CrossRef] [PubMed]
- Strong, R.; Miller, R.A.; Antebi, A.; Astle, C.M.; Bogue, M.; Denzel, M.S.; Fernandez, E.; Flurkey, K.; Hamilton, K.L.; Lamming, D.W.; et al. Longer lifespan in male mice treated with a weakly estrogenic agonist, an antioxidant, an α-glucosidase inhibitor or a Nrf2-inducer. Aging Cell 2016, 15, 872–884. [Google Scholar] [CrossRef]
- Sorrenti, V.; Benedetti, F.; Buriani, A.; Fortinguerra, S.; Caudullo, G.; Davinelli, S.; Zella, D.; Scapagnini, G. Immunomodulatory and Antiaging Mechanisms of Resveratrol, Rapamycin, and Metformin: Focus on mTOR and AMPK Signaling Networks. Pharmaceuticals 2022, 15, 912. [Google Scholar] [CrossRef] [PubMed]
- Borriello, A.; Bencivenga, D.; Caldarelli, I.; Tramontano, A.; Borgia, A.; Zappia, V.; Della Ragione, F. Resveratrol: From basic studies to bedside. Adv. Nutr. Cancer 2014, 159, 167–184. [Google Scholar] [CrossRef]
- Zhang, H.; Alsaleh, G.; Feltham, J.; Sun, Y.; Napolitano, G.; Riffelmacher, T.; Charles, P.; Frau, L.; Hublitz, P.; Yu, Z.; et al. Polyamines Control eIF5A Hypusination, TFEB Translation, and Autophagy to Reverse B Cell Senescence. Mol. Cell 2019, 76, 110–125. [Google Scholar] [CrossRef]
- Allan, R.S.; Zueva, E.; Cammas, F.; Schreiber, H.A.; Masson, V.; Belz, G.T.; Roche, D.; Maison, C.; Quivy, J.P.; Almouzni, G.; et al. An epigenetic silencing pathway controlling T helper 2 cell lineage commitment. Nature 2012, 487, 249–253. [Google Scholar] [CrossRef]
- Pace, L.; Goudot, C.; Zueva, E.; Gueguen, P.; Burgdorf, N.; Waterfall, J.J.; Quivy, J.P.; Almouzni, G.; Amigorena, S. The epigenetic control of stemness in CD8. Science 2018, 359, 177–186. [Google Scholar] [CrossRef]
- Schönheit, J.; Leutz, A.; Rosenbauer, F. Chromatin dynamics during differentiation of myeloid cells. J. Mol. Biol. 2015, 427, 670–687. [Google Scholar] [CrossRef]
- Sciumè, G.; Shih, H.Y.; Mikami, Y.; O’Shea, J.J. Epigenomic Views of Innate Lymphoid Cells. Front. Immunol. 2017, 8, 1579. [Google Scholar] [CrossRef]
- Keenan, C.R.; Allan, R.S. Epigenomic drivers of immune dysfunction in aging. Aging Cell 2019, 18, e12878. [Google Scholar] [CrossRef]
- Ganesan, A.; Arimondo, P.B.; Rots, M.G.; Jeronimo, C.; Berdasco, M. The timeline of epigenetic drug discovery: From reality to dreams. Clin. Epigenet. 2019, 11, 174. [Google Scholar] [CrossRef]
- Yates, K.B.; Tonnerre, P.; Martin, G.E.; Gerdemann, U.; Al Abosy, R.; Comstock, D.E.; Weiss, S.A.; Wolski, D.; Tully, D.C.; Chung, R.T.; et al. Epigenetic scars of CD8. Nat. Immunol. 2021, 22, 1020–1029. [Google Scholar] [CrossRef]
- Tonnerre, P.; Wolski, D.; Subudhi, S.; Aljabban, J.; Hoogeveen, R.C.; Damasio, M.; Drescher, H.K.; Bartsch, L.M.; Tully, D.C.; Sen, D.R.; et al. Differentiation of exhausted CD8. Nat. Immunol. 2021, 22, 1030–1041. [Google Scholar] [CrossRef]
- Kanfi, Y.; Naiman, S.; Amir, G.; Peshti, V.; Zinman, G.; Nahum, L.; Bar-Joseph, Z.; Cohen, H.Y. The sirtuin SIRT6 regulates lifespan in male mice. Nature 2012, 483, 218–221. [Google Scholar] [CrossRef]
- Serrano, M.; Barzilai, N. Targeting senescence. Nat. Med. 2018, 24, 1092–1094. [Google Scholar] [CrossRef]
- Bellon, M.; Nicot, C. Telomere Dynamics in Immune Senescence and Exhaustion Triggered by Chronic Viral Infection. Viruses 2017, 9, 289. [Google Scholar] [CrossRef]
- Dyavar Shetty, R.; Velu, V.; Titanji, K.; Bosinger, S.E.; Freeman, G.J.; Silvestri, G.; Amara, R.R. PD-1 blockade during chronic SIV infection reduces hyperimmune activation and microbial translocation in rhesus macaques. J. Clin. Investig. 2012, 122, 1712–1716. [Google Scholar] [CrossRef]
- Palmer, C.S.; Henstridge, D.C.; Yu, D.; Singh, A.; Balderson, B.; Duette, G.; Cherry, C.L.; Anzinger, J.J.; Ostrowski, M.; Crowe, S.M. Emerging Role and Characterization of Immunometabolism: Relevance to HIV Pathogenesis, Serious Non-AIDS Events, and a Cure. J. Immunol. 2016, 196, 4437–4444. [Google Scholar] [CrossRef]
- Gay, C.L.; Bosch, R.J.; Ritz, J.; Hataye, J.M.; Aga, E.; Tressler, R.L.; Mason, S.W.; Hwang, C.K.; Grasela, D.M.; Ray, N.; et al. Clinical Trial of the Anti-PD-L1 Antibody BMS-936559 in HIV-1 Infected Participants on Suppressive Antiretroviral Therapy. J. Infect. Dis. 2017, 215, 1725–1733. [Google Scholar] [CrossRef]
- Tzeng, H.T.; Tsai, H.F.; Liao, H.J.; Lin, Y.J.; Chen, L.; Chen, P.J.; Hsu, P.N. PD-1 blockage reverses immune dysfunction and hepatitis B viral persistence in a mouse animal model. PLoS ONE 2012, 7, e39179. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, D.; Lalezari, J.; Lawitz, E.; DiMicco, M.; Ghalib, R.; Reddy, K.R.; Chang, K.M.; Sulkowski, M.; Marro, S.O.; Anderson, J.; et al. A randomized, double-blind, placebo-controlled assessment of BMS-936558, a fully human monoclonal antibody to programmed death-1 (PD-1), in patients with chronic hepatitis C virus infection. PLoS ONE 2013, 8, e63818. [Google Scholar] [CrossRef]
- Kaufmann, D.E.; Walker, B.D. PD-1 and CTLA-4 inhibitory cosignaling pathways in HIV infection and the potential for therapeutic intervention. J. Immunol. 2009, 182, 5891–5897. [Google Scholar] [CrossRef] [PubMed]
- Cecchinato, V.; Tryniszewska, E.; Ma, Z.M.; Vaccari, M.; Boasso, A.; Tsai, W.P.; Petrovas, C.; Fuchs, D.; Heraud, J.M.; Venzon, D.; et al. Immune activation driven by CTLA-4 blockade augments viral replication at mucosal sites in simian immunodeficiency virus infection. J. Immunol. 2008, 180, 5439–5447. [Google Scholar] [CrossRef] [PubMed]
- Nakamoto, N.; Kaplan, D.E.; Coleclough, J.; Li, Y.; Valiga, M.E.; Kaminski, M.; Shaked, A.; Olthoff, K.; Gostick, E.; Price, D.A.; et al. Functional restoration of HCV-specific CD8 T cells by PD-1 blockade is defined by PD-1 expression and compartmentalization. Gastroenterology 2008, 134, 1927–1937. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.D.; Xu, X.; Jones, R.; Delecluse, H.J.; Zumwalde, N.A.; Sharma, A.; Gumperz, J.E.; Kenney, S.C. PD-1/CTLA-4 Blockade Inhibits Epstein-Barr Virus-Induced Lymphoma Growth in a Cord Blood Humanized-Mouse Model. PLoS Pathog. 2016, 12, e1005642. [Google Scholar] [CrossRef]
- Tinoco, R.; Carrette, F.; Barraza, M.L.; Otero, D.C.; Magaña, J.; Bosenberg, M.W.; Swain, S.L.; Bradley, L.M. PSGL-1 Is an Immune Checkpoint Regulator that Promotes T Cell Exhaustion. Immunity 2016, 44, 1190–1203. [Google Scholar] [CrossRef]
- Cook, K.D.; Whitmire, J.K. LAG-3 Confers a Competitive Disadvantage upon Antiviral CD8+ T Cell Responses. J. Immunol. 2016, 197, 119–127. [Google Scholar] [CrossRef]
- Blackburn, S.D.; Shin, H.; Haining, W.N.; Zou, T.; Workman, C.J.; Polley, A.; Betts, M.R.; Freeman, G.J.; Vignali, D.A.; Wherry, E.J. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat. Immunol. 2009, 10, 29–37. [Google Scholar] [CrossRef]
- Ye, B.; Li, X.; Dong, Y.; Wang, Y.; Tian, L.; Lin, S.; Liu, X.; Kong, H.; Chen, Y. Increasing LAG-3 expression suppresses T-cell function in chronic hepatitis B: A balance between immunity strength and liver injury extent. Medicine 2017, 96, e5275. [Google Scholar] [CrossRef]
- Sada-Ovalle, I.; Chávez-Galán, L.; Torre-Bouscoulet, L.; Nava-Gamiño, L.; Barrera, L.; Jayaraman, P.; Torres-Rojas, M.; Salazar-Lezama, M.A.; Behar, S.M. The Tim3-galectin 9 pathway induces antibacterial activity in human macrophages infected with Mycobacterium tuberculosis. J. Immunol. 2012, 189, 5896–5902. [Google Scholar] [CrossRef]
- Blackburn, S.D.; Wherry, E.J. IL-10, T cell exhaustion and viral persistence. Trends Microbiol. 2007, 15, 143–146. [Google Scholar] [CrossRef]
- Brockman, M.A.; Kwon, D.S.; Tighe, D.P.; Pavlik, D.F.; Rosato, P.C.; Sela, J.; Porichis, F.; Le Gall, S.; Waring, M.T.; Moss, K.; et al. IL-10 is up-regulated in multiple cell types during viremic HIV infection and reversibly inhibits virus-specific T cells. Blood 2009, 114, 346–356. [Google Scholar] [CrossRef]
- Porichis, F.; Hart, M.G.; Zupkosky, J.; Barblu, L.; Kwon, D.S.; McMullen, A.; Brennan, T.; Ahmed, R.; Freeman, G.J.; Kavanagh, D.G.; et al. Differential impact of PD-1 and/or interleukin-10 blockade on HIV-1-specific CD4 T cell and antigen-presenting cell functions. J. Virol. 2014, 88, 2508–2518. [Google Scholar] [CrossRef]
- Brooks, D.G.; Ha, S.J.; Elsaesser, H.; Sharpe, A.H.; Freeman, G.J.; Oldstone, M.B. IL-10 and PD-L1 operate through distinct pathways to suppress T-cell activity during persistent viral infection. Proc. Natl. Acad. Sci. USA 2008, 105, 20428–20433. [Google Scholar] [CrossRef]
- Grint, D.; Tedaldi, E.; Peters, L.; Mocroft, A.; Edlin, B.; Gallien, S.; Klinker, H.; Boesecke, C.; Kokordelis, P.; Rockstroh, J.K. Hepatitis C virus (HCV) RNA profiles among chronic HIV/HCV-coinfected individuals in ESPRIT; spontaneous HCV RNA clearance observed in nine individuals. HIV Med. 2017, 18, 430–434. [Google Scholar] [CrossRef]
- Zhen, A.; Rezek, V.; Youn, C.; Lam, B.; Chang, N.; Rick, J.; Carrillo, M.; Martin, H.; Kasparian, S.; Syed, P.; et al. Targeting type I interferon-mediated activation restores immune function in chronic HIV infection. J. Clin. Investig. 2017, 127, 260–268. [Google Scholar] [CrossRef]
- Bengsch, B.; Johnson, A.L.; Kurachi, M.; Odorizzi, P.M.; Pauken, K.E.; Attanasio, J.; Stelekati, E.; McLane, L.M.; Paley, M.A.; Delgoffe, G.M.; et al. Bioenergetic Insufficiencies Due to Metabolic Alterations Regulated by the Inhibitory Receptor PD-1 Are an Early Driver of CD8+ T Cell Exhaustion. Immunity 2016, 45, 358–373. [Google Scholar] [CrossRef]
- Schurich, A.; Pallett, L.J.; Jajbhay, D.; Wijngaarden, J.; Otano, I.; Gill, U.S.; Hansi, N.; Kennedy, P.T.; Nastouli, E.; Gilson, R.; et al. Distinct Metabolic Requirements of Exhausted and Functional Virus-Specific CD8 T Cells in the Same Host. Cell Rep. 2016, 16, 1243–1252. [Google Scholar] [CrossRef]
- Nehme, J.; Borghesan, M.; Mackedenski, S.; Bird, T.G.; Demaria, M. Cellular senescence as a potential mediator of COVID-19 severity in the elderly. Aging Cell 2020, 19, e13237. [Google Scholar] [CrossRef]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef]
- Kakkola, L.; Denisova, O.V.; Tynell, J.; Viiliäinen, J.; Ysenbaert, T.; Matos, R.C.; Nagaraj, A.; Ohman, T.; Kuivanen, S.; Paavilainen, H.; et al. Anticancer compound ABT-263 accelerates apoptosis in virus-infected cells and imbalances cytokine production and lowers survival rates of infected mice. Cell Death Dis. 2013, 4, e742. [Google Scholar] [CrossRef]
- Singh, A.K.; McGuirk, J.P. CAR T cells: Continuation in a revolution of immunotherapy. Lancet Oncol. 2020, 21, e168–e178. [Google Scholar] [CrossRef]
- Ellebrecht, C.T.; Bhoj, V.G.; Nace, A.; Choi, E.J.; Mao, X.; Cho, M.J.; Di Zenzo, G.; Lanzavecchia, A.; Seykora, J.T.; Cotsarelis, G.; et al. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science 2016, 353, 179–184. [Google Scholar] [CrossRef]
- Kansal, R.; Richardson, N.; Neeli, I.; Khawaja, S.; Chamberlain, D.; Ghani, M.; Ghani, Q.U.; Balazs, L.; Beranova-Giorgianni, S.; Giorgianni, F.; et al. Sustained B cell depletion by CD19-targeted CAR T cells is a highly effective treatment for murine lupus. Sci. Transl. Med. 2019, 11, eaav1648. [Google Scholar] [CrossRef] [PubMed]
- Amor, C.; Feucht, J.; Leibold, J.; Ho, Y.J.; Zhu, C.; Alonso-Curbelo, D.; Mansilla-Soto, J.; Boyer, J.A.; Li, X.; Giavridis, T.; et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature 2020, 583, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Schumann, K.; Lin, S.; Boyer, E.; Simeonov, D.R.; Subramaniam, M.; Gate, R.E.; Haliburton, G.E.; Ye, C.J.; Bluestone, J.A.; Doudna, J.A.; et al. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proc. Natl. Acad. Sci. USA 2015, 112, 10437–10442. [Google Scholar] [CrossRef] [PubMed]
- Pereira, B.I.; Devine, O.P.; Vukmanovic-Stejic, M.; Chambers, E.S.; Subramanian, P.; Patel, N.; Virasami, A.; Sebire, N.J.; Kinsler, V.; Valdovinos, A.; et al. Senescent cells evade immune clearance via HLA-E-mediated NK and CD8. Nat. Commun. 2019, 10, 2387. [Google Scholar] [CrossRef]
- Sagiv, A.; Burton, D.G.; Moshayev, Z.; Vadai, E.; Wensveen, F.; Ben-Dor, S.; Golani, O.; Polic, B.; Krizhanovsky, V. NKG2D ligands mediate immunosurveillance of senescent cells. Aging 2016, 8, 328–344. [Google Scholar] [CrossRef]
- Childs, B.G.; Gluscevic, M.; Baker, D.J.; Laberge, R.M.; Marquess, D.; Dananberg, J.; van Deursen, J.M. Senescent cells: An emerging target for diseases of ageing. Nat. Rev. Drug Discov. 2017, 16, 718–735. [Google Scholar] [CrossRef]
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 Years of NF-κB: A Blossoming of Relevance to Human Pathobiology. Cell 2017, 168, 37–57. [Google Scholar] [CrossRef]
- Horwitz, B.H.; Scott, M.L.; Cherry, S.R.; Bronson, R.T.; Baltimore, D. Failure of lymphopoiesis after adoptive transfer of NF-kappaB-deficient fetal liver cells. Immunity 1997, 6, 765–772. [Google Scholar] [CrossRef]
- Park, J.M.; Greten, F.R.; Wong, A.; Westrick, R.J.; Arthur, J.S.; Otsu, K.; Hoffmann, A.; Montminy, M.; Karin, M. Signaling pathways and genes that inhibit pathogen-induced macrophage apoptosis--CREB and NF-kappaB as key regulators. Immunity 2005, 23, 319–329. [Google Scholar] [CrossRef]
- Nagashima, K.; Sasseville, V.G.; Wen, D.; Bielecki, A.; Yang, H.; Simpson, C.; Grant, E.; Hepperle, M.; Harriman, G.; Jaffee, B.; et al. Rapid TNFR1-dependent lymphocyte depletion in vivo with a selective chemical inhibitor of IKKbeta. Blood 2006, 107, 4266–4273. [Google Scholar] [CrossRef]
- Schmidt-Supprian, M.; Tian, J.; Ji, H.; Terhorst, C.; Bhan, A.K.; Grant, E.P.; Pasparakis, M.; Casola, S.; Coyle, A.J.; Rajewsky, K. I kappa B kinase 2 deficiency in T cells leads to defects in priming, B cell help, germinal center reactions, and homeostatic expansion. J. Immunol. 2004, 173, 1612–1619. [Google Scholar] [CrossRef]
- Lavon, I.; Goldberg, I.; Amit, S.; Landsman, L.; Jung, S.; Tsuberi, B.Z.; Barshack, I.; Kopolovic, J.; Galun, E.; Bujard, H.; et al. High susceptibility to bacterial infection, but no liver dysfunction, in mice compromised for hepatocyte NF-kappaB activation. Nat. Med. 2000, 6, 573–577. [Google Scholar] [CrossRef]
- Justice, J.N.; Nambiar, A.M.; Tchkonia, T.; LeBrasseur, N.K.; Pascual, R.; Hashmi, S.K.; Prata, L.; Masternak, M.M.; Kritchevsky, S.B.; Musi, N.; et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine 2019, 40, 554–563. [Google Scholar] [CrossRef]
- Hickson, L.J.; Langhi Prata, L.G.P.; Bobart, S.A.; Evans, T.K.; Giorgadze, N.; Hashmi, S.K.; Herrmann, S.M.; Jensen, M.D.; Jia, Q.; Jordan, K.L.; et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine 2019, 47, 446–456. [Google Scholar] [CrossRef]
- Friedman, A.A.; Letai, A.; Fisher, D.E.; Flaherty, K.T. Precision medicine for cancer with next-generation functional diagnostics. Nat. Rev. Cancer 2015, 15, 747–756. [Google Scholar] [CrossRef]
- Soukas, A.A.; Hao, H.; Wu, L. Metformin as Anti-Aging Therapy: Is It for Everyone? Trends Endocrinol. Metab. 2019, 30, 745–755. [Google Scholar] [CrossRef]


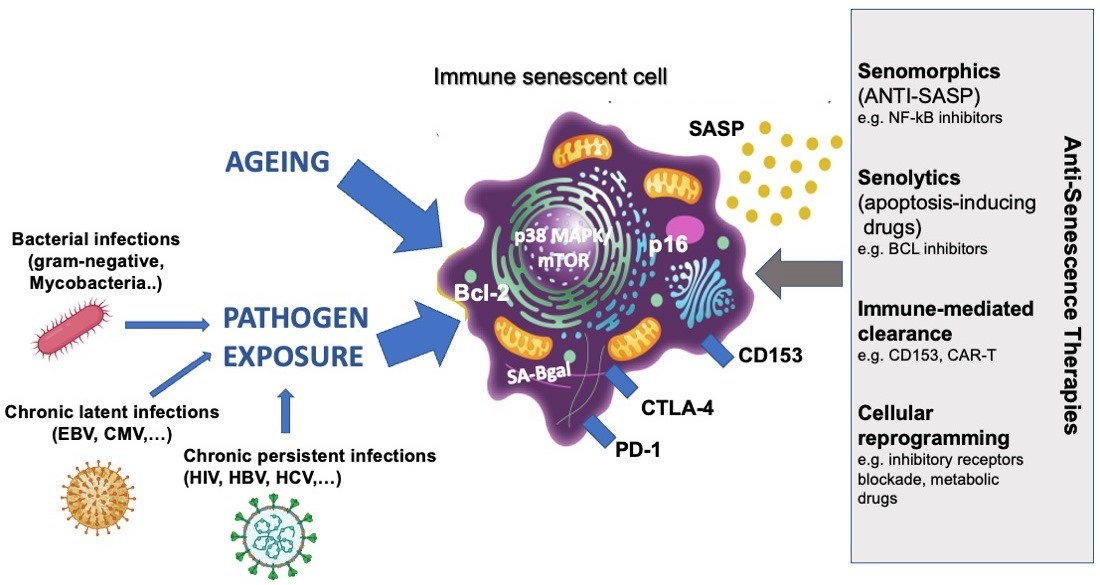{kind=link}
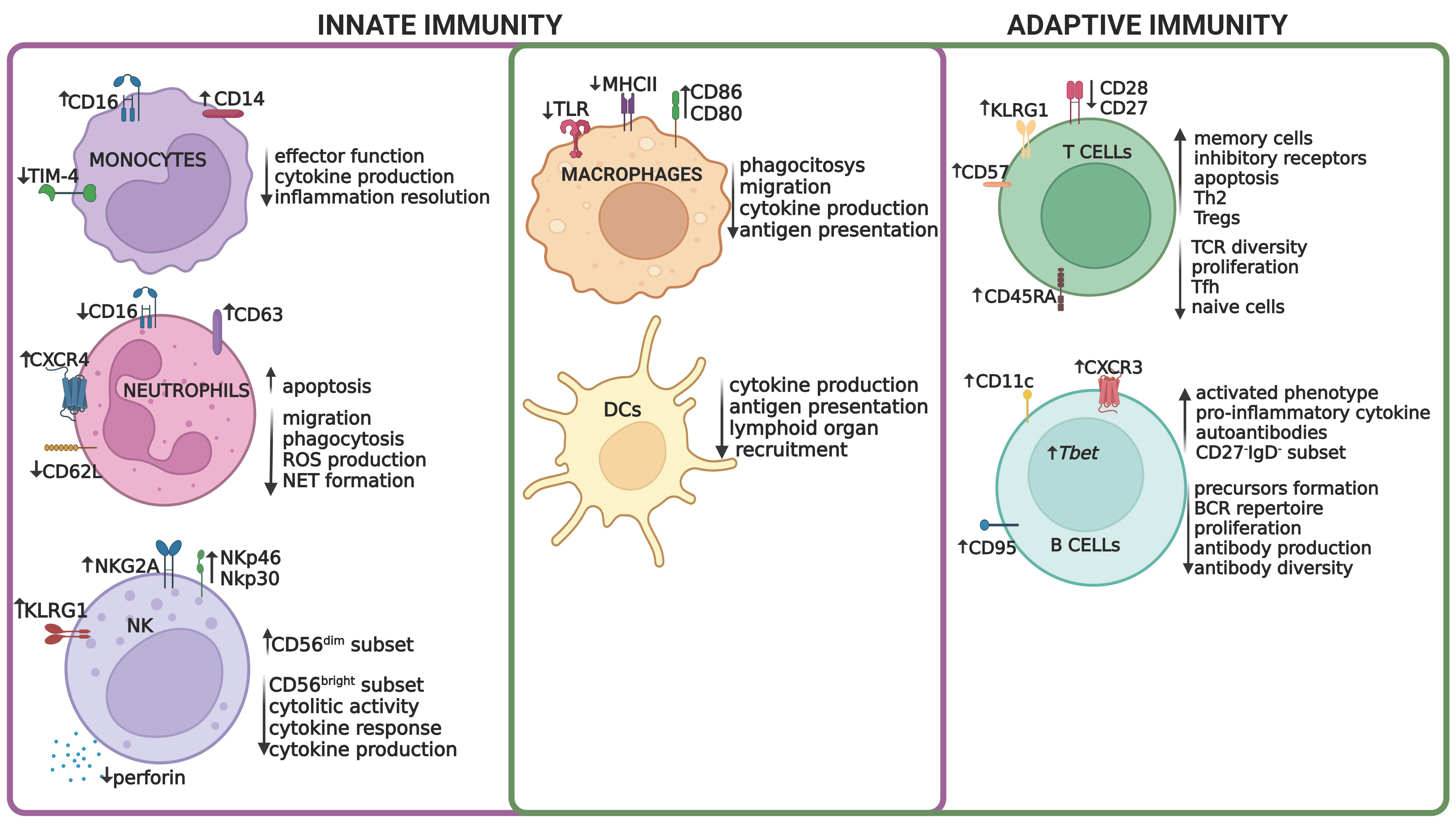{kind=link}
| Approach | Target (molecular/cellular) | Experimental outcome | Benefits | Limitations | Ref. |
|---|---|---|---|---|---|
| Senolytics | |||||
| CD153 peptide (vaccination) | CD4+ CD44high CD62Llow PD-1+ CD153+ cells in VAT tissue | Elimination of infiltrating senescent T cells in adipose tissue in obese mice | Improved glucose tolerance and insulin resistance | Contraindicated in case of mycobacterial infections | [226,227] |
| Prodrug SSK1 (oral administration) | SA- β -gal positive cells | Clearance of senescent macrophages | Dampened inflammation and restored physical function in aged mice | Off-target effects | [224] |
| Genetic deletion | p16(INK4a) in lymphocytes | Improved T cell immune function | Ameliorated several aging phenotypes | B lineage-specific ablation was associated with a markedly increased incidence of systemic, high-grade B-cell neoplasms | [229] |
| Senomorphics | |||||
| Leniolisib (oral administration) | PI3Kd | Reduction in PD-1+CD4+ and senescent CD57+CD4− T cells | Improved immune dysregulation and decreased fatigue in ongoing clinical trials | Potential genomic instability by augmenting off-target activity of activation-induced cytidine deaminase | [231,232] |
| Genetic inhibition | Sestrins in T cells | Restored T cell proliferation and cytokine production via p38 MAPK inhibition in senescent-like CD27−CD28−CD8+ T cells. | Enhanced vaccine responsiveness in old mice; enhanced immune function in primary human T cells | Prolonged inhibition of sestrins may result in malignancy | [29,30] |
| Losmapimod (oral administration) | p38 MAPK | Reduced systemic symptoms of inflamm-ageingr; escued TIM-4 expression; cleared apoptotic bodies and restored efferocytosis in macrophages | Improved skin inflammation resolution in aged people | Tested on a small cohort of individuals | [122] |
| BEZ235 (oral administration) | PI3K/mTOR | Augment the type I IFN response; reversed infection-induced changes in metabolism | Increased lifespan and health and reduced the incidence of respiratory tract infections in mice | Controversial results obtained in humans | [239,240,241,242] |
| Metformin (oral administration) | AMPK | Enhanced T cell autophagy, normalized mitochondrial function, and alleviated senes-cence-associated inflammation | Extended health span and lifespan in multiple animal models | Focus on CD4 T cells as sources of inflammageing; studies in humans not conclusive | [246,247,248] |
| Resveratrol (oral administration) | SIRT1 | Reduced ROS, inhibited COX, and activated anti-inflammatory pathways (Sirt1) | Anti-ageing in human trials; Restored T-cell function and NK cell activities | Possible risks related to nephrotoxicity | [249] |
| Spermidine (oral administration) | eIF5A | Reduced B cells senescence; improved autophagy in T cells | Restored response to vaccination and infection of CD8+ T cells in old mice | Toxic at high dose | [251] |
| Antibodies against cytokines | |||||
| Anti-IL10 | IL-10 | Enhanced T-cell functions | Inhibited viral persistence during LCMC infection in mice | Potential side-effects due to its role in immune tolerance | [278] |
| Anti-IFN-I | IFN receptor 2 | Decreased T cell exhaustion marker expression, restored viral-specific CD8 T cell function | Decreased viral replication in conjunction with ART treatment in HIV-infected humanized mice | Potential side-effect due to different role of IFN-I signaling during acute and chronic infection | [282] |
| Inhibitory receptors blockade | |||||
| Anti-PD-1 | PD-1 | Reduced exhaustion, expanded and increased functionality of virus-specific CD8+ T cells, | Significantly reduced plasma SIV RNA and prolonged survival in SIV-infected macaques | Potential side-effect related to break of tolerance | [157,263] |
| Reversed the exhausted phenotype, Increased IFN-g effector functions | Clearance of HBV virus in an in vivo model of HBV | [266] | |||
| BMS-936558 | No substantial changes in immune phenotype | Reduced viral load in a subset of HCV patients enrolled in the trial | Immune-related adverse events of mild-to-moderate intensity | [267] | |
| Anti-PD-L1 | PD-L1 | Restored viral-specific T cell functions | Reduced HIV-1 replication in HIV-infected humanized mice | No effect on HIV viral load in humans, likely due to low dosage used. | [265] |
| Anti-CTLA-4 | CTLA-4 | Enhanced viral specific responses | Better control of EBV and HIV infections in combination with anti-PD-1 | No effect in viral infection when used as monotherapy | [270,271] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marrella, V.; Facoetti, A.; Cassani, B. Cellular Senescence in Immunity against Infections. Int. J. Mol. Sci. 2022, 23, 11845. https://doi.org/10.3390/ijms231911845
Marrella V, Facoetti A, Cassani B. Cellular Senescence in Immunity against Infections. International Journal of Molecular Sciences. 2022; 23(19):11845. https://doi.org/10.3390/ijms231911845
Chicago/Turabian StyleMarrella, Veronica, Amanda Facoetti, and Barbara Cassani. 2022. "Cellular Senescence in Immunity against Infections" International Journal of Molecular Sciences 23, no. 19: 11845. https://doi.org/10.3390/ijms231911845
APA StyleMarrella, V., Facoetti, A., & Cassani, B. (2022). Cellular Senescence in Immunity against Infections. International Journal of Molecular Sciences, 23(19), 11845. https://doi.org/10.3390/ijms231911845