
New Oxazolo[5,4-d]pyrimidines as Potential Anticancer Agents: Their Design, Synthesis, and In Vitro Biological Activity Research

 , ,
, ,  ,
,  ,
,
Abstract
1. Introduction
2. Results
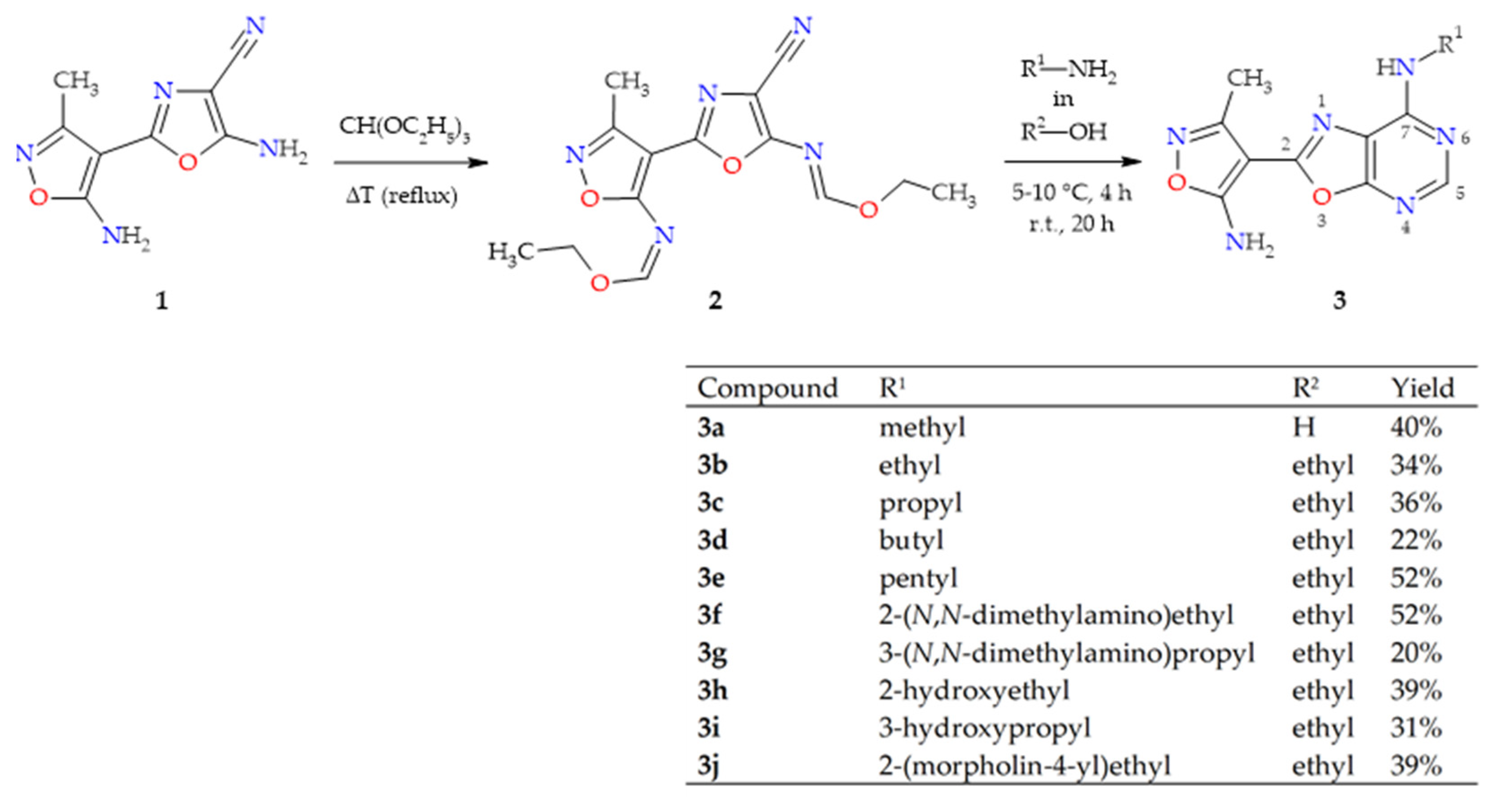
2.1. Synthesis of Oxazolo[5,4-d]pyrimidines 3a–j
2.2. In Vitro Cytotoxicity
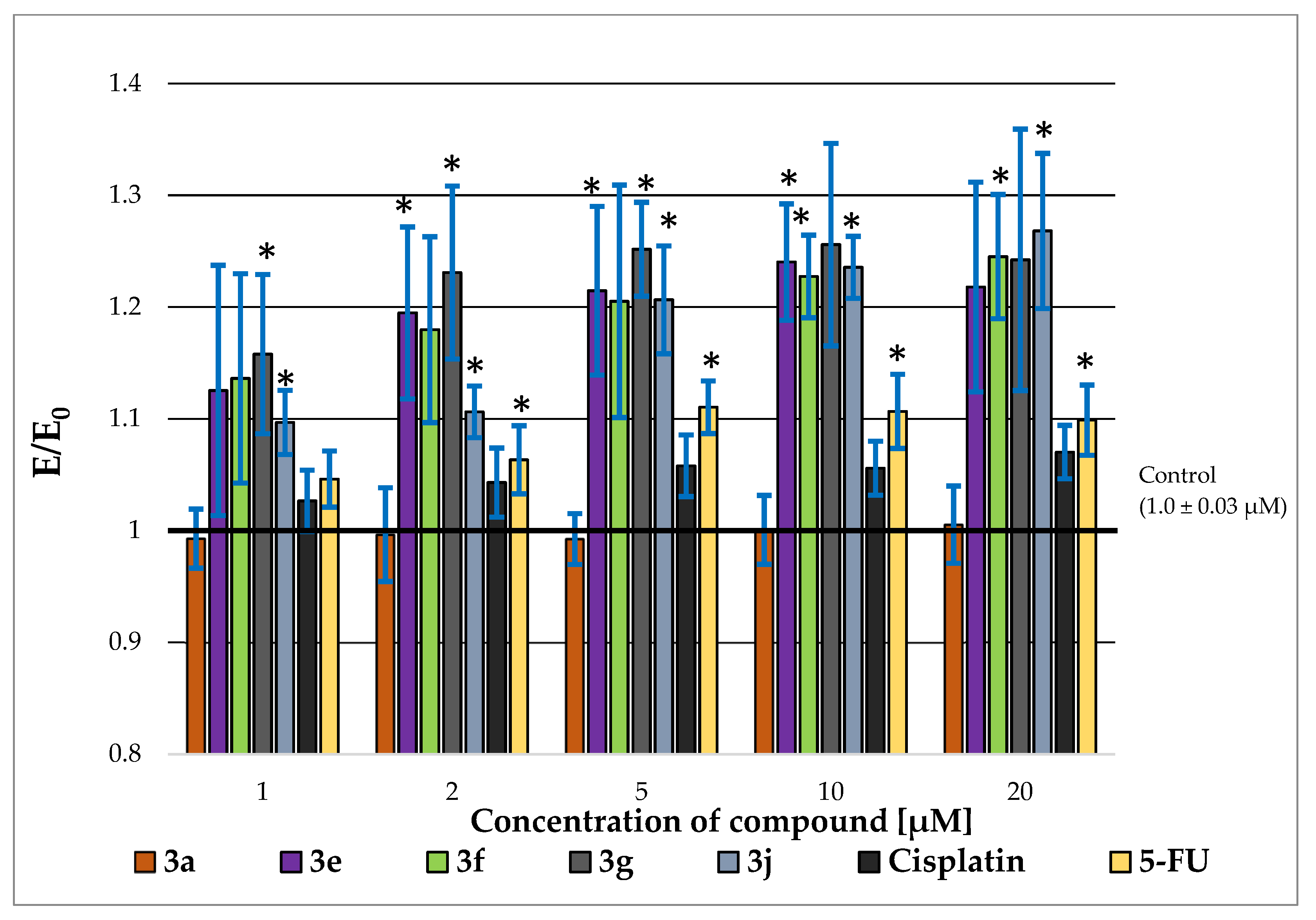
2.3. P-glycoprotein-Inhibitory Ability
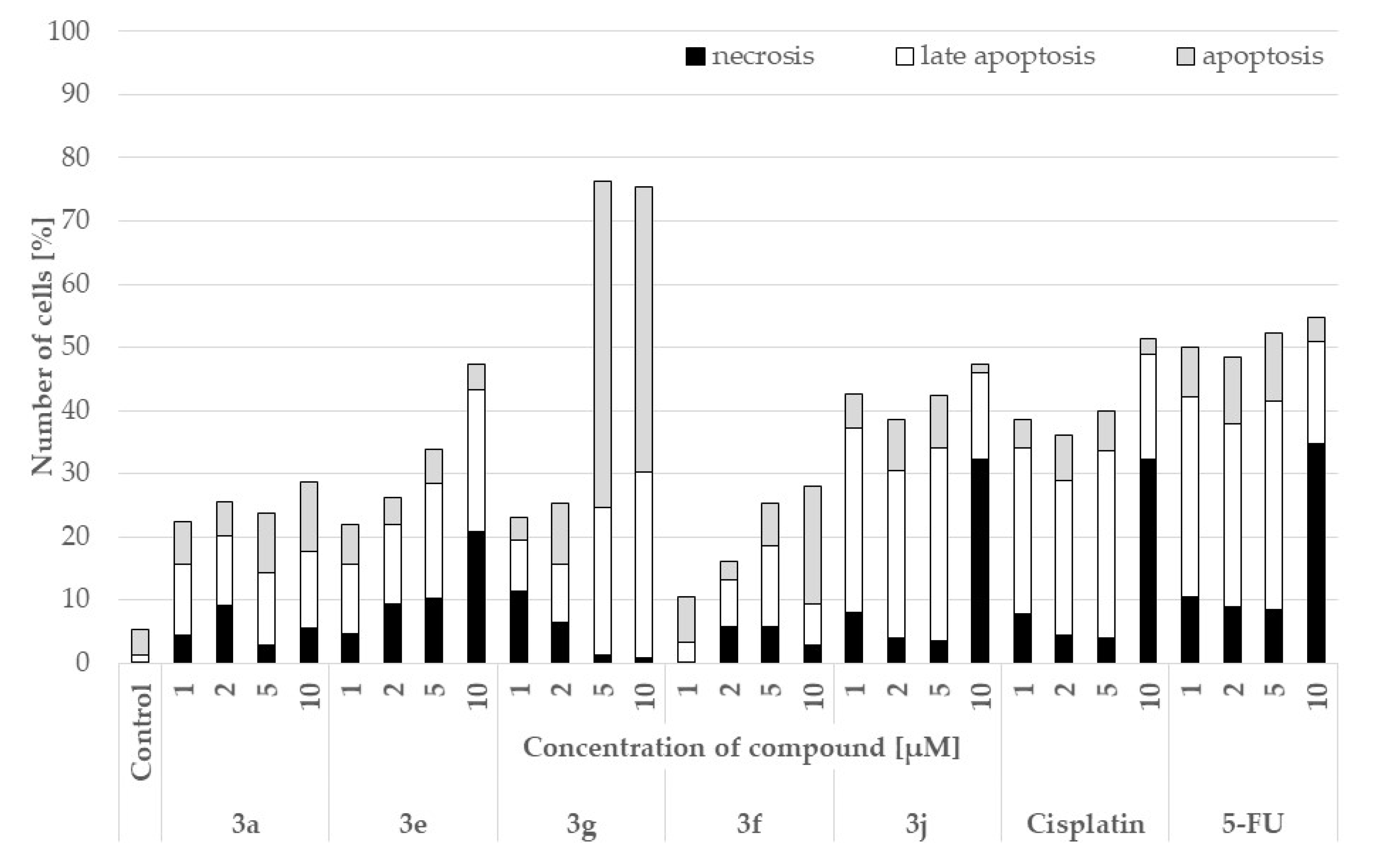
2.4. Pro-Apoptotic Activity
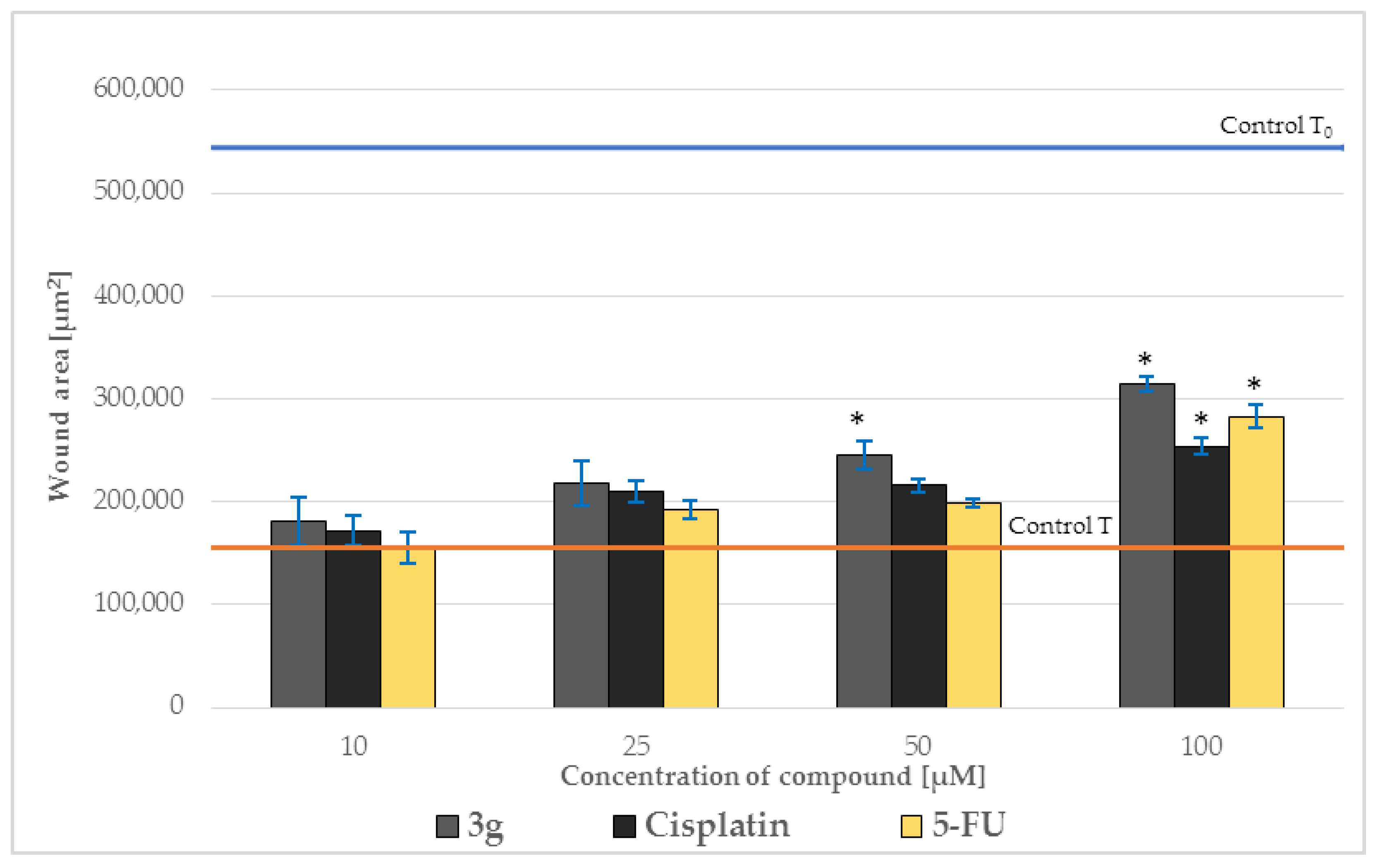
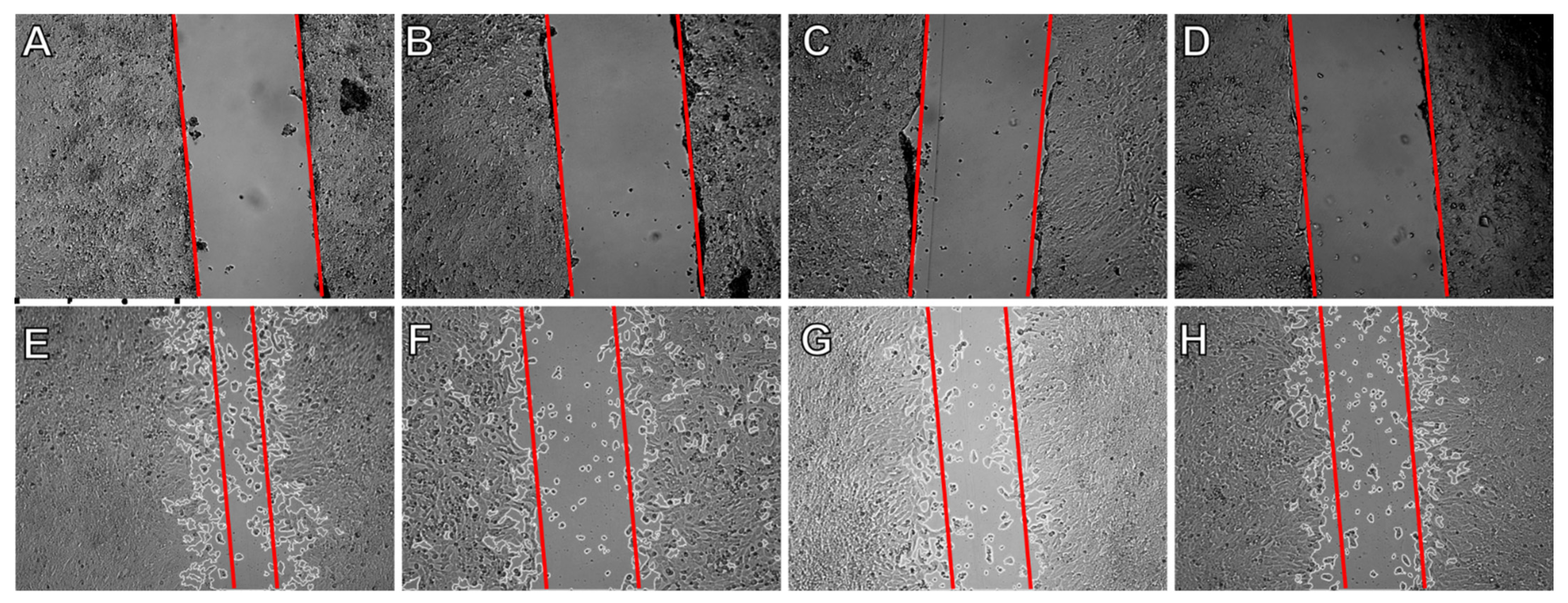
2.5. Effect on Cell Migration
2.6. Effects on Levels of p53, Caspase-3, and BCL-2
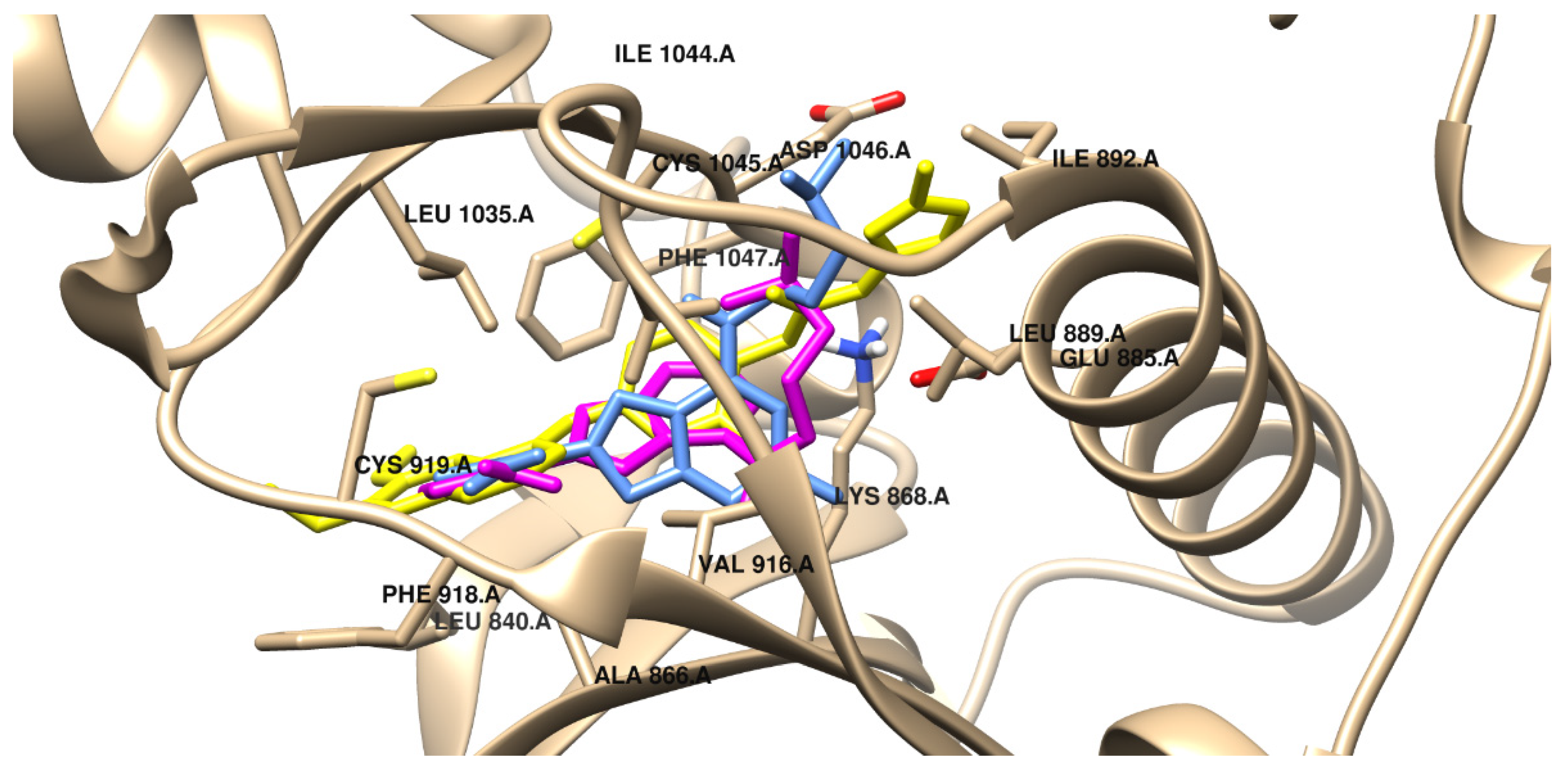
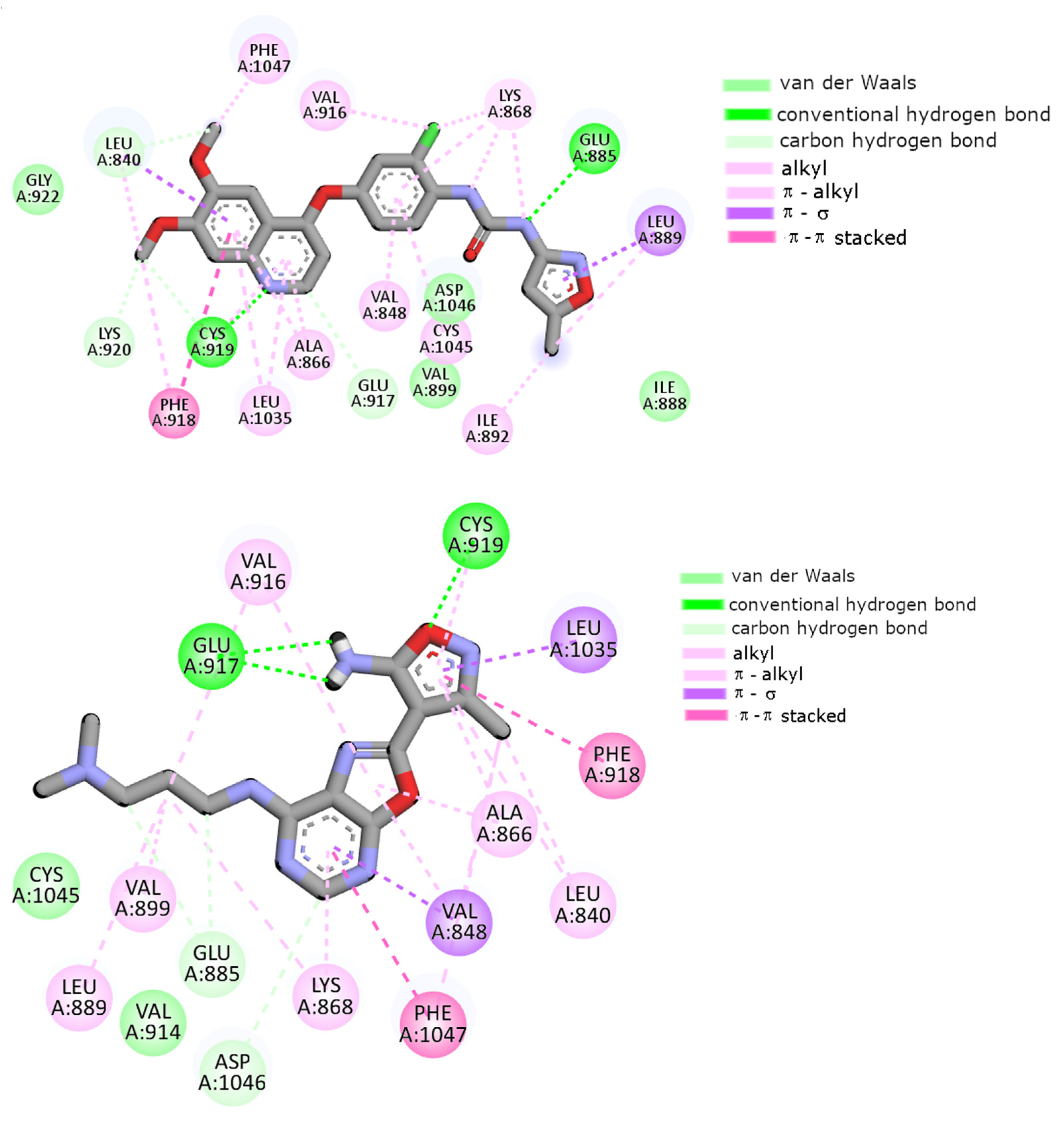
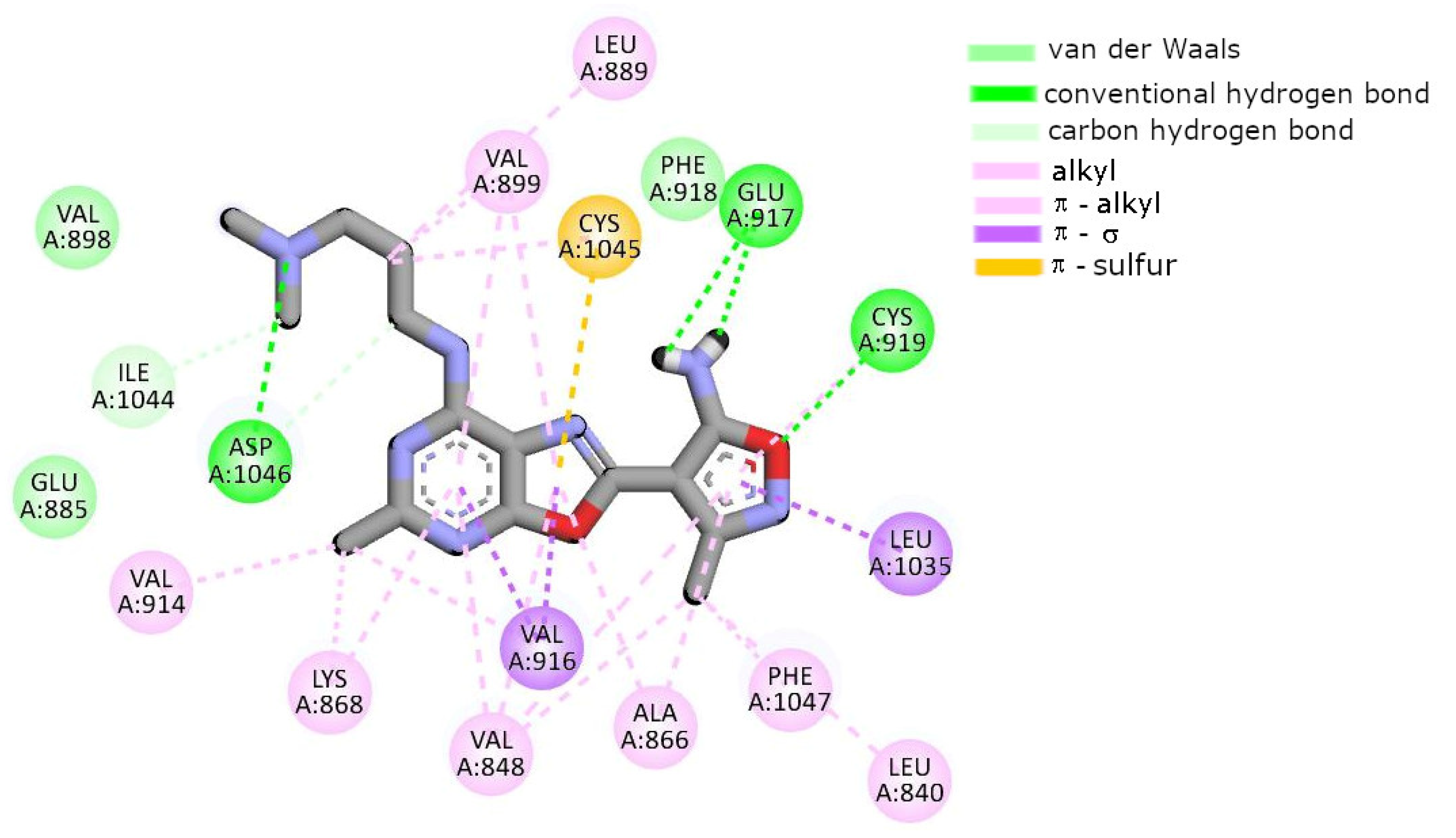
2.7. Molecular Docking
2.8. Physicochemical Properties, Pharmacokinetics, and ADME Activity
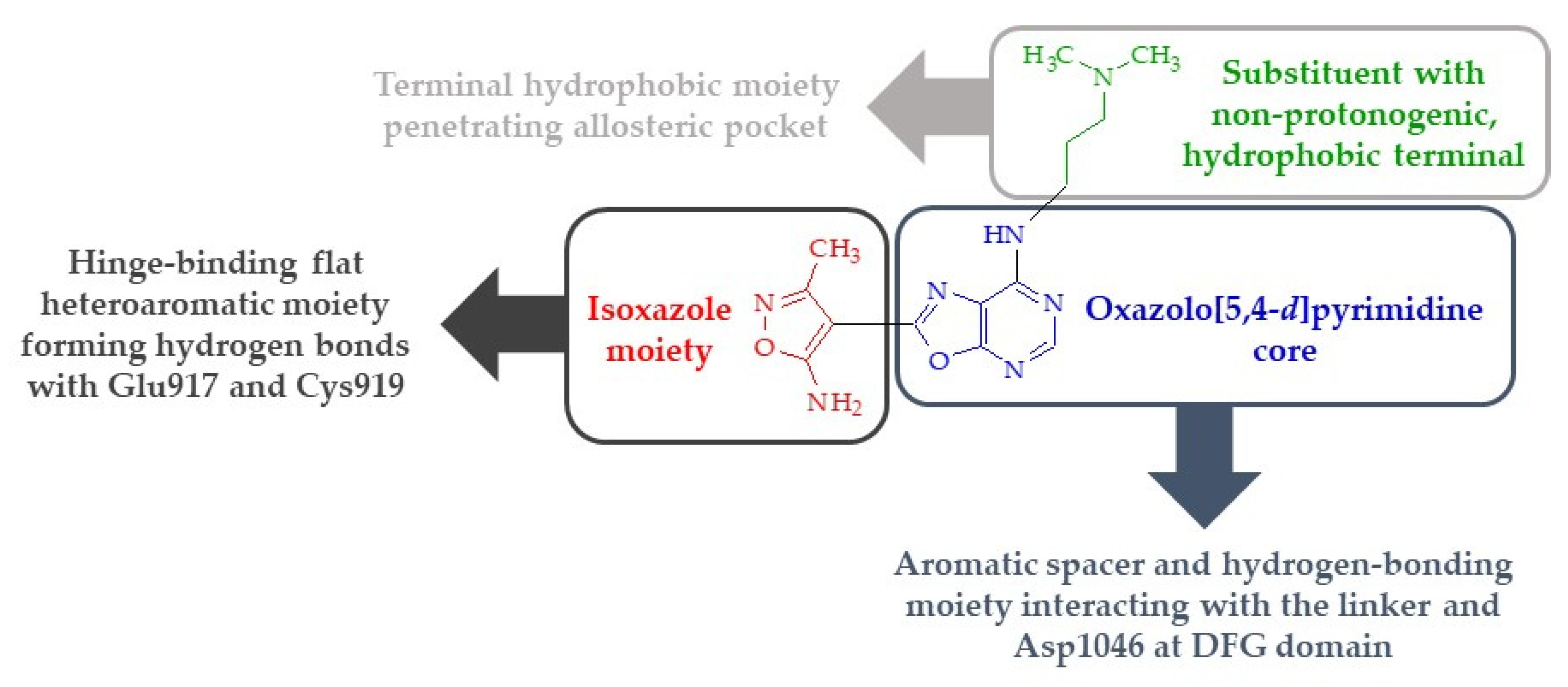
3. Discussion and Conclusions
- The presence of a substituent of appropriate size (i.e., a molecular tail with an optimal length of 6–7 atoms other than protons) and moderate lipophilicity at position 7 of the oxazolo[5,4-d]pyrimidine system.
- The absence of proton donor groups (such as OH) at the end of this substituent at position 7 of the oxazolo[5,4-d]pyrimidine system. If there is a strongly electronegative element (such as oxygen or nitrogen) at the terminal of this tail, then protons connected to this atom should be replaced with lipophilic substituents (such as methyl groups).
- The presence of an acidic proton at position 5 of the oxazolo[5,4-d]pyrimidine system, which will allow hydrogen bonds to form at this position with receptor structures.
- The ability of the substituent (except its terminal lipophilic fragment) at position 7 of the oxazolo[5,4-d]pyrimidine system to strongly interact with the Glu885 residue of the receptor active site (e.g., by forming hydrogen bonds).
4. Materials and Methods
4.1. Chemistry
4.2. Preparation and Experimental Properties of Compounds 2 and 3a–j
4.2.1. N-{4-cyano-2-[5-(ethoxymethylidene)amino-3-methylisoxazol-4-yl]oxazol-5-yl}methanimidate (2)
4.2.2. General Procedure for the Preparation of oxazolo[5,4-d]pyrimidines (3a–j)
4.3. Biology
4.3.1. Cell Lines and Conditions
4.3.2. Preparation of Solutions of the Tested Compounds
4.3.3. Viability Assay
4.3.4. Rh-123 Assay
4.3.5. Detection of Apoptosis
4.3.6. Migration Assay
4.3.7. ELISA
Human Caspase-3 (Active) ELISA Kit (KHO1091)
p53 Human ELISA Kit (BMS256)
BCL-2 Human ELISA Kit (BMS244-3)
4.3.8. Statistical Analysis
4.4. Molecular Modeling
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sochacka-Ćwikła, A.; Mączyński, M.; Regiec, A. FDA-approved small molecule compounds as drugs for solid cancers from early 2011 to the end of 2021. Molecules 2022, 27, 2259. [Google Scholar] [CrossRef]
- Sochacka-Ćwikła, A.; Mączyński, M.; Regiec, A. FDA-approved drugs for hematological malignancies—The last decade review. Cancers 2022, 14, 87. [Google Scholar] [CrossRef]
- Zhang, C.; Tan, C.; Ding, H.; Xin, T.; Jiang, Y. Selective VEGFR inhibitors for anticancer therapeutics in clinical use and clinical trials. Curr. Pharm. Des. 2012, 18, 2921–2935. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Bove, A.M.; Simone, G.; Ma, B. Molecular bases of VEGFR-2-mediated physiological function and pathological role. Front. Cell Dev. Biol. 2020, 8, 599281. [Google Scholar] [CrossRef]
- Modi, S.J.; Kulkarni, V.M. Vascular endothelial growth factor receptor (VEGFR-2)/KDR inhibitors: Medicinal chemistry perspective. Med. Drug Discov. 2019, 2, 100009. [Google Scholar] [CrossRef]
- Velihina, Y.; Scattolin, T.; Bondar, D.; Pilo, S.; Obernikhina, N.; Kachkovskyi, O.; Semenyuta, I.; Caligiuri, I.; Rizzolio, F.; Brovarets, V.; et al. Synthesis in silico and in vitro evaluation of novel oxazolopyrimidines as promising anticancer agents. Helv. Chim. Acta 2020, 103, e2000169. [Google Scholar] [CrossRef]
- Deng, Y.-H.; Xu, D.; Su, Y.-X.; Cheng, Y.-J.; Yang, Y.-L.; Wang, X.-Y.; Zhang, J.; You, Q.-D.; Sun, L.-P. Synthesis and biological evaluation of novel oxazolo[5,4-d]pyrimidines as potent VEGFR-2 inhibitors. Chem. Biodivers. 2015, 12, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Martin-Kohler, A.; Widmer, J.; Bold, G.; Meyer, T.; Sequin, U.; Traxler, P. Furo[2,3-d]pyrimidines and oxazolo[5,4-d]pyrimidines as inhibitors of receptor tyrosine kinases (RTK). Helv. Chim. Acta 2004, 87, 956–975. [Google Scholar] [CrossRef]
- Bauser, M.; Delapierre, G.; Hauswald, M.; Flessner, T.; D’Urso, D.; Hermann, A.; Beyreuther, B.; De Vry, J.; Spreyer, P.; Reissmüller, E.; et al. Discovery and opitimalization of 2-aryl oxazolo-pyrimidines as adenosine kinase inhibitors using liquid phase paralel synthesis. Bioorg. Med. Chem. Lett. 2004, 14, 1997–2000. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, H.-P.; Coumar, S.M.; Chao, Y.-S. Preparation of Fused Multicyclic Compounds as Protein Kinase Inhibitors. WIPO Patent WO2010036629A, 1 April 2010. [Google Scholar]
- Rodgers, J.D.; Shepard, S.; Arvanitis, A.G.; Wang, H.; Storace, L.; Folmer, B.; Shao, L.; Zhu, W.; Glenn, J.P. Preparation of N-(Hetero)arylpyrrolidine Derivatives of Pyrazol-4-ylpyrrolo[2,3-D]pyrimidines and Pyrrol-3-ylpyrrolo[2,3-D]-pyrimidines as Janus Kinase JAK1 Inhibitors. WIPO Patent WO2010135650A1, 25 November 2010. [Google Scholar]
- Ioannidis, S.; Lamb, M.; Su, M. N-Pyrazolyl Fused Pyrimidinamine Derivativesas JAK Inhibitors and Their Preparation, Pharmaceutical Compositions and Use in the Treatment of Myeloproliferative Disorders and Cancer. WIPO Patent WO2009013545A2, 29 January 2009. [Google Scholar]
- Claiborne, C.F.; Critchley, S.; Langston, S.P.; Olhava, E.J.; Peluso, S.; Weatherhead, G.S.; Vyskocil, S.; Visiers, I.; Mizutani, H.; Cullis, C. Heteroaryl Compounds Useful as Inhibitors of E1 Activating Enzymes. WIPO Patent WO2008019124A1, 14 February 2008. [Google Scholar]
- Cai, S.X.; Kemnitzer, W.E.; Sirisoma, S.; Zhang, H.-Z. N-Aryl-Isoxazolopyrimidin-4-Amines and Related Compounds as Activators of Caspases and Inducers of Apoptosis and the Use Thereof. WIPO Patent WO2008057402A2, 15 May 2008. [Google Scholar]
- Liu, J.; Deng, Y.H.; Yang, L.; Chen, Y.; Lawali, M.; Sun, L.P.; Liu, Y. CPU-12, a novel synthesized oxazolo[5,4-d]pyrimidine derivative, showed superior anti-angiogenic activity. J. Pharmacol. Sci. 2015, 129, 9–17. [Google Scholar] [CrossRef]
- Deng, Y.H.; Liu, J.P.; Cheng, Y.J.; Liu, Y.; Sun, L.P. Diarylureas and diarylamides with oxazolo[5,4-d]pyrimidine scaffold as angiogenesis inhibitors. Chem. Biodivers. 2016, 13, 1230–1239. [Google Scholar] [CrossRef] [PubMed]
- Perupogu, N.; Kumar, R.D.; Ramachandran, D. Anticancer activity of newly synthesized 1,2,4-oxadiazole linked 4-oxazolo[5,4-d]pyrimidine derivatives. Chem. Data Collect. 2020, 27, 100363. [Google Scholar] [CrossRef]
- Sochacka-Ćwikła, A.; Regiec, A.; Zimecki, M.; Artym, J.; Zaczyńska, E.; Kocięba, M.; Kochanowska, I.; Bryndal, I.; Pyra, A.; Mączyński, M. Synthesis and biological activity of new 7-amino-oxazolo[5,4-d]pyrimidine derivatives. Molecules 2020, 25, 3558. [Google Scholar] [CrossRef]
- Chen, D.; Shen, A.; Li, J.; Shi, F.; Chen, W.; Ren, J.; Liu, H.; Xu, Y.; Wang, X.; Yang, X.; et al. Discovery of potent N-(isoxazol-5-yl)amides as HSP90 inhibitors. Eur. J. Med. Chem. 2014, 87, 765–781. [Google Scholar] [CrossRef]
- Bargiotti, A.; Musso, L.; Dallavalle, S.; Merlini, L.; Gallo, G.; Ciacci, A.; Giannini, G.; Cabri, W.; Penco, S.; Vesci, L.; et al. Isoxazolo(aza)naphthoquinones: A new class of cytotoxic Hsp90 inhibitors. Eur. J. Med. Chem. 2012, 53, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Eccles, S.A.; Massey, A.; Raynaud, F.I.; Sharp, S.Y.; Box, G.; Valenti, M.; Patterson, L.; de Haven Brandon, A.; Gowan, S.; Boxall, F.; et al. NVP-AUY922: A novel heat shock protein 90 inhibitor active against xenograft tumor growth, angiogenesis, and metastasis. Cancer Res. 2008, 68, 2850–2860. [Google Scholar] [CrossRef]
- Sharp, S.Y.; Prodromou, C.; Boxall, K.; Powers, M.V.; Holmes, J.L.; Box, G.; Matthews, T.P.; Cheung, K.M.; Kalusa, A.; James, K.; et al. Inhibition of the heat shock protein 90 molecular chaperone in vitro and in vivo by novel, synthetic, potent resorcinylic pyrazole/isoxazole amide analogues. Mol. Cancer Ther. 2007, 6, 1198–1211. [Google Scholar] [CrossRef]
- Yi, X.J.; El-Idreesy, T.T.; Eldebss, T.M.; Farag, A.M.; Abdulla, M.M.; Hassan, S.A.; Mabkhot, Y.N. Synthesis, biological evaluation, and molecular docking studies of new pyrazol-3-one derivatives with aromatase inhibition activities. Chem. Biol. Drug Des. 2016, 88, 832–843. [Google Scholar] [CrossRef] [PubMed]
- Di Matteo, M.; Ammazzalorso, A.; Andreoli, F.; Caffa, I.; De Filippis, B.; Fantacuzzi, M.; Giampietro, L.; Maccallini, C.; Nencioni, A.; Parenti, M.D.; et al. Synthesis and biological characterization of 3-(imidazole-1-ylmethyl) piperidine sulfonamides as aromatase inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 3192–3194. [Google Scholar] [CrossRef]
- Ling, Y.-Z.; Li, J.-S.; Liu, Y.; Kato, K.; Klus, G.T.; Brodie, A. 17-imidazolyl, pyrazolyl, and isoxazolyl androstene derivatives. Novel steroidal inhibitors of human cytochrome Cl7, 20-lyase (P45017a). J. Med. Chem. 1997, 40, 3297–3304. [Google Scholar] [CrossRef]
- Conti, P.; Tamborini, L.; Pinto, A.; Sola, L.; Ettari, R.; Mercurio, C.; De Micheli, C. Design and synthesis of novel isoxazole-based HDAC inhibitors. Eur. J. Med. Chem. 2010, 45, 4331–4338. [Google Scholar] [CrossRef] [PubMed]
- Pedada, S.R.; Yarla, N.S.; Tambade, P.J.; Dhananjaya, B.L.; Bishayee, A.; Arunasree, K.M.; Philip, G.H.; Dharmapuri, G.; Aliev, G.; Putta, S.; et al. Synthesis of new secretory phospholipase A2-inhibitory indole containing isoxazole derivatives as anti-inflammatory and anticancer agents. Eur. J. Med. Chem. 2016, 112, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Ravula, S.; Bobbala, R.R.; Kolli, B. Synthesis of novel isoxazole functionalized pyrazolo[3,4-b]pyridine derivatives; their anticancer activity. J. Heterocycl. Chem. 2020, 57, 2535–2538. [Google Scholar] [CrossRef]
- Fahim, A.M.; Shalaby, M.A. Synthesis, biological evaluation, molecular docking and DFT calculations of novel benzenesulfonamide derivatives. J. Mol. Struct. 2019, 1176, 408–421. [Google Scholar] [CrossRef]
- Warda, E.T.; Shehata, I.A.; El-Ashmawy, M.B.; El-Gohary, N.S. New series of isoxazole derivatives targeting EGFR-TK: Synthesis, molecular modeling and antitumor evaluation. Bioorg. Med. Chem. 2020, 28, 115674. [Google Scholar] [CrossRef]
- Im, D.; Jung, K.; Yang, S.; Aman, W.; Hah, J.-M. Discovery of 4-arylamido 3- methyl isoxazole derivatives as novel FMS kinase inhibitors. Eur. J. Med. Chem. 2015, 102, 600–610. [Google Scholar] [CrossRef]
- He, H.; Ge, Y.; Dai, H.; Cui, S.; Ye, F.; Jin, J.; Shi, Y. Design, synthesis and biological evaluation of stilbene derivatives as novel inhibitors of protein tyrosine phosphatase 1B. Molecules 2016, 21, 1722. [Google Scholar] [CrossRef]
- Modi, S.J.; Kulkarni, V.M. Exploration of structural requirements for the inhibition of VEGFR-2 tyrosine kinase: Binding site analysis of type II, ‘DFG-out’ inhibitors. J. Biomol. Struct. Dyn. 2021, 40, 5712–5727. [Google Scholar] [CrossRef]
- Lampronti, I.; Simoni, D.; Rondanin, R.; Baruchello, R.; Scapoli, C.; Finotti, A.; Borgatti, M.; Tupini, C.; Gambari, R. Pro-apoptotic activity of novel synthetic isoxazole derivatives exhibiting inhibitory activity against tumor cell growth in vitro. Oncol. Lett. 2020, 20, 151. [Google Scholar] [CrossRef]
- Bernal, C.C.; Vesga, L.C.; Mendez-Sanchez, S.C.; Bohorquez, A.R.R. Synthesis and anticancer activity of new tetrahydroquinoline hybrid derivatives tethered to isoxazoline moiety. Med. Chem. Res. 2020, 29, 675–689. [Google Scholar] [CrossRef]
- Çalıskan, B.; Sinoplu, E.; Ibis, K.; Akhan Güzelcan, E.; Çetin Atalay, R.; Banoglu, E. Synthesis and cellular bioactivities of novel isoxazole derivatives incorporating an arylpiperazine moiety as anticancer agents. J. Enzym. Inhib. Med. Chem. 2018, 33, 1352–1361. [Google Scholar] [CrossRef] [PubMed]
- Shin, K.D.; Lee, M.Y.; Shin, D.S.; Lee, S.; Son, K.H.; Koh, S.; Paik, Y.K.; Kwon, B.M.; Han, D.C. Blocking tumor cell migration and invasion with biphenyl isoxazole derivative KRIBB3, a synthetic molecule that inhibits Hsp27 phosphorylation. J. Biol. Chem. 2005, 280, 41439–44148. [Google Scholar] [CrossRef] [PubMed]
- Mączyński, M.; Regiec, A.; Sochacka-Ćwikła, A.; Kochanowska, I.; Kocięba, M.; Zaczyńska, E.; Artym, J.; Kałas, W.; Zimecki, M. Synthesis, physicochemical characteristics and plausible mechanism of action of an immunosuppressive isoxazolo[5,4-e]-1,2,4-triazepine derivative (RM33). Pharmaceuticals 2021, 14, 468. [Google Scholar] [CrossRef] [PubMed]
- Bąchor, U.; Ryng, S.; Mączyński, M.; Artym, J.; Kocięba, M.; Zaczyńska, E.; Kochanowska, I.; Tykarska, E.; Zimecki, M. Synthesis, immunosuppressive properties, mechanism of action and X-ray analysis of a new class of isoxazole derivatives. Acta Pol. Pharm. Drug Res. 2019, 76, 251–263. [Google Scholar] [CrossRef]
- Mączyński, M.; Artym, J.; Kocięba, M.; Sochacka-Ćwikła, A.; Drozd-Szczygieł, E.; Ryng, S.; Zimecki, M. Synthesis and immunoregulatory properties of selected 5-amino-3-methyl-4 isoxazolecarboxylic acid benzylamides. Acta Pol. Pharm. 2016, 73, 1201–1211. [Google Scholar]
- Płoszaj, A.; Regiec, A.; Ryng, S.; Piwowar, A.; Kruzel, M. Influence of 5-amino-3-methyl-4-isoxazolecarbohydrazide on selective gene expression in Caco-2 cultured cells. Immunopharmacol. Immunotoxicol. 2016, 38, 486–494. [Google Scholar] [CrossRef]
- Drynda, A.; Obmińska-Mrukowicz, B.; Mączyński, M.; Ryng, S. The effect of 5-amino-3-methyl-4-isoxazolecarboxylic acid hydrazide on lymphocyte subsets and humoral immune response in SRBC-immunized mice. Immunopharmacol. Immunotoxicol. 2015, 37, 148–157. [Google Scholar] [CrossRef]
- Maczyński, M.; Ryng, S.; Artym, J.; Kocięba, M.; Zimecki, M.; Brudnik, K.; Jodkowski, J.T. New lead structures in the isoxazole system: Relationship between quantum chemical parameters and immunological activity. Acta Pol. Pharm. 2014, 71, 71–83. [Google Scholar]
- Drynda, A.; Mączyński, M.; Ryng, S.; Obmińska-Mrukowicz, B. In vitro immunomodulatory effects of 5-amino-3-methyl-4-isoxazolecarboxylic acid hydrazide on the cellular immune response. Immunopharmacol. Immunotoxicol. 2014, 36, 150–157. [Google Scholar] [CrossRef]
- Zimecki, M.; Artym, J.; Kocięba, M.; Obmińska-Mrukowicz, B.; Mączyński, M.; Ryng, S. Restoration of immune system function is accelerated in immunocompromised mice by the B-cell-tropic isoxazole R-11. Pharmacol. Rep. 2012, 64, 403–411. [Google Scholar] [CrossRef]
- Zimecki, M.; Maczyński, M.; Artym, J.; Ryng, S. Closely related isoxazoles may exhibit opposite immunological activities. Acta Pol. Pharm. 2008, 65, 793–794. [Google Scholar] [PubMed]
- Basaki, Y.; Chikahisa, L.; Aoyagi, K.; Miyadera, K.; Yonekura, K.; Hashimoto, A.; Okabe, S.; Wierzba, K.; Yamada, Y. γ-Hydroxybutyric acid and 5-fluorouracil, metabolites of UFT, inhibit the angiogenesis induced by vascular endothelial growth factor. Angiogenesis 2001, 4, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Reddy, N.N.; Hung, S.-J.; Swamy, M.K.; Sanjeev, A.; Rao, V.S.; Rohini, R.; Raju, A.K.; Bhaskar, K.; Hu, A.; Reddy, P.M. Synthesis and rational design of new appended 1,2,3-triazole-uracil ensembles as promising anti-tumor agents via in silico VEGFR-2 transferase inhibition. Molecules 2021, 26, 1952. [Google Scholar] [CrossRef] [PubMed]
- Murray, N.; Turrisi, A.T. A review of first-line treatment for small-cell lung cancer. J. Thorac. Oncol. 2006, 1, 270–278. [Google Scholar] [CrossRef]
- Jouan, E.; Le Vée, M.; Mayati, A.; Denizot, C.; Parmentier, Y.; Fardel, O. Evaluation of P-glycoprotein inhibitory potential using a rhodamine 123 accumulation assay. Pharmaceutics 2016, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Cheraghi, O.; Dehghan, G.; Mahdavi, M.; Rahbarghazi, R.; Rezabakhsh, A.; Charoudeh, H.N.; Iranshahi, M.; Montazersaheb, S. Potent anti-angiogenic and cytotoxic effect of conferone on human colorectal adenocarcinoma HT-29 cells. Phytomedicine 2016, 23, 398–405. [Google Scholar] [CrossRef]
- Papanikolaou, X.; Johnson, S.; Garg, T.; Tian, E.; Tytarenko, R.; Zhang, Q.; Stein, C.; Barlogie, B.; Epstein, J.; Heuck, C. Artesunate overcomes drug resistance in multiple myeloma by inducing mitochondrial stress and non-caspase apoptosis. Oncotarget 2014, 5, 4118–4128. [Google Scholar] [CrossRef]
- Sato, A.; Hiramoto, A.; Kim, H.-S.; Wataya, Y. Anticancer strategy targeting cell death regulators: Switching the mechanism of anticancer floxuridine-induced cell death from necrosis to apoptosis. Int. J. Mol. Sci. 2020, 21, 5876. [Google Scholar] [CrossRef]
- Igney, F.H.; Krammer, P.H. Death and anti-death: Tumour resistance to apoptosis. Nat. Rev. Cancer 2002, 2, 277–288. [Google Scholar] [CrossRef]
- Lopez, J.; Tait, S.W.G. Mitochondrial apoptosis: Killing cancer using the enemy within. Br. J. Cancer 2015, 112, 957–962. [Google Scholar] [CrossRef]
- Krammer, P.H.; Arnold, R.; Lavrik, I.N. Life and death in peripheral T cells. Nat. Rev. Immunol. 2007, 7, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Croft, M. Co-stimulatory members of the TNFR family: Keys to effective T-cell immunity? Nat. Rev. Immunol. 2003, 3, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Bąchor, R. Peptidyl-resin substrates as a tool in the analysis of caspase activity. Molecules 2022, 27, 4107. [Google Scholar] [CrossRef] [PubMed]
- Kale, J.; Osterlund, E.; Andrews, D. BCL-2 family proteins: Changing partners in the dance towards death. Cell Death Differ. 2018, 25, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Cuartas, V.; Aragón-Muriel, A.; Liscano, Y.; Polo-Cerón, D.; Crespo-Ortiz, M.D.P.; Quiroga, J.; Abonia, R.; Insuasty, B. Anticancer activity of pyrimidodiazepines based on 2-chloro-4-anilinoquinazoline: Synthesis, DNA binding and molecular docking. RSC Adv. 2021, 11, 23310–23329. [Google Scholar] [CrossRef]
- Bannu, S.M.; Lomada, D.; Varala, S.; Reddy, M.C. Molecular docking analysis of C-phycocyanin with VEGFR2. Bioinformation 2020, 16, 869–877. [Google Scholar] [CrossRef]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef]
- Vuppalanchi, R. Metabolism of drugs and xenobiotics. In Practical Hepatic Pathology: A Diagnostic Approach; Saxena, R., Ed.; Elsevier Saunders: Philadelphia, PA, USA, 2011; pp. 45–52. [Google Scholar]
- Lu, L.; Zhao, T.T.; Liu, T.B.; Sun, W.X.; Xu, C.; Li, D.D.; Zhu, H.L. Synthesis, molecular modeling and biological evaluation of 4-alkoxyquinazoline derivatives as novel inhibitors of VEGFR2. Chem. Pharm. Bull. 2016, 64, 1570–1575. [Google Scholar] [CrossRef]
- Belli, V.; Sforza, V.; Cardone, C.; Martinelli, E.; Barra, G.; Matrone, N.; Napolitano, S.; Morgillo, F.; Tuccillo, C.; Federico, A.; et al. Regorafenib in combination with silybin as a novel potential strategy for the treatment of metastatic colorectal cancer. Oncotarget 2017, 8, 68305–68316. [Google Scholar] [CrossRef]
- Zhou, Y.; Xu, X.; Wang, F.; He, H.; Qi, B. Discovery of 4-((4-(4-(3-(2-(2,6-difluorophenyl)-4-oxothiazolidin-3-yl)ureido)-2-fluorophenoxy)-6-methoxyquinolin-7-yl)oxy)-N,N-diethylpiperidine-1-carboxamide as kinase inhibitor for the treatment of colorectal cancer. Bioorg. Chem. 2021, 106, 104511. [Google Scholar] [CrossRef]
- Qi, B.; Yang, Y.; Gong, G.; He, H.; Yue, X.; Xu, X.; Hu, Y.; Li, J.; Chen, T.; Wan, X.; et al. Discovery of N1-(4-((7-(3-(4-ethylpiperazin-1-yl)propoxy)-6-methoxyquinolin-4-yl)oxy)-3,5-difluorophenyl)-N3-(2-(2,6-difluorophenyl)-4-oxothiazolidin-3-yl)urea as a multi-tyrosine kinase inhibitor for drug-sensitive and drug-resistant cancers treatment. Eur. J. Med. Chem. 2019, 163, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Wu, W.; Zha, D.; Zhou, W.; Wang, C.; Huang, J.; Chen, S.; Yu, L.; Li, Y.; Huang, Q.; et al. Synthesis and biological evaluation of novel ligustrazine-chalcone derivatives as potential anti-triple negative breast cancer agents. Bioorg. Med. Chem. Lett. 2021, 47, 128230. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Kodama, K.; Takase, K.; Sugi, N.H.; Yamamoto, Y.; Iwata, M.; Tsuruoka, A. Antitumor activities of the targeted multi-tyrosine kinase inhibitor lenvatinib (E7080) against RET gene fusion-driven tumor models. Cancer Lett. 2013, 340, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Wiegering, A.; Korb, D.; Thalheimer, A.; Kämmerer, U.; Allmanritter, J.; Matthes, N.; Linnebacher, M.; Schlegel, N.; Klein, I.; Ergün, S.; et al. E7080 (lenvatinib), a multi-targeted tyrosine kinase inhibitor, demonstrates antitumor activities against colorectal cancer xenografts. Neoplasia 2014, 16, 972–981. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.G.; Reddy, T.S.; Jadala, C.; Reddy, M.S.; Sultana, F.; Akunuri, R.; Bhargava, S.K.; Wlodkowic, D.; Srihari, P.; Kamal, A. Pyrazolo-benzothiazole hybrids: Synthesis, anticancer properties and evaluation of antiangiogenic activity using in vitro VEGFR-2 kinase and in vivo transgenic zebrafish model. Eur. J. Med. Chem. 2019, 182, 111609. [Google Scholar] [CrossRef]
- Wedge, S.R.; Ogilvie, D.J.; Dukes, M.; Kendrew, J.; Chester, R.; Jackson, J.A.; Boffey, S.J.; Valentine, P.J.; Curwen, J.O.; Musgrove, H.L.; et al. ZD6474 inhibits vascular endothelial growth factor signaling, angiogenesis, and tumor growth following oral administration. Cancer Res. 2002, 62, 4645–4655. [Google Scholar]
- Sarkar, S.; Mazumdar, A.; Dash, R.; Sarkar, D.; Fisher, P.B.; Mandal, M. ZD6474 enhances paclitaxel antiproliferative and apoptotic effects in breast carcinoma cells. J. Cell. Physiol. 2011, 226, 375–384. [Google Scholar] [CrossRef]
- Azzariti, A.; Porcelli, L.; Xu, J.M.; Simone, G.M.; Paradiso, A. Prolonged exposure of colon cancer cells to the epidermal growth factor receptor inhibitor gefitinib (Iressa(TM)) and to the antiangiogenic agent ZD6474: Cytotoxic and biomolecular effects. World J. Gastroenterol. 2006, 12, 5140–5147. [Google Scholar] [CrossRef]
- Zhao, H.; Yang, F.; Shen, W.; Wang, Y.; Li, X.; You, J.; Zhou, Q. Pazopanib diminishes non-small cell lung cancer (NSCLC) growth and metastases in vivo. Thorac. Cancer 2015, 6, 133–140. [Google Scholar] [CrossRef]
- Gril, B.; Palmieri, D.; Qian, Y.; Anwar, T.; Ileva, L.; Bernardo, M.; Choyke, P.; Liewehr, D.J.; Steinberg, S.M.; Steeg, P.S. The B-Raf status of tumor cells may be a significant determinant of both antitumor and anti-angiogenic effects of pazopanib in xenograft tumor models. PLoS ONE 2011, 6, e25625. [Google Scholar] [CrossRef]
- Zang, J.; Liang, X.; Huang, Y.; Jia, Y.; Li, X.; Xu, W.; Chou, C.J.; Zhang, Y. Discovery of novel pazopanib-based HDAC and VEGFR dual inhibitors targeting cancer epigenetics and angiogenesis simultaneously. J. Med. Chem. 2018, 61, 5304–5322. [Google Scholar] [CrossRef]
- Shah, S.; Lee, C.; Choi, H.; Gautam, J.; Jang, H.; Kim, G.J.; Lee, Y.J.; Chaudhary, C.L.; Park, S.W.; Nam, T.G.; et al. 5-Hydroxy-7-azaindolin-2-one, a novel hybrid of pyridinol and sunitinib: Design, synthesis and cytotoxicity against cancer cells. Org. Biomol. Chem. 2016, 14, 4829–4841. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Zhang, Y.; Zhang, L.; Xu, W.; Ma, W.; Sun, R.; Zeng, H. Synergistic inhibition of sunitinib and ethaselen against human colorectal cancer cells proliferation. Biomed. Pharmacother. 2016, 83, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Azimian, F.; Hamzeh-Mivehroud, M.; Shahbazi Mojarrad, J.; Hemmati, S.; Dastmalchi, S. Facile one-pot sequential synthesis of novel diaryl urea derivatives and evaluation of their in vitro cytotoxicity on adenocarcinoma cells. Med. Chem. Res. 2021, 30, 672–684. [Google Scholar] [CrossRef]
- Saleh, N.M.; El-Gaby, M.S.A.; El-Adl, K.; Abd El-Sattar, N.E.A. Design, green synthesis, molecular docking and anticancer evaluations of diazepam bearing sulfonamide moieties as VEGFR-2 inhibitors. Bioorg. Chem. 2020, 104, 104350. [Google Scholar] [CrossRef]
- Hassan, R.A.; Hamed, M.I.A.; Abdou, A.M.; El-Dash, Y. Novel antiproliferative agents bearing substituted thieno[2,3-d]pyrimidine scaffold as dual VEGFR-2 and BRAF kinases inhibitors and apoptosis inducers; design, synthesis and molecular docking. Bioorg. Chem. 2022, 125, 105861. [Google Scholar] [CrossRef]
- Hu, J.M.; Chang, Y.L.; Hsieh, C.C.; Huang, S.M. The synergistic cytotoxic effects of GW5074 and sorafenib by impacting mitochondrial functions in human colorectal cancer cell lines. Front. Oncol. 2022, 12, 925653. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Citation; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Mctigue, M.; Murray, B.W.; Chen, J.H.; Deng, Y.; Solowiej, J.; Kania, R.S. Molecular conformations, interactions, and properties associated with drug efficiency and clinical performance among vegfr Tk inhibitors. Proc. Natl. Acad. Sci. USA 2012, 109, 18281. [Google Scholar] [CrossRef]
- Velázquez-Libera, J.L.; Durán-Verdugo, F.; Valdés-Jiménez, A.; Núñez-Vivanco, G.; Caballero, J. LigRMSD: A web server for automatic structure matching and RMSD calculations among identical and similar compounds in protein-ligand docking. Bioinformatics 2020, 36, 2912–2914. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera-A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- BIOVIA, Dassault Systèmes. Biovia Diccovery Studio Vizualizer, v21.1.0.20298; Dassault Systèmes: San Diego, CA, USA, 2020. [Google Scholar]
- IUPAC. Compendium of Chemical Terminology Gold Book (Version 2.3.3); International Union of Pure and Applied Chemistry: Research Triangle Park, NC, USA, 2014; p. 1296. [Google Scholar]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | L929 | NHDF | A549 | MCF7 | LoVo | HT29 | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| CC50 ± SD a (µM) | CC50 ± SD a (µM) | CC50 ± SD a (µM) | SI | CC50 ± SD a (µM) | SI | CC50 ± SD a (µM) | SI | CC50 ± SD a (µM) | SI | |
| 3a | Non-toxic b | Non-toxic | 245.15 ± 19.02 | >>1 c | N/A | - | N/A | - | 158.76 ± 12.07 | >>1 |
| 3b | 235.08 ± 11.54 | Non-toxic | N/A | - | 192.79 ± 16.90 | >>1 | N/A | - | 194.20 ± 15.36 | >>1 |
| 3c | 90.64 ± 8.75 | 171.81 ± 14.47 | N/A | - | 199.26 ± 16.06 | 0.862 | N/A | - | 224.32 ± 23.05 | 0.766 |
| 3d | Non-toxic | 126.17 ± 9.87 | 178.95 ± 14.85 | 0.705 | 133.63 ± 14.03 | 0.944 | 226.05 ± 8.28 | 0.558 | 153.79 ± 13.38 | 0.820 |
| 3e | 89.20 ± 6.04 | 124.65 ± 11.46 | 124.77 ± 12.05 | 0.999 | 155.03 ± 15.54 | 0.804 | 177.52 ± 6.65 | 0.702 | 129.41 ± 10.04 | 0.963 |
| 3f | 207.49 ± 16.47 | 171.62 ± 20.70 | 147.81 ± 13.80 | 1.161 | 194.73 ± 11.97 | 0.881 | 235.26 ± 13.02 | 0.729 | 152.69 ± 15.02 | 1.124 |
| 3g | 83.67 ± 4.42 | 155.58 ± 15.49 | 138.50 ± 12.86 | 1.123 | 208.65 ± 9.67 | - | 223.93 ± 17.09 | 0.695 | 58.44 ± 8.75 | 2.662 |
| 3h | 145.75 ± 11.11 | Non-toxic | N/A | - | N/A | - | N/A | - | N/A | - |
| 3i | 171.36 ± 22.77 | Non-toxic | N/A | - | 251.20 ± 22.05 | >>1 | N/A | - | N/A | - |
| 3j | 52.85 ± 9.51 | Non-toxic | N/A | - | 197.49 ± 13.08 | >>1 | N/A | - | 99.87 ± 10.90 | >>1 |
| Cisplatin | 30.05 ± 5.07 | 11.49 ± 3.02 | 8.17 ± 2.39 | 1.406 | 29.72 ± 5.25 | 0.387 | 27.39 ± 8.01 | 0.419 | 47.17 ± 7.43 | 0.244 |
| 5-FU | 38.15 ± 7.13 | 20.37 ± 2.97 | 269.45 ± 17.15 | 0.076 | 217.47 ± 20.01 | 0.094 | 72.20 ± 14.43 | 0.282 | 381.16 ± 25.51 | 0.053 |
| Signaling Molecules | Compound 3g | Standard Deviation (SD) |
|---|---|---|
| BCL-2 | 0.41 [pg/mL] | ±0.11 |
| Caspase-3 | 12.27 [ng/mL] | ±3.03 |
| p53 | 4.22 [U/mL] | ±0.97 |
| Compound 3a | Compound 3e | Compound 3f | Compound 3g | Compound 3j | Tivozanib | |
|---|---|---|---|---|---|---|
| ∆Gbinding (kJ/mol) | −38.5 | −39.3 | −40.0 | −41.0 | −47.3 | −48.1 |
| Ki (μM) | 0.16 | 0.11 | 0.09 | 0.06 | 0.04 | 0.02 |
| Conventional HB | Glu917 Cys919 | Glu917 Cys919 | Glu917 | Glu917 Cys919 | Glu917 Cys919 | Glu885 Cys919 |
| Carbon HB | Ala866 Val914 Asp1046 | - | Ala866 Val914 | Glu885 Asp1046 | Glu885 Val914 Cys1045 | Leu840 Glu917 Cys919 Lys920 |
| π–π stacking | Phe918 | Phe918 Phe1047 | Phe918 Phe1047 | Phe918 Phe1047 | Phe918 | Phe918 |
| Alkyl, π–alkyl | Leu840 Val848 Ala866 Lys868 Val916 Cys919 Leu1035 Cys1045 Phe1047 | Leu840 Val848 Ala866 Lys868 Val914 Val916 Cys919 Phe1047 | Leu840 Val848 Ala866 Leu889 Val916 Cys919 Phe1047 | Leu840 Val848 Ala866 Lys868 Leu889 Val899 Val916 Cys919 Phe1047 | Leu840 Ala866 Lys868 Leu1035 Phe1047 | Val848 Ala866 Lys868 Leu889 Ile892 Val916 Phe918 Leu1035 Cys1045 Phe1047 |
| π–σ | Leu1035 | Leu1035 | Leu1035 | Val848 Leu1035 | Val848 Leu1035 | Leu889 |
| Van der Waals | Val867 Glu885 Val899 | Val867 Glu885 Leu889 Val899 Cys1045 Asp1046 Ala1050 | Val867 Lys868 Glu885 Leu889 Ile915 Asp1046 | Val914 Cys1045 | Val867 Leu889 Val899 Val916 Gly922 Cys1045 | Ile888 Val899 Gly992 Asp1046 |
| π–Sulfur | - | - | Cys1045 | - | - | - |
| Drug Name | IC50 (µM) | |||
|---|---|---|---|---|
| A549 | MCF7 | LoVo | HT29 | |
| Tivozanib | 0.19 ± 0.04 a | 0.25 ± 0.03 a | No data | No data |
| Regorafenib | No data | No data | 2 b | 2.75 ± 0.18 c |
| Cabozantinib | 9.52 ± 0.91 d 15.23 ± 0.6 e | 14.26 ± 0.87 e | No data | 11.5 ± 1.1 d |
| Lenvatinib | No effect f | No data | No data | 431 g |
| Axitinib | 4.88 ± 0.83 h | No data | No data | 13.12 ± 1.84 h |
| Vandetanib | 2.7 ± 0.5 i | 12.23 ± 0.28 j | 3.5 ± 0.9 k | 10 ± 0.4 k |
| Pazopanib | 4–6 l | 6.29 m | No data | >5 n |
| Sunitinib | 6.98 o | 7.30 o | 4.18 ± 0.27 p | 4.89 o 2.25 ± 0.27 p |
| Sorafenib | 5.62 ± 2.13 r | 7.26 ± 0.3 s 2.0 t | 31 u | 17.28 ± 3.8 r |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sochacka-Ćwikła, A.; Mączyński, M.; Czyżnikowska, Ż.; Wiatrak, B.; Jęśkowiak, I.; Czerski, A.; Regiec, A. New Oxazolo[5,4-d]pyrimidines as Potential Anticancer Agents: Their Design, Synthesis, and In Vitro Biological Activity Research. Int. J. Mol. Sci. 2022, 23, 11694. https://doi.org/10.3390/ijms231911694
Sochacka-Ćwikła A, Mączyński M, Czyżnikowska Ż, Wiatrak B, Jęśkowiak I, Czerski A, Regiec A. New Oxazolo[5,4-d]pyrimidines as Potential Anticancer Agents: Their Design, Synthesis, and In Vitro Biological Activity Research. International Journal of Molecular Sciences. 2022; 23(19):11694. https://doi.org/10.3390/ijms231911694
Chicago/Turabian StyleSochacka-Ćwikła, Aleksandra, Marcin Mączyński, Żaneta Czyżnikowska, Benita Wiatrak, Izabela Jęśkowiak, Albert Czerski, and Andrzej Regiec. 2022. "New Oxazolo[5,4-d]pyrimidines as Potential Anticancer Agents: Their Design, Synthesis, and In Vitro Biological Activity Research" International Journal of Molecular Sciences 23, no. 19: 11694. https://doi.org/10.3390/ijms231911694
APA StyleSochacka-Ćwikła, A., Mączyński, M., Czyżnikowska, Ż., Wiatrak, B., Jęśkowiak, I., Czerski, A., & Regiec, A. (2022). New Oxazolo[5,4-d]pyrimidines as Potential Anticancer Agents: Their Design, Synthesis, and In Vitro Biological Activity Research. International Journal of Molecular Sciences, 23(19), 11694. https://doi.org/10.3390/ijms231911694