
Hepatocyte-Specific Smad4 Deficiency Alleviates Liver Fibrosis via the p38/p65 Pathway


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Smad4 Expression Is Upregulated in Hepatocytes during Liver Fibrosis
2.2. Hepatocyte-Specific Smad4 Deficiency Attenuates Liver Fibrosis
2.3. Hepatocyte-Specific Smad4 Deficiency Reduces Cell Proliferation and EMT
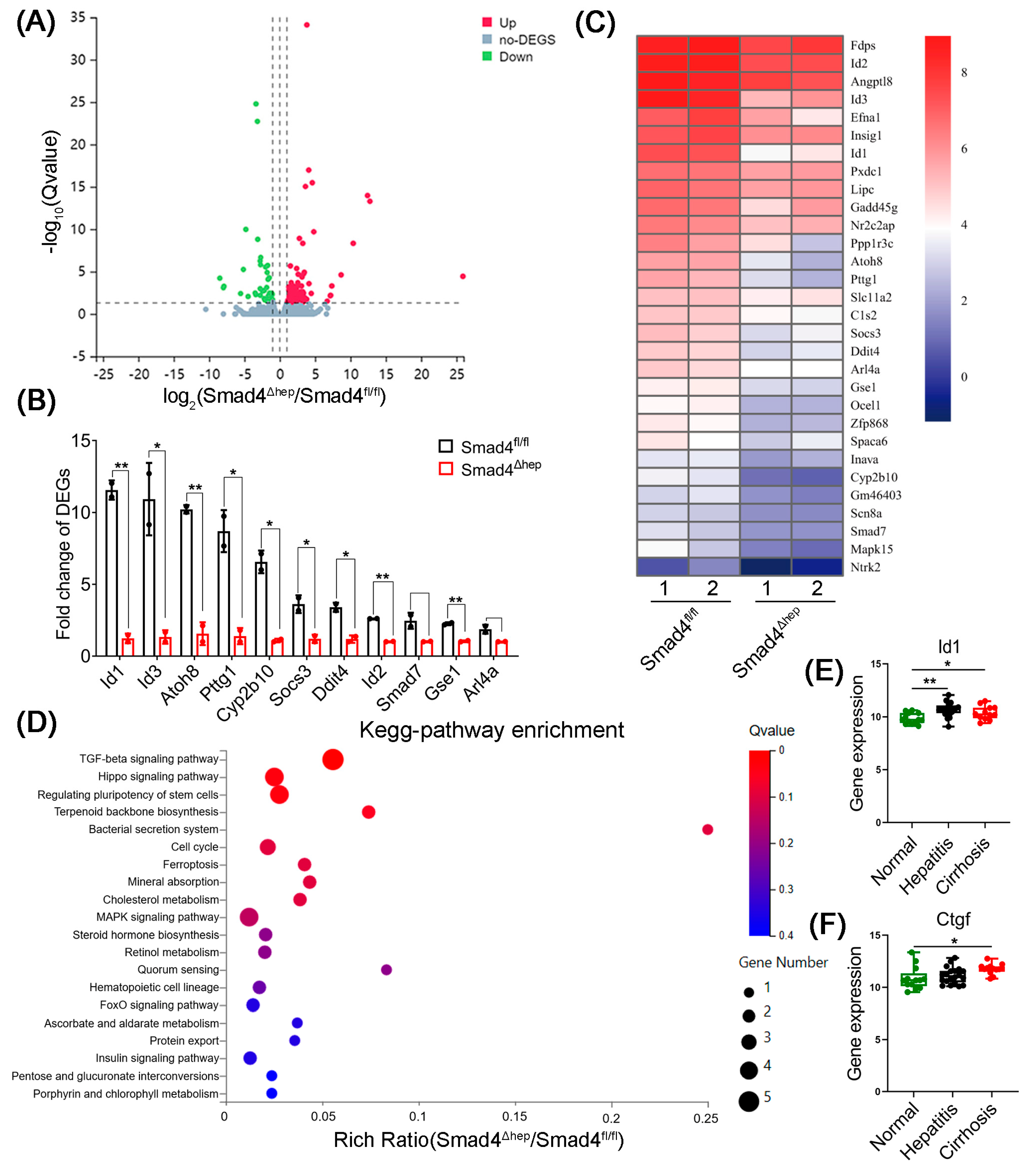
2.4. Hepatocyte-Specific Smad4 Deficiency Reduced ID1 and CTGF Expression
2.5. CTGF Promotes HSCs Activation via p38/p65 Signaling
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Carbon Tetrachloride (CCl4)-Induced Acute Liver Fibrosis Model
4.3. Immunohistochemistry and Immunofluorescence Analysis
4.4. Western Blotting Analysis
4.5. Isolation of Mouse Primary Hepatocytes
4.6. Cell Culture
4.7. Small Interfering RNA (siRNA) Interference
4.8. Quantitative Real-Time Polymerase Chain Reaction (qPCR)
4.9. Cell Viability Analysis
4.10. Wound-Healing Assay
4.11. Flow Cytometry Analysis
4.12. RNA Sequencing Analysis
4.13. Public Database Analysis
4.14. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Kisseleva, T.; Brenner, D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Li, J.; Zhang, J.; Dai, C.; Liu, X.; Wang, J.; Gao, Z.; Guo, H.; Wang, R.; Lu, S.; et al. S100A4 promotes liver fibrosis via activation of hepatic stellate cells. J. Hepatol. 2015, 62, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Seki, E.; Schwabe, R.F. Hepatic inflammation and fibrosis: Functional links and key pathways. Hepatology 2015, 61, 1066–1079. [Google Scholar] [CrossRef]
- Chen, Y.; Fan, Y.; Guo, D.Y.; Xu, B.; Shi, X.Y.; Li, J.T.; Duan, L.F. Study on the relationship between hepatic fibrosis and epithelial-mesenchymal transition in intrahepatic cells. Biomed. Pharmacother. 2020, 129, 110413–110424. [Google Scholar] [CrossRef]
- Zeisberg, M.; Yang, C.; Martino, M.; Duncan, M.B.; Rieder, F.; Tanjore, H.; Kalluri, R. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J. Biol. Chem. 2007, 282, 23337–23347. [Google Scholar] [CrossRef]
- Oh, S.H.; Swiderska-Syn, M.; Jewell, M.L.; Premont, R.T.; Diehl, A.M. Liver regeneration requires Yap1-TGFbeta-dependent epithelial-mesenchymal transition in hepatocytes. J. Hepatol. 2018, 69, 359–367. [Google Scholar] [CrossRef]
- Mooring, M.; Fowl, B.H.; Lum, S.Z.C.; Liu, Y.; Yao, K.; Softic, S.; Kirchner, R.; Bernstein, A.; Singhi, A.D.; Jay, D.G.; et al. Hepatocyte Stress Increases Expression of Yes-Associated Protein and Transcriptional Coactivator With PDZ-Binding Motif in Hepatocytes to Promote Parenchymal Inflammation and Fibrosis. Hepatology 2020, 71, 1813–1830. [Google Scholar] [CrossRef]
- Borkham-Kamphorst, E.; Weiskirchen, R. The PDGF system and its antagonists in liver fibrosis. Cytokine Growth Factor Rev. 2016, 28, 53–61. [Google Scholar] [CrossRef]
- Brenner, C.; Galluzzi, L.; Kepp, O.; Kroemer, G. Decoding cell death signals in liver inflammation. J. Hepatol. 2013, 59, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Liu, C.; Zhou, D.; Zhang, L. TGF-beta/SMAD Pathway and Its Regulation in Hepatic Fibrosis. J. Histochem. Cytochem. 2016, 64, 157–167. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, A.J.; Chetty, R. Smad4/DPC4. J. Clin. Pathol. 2018, 71, 661–664. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Mishra, L.; Deng, C.X. The role of TGF-beta/SMAD4 signaling in cancer. Int. J. Biol. Sci. 2018, 14, 111–123. [Google Scholar] [CrossRef]
- Moussa, M.M.; Helal, N.S.; Youssef, M.M. Significance of pSmad2/3 and Smad4 in hepatitis C virus-related liver fibrosis and hepatocellular carcinoma. APMIS 2018, 126, 477–485. [Google Scholar] [CrossRef]
- Qin, G.; Wang, G.Z.; Guo, D.D.; Bai, R.X.; Wang, M.; Du, S.Y. Deletion of Smad4 attenuates the hepatic inflammation and fibrogenesis during nonalcoholic steatohepatitis progression. J. Dig. Dis. 2018, 19, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Inokuchi, S.; Roh, Y.S.; Song, J.; Loomba, R.; Park, E.J.; Seki, E. Transforming growth factor-beta signaling in hepatocytes promotes hepatic fibrosis and carcinogenesis in mice with hepatocyte-specific deletion of TAK1. Gastroenterology 2013, 144, 1042–1054.e4. [Google Scholar] [CrossRef]
- Khanizadeh, S.; Ravanshad, M.; Hosseini, S.; Davoodian, P.; Zadeh, A.N.; Sarvari, J. Blocking of SMAD4 expression by shRNA effectively inhibits fibrogenesis of human hepatic stellate cells. Gastroenterol. Hepatol. Bed Bench 2015, 8, 262–269. [Google Scholar]
- Wang, R.H.; Li, C.; Xu, X.; Zheng, Y.; Xiao, C.; Zerfas, P.; Cooperman, S.; Eckhaus, M.; Rouault, T.; Mishra, L.; et al. A role of SMAD4 in iron metabolism through the positive regulation of hepcidin expression. Cell Metab. 2005, 2, 399–409. [Google Scholar] [CrossRef]
- Hernanda, P.Y.; Chen, K.; Das, A.M.; Sideras, K.; Wang, W.; Li, J.; Cao, W.; Bots, S.J.; Kodach, L.L.; de Man, R.A.; et al. SMAD4 exerts a tumor-promoting role in hepatocellular carcinoma. Oncogene 2015, 34, 5055–5068. [Google Scholar] [CrossRef]
- Zhao, H.; Wang, Z.; Tang, F.; Zhao, Y.; Feng, D.; Li, Y.; Hu, Y.; Wang, C.; Zhou, J.; Tian, X.; et al. Carnosol-mediated Sirtuin 1 activation inhibits Enhancer of Zeste Homolog 2 to attenuate liver fibrosis. Pharmacol. Res. 2018, 128, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Wang, D.; Song, J. Cortactin is involved in transforming growth factor-beta1-induced epithelial-mesenchymal transition in AML-12 cells. Acta Biochim. Biophys. Sin. 2009, 41, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Bu, Y.; Jiang, S.; Chang, F.; Jia, F.; Xiao, X.; Song, G.; Zhang, M.; Ning, P.; Jia, Q. CCN2-MAPK-Id-1 loop feedback amplification is involved in maintaining stemness in oxaliplatin-resistant hepatocellular carcinoma. Hepatol. Int. 2019, 13, 440–453. [Google Scholar] [CrossRef] [PubMed]
- Weng, H.-L.; Ciuclan, L.; Liu, Y.; Hamzavi, J.; Godoy, P.; Gaitantzi, H.; Kanzler, S.; Heuchel, R.; Ueberham, U.; Gebhardt, R.; et al. Profibrogenic transforming growth factor-β/activin receptor-like kinase 5 signaling via connective tissue growth factor expression in hepatocytes. Hepatology 2007, 46, 1257–1270. [Google Scholar] [CrossRef]
- Rayego-Mateos, S.; Morgado-Pascual, J.L.; Rodrigues-Diez, R.R.; Rodrigues-Diez, R.; Falke, L.L.; Mezzano, S.; Ortiz, A.; Egido, J.; Goldschmeding, R.; Ruiz-Ortega, M. Connective tissue growth factor induces renal fibrosis via epidermal growth factor receptor activation. J. Pathol. 2018, 244, 227–241. [Google Scholar] [CrossRef]
- Yuan, H.H.; Liang, Z.; Yi, J.; Chen, X.J.; Li, R.F.; Wu, J.; Sun, Z.L. Koumine Promotes ROS Production to Suppress Hepatocellular Carcinoma Cell Proliferation Via NF-κB and ERK/p38 MAPK Signaling. Biomolecules 2019, 9, 559–571. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; Pan, L.; Qi, S.; Liu, F.; Wang, Z.; Qian, C.; Chen, L.; Du, J. RNF2 Mediates Hepatic Stellate Cells Activation by Regulating ERK/p38 Signaling Pathway in LX-2 Cells. Front. Cell Dev. Biol. 2021, 9, 634902–634912. [Google Scholar] [CrossRef]
- Mu, M.; Zuo, S.; Wu, R.M.; Deng, K.S.; Lu, S.; Zhu, J.J.; Zou, G.L.; Yang, J.; Cheng, M.L.; Zhao, X.K. Ferulic acid attenuates liver fibrosis and hepatic stellate cell activation via inhibition of TGF-beta/Smad signaling pathway. Drug Des. Devel. Ther. 2018, 12, 4107–4115. [Google Scholar] [CrossRef]
- Xia, Y.; Yu, B.; Ma, C.; Tu, Y.; Zhai, L.; Yang, Y.; Liu, D.; Liu, Y.; Wu, H.; Dan, H.; et al. Yu Gan Long reduces rat liver fibrosis by blocking TGF-beta1/Smad pathway and modulating the immunity. Biomed. Pharmacother. 2018, 106, 1332–1338. [Google Scholar] [CrossRef]
- Tee, J.K.; Peng, F.; Tan, Y.L.; Yu, B.; Ho, H.K. Magnesium Isoglycyrrhizinate Ameliorates Fibrosis and Disrupts TGF-beta-Mediated SMAD Pathway in Activated Hepatic Stellate Cell Line LX2. Front. Pharmacol. 2018, 9, 1018–1032. [Google Scholar] [CrossRef]
- Wang, C.Y.; Xiao, X.; Bayer, A.; Xu, Y.; Dev, S.; Canali, S.; Nair, A.V.; Masia, R.; Babitt, J.L. Ablation of Hepatocyte Smad1, Smad5, and Smad8 Causes Severe Tissue Iron Loading and Liver Fibrosis in Mice. Hepatology 2019, 70, 1986–2002. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; He, Z.; Leng, X.; Liang, Z.; Peng, J.; Zhang, H.; Xiao, M.; Zhang, H.; Liu, C.; Zhang, X. Effects of Smad4 on liver fibrosis and hepatocarcinogenesis in mice treated with CCl4/ethanol. Zhonghua Gan Zang Bing Za Zhi 2010, 18, 119–123. [Google Scholar] [PubMed]
- Huda, N.; Liu, G.; Hong, H.; Yan, S.; Khambu, B.; Yin, X.M. Hepatic senescence, the good and the bad. World J. Gastroenterol. 2019, 25, 5069–5081. [Google Scholar] [CrossRef]
- Kong, D.; Zhang, Z.; Chen, L.; Huang, W.; Zhang, F.; Wang, L.; Wang, Y.; Cao, P.; Zheng, S. Curcumin blunts epithelial-mesenchymal transition of hepatocytes to alleviate hepatic fibrosis through regulating oxidative stress and autophagy. Redox Biol. 2020, 36, 101600–101610. [Google Scholar] [CrossRef]
- Kaimori, A.; Potter, J.; Kaimori, J.Y.; Wang, C.; Mezey, E.; Koteish, A. Transforming growth factor-beta1 induces an epithelial-to-mesenchymal transition state in mouse hepatocytes in vitro. J. Biol. Chem. 2007, 282, 22089–22101. [Google Scholar] [CrossRef] [PubMed]
- Taura, K.; Miura, K.; Iwaisako, K.; Osterreicher, C.H.; Kodama, Y.; Penz-Osterreicher, M.; Brenner, D.A. Hepatocytes do not undergo epithelial-mesenchymal transition in liver fibrosis in mice. Hepatology 2010, 51, 1027–1036. [Google Scholar] [CrossRef]
- Makino, Y.; Hikita, H.; Kodama, T.; Shigekawa, M.; Yamada, R.; Sakamori, R.; Eguchi, H.; Morii, E.; Yokoi, H.; Mukoyama, M.; et al. CTGF Mediates Tumor-Stroma Interactions between Hepatoma Cells and Hepatic Stellate Cells to Accelerate HCC Progression. Cancer Res. 2018, 78, 4902–4914. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Bo, Z.; Gong, W.; Guo, Y. Inhibitor of Differentiation 1 (Id1) in Cancer and Cancer Therapy. Int. J. Med. Sci. 2020, 17, 995–1005. [Google Scholar] [CrossRef]
- Kwon, Y.C.; Sasaki, R.; Meyer, K.; Ray, R. Hepatitis C Virus Core Protein Modulates Endoglin (CD105) Signaling Pathway for Liver Pathogenesis. J. Virol. 2017, 91, e01235-17. [Google Scholar] [CrossRef]
- Yin, X.; Tang, B.; Li, J.H.; Wang, Y.; Zhang, L.; Xie, X.Y.; Zhang, B.H.; Qiu, S.J.; Wu, W.Z.; Ren, Z.G. ID1 promotes hepatocellular carcinoma proliferation and confers chemoresistance to oxaliplatin by activating pentose phosphate pathway. J. Exp. Clin. Cancer Res. 2017, 36, 166–179. [Google Scholar] [CrossRef]
- Kautz, L.; Meynard, D.; Monnier, A.; Darnaud, V.; Bouvet, R.; Wang, R.H.; Deng, C.; Vaulont, S.; Mosser, J.; Coppin, H.; et al. Iron regulates phosphorylation of Smad1/5/8 and gene expression of Bmp6, Smad7, Id1, and Atoh8 in the mouse liver. Blood 2008, 112, 1503–1509. [Google Scholar] [CrossRef] [PubMed]
- Kodama, T.; Takehara, T.; Hikita, H.; Shimizu, S.; Shigekawa, M.; Tsunematsu, H.; Li, W.; Miyagi, T.; Hosui, A.; Tatsumi, T.; et al. Increases in p53 expression induce CTGF synthesis by mouse and human hepatocytes and result in liver fibrosis in mice. J. Clin. Invest. 2011, 121, 3343–3356. [Google Scholar] [CrossRef] [PubMed]
- Rachfal, A. Connective tissue growth factor (CTGF/CCN2) in hepatic fibrosis. Hepatol. Res. 2003, 26, 1–9. [Google Scholar] [CrossRef]
- Huang, G.; Brigstock, D.R. Regulation of hepatic stellate cells by connective tissue growth factor. Front. Biosci. 2012, 17, 2495–2507. [Google Scholar] [CrossRef] [PubMed]
- Rayego-Mateos, S.; Rodrigues-Diez, R.; Morgado-Pascual, J.L.; Valentijn, F.; Valdivielso, J.M.; Goldschmeding, R.; Ruiz-Ortega, M. Role of Epidermal Growth Factor Receptor (EGFR) and Its Ligands in Kidney Inflammation and Damage. Mediat. Inflamm. 2018, 2018, 8739473–8739495. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, B.C.; Hoshida, Y.; Fujii, T.; Wei, L.; Yamada, S.; Lauwers, G.Y.; McGinn, C.M.; DePeralta, D.K.; Chen, X.; Kuroda, T.; et al. Epidermal growth factor receptor inhibition attenuates liver fibrosis and development of hepatocellular carcinoma. Hepatology 2014, 59, 1577–1590. [Google Scholar] [CrossRef] [PubMed]
- Tormos, A.M.; Arduini, A.; Talens-Visconti, R.; del Barco Barrantes, I.; Nebreda, A.R.; Sastre, J. Liver-specific p38alpha deficiency causes reduced cell growth and cytokinesis failure during chronic biliary cirrhosis in mice. Hepatology 2013, 57, 1950–1961. [Google Scholar] [CrossRef]
- Steins, M.; Thomas, M.; Geissler, M. Erlotinib. Recent Results Cancer Res. 2014, 201, 109–123. [Google Scholar]
- Goffin, J.R.; Zbuk, K. Epidermal growth factor receptor: Pathway, therapies, and pipeline. Clin. Ther. 2013, 35, 1282–1303. [Google Scholar] [CrossRef]
- Tan, S.; Liu, X.; Chen, L.; Wu, X.; Tao, L.; Pan, X.; Tan, S.; Liu, H.; Jiang, J.; Wu, B. Fas/FasL mediates NF-kappaBp65/PUMA-modulated hepatocytes apoptosis via autophagy to drive liver fibrosis. Cell Death Dis. 2021, 12, 474–492. [Google Scholar] [CrossRef]
- Czerkies, M.; Korwek, Z.; Prus, W.; Kochanczyk, M.; Jaruszewicz-Blonska, J.; Tudelska, K.; Blonski, S.; Kimmel, M.; Brasier, A.R.; Lipniacki, T. Cell fate in antiviral response arises in the crosstalk of IRF, NF-kappaB and JAK/STAT pathways. Nat. Commun. 2018, 9, 493–507. [Google Scholar] [CrossRef] [PubMed]
- Feng, G.S. Conflicting roles of molecules in hepatocarcinogenesis: Paradigm or paradox. Cancer Cell 2012, 21, 150–154. [Google Scholar] [CrossRef] [PubMed]
- Postic, C.; Shiota, M.; Niswender, K.D.; Jetton, T.L.; Chen, Y.; Moates, J.M.; Shelton, K.D.; Lindner, J.; Cherrington, A.D.; Magnuson, M.A. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J. Biol. Chem. 1999, 274, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Zhang, J.; Liu, Y.; Chen, H.; Liu, H.; Wang, J.; Niu, M.; Hou, L.; Wu, Z.; Chen, Z.; et al. MyD88 in myofibroblasts regulates aerobic glycolysis-driven hepatocarcinogenesis via ERK-dependent PKM2 nuclear relocalization and activation. J. Pathol. 2022, 256, 414–426. [Google Scholar] [CrossRef]
- Li, W.C.; Ralphs, K.L.; Tosh, D. Isolation and culture of adult mouse hepatocytes. Methods Mol. Biol. 2010, 633, 185–196. [Google Scholar]
- Zhang, J.; Song, K.; Wang, J.; Li, Y.; Liu, S.; Dai, C.; Chen, L.; Wang, S.; Qin, Z. S100A4 blockage alleviates agonistic anti-CD137 antibody-induced liver pathology without disruption of antitumor immunity. Oncoimmunology 2018, 7, e1296996–e1297011. [Google Scholar] [CrossRef]
- Ge, S.; Yang, W.; Chen, H.; Yuan, Q.; Liu, S.; Zhao, Y.; Zhang, J. MyD88 in Macrophages Enhances Liver Fibrosis by Activation of NLRP3 Inflammasome in HSCs. Int. J. Mol. Sci. 2021, 22, 12413–12430. [Google Scholar] [CrossRef]
- Shen, Q.; Eun, J.W.; Lee, K.; Kim, H.S.; Yang, H.D.; Kim, S.Y.; Lee, E.K.; Kim, T.; Kang, K.; Kim, S.; et al. Barrier to autointegration factor 1, procollagen-lysine, 2-oxoglutarate 5-dioxygenase 3, and splicing factor 3b subunit 4 as early-stage cancer decision markers and drivers of hepatocellular carcinoma. Hepatology 2018, 67, 1360–1377. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, M.; Yan, X.; Xin, X.; Chen, H.; Hou, L.; Zhang, J. Hepatocyte-Specific Smad4 Deficiency Alleviates Liver Fibrosis via the p38/p65 Pathway. Int. J. Mol. Sci. 2022, 23, 11696. https://doi.org/10.3390/ijms231911696
Wei M, Yan X, Xin X, Chen H, Hou L, Zhang J. Hepatocyte-Specific Smad4 Deficiency Alleviates Liver Fibrosis via the p38/p65 Pathway. International Journal of Molecular Sciences. 2022; 23(19):11696. https://doi.org/10.3390/ijms231911696
Chicago/Turabian StyleWei, Miaomiao, Xinlong Yan, Xin Xin, Haiqiang Chen, Lingling Hou, and Jinhua Zhang. 2022. "Hepatocyte-Specific Smad4 Deficiency Alleviates Liver Fibrosis via the p38/p65 Pathway" International Journal of Molecular Sciences 23, no. 19: 11696. https://doi.org/10.3390/ijms231911696
APA StyleWei, M., Yan, X., Xin, X., Chen, H., Hou, L., & Zhang, J. (2022). Hepatocyte-Specific Smad4 Deficiency Alleviates Liver Fibrosis via the p38/p65 Pathway. International Journal of Molecular Sciences, 23(19), 11696. https://doi.org/10.3390/ijms231911696