
ALDH3A2, ODF2, QSOX2, and MicroRNA-503-5p Expression to Forecast Recurrence in TMPRSS2-ERG-Positive Prostate Cancer
 ,
,  , ,
, ,  , , , ,
, , , , 
Abstract
1. Introduction
2. Results
2.1. Differentially Expressed miRNAs
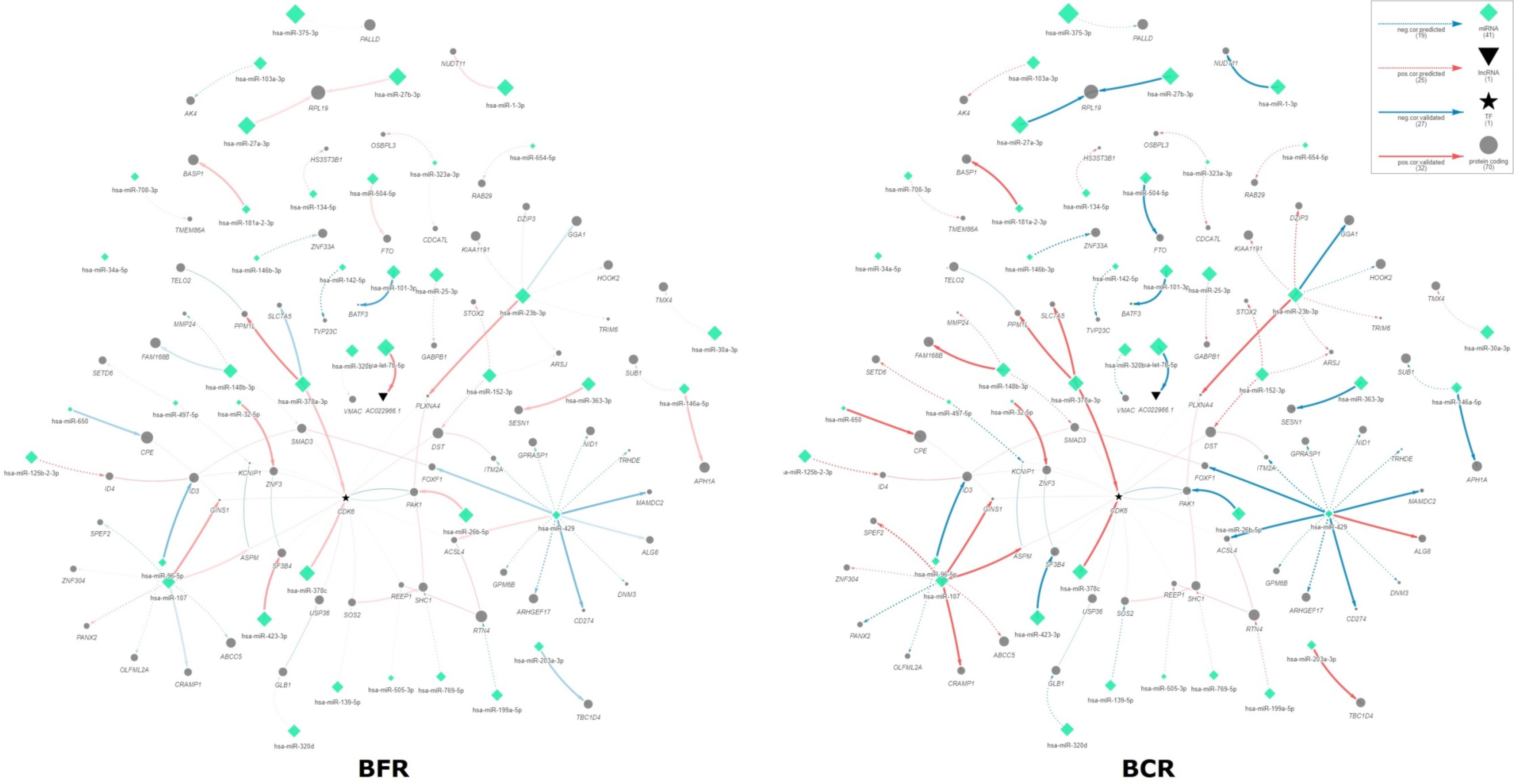
2.2. Interactome Network miRNA–Target Correlations Associated with BCR
2.3. Predictive Model of BCR Based on mRNA and miRNA Expression
2.4. mRNA and miRNA Expression Validation by qPCR
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Methods
4.2.1. Isolation of RNA and Reverse Transcription
4.2.2. miRNA Sequencing
4.2.3. Quantitative PCR (qPCR)
4.2.4. Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cucchiara, V.; Cooperberg, M.R.; Dall’Era, M.; Lin, D.W.; Montorsi, F.; Schalken, J.A.; Evans, C.P. Genomic Markers in Prostate Cancer Decision Making. Eur. Urol. 2018, 73, 572–582. [Google Scholar] [CrossRef] [PubMed]
- Rebello, R.J.; Oing, C.; Knudsen, K.E.; Loeb, S.; Johnson, D.C.; Reiter, R.E.; Gillessen, S.; Van der Kwast, T.; Bristow, R.G. Prostate cancer. Nat. Rev. Dis. Prim. 2021, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Pettersson, A.; Graff, R.E.; Bauer, S.R.; Pitt, M.J.; Lis, R.T.; Stack, E.C.; Martin, N.E.; Kunz, L.; Penney, K.L.; Ligon, A.H.; et al. The TMPRSS2:ERG rearrangement, ERG expression, and prostate cancer outcomes: A cohort study and meta-analysis. Cancer Epidemiol. Biomark. Prev. 2012, 21, 1497–1509. [Google Scholar] [CrossRef]
- D’Amico, A.V.; Whittington, R.; Malkowicz, S.B.; Schultz, D.; Blank, K.; Broderick, G.A.; Tomaszewski, J.E.; Renshaw, A.A.; Kaplan, I.; Beard, C.J.; et al. Biochemical outcome after radical prostatectomy, external beam radiation therapy, or interstitial radiation therapy for clinically localized prostate cancer. J. Am. Med Assoc. 1998, 280, 969–974. [Google Scholar] [CrossRef] [PubMed]
- Moschini, M.; Spahn, M.; Mattei, A.; Cheville, J.; Karnes, R.J. Incorporation of tissue-based genomic biomarkers into localized prostate cancer clinics. BMC Med. 2016, 14, 67. [Google Scholar] [CrossRef] [PubMed]
- Sternberg, I.A.; Vela, I.; Scardino, P.T. Molecular Profiles of Prostate Cancer: To Treat or Not to Treat. Annu. Rev. Med. 2016, 67, 119–135. [Google Scholar] [CrossRef]
- Kretschmer, A.; Tilki, D. Biomarkers in prostate cancer—Current clinical utility and future perspectives. Crit. Rev. Oncol. Hematol. 2017, 120, 180–193. [Google Scholar] [CrossRef]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef]
- Arora, K.; Barbieri, C.E. Molecular Subtypes of Prostate Cancer. Curr. Oncol. Rep. 2018, 20, 58. [Google Scholar] [CrossRef] [PubMed]
- Carver, B.S.; Tran, J.; Chen, Z.; Carracedo-Perez, A.; Alimonti, A.; Nardella, C.; Gopalan, A.; Scardino, P.T.; Cordon-Cardo, C.; Gerald, W.; et al. ETS rearrangements and prostate cancer initiation. Nature 2009, 457, E1, discussion E2–3. [Google Scholar] [CrossRef] [PubMed]
- Perner, S.; Mosquera, J.M.; Demichelis, F.; Hofer, M.D.; Paris, P.L.; Simko, J.; Collins, C.; Bismar, T.A.; Chinnaiyan, A.M.; De Marzo, A.M.; et al. TMPRSS2-ERG fusion prostate cancer: An early molecular event associated with invasion. Am. J. Surg. Pathol. 2007, 31, 882–888. [Google Scholar] [CrossRef] [PubMed]
- Adamo, P.; Ladomery, M.R. The oncogene ERG: A key factor in prostate cancer. Oncogene 2016, 35, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Hagglof, C.; Hammarsten, P.; Stromvall, K.; Egevad, L.; Josefsson, A.; Stattin, P.; Granfors, T.; Bergh, A. TMPRSS2-ERG expression predicts prostate cancer survival and associates with stromal biomarkers. PLoS ONE 2014, 9, e86824. [Google Scholar] [CrossRef]
- Hill, M.; Tran, N. miRNA interplay: Mechanisms and consequences in cancer. Dis. Model. Mech. 2021, 14, dmm047662. [Google Scholar] [CrossRef]
- He, B.; Zhao, Z.; Cai, Q.; Zhang, Y.; Zhang, P.; Shi, S.; Xie, H.; Peng, X.; Yin, W.; Tao, Y.; et al. miRNA-based biomarkers, therapies, and resistance in Cancer. Int. J. Biol. Sci. 2020, 16, 2628–2647. [Google Scholar] [CrossRef]
- Zhang, X.; Pan, Y.; Fu, H.; Zhang, J. miRNAs-205 and miRNAs-338-3p Reduces Cell Apoptosis in Prostate Carcinoma Tissue and LNCaP Prostate Carcinoma Cells by Directly Targeting the B-Cell Lymphoma 2 (Bcl-2) Gene. Med. Sci. Monit. 2019, 25, 1122–1132. [Google Scholar] [CrossRef]
- Wang, X.; Ruan, Y.; Wang, X.; Zhao, W.; Jiang, Q.; Jiang, C.; Zhao, Y.; Xu, Y.; Sun, F.; Zhu, Y.; et al. Long intragenic non-coding RNA lincRNA-p21 suppresses development of human prostate cancer. Cell Prolif. 2017, 50, e12318. [Google Scholar] [CrossRef]
- Sindhu, K.J.; Venkatesan, N.; Karunagaran, D. MiRNAs Interactome Multiomics Characterization for Cancer Research and Personalized Medicine: An Expert Review. OMICS A J. Integr. Biol. 2021, 25, 545–566. [Google Scholar] [CrossRef]
- Kobelyatskaya, A.A.; Pudova, E.A.; Snezhkina, A.V.; Fedorova, M.S.; Pavlov, V.S.; Guvatova, Z.G.; Savvateeva, M.V.; Melnikova, N.V.; Dmitriev, A.A.; Trofimov, D.Y.; et al. Impact TMPRSS2-ERG Molecular Subtype on Prostate Cancer Recurrence. Life 2021, 11, 588. [Google Scholar] [CrossRef] [PubMed]
- Tan, G.; Jiang, L.; Li, G.; Bai, K. ESTAT3 Inhibitor AG-490 Inhibits the Growth of Prostate Cancer by miR-503-5p Both In Vivo and In Vitro. Technol. Cancer Res. Treat. 2020, 19, 1533033820948062. [Google Scholar] [CrossRef] [PubMed]
- Bjorklund, M.; Koivunen, E. Gelatinase-mediated migration and invasion of cancer cells. Biochim. Biophys. Acta 2005, 1755, 37–69. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Han, Q.; Chi, C.; Zhu, Y.; Pan, J.; Dong, B.; Huang, Y.; Xia, W.; Xue, W.; Sha, J. Transcriptional regulation of PRKAR2B by miR-200b-3p/200c-3p and XBP1 in human prostate cancer. Biomed. Pharmacother. 2020, 124, 109863. [Google Scholar] [CrossRef]
- Pelka, K.; Klicka, K.; Grzywa, T.M.; Gondek, A.; Marczewska, J.M.; Garbicz, F.; Szczepaniak, K.; Paskal, W.; Wlodarski, P.K. miR-96-5p, miR-134-5p, miR-181b-5p and miR-200b-3p heterogenous expression in sites of prostate cancer versus benign prostate hyperplasia-archival samples study. Histochem. Cell Biol. 2021, 155, 423–433. [Google Scholar] [CrossRef]
- Janiak, M.; Paskal, W.; Rak, B.; Garbicz, F.; Jarema, R.; Sikora, K.; Wlodarski, P. TIMP4 expression is regulated by miR-200b-3p in prostate cancer cells. APMIS 2017, 125, 101–105. [Google Scholar] [CrossRef]
- Kai, H.S.; Butler, G.S.; Morrison, C.J.; King, A.E.; Pelman, G.R.; Overall, C.M. Utilization of a novel recombinant myoglobin fusion protein expression system to characterize the tissue inhibitor of metalloproteinase (TIMP)-4 and TIMP-2 C-terminal domain and tails by mutagenesis. The importance of acidic residues in binding the MMP-2 hemopexin C-domain. J. Biol. Chem. 2002, 277, 48696–48707. [Google Scholar] [CrossRef]
- Long, N.P.; Lee, W.J.; Huy, N.T.; Lee, S.J.; Park, J.H.; Kwon, S.W. Novel Biomarker Candidates for Colorectal Cancer Metastasis: A Meta-analysis of In Vitro Studies. Cancer Inform. 2016, 15, 11–17. [Google Scholar] [CrossRef]
- Antonowicz, S.; Bodai, Z.; Wiggins, T.; Markar, S.R.; Boshier, P.R.; Goh, Y.M.; Adam, M.E.; Lu, H.; Kudo, H.; Rosini, F.; et al. Endogenous aldehyde accumulation generates genotoxicity and exhaled biomarkers in esophageal adenocarcinoma. Nat. Commun. 2021, 12, 1454. [Google Scholar] [CrossRef]
- Soung, N.K.; Park, J.E.; Yu, L.R.; Lee, K.H.; Lee, J.M.; Bang, J.K.; Veenstra, T.D.; Rhee, K.; Lee, K.S. Plk1-dependent and -independent roles of an ODF2 splice variant, hCenexin1, at the centrosome of somatic cells. Dev. Cell. 2009, 16, 539–550. [Google Scholar] [CrossRef]
- Li, Y.; Liu, M.; Zhang, Z.; Deng, L.; Zhai, Z.; Liu, H.; Wang, Y.; Zhang, C.; Xiong, J.; Shi, C. QSOX2 Is an E2F1 Target Gene and a Novel Serum Biomarker for Monitoring Tumor Growth and Predicting Survival in Advanced NSCLC. Front. Cell Dev. Biol. 2021, 9, 688798. [Google Scholar] [CrossRef] [PubMed]
- Chai, W.X.; Sun, L.G.; Dai, F.H.; Shao, H.S.; Zheng, N.G.; Cai, H.Y. Inhibition of PRRX2 suppressed colon cancer liver metastasis via inactivation of Wnt/beta-catenin signaling pathway. Pathol. Res. Pract. 2019, 215, 152593. [Google Scholar] [CrossRef] [PubMed]
- Nazempour, N.; Taleqani, M.H.; Taheri, N.; Haji Ali Asgary Najafabadi, A.H.; Shokrollahi, A.; Zamani, A.; Fattahi Dolatabadi, N.; Peymani, M.; Mahdevar, M. The role of cell surface proteins gene expression in diagnosis, prognosis, and drug resistance of colorectal cancer: In silico analysis and validation. Exp. Mol. Pathol. 2021, 123, 104688. [Google Scholar] [CrossRef]
- Jiang, T.; Zheng, L.; Li, X.; Liu, J.; Song, H.; Xu, Y.; Dong, C.; Liu, L.; Wang, H.; Wang, S.; et al. Quiescin Sulfhydryl Oxidase 2 Overexpression Predicts Poor Prognosis and Tumor Progression in Patients With Colorectal Cancer: A Study Based on Data Mining and Clinical Verification. Front. Cell Dev. Biol. 2021, 9, 678770. [Google Scholar] [CrossRef] [PubMed]
- Krasnov, G.S.; Kudryavtseva, A.V.; Snezhkina, A.V.; Lakunina, V.A.; Beniaminov, A.D.; Melnikova, N.V.; Dmitriev, A.A. Pan-Cancer Analysis of TCGA Data Revealed Promising Reference Genes for qPCR Normalization. Front. Genet. 2019, 10, 97. [Google Scholar] [CrossRef] [PubMed]
- Krasnov, G.S.; Oparina, N.; Dmitriev, A.A.; Kudriavtsev, A.V.; Anedchenko, E.A.; Kondrat’eva, T.T.; Zabarovskii, E.R.; Senchenko, V.N. Novel reference gene RPN1 for normalization of quantitative data in lung and kidney cancer. Mol. Biol. 2011, 45, 238–248. [Google Scholar] [CrossRef]
- Patil, A.H.; Halushka, M.K. miRge3.0: A comprehensive miRNAs and tRF sequencing analysis pipeline. NAR Genom. Bioinform. 2021, 3, lqab068. [Google Scholar] [CrossRef]
- R: The R Project for Statistical Computing/February 2021. Available online: https://www.r-project.org/ (accessed on 5 June 2022).
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Ru, Y.; Kechris, K.J.; Tabakoff, B.; Hoffman, P.; Radcliffe, R.A.; Bowler, R.; Mahaffey, S.; Rossi, S.; Calin, G.A.; Bemis, L.; et al. The multiMiR R package and database: Integration of miRNAs-target interactions along with their disease and drug associations. Nucleic Acids Res. 2014, 42, e133. [Google Scholar] [CrossRef]
- Li, J.H.; Liu, S.; Zhou, H.; Qu, L.H.; Yang, J.H. starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014, 42, D92–D97. [Google Scholar] [CrossRef]
- Oki, S.; Ohta, T.; Shioi, G.; Hatanaka, H.; Ogasawara, O.; Okuda, Y.; Kawaji, H.; Nakaki, R.; Sese, J.; Meno, C. ChIP-Atlas: A data-mining suite powered by full integration of public ChIP-seq data. EMBO Rep. 2018, 19, e46255. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef] [PubMed]
- The Gene Ontology, C. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Yu, G.; He, Q.Y. ReactomePA: An R/Bioconductor package for reactome pathway analysis and visualization. Mol. Biosyst. 2016, 12, 477–479. [Google Scholar] [CrossRef]
- Sticht, C.; De La Torre, C.; Parveen, A.; Gretz, N. miRWalk: An online resource for prediction of miRNAs binding sites. PLoS ONE 2018, 13, e0206239. [Google Scholar] [CrossRef]
- Almende, B.; Thieurmel, B.; Robert, T. visNetwork: Network Visualization Using ‘vis.js’ Library. R Package. Available online: https://CRAN.R-project.org/package=visNetwork (accessed on 5 June 2022).
- Chollet, F.; Allaire, J. “Keras”. 2015/February 2021. Available online: https://keras.io (accessed on 5 June 2022).
- Abadi, M.; Agarwal, A.; Barham, P.; Brevdo, E.; Chen, Z.; Citro, C.; Corrado, G.; Davis, A.; Dean, J.; Devin, M.; et al. “Tensorflow: A System for Large-Scale Machine Learning”, 2016/February 2021. Available online: https://tensorflow.rstudio.com/ (accessed on 5 June 2022).
- Falbel, D.; Allaire, J.J.; Chollet, F. R Interface to ‘Keras’/February 2021. Available online: https://keras.rstudio.com/index.html (accessed on 5 June 2022).
- Kingma, D.P.; Ba, J. Adam: A Method for Stochastic Optimization/February 2021. Available online: https://arxiv.org/abs/1412.6980 (accessed on 5 June 2022).
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.C.; Muller, M. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNAs | FC | log2CPM | QLF p-Value | MW p-Value |
|---|---|---|---|---|
| Cohort-A | ||||
| hsa-miR-503-5p | ↑3.75 | 2.36 | 4.9 × 10−2 | 8.5 × 10−4 |
| hsa-miR-200b-3p | ↑1.52 | 11.89 | 4.5 × 10−2 | 4.9 × 10−2 |
| Cohort-B | ||||
| hsa-miR-503-5p | ↑2.12 | 2.64 | 2.3 × 10−2 | 2.6 × 10−2 |
| hsa-miR-200b-3p | ↑1.41 | 9.61 | 1.8 × 10−2 | 2.7 × 10−2 |
| Pathway Name (ID, Ontology): Genes, miRNAs | p-Value |
|---|---|
| KEGG | |
| TGF-beta signaling pathway (hsa04350): ID3 ID4 SMAD3 miR-27a-3p | 5.5 × 10−3 |
| ErbB signaling pathway (hsa04012): PAK1 SHC1 SOS2 let-7b-5p miR-26b-5p miR-429 | 4.1 × 10−3 |
| Natural killer cell mediated cytotoxicity (hsa04650): PAK1 SHC1 SOS2 let-7b-5p miR-26b-5p miR-429 | 1.2 × 10−2 |
| Chemokine signaling pathway (hsa04062): PAK1 SHC1 SOS2 let-7b-5p miR-26b-5p miR-429 | 3.7 × 10−2 |
| Focal adhesion (hsa04510): PAK1 SHC1 SOS2 let-7b-5p miR-26b-5p miR-429 | 4.1 × 10−2 |
| Gene Ontology | |
| Negative regulation of neuron differentiation (GO:0045665, BP): ASPM DNM3 ID3 ID4 RAB29 RTN4 miR-101-3p miR-148b-3p miR-26b-5p miR-27a-3p miR-34a-5p | 2.0 × 10−4 |
| Negative regulation of osteoblast differentiation (GO:0045668, BP): CDK6 ID3 SMAD3 let-7b-5p miR-103a-3p miR-107 miR-26b-5p miR-30a-3p miR-34a-5p | 7.4 × 10−4 |
| Extracellular matrix organization (GO:0030198, BP): FOXF1 GPM6B MMP24 NID1 OLFML2A SMAD3 miR-146a-5p | 2.5 × 10−3 |
| Ras protein signal transduction (GO:0007265, BP): ARHGEF17 RAB29 SHC1 SOS2 ZNF304 miR-429 | 2.7 × 10−2 |
| Regulation of axonogenesis (GO:0050770, BP): PAK1 PLXNA4 RTN4 miR-101-3p miR-148b-3p miR-34a-5p | 3.2 × 10−2 |
| Negative regulation of cell growth (GO:0030308, BP): PAK1 RTN4 SMAD3 miR-101-3p miR-148b-3p miR-34a-5p | 3.3 × 10−2 |
| Cellular response to insulin stimulus (GO:0032869, BP): PAK1 SHC1 TBC1D4 let-7b-5p miR-26b-5p | 4.9 × 10−2 |
| Z disc (GO:0030018, CC): DST PAK1 PALLD let-7b-5p miR-26b-5p miR-32-5p | 1.4 × 10−2 |
| Ruffle (GO:0001726, CC): CDK6 PAK1 PALLD let-7b-5p miR-103a-3p miR-107 miR-26b-5p miR-30a-3p miR-34a-5p | 3.0 × 10−2 |
| Nuclear envelope (GO:0005635, CC): DST OSBPL3 PAK1 RTN4 SMAD3 miR-101-3p miR-148b-3p miR-32-5p miR-34a-5p | 3.3 × 10−2 |
| Collagen binding (GO:0005518, MF): NID1 PAK1 SMAD3 let-7b-5p miR-26b-5p | 2.1 × 10−3 |
| Transcription corepressor activity (GO:0003714, MF): BASP1 BATF3 ID3 ID4 miR-26b-5p miR-27a-3p | 1.3 × 10−2 |
| RNA polymerase II transcription factor binding (GO:0001085, MF): ID3 ID4 SMAD3 miR-27a-3p | 2.1 × 10−2 |
| Models | Test Dataset/Training Dataset | ||||
|---|---|---|---|---|---|
| Se | Sp | Ka | Pr | AUC | |
| Based on clinicopathological parameters | 0.67/0.75 | 0.61/1.00 | 0.60/0.93 | 0.27/1.00 | 0.631/0.875 |
| ALDH3A2 + CHKA + ODF2 + QSOX2 + hsa-miR-503-5p | 1.00/1.00 | 0.93/1.00 | 0.94/1.00 | 0.75/1.00 | 0.963/1.000 |
| ALDH3A2 + CHKA + ODF2 + QSOX2 + hsa-miR-503-5p + ISUP | 1.00/1.00 | 0.96/1.00 | 0.97/1.00 | 0.86/1.00 | 0.982/1.000 |
| mRNA/miRNA | FC | MW p-Value | Spearman Correlation | |
|---|---|---|---|---|
| rs | p-Value | |||
| ALDH3A2 | ↓1.56 | 0.023 * | −0.42 | 0.019 * |
| CHKA | ↑1.22 | 0.611 | 0.10 | 0.605 |
| ODF2 | ↑2.45 | 0.019 * | 0.43 | 0.017 * |
| QSOX2 | ↑1.58 | 0.049 * | 0.35 | 0.045 * |
| hsa-miR-200b-3p | ↑1.73 | 0.025 * | 0.42 | 0.022 * |
| hsa-miR-503-5p | ↑1.72 | 0.035 * | 0.39 | 0.032 * |
| Models | Test Dataset/Training Dataset | ||||
|---|---|---|---|---|---|
| Se | Sp | Ka | Pr | AUC | |
| ALDH3A2 + ODF2 + QSOX2 + hsa-miR-503-5p + ISUP + pT | 0.89/1.00 | 1.00/1.00 | 0.88/1.00 | 1.00/1.00 | 0.944/1.000 |
| ALDH3A2 + ODF2 + QSOX2 + hsa-miR-503-5p + ISUP | 0.89/1.00 | 0.75/1.00 | 0.64/1.00 | 0.80/1.00 | 0.819/1.000 |
| ALDH3A2 + ODF2 + QSOX2 + hsa-miR-503-5p + pT | 0.78/1.00 | 0.62/1.00 | 0.41/1.00 | 0.70/1.00 | 0.701/1.000 |
| ALDH3A2 + ODF2 + QSOX2 + ISUP + pT | 0.89/1.00 | 0.75/1.00 | 0.64/1.00 | 0.80/1.00 | 0.819/1.000 |
| ALDH3A2 + ODF2 + hsa-miR-503-5p + ISUP + pT | 0.89/1.00 | 0.88/1.00 | 0.76/1.00 | 0.89/1.00 | 0.882/1.000 |
| ALDH3A2 + QSOX2 + hsa-miR-503-5p + ISUP + pT | 0.89/0.89 | 0.88/1.00 | 0.76/0.86 | 0.89/1.00 | 0.882/0.944 |
| ODF2 + QSOX2 + hsa-miR-503-5p + ISUP + pT | 0.78/1.00 | 1.00/1.00 | 0.77/1.00 | 1.00/1.00 | 0.889/1.000 |
| Criterion | Cohort-A | Cohort-B | |||
|---|---|---|---|---|---|
| n | % | n | % | ||
| PCa samples | 111 | 100 | 154 | 100 | |
| Age, years | 63 (41–73) | - | 62 (46–78) | - | |
| pT | pT3a | 55 | 50 | 85 | 55 |
| pT3b | 52 | 47 | 65 | 42 | |
| pT4 | 4 | 3 | 4 | 3 | |
| pN | pN0 | 73 | 66 | 102 | 66 |
| pN1 | 38 | 34 | 42 | 27 | |
| cM | cM0 | 111 | 100 | 154 | 100 |
| cM1 | 0 | 0 | 0 | 0 | |
| Gleason score | 6 | 15 | 14 | 6 | 4 |
| 7 | 62 | 56 | 59 | 36 | |
| 8 | 13 | 12 | 23 | 16 | |
| 9 | 20 | 18 | 65 | 43 | |
| 10 | 1 | 0 | 1 | <1 | |
| ISUP | 1 | 15 | 14 | 6 | 4 |
| 2 | 30 | 27 | 28 | 18 | |
| 3 | 32 | 28 | 31 | 20 | |
| 4 | 13 | 12 | 23 | 15 | |
| 5 | 21 | 19 | 66 | 43 | |
| PSA, ng/ml | 13.6 (2.5–61) | - | 8.7 (2.7–87) | - | |
| Biochemical recurrence (PSA ≥ 0.2 ng/mL) | Yes, N0R0 | 22 | 20 | 15 | 10 |
| Yes, N1/R1 | 6 | 5 | 29 | 19 | |
| No, any N/R | 57 | 51 | 104 | 67 | |
| unknown | 26 | 24 | 6 | 4 | |
| TMPRSS2-ERG molecular subtype | Yes | 52 | 47 | 76 | 49 |
| No | 59 | 53 | 78 | 51 | |
| mRNA | Primer Sequence (5′ → 3′) | Product Length, b.p. |
|---|---|---|
| ALDH3A2 | F: GTCTGGACAAGAAGCGAGTAAG R: GACCATGAACGACTGATGAGAG | 110 |
| CHKA | F: GATCCGAACAAGCTCAGAAAGA R: CTGCGAGAATGGCAAACATAAC | 84 |
| ODF2 | F: GGCCTGATTTGTATCTCTGGAA R: GATGAGGACTGGTTGGGTAAAG | 124 |
| QSOX2 | F: CTTAGACCTGATCCCGTATGAAAG R: GTAACACGAAGGGACTGAAGAA | 103 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kobelyatskaya, A.A.; Kudryavtsev, A.A.; Kudryavtseva, A.V.; Snezhkina, A.V.; Fedorova, M.S.; Kalinin, D.V.; Pavlov, V.S.; Guvatova, Z.G.; Naberezhnev, P.A.; Nyushko, K.M.; et al. ALDH3A2, ODF2, QSOX2, and MicroRNA-503-5p Expression to Forecast Recurrence in TMPRSS2-ERG-Positive Prostate Cancer. Int. J. Mol. Sci. 2022, 23, 11695. https://doi.org/10.3390/ijms231911695
Kobelyatskaya AA, Kudryavtsev AA, Kudryavtseva AV, Snezhkina AV, Fedorova MS, Kalinin DV, Pavlov VS, Guvatova ZG, Naberezhnev PA, Nyushko KM, et al. ALDH3A2, ODF2, QSOX2, and MicroRNA-503-5p Expression to Forecast Recurrence in TMPRSS2-ERG-Positive Prostate Cancer. International Journal of Molecular Sciences. 2022; 23(19):11695. https://doi.org/10.3390/ijms231911695
Chicago/Turabian StyleKobelyatskaya, Anastasiya A., Alexander A. Kudryavtsev, Anna V. Kudryavtseva, Anastasiya V. Snezhkina, Maria S. Fedorova, Dmitry V. Kalinin, Vladislav S. Pavlov, Zulfiya G. Guvatova, Pavel A. Naberezhnev, Kirill M. Nyushko, and et al. 2022. "ALDH3A2, ODF2, QSOX2, and MicroRNA-503-5p Expression to Forecast Recurrence in TMPRSS2-ERG-Positive Prostate Cancer" International Journal of Molecular Sciences 23, no. 19: 11695. https://doi.org/10.3390/ijms231911695
APA StyleKobelyatskaya, A. A., Kudryavtsev, A. A., Kudryavtseva, A. V., Snezhkina, A. V., Fedorova, M. S., Kalinin, D. V., Pavlov, V. S., Guvatova, Z. G., Naberezhnev, P. A., Nyushko, K. M., Alekseev, B. Y., Krasnov, G. S., Bulavkina, E. V., & Pudova, E. A. (2022). ALDH3A2, ODF2, QSOX2, and MicroRNA-503-5p Expression to Forecast Recurrence in TMPRSS2-ERG-Positive Prostate Cancer. International Journal of Molecular Sciences, 23(19), 11695. https://doi.org/10.3390/ijms231911695