

Molecular Mechanisms Underlying Pathological and Therapeutic Roles of Pericytes in Atherosclerosis
 , ,
, ,  and
and 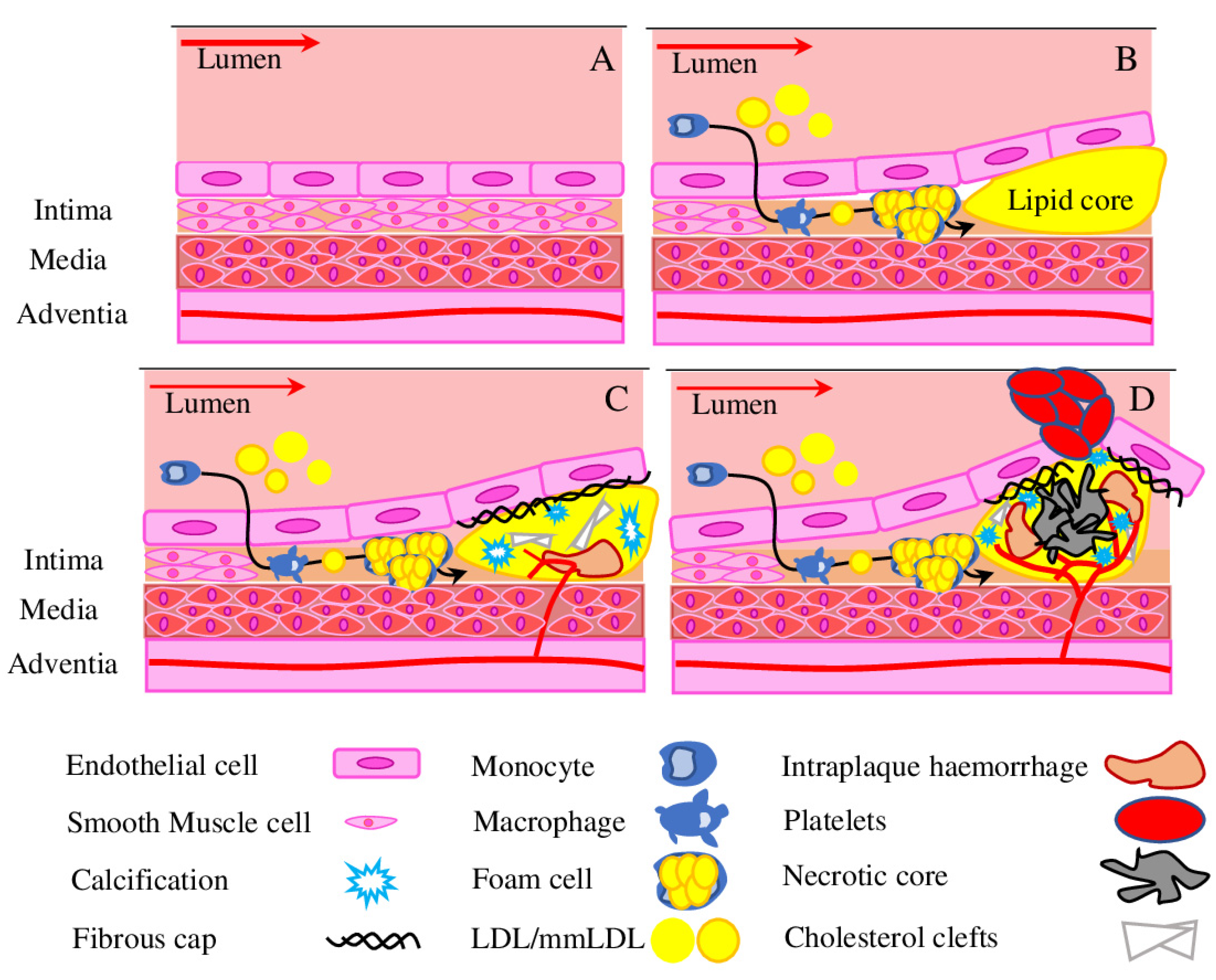{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Pericytes and Atherosclerosis Main Risk Factors
Pericytes in Diabetes
3. Advanced Atherosclerosis
3.1. Hypoxia Drives Angiogenesis
3.2. Angiogenesis in the Atherosclerotic Plaque
3.3. Intraplaque Haemorrhage
3.4. Pericytes in Vascular Calcification
4. Role of Pericytes in Plaque Stabilisation
5. AS Treatments with Pericyte-Mediated Effects
6. Conclusions
- -
- PCs facilitate proper neovessel development and maturation
- -
- PCs prevent matrix degradation and maintain vascular stability
- -
- PC–PC and PC–EC communication, PC recruitment and stabilisation are important for neovessels’ permeability, fragility, and impaired perfusion
- -
- PCs regulate mineralisation and osteogenic differentiation of human mesenchymal stem cells and of SMC
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACS | acute coronary syndrome |
| ADAMTS | disintegrin-like and metalloproteinase with thrombospondin motif |
| AGEs | advanced glycation end products |
| Ang-1 | Angiopoietin 1 |
| AS | atherosclerosis |
| BAX | Bcl-2-associated X protein |
| CD13 | cluster of differentiation 13 |
| CVCs | calcifying vascular cells |
| DLK-1 | delta-like homolog 1 or Endosialin |
| Dll4 | Delta Like Canonical Notch Ligand 4 |
| DM | diabetes mellitus |
| DN | diabetic nephropathy |
| DR | diabetic retinopathy |
| EC | endothelial cells |
| ECM | extracellular matrix |
| eNOS | Endothelia Nitric Oxide Synthase |
| FGF | fibroblast growth factor |
| HbA1c | glycosylated haemoglobin |
| HIF-1 | Hypoxia inducible factor-1 |
| HPSCs | human pluripotent stem cells |
| IPH | intraplaque haemorrhage |
| KLF2 | Kruppel like Factor 2 |
| MMP | matrix metalloproteinases |
| MSCs | multipotent mesenchymal stem cells |
| NF-κB | nuclear factor-kB |
| NG-2 | Neuron-glial antigen 2 |
| Ninj1 | Ninjurin 1 or Nerve injury-induced protein 1 |
| Notch1 | Notch homolog 1 |
| OM | osteoid metaplasia, or bone-like vascular calcification |
| OPG | osteoprotegerin |
| PDGF | platelet-derived growth factor |
| PDGFR-β | platelet-derived growth factor receptor |
| RAAS | Renin-Angiotensin-Aldosterone system |
| RBC | Red blood cells |
| RGS 5 | regulator of G protein signalling 5 |
| SMCs | smooth muscle cells |
| TAZ | transcriptional coactivator with PDZ-binding motif |
| TGF-β | transforming growth factor β |
| Tie 2 | Tyrosine-Protein Kinase Receptor TIE-2 |
| TIMP | tissue inhibitors of matrix metalloproteinase |
| TNF-α | tumor necrosis factor alpha |
| VC | Vascular calcification |
| VEGF | vascular endothelial growth factor |
| VV | vasa vasorum |
| YAP | Yes-associated protein |
| α-SMA | smooth muscle α-actin |
References
- Caporarello, N.; D’Angeli, F.; Cambria, M.T.; Candido, S.; Giallongo, C.; Salmeri, M.; Lombardo, C.; Longo, A.; Giurdanella, G.; Anfuso, C.D.; et al. Pericytes in Microvessels: From “Mural” Function to Brain and Retina Regeneration. Int. J. Mol. Sci. 2019, 20, 6351. [Google Scholar] [CrossRef] [PubMed]
- Craig, D.J.; James, A.W.; Wang, Y.; Tavian, M.; Crisan, M.; Péault, B.M. Blood Vessel Resident Human Stem Cells in Health and Disease. Stem Cells Transl. Med. 2022, 11, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Ayloo, S.; Lazo, C.G.; Sun, S.; Zhang, W.; Cui, B.; Gu, C. Pericyte-to-Endothelial Cell Signaling via Vitronectin-Integrin Regulates Blood-CNS Barrier. Neuron 2022, 110, 1641–1655.e6. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Zhang, H.; Liu, Y.; Adams, S.; Eilken, H.; Stehling, M.; Corada, M.; Dejana, E.; Zhou, B.; Adams, R.H. Endothelial Cells Are Progenitors of Cardiac Pericytes and Vascular Smooth Muscle Cells. Nat. Commun. 2016, 7, 12422. [Google Scholar] [CrossRef] [PubMed]
- Murray, I.R.; Baily, J.E.; Chen, W.C.W.; Dar, A.; Gonzalez, Z.N.; Jensen, A.R.; Petrigliano, F.A.; Deb, A.; Henderson, N.C. Skeletal and Cardiac Muscle Pericytes: Functions and Therapeutic Potential. Pharmacol. Ther. 2017, 171, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Dai, M.; Nuttall, A.; Yang, Y.; Shi, X. Visualization and Contractile Activity of Cochlear Pericytes in the Capillaries of the Spiral Ligament. Hear. Res. 2009, 254, 100–107. [Google Scholar] [CrossRef]
- Meijer, E.M.; van Dijk, C.G.M.; Kramann, R.; Verhaar, M.C.; Cheng, C. Implementation of Pericytes in Vascular Regeneration Strategies. Tissue Eng. Part B Rev. 2022, 28, 1–21. [Google Scholar] [CrossRef]
- Kumar, A.; D’Souza, S.S.; Moskvin, O.V.; Toh, H.; Wang, B.; Zhang, J.; Swanson, S.; Guo, L.-W.; Thomson, J.A.; Slukvin, I.I. Specification and Diversification of Pericytes and Smooth Muscle Cells from Mesenchymoangioblasts. Cell Rep. 2017, 19, 1902–1916. [Google Scholar] [CrossRef]
- Wobma, H.; Satwani, P. Mesenchymal Stromal Cells: Getting Ready for Clinical Primetime. Transfus. Apher. Sci. 2021, 60, 103058. [Google Scholar] [CrossRef]
- Stallcup, W.B. The NG2 Proteoglycan in Pericyte Biology. In Pericyte Biology—Novel Concepts; Birbrair, A., Ed.; (Advances in Experimental Medicine and Biology); Springer International Publishing: Cham, Switzerland, 2018; Volume 1109, pp. 5–19. ISBN 978-3-030-02600-4. [Google Scholar]
- Baek, S.-H.; Maiorino, E.; Kim, H.; Glass, K.; Raby, B.A.; Yuan, K. Single Cell Transcriptomic Analysis Reveals Organ Specific Pericyte Markers and Identities. Front. Cardiovasc. Med. 2022, 9, 876591. [Google Scholar] [CrossRef]
- Billaud, M.; Donnenberg, V.S.; Ellis, B.W.; Meyer, E.M.; Donnenberg, A.D.; Hill, J.C.; Richards, T.D.; Gleason, T.G.; Phillippi, J.A. Classification and Functional Characterization of Vasa Vasorum-Associated Perivascular Progenitor Cells in Human Aorta. Stem Cell Rep. 2017, 9, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Summerhill, V.; Orekhov, A. Pericytes in Atherosclerosis. In Pericyte Biology in Disease; Birbrair, A., Ed.; (Advances in Experimental Medicine and Biology); Springer International Publishing: Cham, Switzerland, 2019; Volume 1147, pp. 279–297. ISBN 978-3-030-16907-7. [Google Scholar]
- Cathery, W.; Faulkner, A.; Maselli, D.; Madeddu, P. Concise Review: The Regenerative Journey of Pericytes Toward Clinical Translation. Stem Cells 2018, 36, 1295–1310. [Google Scholar] [CrossRef] [PubMed]
- Harrell, C.R.; Simovic Markovic, B.; Fellabaum, C.; Arsenijevic, A.; Djonov, V.; Volarevic, V. Molecular Mechanisms Underlying Therapeutic Potential of Pericytes. J. Biomed. Sci. 2018, 25, 21. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Cantrell, A.C.; Zeng, H.; Zhu, S.-H.; Chen, J.-X. Emerging Role of Pericytes and Their Secretome in the Heart. Cells 2021, 10, 548. [Google Scholar] [CrossRef]
- WHO. Fact sheets. In Cardiovascular Diseases (CVDs). Available online: https://www.who.int/health-topics/cardiovascular-diseases#tab=tab_1 (accessed on 11 August 2022).
- Xu, S.; Ilyas, I.; Little, P.J.; Li, H.; Kamato, D.; Zheng, X.; Luo, S.; Li, Z.; Liu, P.; Han, J.; et al. Endothelial Dysfunction in Atherosclerotic Cardiovascular Diseases and Beyond: From Mechanism to Pharmacotherapies. Pharmacol. Rev. 2021, 73, 924–967. [Google Scholar] [CrossRef] [PubMed]
- Glanz, V.; Bezsonov, E.E.; Soldatov, V.; Orekhov, A.N. Thirty-Five-Year History of Desialylated Lipoproteins Discovered by Vladimir Tertov. Biomedicines 2022, 10, 1174. [Google Scholar] [CrossRef] [PubMed]
- Canet-Soulas, E.; Bessueille, L.; Mechtouff, L.; Magne, D. The Elusive Origin of Atherosclerotic Plaque Calcification. Front. Cell Dev. Biol. 2021, 9, 622736. [Google Scholar] [CrossRef]
- Cameron, J.N.; Mehta, O.H.; Michail, M.; Chan, J.; Nicholls, S.J.; Bennett, M.R.; Brown, A.J. Exploring the Relationship between Biomechanical Stresses and Coronary Atherosclerosis. Atherosclerosis 2020, 302, 43–51. [Google Scholar] [CrossRef]
- Cai, X.; Wang, K.-C.; Meng, Z. Mechanoregulation of YAP and TAZ in Cellular Homeostasis and Disease Progression. Front. Cell Dev. Biol. 2021, 9, 673599. [Google Scholar] [CrossRef]
- Dabravolski, S.A.; Sukhorukov, V.N.; Kalmykov, V.A.; Grechko, A.V.; Shakhpazyan, N.K.; Orekhov, A.N. The Role of KLF2 in the Regulation of Atherosclerosis Development and Potential Use of KLF2-Targeted Therapy. Biomedicines 2022, 10, 254. [Google Scholar] [CrossRef]
- Zhao, J.; Huangfu, C.; Chang, Z.; Zhou, W.; Grainger, A.T.; Liu, Z.; Shi, W. Inflammation and Enhanced Atherogenesis in the Carotid Artery with Altered Blood Flow in an Atherosclerosis-resistant Mouse Strain. Physiol. Rep. 2021, 9, e14829. [Google Scholar] [CrossRef] [PubMed]
- Santamaria, S.; de Groot, R. ADAMTS Proteases in Cardiovascular Physiology and Disease. Open Biol. 2020, 10, 200333. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, E.; Hultman, K.; Dunér, P.; Asciutto, G.; Almgren, P.; Orho-Melander, M.; Melander, O.; Nilsson, J.; Hultgårdh-Nilsson, A.; Gonçalves, I. ADAMTS-7 Is Associated with a High-Risk Plaque Phenotype in Human Atherosclerosis. Sci. Rep. 2017, 7, 3753. [Google Scholar] [CrossRef] [PubMed]
- Schrimpf, C.; Koppen, T.; Duffield, J.S.; Böer, U.; David, S.; Ziegler, W.; Haverich, A.; Teebken, O.E.; Wilhelmi, M. TIMP3 Is Regulated by Pericytes upon Shear Stress Detection Leading to a Modified Endothelial Cell Response. Eur. J. Vasc. Endovasc. Surg. 2017, 54, 524–533. [Google Scholar] [CrossRef]
- Ziegler, T.; Abdel Rahman, F.; Jurisch, V.; Kupatt, C. Atherosclerosis and the Capillary Network; Pathophysiology and Potential Therapeutic Strategies. Cells 2019, 9, 50. [Google Scholar] [CrossRef]
- Martinez-Quinones, P.; McCarthy, C.G.; Watts, S.W.; Klee, N.S.; Komic, A.; Calmasini, F.B.; Priviero, F.; Warner, A.; Chenghao, Y.; Wenceslau, C.F. Hypertension Induced Morphological and Physiological Changes in Cells of the Arterial Wall. Am. J. Hypertens. 2018, 31, 1067–1078. [Google Scholar] [CrossRef]
- de Souza-Neto, F.P.; Carvalho Santuchi, M.; de Morais e Silva, M.; Campagnole-Santos, M.J.; da Silva, R.F. Angiotensin-(1–7) and Alamandine on Experimental Models of Hypertension and Atherosclerosis. Curr. Hypertens. Rep. 2018, 20, 17. [Google Scholar] [CrossRef]
- Kostov, K.; Halacheva, L. Role of Magnesium Deficiency in Promoting Atherosclerosis, Endothelial Dysfunction, and Arterial Stiffening as Risk Factors for Hypertension. Int. J. Mol. Sci. 2018, 19, 1724. [Google Scholar] [CrossRef]
- Poznyak, A.V.; Bharadwaj, D.; Prasad, G.; Grechko, A.V.; Sazonova, M.A.; Orekhov, A.N. Renin-Angiotensin System in Pathogenesis of Atherosclerosis and Treatment of CVD. Int. J. Mol. Sci. 2021, 22, 6702. [Google Scholar] [CrossRef]
- Fan, J.; Watanabe, T. Atherosclerosis: Known and Unknown. Pathol. Int. 2022, 72, 151–160. [Google Scholar] [CrossRef]
- Hayes, K.L. Pericytes in Type 2 Diabetes. In Pericyte Biology in Disease; Birbrair, A., Ed.; (Advances in Experimental Medicine and Biology); Springer International Publishing: Cham, Switzerland, 2019; Volume 1147, pp. 265–278. ISBN 978-3-030-16907-7. [Google Scholar]
- Hinkel, R.; Howe, A.; Renner, S.; Ng, J.; Lee, S.; Klett, K.; Kaczmarek, V.; Moretti, A.; Laugwitz, K.-L.; Skroblin, P.; et al. Diabetes Mellitus–Induced Microvascular Destabilization in the Myocardium. J. Am. Coll. Cardiol. 2017, 69, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Hayes, K.L.; Messina, L.M.; Schwartz, L.M.; Yan, J.; Burnside, A.S.; Witkowski, S. Type 2 Diabetes Impairs the Ability of Skeletal Muscle Pericytes to Augment Postischemic Neovascularization in Db/Db Mice. Am. J. Physiol. Cell Physiol. 2018, 314, C534–C544. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.L.; Khakoo, A.Y.; Chintalgattu, V. Cardiac Pericytes Function as Key Vasoactive Cells to Regulate Homeostasis and Disease. FEBS Open Bio 2021, 11, 207–225. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, S.; Seetharaman, S.; Dharmarajan, A.; Kuppan, K. Microvascular Cells: A Special Focus on Heterogeneity of Pericytes in Diabetes Associated Complications. Int. J. Biochem. Cell Biol. 2021, 134, 105971. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.-L.; Chen, C.-H.; Wu, M.-J.; Tsai, S.-F. New Approaches to Diabetic Nephropathy from Bed to Bench. Biomedicines 2022, 10, 876. [Google Scholar] [CrossRef]
- Yadranji Aghdam, S.; Mahmoudpour, A. Proteasome Activators, PA28 α and PA28 β, Govern Development of Microvascular Injury in Diabetic Nephropathy and Retinopathy. Int. J. Nephrol. 2016, 2016, 3846573. [Google Scholar] [CrossRef]
- Huang, H. Pericyte-Endothelial Interactions in the Retinal Microvasculature. Int. J. Mol. Sci. 2020, 21, 7413. [Google Scholar] [CrossRef]
- Mazzoli, V.; Zhong, L.H.; Dang, V.T.; Shi, Y.; Werstuck, G.H. Characterization of Retinal Microvascular Complications and the Effects of Endoplasmic Reticulum Stress in Mouse Models of Diabetic Atherosclerosis. Investig. Opthalmology Vis. Sci. 2020, 61, 49. [Google Scholar] [CrossRef]
- Caolo, V.; Roblain, Q.; Lecomte, J.; Carai, P.; Peters, L.; Cuijpers, I.; Robinson, E.L.; Derks, K.; Sergeys, J.; Noël, A.; et al. Resistance to Retinopathy Development in Obese, Diabetic and Hypertensive ZSF1 Rats: An Exciting Model to Identify Protective Genes. Sci. Rep. 2018, 8, 11922. [Google Scholar] [CrossRef]
- Kim, J.M.; Hong, K.-S.; Song, W.K.; Bae, D.; Hwang, I.-K.; Kim, J.S.; Chung, H.-M. Perivascular Progenitor Cells Derived from Human Embryonic Stem Cells Exhibit Functional Characteristics of Pericytes and Improve the Retinal Vasculature in a Rodent Model of Diabetic Retinopathy. Stem Cells Transl. Med. 2016, 5, 1268–1276. [Google Scholar] [CrossRef]
- Yahagi, K.; Kolodgie, F.D.; Otsuka, F.; Finn, A.V.; Davis, H.R.; Joner, M.; Virmani, R. Pathophysiology of Native Coronary, Vein Graft, and in-Stent Atherosclerosis. Nat. Rev. Cardiol. 2016, 13, 79–98. [Google Scholar] [CrossRef] [PubMed]
- Bäck, M.; Yurdagul, A.; Tabas, I.; Öörni, K.; Kovanen, P.T. Inflammation and Its Resolution in Atherosclerosis: Mediators and Therapeutic Opportunities. Nat. Rev. Cardiol. 2019, 16, 389–406. [Google Scholar] [CrossRef] [PubMed]
- Ruparelia, N.; Choudhury, R. Inflammation and Atherosclerosis: What Is on the Horizon? Heart 2020, 106, 80–85. [Google Scholar] [CrossRef] [PubMed]
- de Vries, M.R.; Quax, P.H.A. Plaque Angiogenesis and Its Relation to Inflammation and Atherosclerotic Plaque Destabilization. Curr. Opin. Lipidol. 2016, 27, 499–506. [Google Scholar] [CrossRef]
- Haasdijk, R.A.; Den Dekker, W.K.; Cheng, C.; Tempel, D.; Szulcek, R.; Bos, F.L.; Hermkens, D.M.A.; Chrifi, I.; Brandt, M.M.; Van Dijk, C.; et al. THSD1 Preserves Vascular Integrity and Protects against Intraplaque Haemorrhaging in ApoE −/− Mice. Cardiovasc. Res. 2016, 110, 129–139. [Google Scholar] [CrossRef]
- Basatemur, G.L.; Jørgensen, H.F.; Clarke, M.C.H.; Bennett, M.R.; Mallat, Z. Vascular Smooth Muscle Cells in Atherosclerosis. Nat. Rev. Cardiol. 2019, 16, 727–744. [Google Scholar] [CrossRef]
- Hutcheson, J.D.; Goettsch, C.; Bertazzo, S.; Maldonado, N.; Ruiz, J.L.; Goh, W.; Yabusaki, K.; Faits, T.; Bouten, C.; Franck, G.; et al. Genesis and Growth of Extracellular-Vesicle-Derived Microcalcification in Atherosclerotic Plaques. Nat. Mater. 2016, 15, 335–343. [Google Scholar] [CrossRef]
- Jain, T.; Nikolopoulou, E.A.; Xu, Q.; Qu, A. Hypoxia Inducible Factor as a Therapeutic Target for Atherosclerosis. Pharmacol. Ther. 2018, 183, 22–33. [Google Scholar] [CrossRef]
- Thomas, C.; Leleu, D.; Masson, D. Cholesterol and HIF-1α: Dangerous Liaisons in Atherosclerosis. Front. Immunol. 2022, 13, 868958. [Google Scholar] [CrossRef]
- Dabravolski, S.A.; Khotina, V.A.; Omelchenko, A.V.; Kalmykov, V.A.; Orekhov, A.N. The Role of the VEGF Family in Atherosclerosis Development and Its Potential as Treatment Targets. Int. J. Mol. Sci. 2022, 23, 931. [Google Scholar] [CrossRef]
- Lin, P.P. Aneuploid Circulating Tumor-Derived Endothelial Cell (CTEC): A Novel Versatile Player in Tumor Neovascularization and Cancer Metastasis. Cells 2020, 9, 1539. [Google Scholar] [CrossRef]
- Milutinović, A.; Šuput, D.; Zorc-Pleskovič, R. Pathogenesis of Atherosclerosis in the Tunica Intima, Media, and Adventitia of Coronary Arteries: An Updated Review. Bosn. J. Basic Med. Sci. 2019, 20, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Payne, L.B.; Zhao, H.; James, C.C.; Darden, J.; McGuire, D.; Taylor, S.; Smyth, J.W.; Chappell, J.C. The Pericyte Microenvironment during Vascular Development. Microcirculation 2019, 26, e12554. [Google Scholar] [CrossRef]
- Kang, T.-Y.; Bocci, F.; Jolly, M.K.; Levine, H.; Onuchic, J.N.; Levchenko, A. Pericytes Enable Effective Angiogenesis in the Presence of Proinflammatory Signals. Proc. Natl. Acad. Sci. USA 2019, 116, 23551–23561. [Google Scholar] [CrossRef] [PubMed]
- Kowara, M.; Cudnoch-Jedrzejewska, A. Pathophysiology of Atherosclerotic Plaque Development-Contemporary Experience and New Directions in Research. Int. J. Mol. Sci. 2021, 22, 3513. [Google Scholar] [CrossRef] [PubMed]
- Tomaniak, M.; Katagiri, Y.; Modolo, R.; de Silva, R.; Khamis, R.Y.; Bourantas, C.V.; Torii, R.; Wentzel, J.J.; Gijsen, F.J.H.; van Soest, G.; et al. Vulnerable Plaques and Patients: State-of-the-Art. Eur. Heart J. 2020, 41, 2997–3004. [Google Scholar] [CrossRef]
- Parma, L.; Baganha, F.; Quax, P.H.A.; de Vries, M.R. Plaque Angiogenesis and Intraplaque Hemorrhage in Atherosclerosis. Eur. J. Pharmacol. 2017, 816, 107–115. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Melnichenko, A.A.; Myasoedova, V.A.; Grechko, A.V.; Orekhov, A.N. Role of Lipids and Intraplaque Hypoxia in the Formation of Neovascularization in Atherosclerosis. Ann. Med. 2017, 49, 661–677. [Google Scholar] [CrossRef]
- Omorphos, N.P.; Gao, C.; Tan, S.S.; Sangha, M.S. Understanding Angiogenesis and the Role of Angiogenic Growth Factors in the Vascularisation of Engineered Tissues. Mol. Biol. Rep. 2021, 48, 941–950. [Google Scholar] [CrossRef]
- Teichert, M.; Milde, L.; Holm, A.; Stanicek, L.; Gengenbacher, N.; Savant, S.; Ruckdeschel, T.; Hasanov, Z.; Srivastava, K.; Hu, J.; et al. Pericyte-Expressed Tie2 Controls Angiogenesis and Vessel Maturation. Nat. Commun. 2017, 8, 16106. [Google Scholar] [CrossRef]
- Van der Veken, B.; De Meyer, G.R.; Martinet, W. Intraplaque Neovascularization as a Novel Therapeutic Target in Advanced Atherosclerosis. Expert Opin. Ther. Targets 2016, 20, 1247–1257. [Google Scholar] [CrossRef] [PubMed]
- Michel, J.-B.; Martin-Ventura, J.L. Red Blood Cells and Hemoglobin in Human Atherosclerosis and Related Arterial Diseases. Int. J. Mol. Sci. 2020, 21, 6756. [Google Scholar] [CrossRef] [PubMed]
- Tecellioglu, M.; Alan, S.; Kamisli, S.; Tecellioglu, F.S.; Kamisli, O.; Ozcan, C. Hemoglobin A1c-Related Histologic Characteristics of Symptomatic Carotid Plaques. Niger. J. Clin. Pract. 2019, 22, 393–398. [Google Scholar] [CrossRef]
- Xia, S.; Qiu, W.; Cai, A.; Kong, B.; Xu, L.; Wu, Z.; Li, L. The Association of Lipoprotein(a) and Intraplaque Neovascularization in Patients with Carotid Stenosis: A Retrospective Study. BMC Cardiovasc. Disord. 2021, 21, 285. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Sun, Z.; Zang, G.; Zhang, L.; Hou, L.; Shao, C.; Wang, Z. Epidemiological Research Advances in Vascular Calcification in Diabetes. J. Diabetes Res. 2021, 2021, 4461311. [Google Scholar] [CrossRef] [PubMed]
- Bardeesi, A.S.A.; Gao, J.; Zhang, K.; Yu, S.; Wei, M.; Liu, P.; Huang, H. A Novel Role of Cellular Interactions in Vascular Calcification. J. Transl. Med. 2017, 15, 95. [Google Scholar] [CrossRef]
- Leszczynska, A.; Murphy, J.M. Vascular Calcification: Is It Rather a Stem/Progenitor Cells Driven Phenomenon? Front. Bioeng. Biotechnol. 2018, 6, 10. [Google Scholar] [CrossRef]
- Davaine, J.-M.; Quillard, T.; Brion, R.; Lapérine, O.; Guyomarch, B.; Merlini, T.; Chatelais, M.; Guilbaud, F.; Brennan, M.Á.; Charrier, C.; et al. Osteoprotegerin, Pericytes and Bone-Like Vascular Calcification Are Associated with Carotid Plaque Stability. PLoS ONE 2014, 9, e107642. [Google Scholar] [CrossRef]
- Davaine, J.-M.; Quillard, T.; Chatelais, M.; Guilbaud, F.; Brion, R.; Guyomarch, B.; Brennan, M.Á.; Heymann, D.; Heymann, M.-F.; Gouëffic, Y. Bone Like Arterial Calcification in Femoral Atherosclerotic Lesions: Prevalence and Role of Osteoprotegerin and Pericytes. Eur. J. Vasc. Endovasc. Surg. 2016, 51, 259–267. [Google Scholar] [CrossRef]
- Strobescu-Ciobanu, C.; Giuşcă, S.E.; Căruntu, I.-D.; Amălinei, C.; Rusu, A.; Cojocaru, E.; Popa, R.F.; Lupaşcu, C.D. Osteopontin and Osteoprotegerin in Atherosclerotic Plaque—Are They Significant Markers of Plaque Vulnerability? Rom. J. Morphol. Embryol. 2021, 61, 793–801. [Google Scholar] [CrossRef]
- Leszczynska, A.; O’Doherty, A.; Farrell, E.; Pindjakova, J.; O’Brien, F.J.; O’Brien, T.; Barry, F.; Murphy, M. Differentiation of Vascular Stem Cells Contributes to Ectopic Calcification of Atherosclerotic Plaque. Stem Cells 2016, 34, 913–923. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Pasterkamp, G.; Crea, F.; Jang, I.-K. Reassessing the Mechanisms of Acute Coronary Syndromes: The “Vulnerable Plaque” and Superficial Erosion. Circ. Res. 2019, 124, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Hafiane, A. Vulnerable Plaque, Characteristics, Detection, and Potential Therapies. J. Cardiovasc. Dev. Dis. 2019, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Lahdentausta, L.; Leskelä, J.; Winkelmann, A.; Tervahartiala, T.; Sorsa, T.; Pesonen, E.; Pussinen, P.J. Serum MMP-9 Diagnostics, Prognostics, and Activation in Acute Coronary Syndrome and Its Recurrence. J Cardiovasc. Transl. Res. 2018, 11, 210–220. [Google Scholar] [CrossRef]
- Mao, Y.; Liu, X.Q.; Song, Y.; Zhai, C.G.; Xu, X.L.; Zhang, L.; Zhang, Y. Fibroblast Growth Factor-2/Platelet-derived Growth Factor Enhances Atherosclerotic Plaque Stability. J. Cell. Mol. Med. 2020, 24, 1128–1140. [Google Scholar] [CrossRef]
- Song, K.; Wu, H.; Rahman, H.N.A.; Dong, Y.; Wen, A.; Brophy, M.L.; Wong, S.; Kwak, S.; Bielenberg, D.R.; Chen, H. Endothelial Epsins as Regulators and Potential Therapeutic Targets of Tumor Angiogenesis. Cell. Mol. Life Sci. 2017, 74, 393–398. [Google Scholar] [CrossRef]
- de Vries, M.R.; Parma, L.; Peters, H.A.B.; Schepers, A.; Hamming, J.F.; Jukema, J.W.; Goumans, M.J.T.H.; Guo, L.; Finn, A.V.; Virmani, R.; et al. Blockade of Vascular Endothelial Growth Factor Receptor 2 Inhibits Intraplaque Haemorrhage by Normalization of Plaque Neovessels. J. Intern. Med. 2019, 285, 59–74. [Google Scholar] [CrossRef]
- Payne, L.B.; Tewari, B.P.; Dunkenberger, L.; Bond, S.; Savelli, A.; Darden, J.; Zhao, H.; Willi, C.; Kanodia, R.; Gude, R.; et al. Pericyte Progenitor Coupling to the Emerging Endothelium During Vasculogenesis via Connexin 43. Arterioscler. Thromb. Vasc. Biol. 2022, 42, e96–e114. [Google Scholar] [CrossRef]
- Andreeva, E.R.; Pugach, I.M.; Gordon, D.; Orekhov, A.N. Continuous Subendothelial Network Formed by Pericyte-like Cells in Human Vascular Bed. Tissue Cell 1998, 30, 127–135. [Google Scholar] [CrossRef]
- Rekhter, M.D.; Andreeva, E.R.; Mironov, A.A.; Orekhov, A.N. Three-Dimensional Cytoarchitecture of Normal and Atherosclerotic Intima of Human Aorta. Am. J. Pathol. 1991, 138, 569–580. [Google Scholar]
- Andreeva, E.R.; Serebryakov, V.N.; Orekhov, A.N. Gap Junctional Communication in Primary Culture of Cells Derived from Human Aortic Intima. Tissue Cell 1995, 27, 591–597. [Google Scholar] [CrossRef]
- Orekhov, A.N.; Andreeva, E.R.; Bobryshev, Y.V. Cellular Mechanisms of Human Atherosclerosis: Role of Cell-to-Cell Communications in Subendothelial Cell Functions. Tissue Cell 2016, 48, 25–34. [Google Scholar] [CrossRef]
- Armulik, A.; Genové, G.; Mäe, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes Regulate the Blood–Brain Barrier. Nature 2010, 468, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Torres, J.; San José, E. Src Tyrosine Kinase Inhibitors: New Perspectives on Their Immune, Antiviral, and Senotherapeutic Potential. Front. Pharmacol. 2019, 10, 1011. [Google Scholar] [CrossRef] [PubMed]
- Bartoschek, M.; Pietras, K. PDGF Family Function and Prognostic Value in Tumor Biology. Biochem. Biophys. Res. Commun. 2018, 503, 984–990. [Google Scholar] [CrossRef] [PubMed]
- Tillie, R.J.H.A.; Theelen, T.L.; van Kuijk, K.; Temmerman, L.; de Bruijn, J.; Gijbels, M.; Betsholtz, C.; Biessen, E.A.L.; Sluimer, J.C. A Switch from Cell-Associated to Soluble PDGF-B Protects against Atherosclerosis, despite Driving Extramedullary Hematopoiesis. Cells 2021, 10, 1746. [Google Scholar] [CrossRef]
- Mao, Y.; Liu, X.; Song, Y.; Zhai, C.; Zhang, L. VEGF-A/VEGFR-2 and FGF-2/FGFR-1 but Not PDGF-BB/PDGFR-β Play Important Roles in Promoting Immature and Inflammatory Intraplaque Angiogenesis. PLoS ONE 2018, 13, e0201395. [Google Scholar] [CrossRef]
- Pandey, A.; Shen, C.; Feng, S.; Man, S.M. Cell Biology of Inflammasome Activation. Trends Cell Biol. 2021, 31, 924–939. [Google Scholar] [CrossRef]
- Kim, S.-W.; Lee, H.-K.; Seol, S.-I.; Davaanyam, D.; Lee, H.; Lee, J.-K. Ninjurin 1 Dodecamer Peptide Containing the N-Terminal Adhesion Motif (N-NAM) Exerts Proangiogenic Effects in HUVECs and in the Postischemic Brain. Sci. Rep. 2020, 10, 16656. [Google Scholar] [CrossRef]
- Matsuki, M.; Kabara, M.; Saito, Y.; Shimamura, K.; Minoshima, A.; Nishimura, M.; Aonuma, T.; Takehara, N.; Hasebe, N.; Kawabe, J. Ninjurin1 Is a Novel Factor to Regulate Angiogenesis Through the Function of Pericytes. Circ. J. 2015, 79, 1363–1371. [Google Scholar] [CrossRef]
- Minoshima, A.; Kabara, M.; Matsuki, M.; Yoshida, Y.; Kano, K.; Tomita, Y.; Hayasaka, T.; Horiuchi, K.; Saito, Y.; Aonuma, T.; et al. Pericyte-Specific Ninjurin1 Deletion Attenuates Vessel Maturation and Blood Flow Recovery in Hind Limb Ischemia. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2358–2370. [Google Scholar] [CrossRef]
- Horiuchi, K.; Kano, K.; Minoshima, A.; Hayasaka, T.; Yamauchi, A.; Tatsukawa, T.; Matsuo, R.; Yoshida, Y.; Tomita, Y.; Kabara, M.; et al. Pericyte-Specific Deletion of Ninjurin-1 Induces Fragile Vasa Vasorum Formation and Enhances Intimal Hyperplasia of Injured Vasculature. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H2438–H2447. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.; Kim, T.K.; Jeong, S.-J.; Jung, I.-H.; Kim, N.; Lee, M.-N.; Sonn, S.-K.; Seo, S.; Jin, J.; Kweon, H.Y.; et al. Anti-Inflammatory Actions of Soluble Ninjurin-1 Ameliorate Atherosclerosis. Circulation 2020, 142, 1736–1751. [Google Scholar] [CrossRef] [PubMed]
- Mahjoubin-Tehran, M.; Teng, Y.; Jalili, A.; Aghaee-Bakhtiari, S.H.; Markin, A.M.; Sahebkar, A. Decoy Technology as a Promising Therapeutic Tool for Atherosclerosis. Int. J. Mol. Sci. 2021, 22, 4420. [Google Scholar] [CrossRef]
- Ou, L.; Zhong, S.; Ou, J.; Tian, J. Application of Targeted Therapy Strategies with Nanomedicine Delivery for Atherosclerosis. Acta Pharmacol. Sin. 2021, 42, 10–17. [Google Scholar] [CrossRef]
- Muruganantham, S.; Krishnaswami, V.; Alagarsamy, S.; Kandasamy, R. Anti-Platelet Drug-Loaded Targeted Technologies for the Effective Treatment of Atherothrombosis. Curr. Drug Targets 2021, 22, 399–419. [Google Scholar] [CrossRef] [PubMed]
- Deroissart, J.; Porsch, F.; Koller, T.; Binder, C.J. Anti-Inflammatory and Immunomodulatory Therapies in Atherosclerosis. In Prevention and Treatment of Atherosclerosis; von Eckardstein, A., Binder, C.J., Eds.; (Handbook of Experimental Pharmacology); Springer International Publishing: Cham, Switzerland, 2021; Volume 270, pp. 359–404. ISBN 978-3-030-86075-2. [Google Scholar]
- Ali, A.H.; Younis, N.; Abdallah, R.; Shaer, F.; Dakroub, A.; Ayoub, M.A.; Iratni, R.; Yassine, H.M.; Zibara, K.; Orekhov, A.; et al. Lipid-Lowering Therapies for Atherosclerosis: Statins, Fibrates, Ezetimibe and PCSK9 Monoclonal Antibodies. Curr. Med. Chem. 2021, 28, 7427–7445. [Google Scholar] [CrossRef]
- Baganha, F.; de Jong, R.C.M.; Peters, E.A.; Voorham, W.; Jukema, J.W.; Delibegovic, M.; de Vries, M.R.; Quax, P.H.A. Atorvastatin Pleiotropically Decreases Intraplaque Angiogenesis and Intraplaque Haemorrhage by Inhibiting ANGPT2 Release and VE-Cadherin Internalization. Angiogenesis 2021, 24, 567–581. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Deng, L.; Guo, H.; Zhao, Y.; Peng, F.; Wang, G.; Yu, C. The Anti-Colon Cancer Effects of Essential Oil of Curcuma Phaeocaulis Through Tumour Vessel Normalisation. Front. Oncol. 2021, 11, 728464. [Google Scholar] [CrossRef]
- Herrera, J.L.; Komatsu, M. R-Ras Deficiency in Pericytes Causes Frequent Microphthalmia and Perturbs Retinal Vascular Development. J. Vasc. Res. 2021, 58, 252–266. [Google Scholar] [CrossRef]
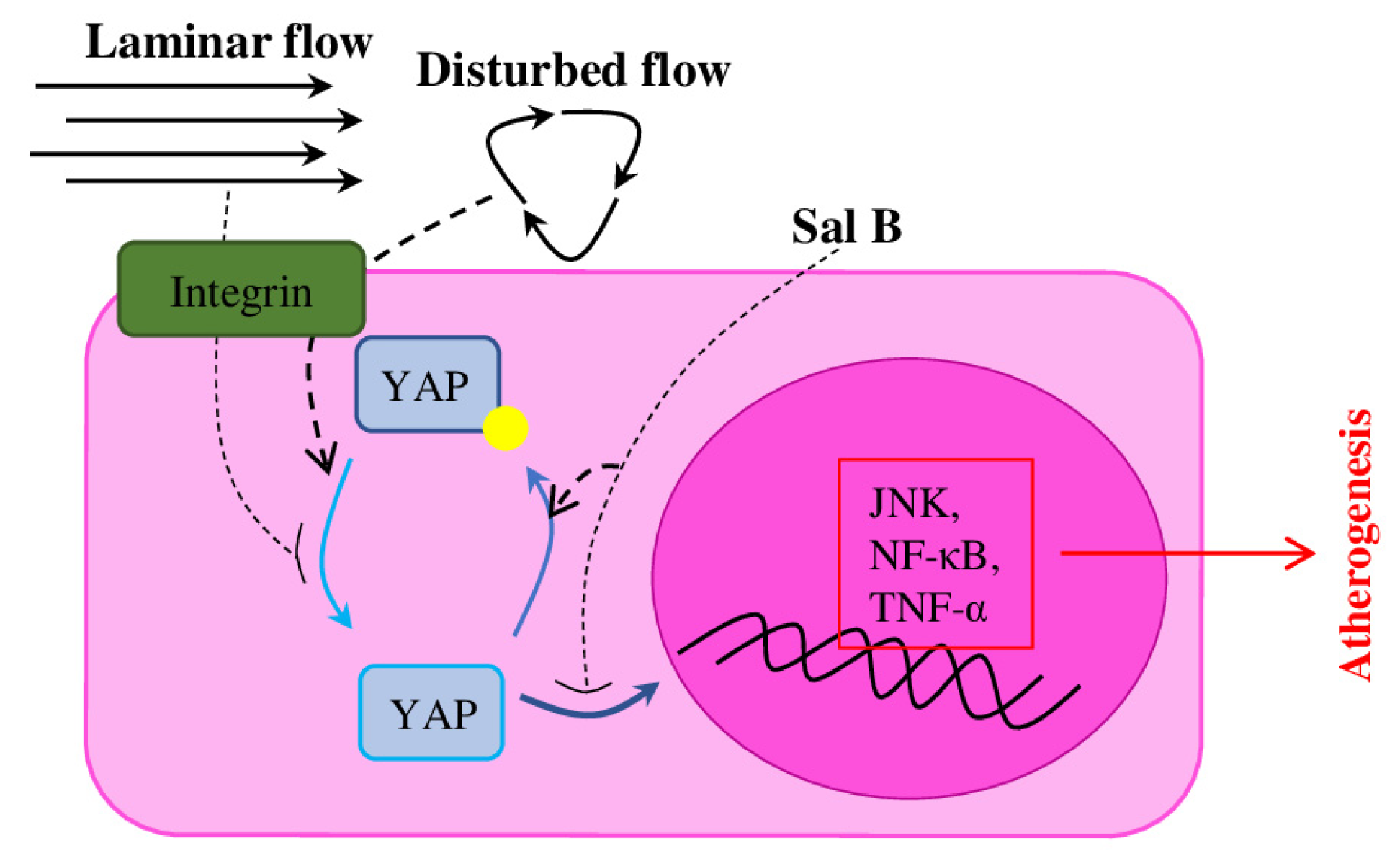
- Yang, Y.; Pei, K.; Zhang, Q.; Wang, D.; Feng, H.; Du, Z.; Zhang, C.; Gao, Z.; Yang, W.; Wu, J.; et al. Salvianolic Acid B Ameliorates Atherosclerosis via Inhibiting YAP/TAZ/JNK Signaling Pathway in Endothelial Cells and Pericytes. Biochim. Biophys. Acta 2020, 1865, 158779. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Luo, J.-Y.; Li, B.; Tian, X.Y.; Chen, L.-J.; Huang, Y.; Liu, J.; Deng, D.; Lau, C.W.; Wan, S.; et al. Integrin-YAP/TAZ-JNK Cascade Mediates Atheroprotective Effect of Unidirectional Shear Flow. Nature 2016, 540, 579–582. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Luo, Q.; Wei, J.; Lin, R.; Lin, L.; Li, Y.; Chen, Z.; Lin, W.; Chen, Q. Mechanism of Salvianolic Acid B Neuroprotection against Ischemia/Reperfusion Induced Cerebral Injury. Brain Res. 2018, 1679, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Habtemariam, S. Molecular Pharmacology of Rosmarinic and Salvianolic Acids: Potential Seeds for Alzheimer’s and Vascular Dementia Drugs. Int. J. Mol. Sci. 2018, 19, 458. [Google Scholar] [CrossRef]
- Wu, C.-R.; Yang, Q.-Y.; Chen, Q.-W.; Li, C.-Q.; He, W.-Y.; Zhao, Y.-P.; Wang, L. Ghrelin Attenuate Cerebral Microvascular Leakage by Regulating Inflammation and Apoptosis Potentially via a P38 MAPK-JNK Dependent Pathway. Biochem. Biophys. Res. Commun. 2021, 552, 37–43. [Google Scholar] [CrossRef]
- Liao, P.; Yang, D.; Liu, D.; Zheng, Y. GLP-1 and Ghrelin Attenuate High Glucose/High Lipid-Induced Apoptosis and Senescence of Human Microvascular Endothelial Cells. Cell. Physiol. Biochem. 2017, 44, 1842–1855. [Google Scholar] [CrossRef]
- Zanetti, M.; Gortan Cappellari, G.; Graziani, A.; Barazzoni, R. Unacylated Ghrelin Improves Vascular Dysfunction and Attenuates Atherosclerosis during High-Fat Diet Consumption in Rodents. Int. J. Mol. Sci. 2019, 20, 499. [Google Scholar] [CrossRef]
- Li, M.; Qi, Z.; Zhang, J.; Zhu, K.; Wang, Y. Effect and Mechanism of Si-Miao-Yong-An on Vasa Vasorum Remodeling in ApoE−/− Mice with Atherosclerosis Vulnerable Plague. Front. Pharmacol. 2021, 12, 634611. [Google Scholar] [CrossRef]
- Qi, Z.; Li, M.; Zhu, K.; Zhang, J. Si-Miao-Yong-An on Promoting the Maturation of Vasa Vasorum and Stabilizing Atherosclerotic Plaque in ApoE-/- Mice: An Experimental Study. Biomed. Pharmacother. 2019, 114, 108785. [Google Scholar] [CrossRef]
- López-Mateo, I.; Arruabarrena-Aristorena, A.; Artaza-Irigaray, C.; López, J.A.; Calvo, E.; Belandia, B. HEY1 Functions Are Regulated by Its Phosphorylation at Ser-68. Biosci. Rep. 2016, 36, e00343. [Google Scholar] [CrossRef]
- Zhu, G.; Lin, Y.; Liu, H.; Jiang, D.; Singh, S.; Li, X.; Yu, Z.; Fan, L.; Wang, S.; Rhen, J.; et al. Dll4-Notch1 Signaling but Not VEGF-A Is Essential for Hyperoxia Induced Vessel Regression in Retina. Biochem. Biophys. Res. Commun. 2018, 507, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Jin, H.; Tong, M.; Zheng, G.; Xie, Z.; Tang, S.; Jin, J.; Shang, P.; Xu, H.; Shen, L.; et al. Inhibition of Dll4/Notch1 Pathway Promotes Angiogenesis of Masquelet’s Induced Membrane in Rats. Exp. Mol. Med. 2018, 50, 41. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, E.; Okamoto, T.; Ito, A.; Kawamoto, E.; Asanuma, K.; Wada, K.; Shimaoka, M.; Takao, M.; Shimamoto, A. Substrate Stiffness Modulates Endothelial Cell Function via the YAP-Dll4-Notch1 Pathway. Exp. Cell Res. 2021, 408, 112835. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dabravolski, S.A.; Markin, A.M.; Andreeva, E.R.; Eremin, I.I.; Orekhov, A.N.; Melnichenko, A.A. Molecular Mechanisms Underlying Pathological and Therapeutic Roles of Pericytes in Atherosclerosis. Int. J. Mol. Sci. 2022, 23, 11663. https://doi.org/10.3390/ijms231911663
Dabravolski SA, Markin AM, Andreeva ER, Eremin II, Orekhov AN, Melnichenko AA. Molecular Mechanisms Underlying Pathological and Therapeutic Roles of Pericytes in Atherosclerosis. International Journal of Molecular Sciences. 2022; 23(19):11663. https://doi.org/10.3390/ijms231911663
Chicago/Turabian StyleDabravolski, Siarhei A., Alexander M. Markin, Elena R. Andreeva, Ilya I. Eremin, Alexander N. Orekhov, and Alexandra A. Melnichenko. 2022. "Molecular Mechanisms Underlying Pathological and Therapeutic Roles of Pericytes in Atherosclerosis" International Journal of Molecular Sciences 23, no. 19: 11663. https://doi.org/10.3390/ijms231911663
APA StyleDabravolski, S. A., Markin, A. M., Andreeva, E. R., Eremin, I. I., Orekhov, A. N., & Melnichenko, A. A. (2022). Molecular Mechanisms Underlying Pathological and Therapeutic Roles of Pericytes in Atherosclerosis. International Journal of Molecular Sciences, 23(19), 11663. https://doi.org/10.3390/ijms231911663