
TRIM26 Maintains Cell Survival in Response to Oxidative Stress through Regulating DNA Glycosylase Stability


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
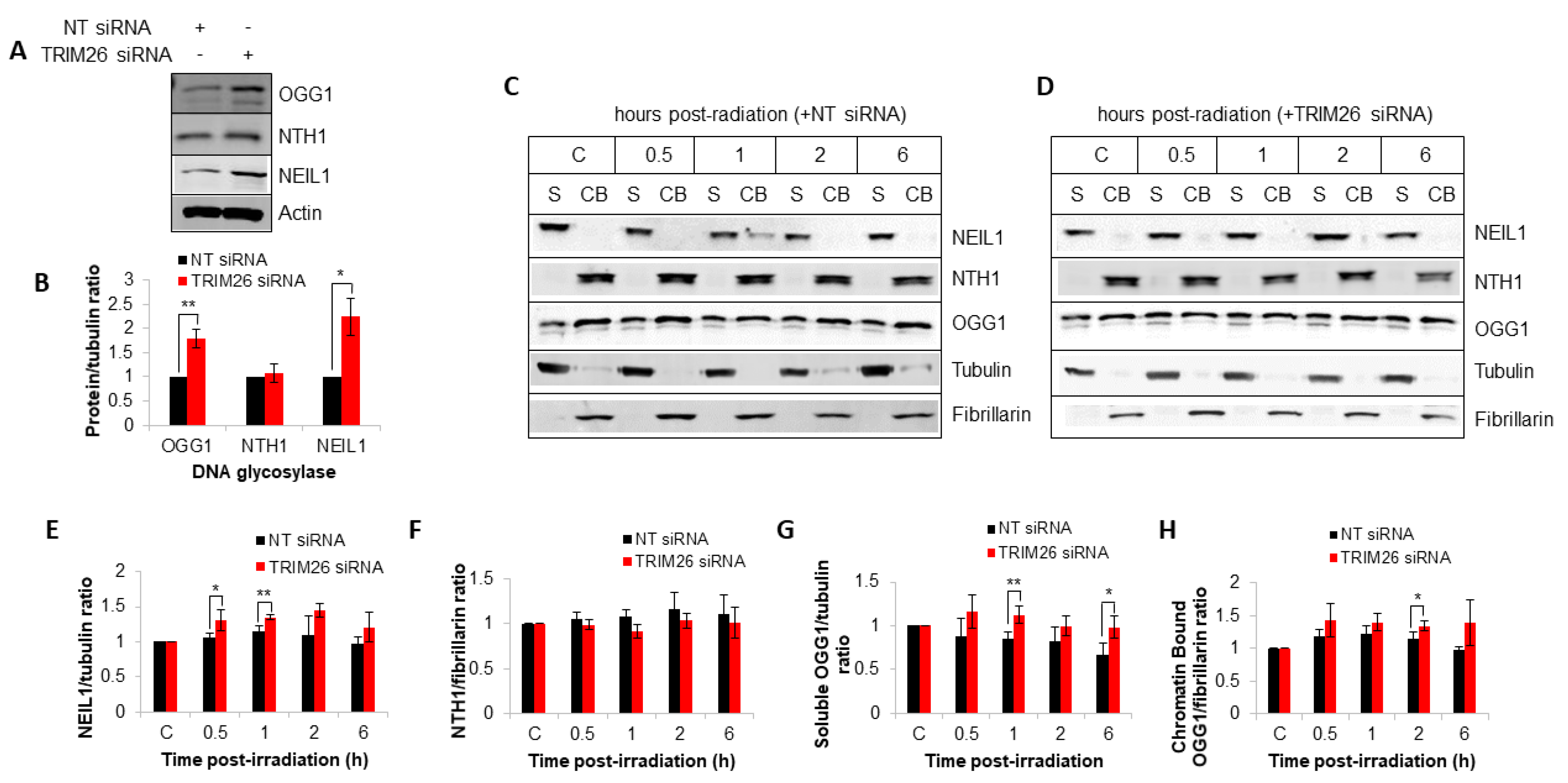
2.1. TRIM26 Can Ubiquitylate Multiple Oxidative DNA Glycosylases In Vitro
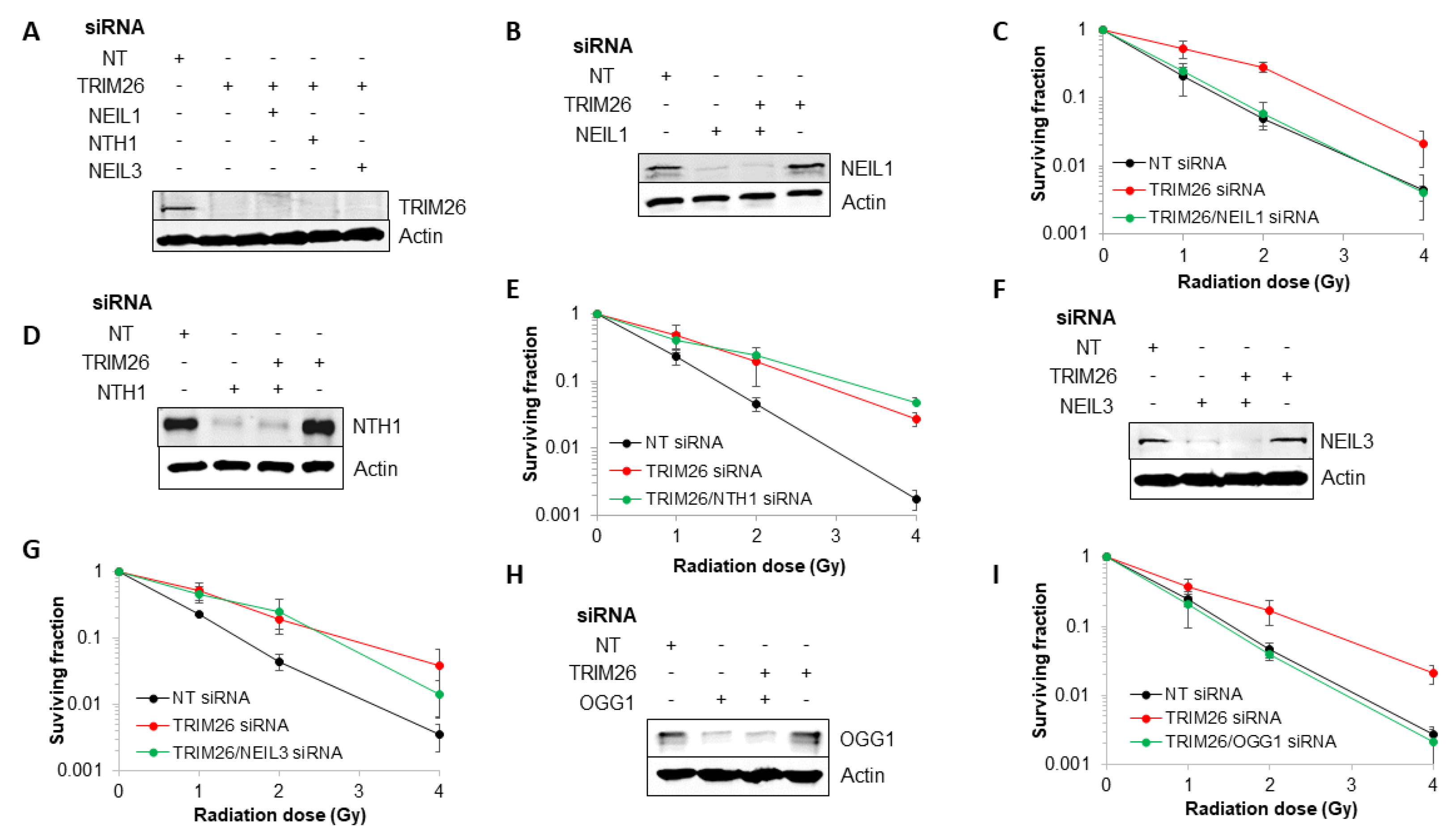
2.2. Acquired Resistance of TRIM26 Knockdown Cells to Ionizing Radiation Can Be Overcome by Targeting NEIL1 and OGG1
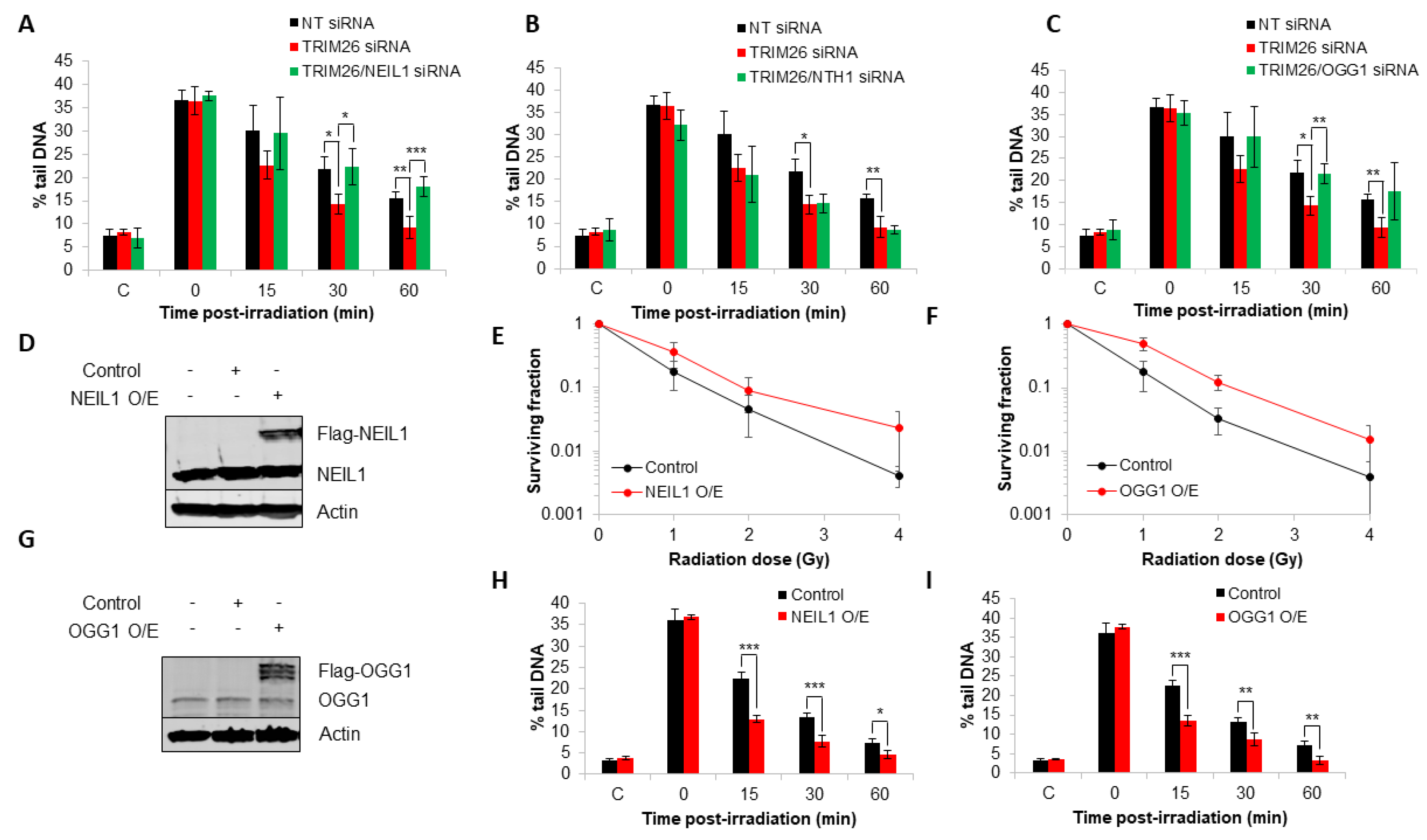
2.3. TRIM26 Controls the Rates of Repair of Radiation-Induced DNA Damage in a NEIL1 and OGG1-Dependent Manner
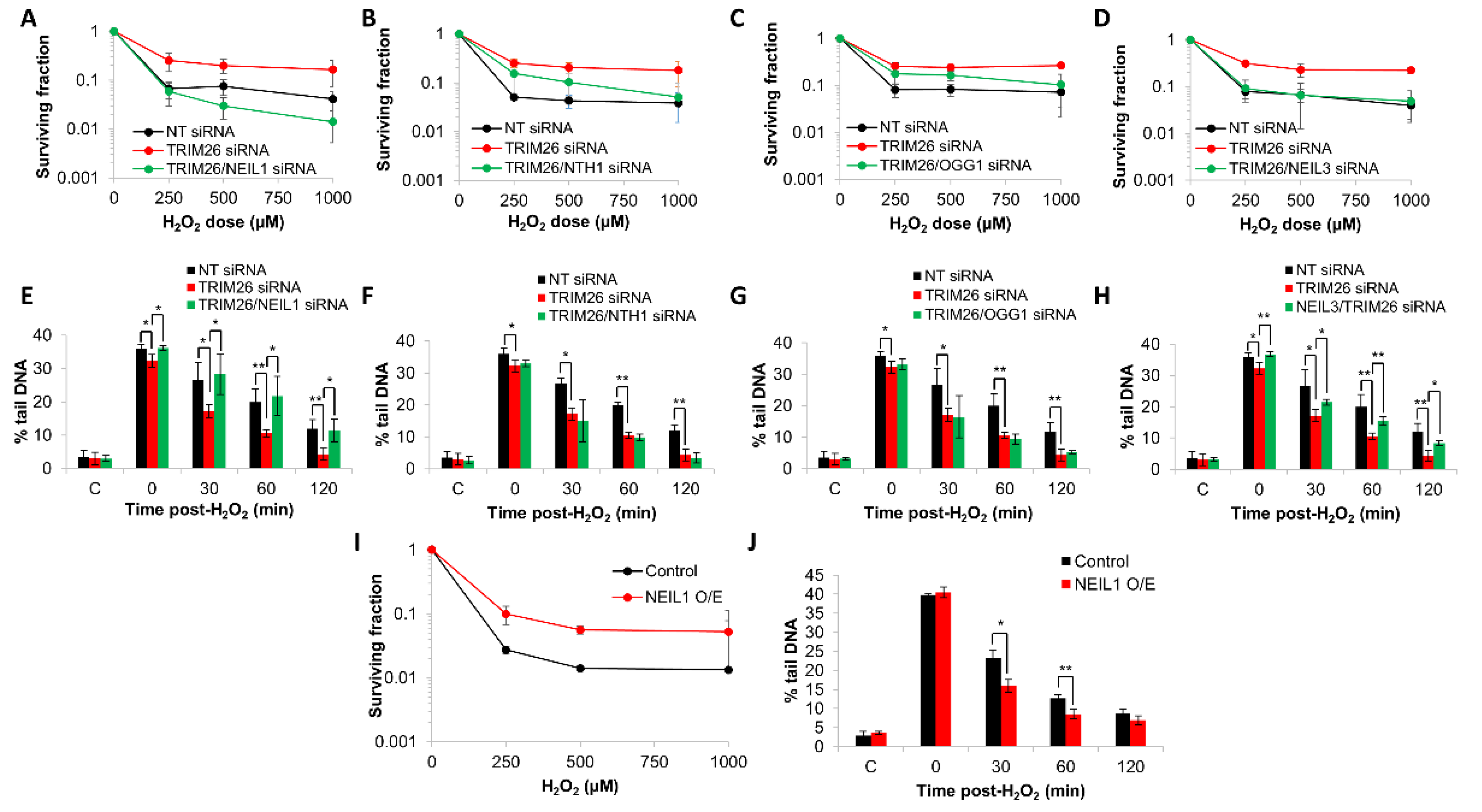
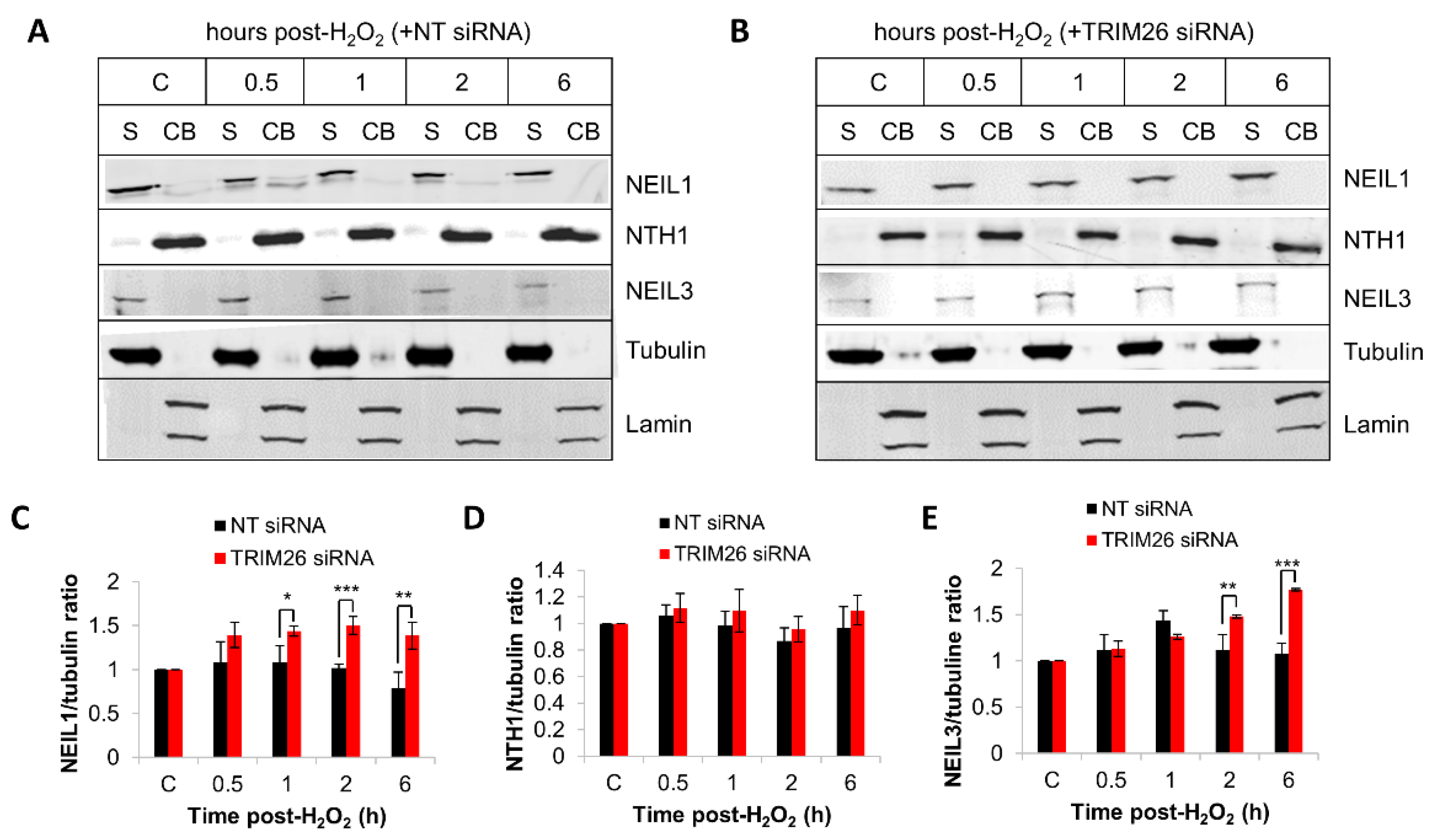
2.4. Acquired Resistance of TRIM26 Knockdown Cells to Hydrogen Peroxide Can Be Overcome by Targeting NEIL1 and NEIL3
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture, siRNA Knockdowns, and Plasmid Overexpressions
4.3. Cell Treatments and Clonogenic Assays
4.4. Whole-Cell Extract Preparation and Cell Fractionation
4.5. In Vitro Ubiquitylation Assay
4.6. Alkaline Single-Cell Gel Electrophoresis (Comet) Assay
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef]
- Jacobs, A.L.; Schar, P. DNA glycosylases: In DNA repair and beyond. Chromosoma 2012, 121, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Wallace, S.S. DNA glycosylases search for and remove oxidized DNA bases. Environ. Mol. Mutagen. 2013, 54, 691–704. [Google Scholar] [CrossRef] [PubMed]
- Grundy, G.J.; Parsons, J.L. Base excision repair and its implications to cancer therapy. Essays Biochem. 2020, 64, 831–843. [Google Scholar] [CrossRef] [PubMed]
- Boiteux, S.; Radicella, J.P. The human OGG1 gene: Structure, functions, and its implication in the process of carcinogenesis. Arch. Biochem. Biophys. 2000, 377, 1–8. [Google Scholar] [CrossRef]
- Aspinwall, R.; Rothwell, D.G.; Roldan-Arjona, T.; Anselmino, C.; Ward, C.J.; Cheadle, J.P.; Sampson, J.R.; Lindahl, T.; Harris, P.C.; Hickson, I.D. Cloning and characterization of a functional human homolog of Escherichia coli endonuclease III. Proc. Natl. Acad. Sci. USA 1997, 94, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Dou, H.; Theriot, C.A.; Das, A.; Hegde, M.L.; Matsumoto, Y.; Boldogh, I.; Hazra, T.K.; Bhakat, K.K.; Mitra, S. Interaction of the human DNA glycosylase NEIL1 with proliferating cell nuclear antigen. The potential for replication-associated repair of oxidized bases in mammalian genomes. J. Biol. Chem. 2008, 283, 3130–3140. [Google Scholar] [CrossRef]
- Hegde, M.L.; Hegde, P.M.; Bellot, L.J.; Mandal, S.M.; Hazra, T.K.; Li, G.M.; Boldogh, I.; Tomkinson, A.E.; Mitra, S. Prereplicative repair of oxidized bases in the human genome is mediated by NEIL1 DNA glycosylase together with replication proteins. Proc. Natl. Acad. Sci. USA 2013, 110, E3090–E3099. [Google Scholar] [CrossRef]
- Liu, M.; Bandaru, V.; Bond, J.P.; Jaruga, P.; Zhao, X.; Christov, P.P.; Burrows, C.J.; Rizzo, C.J.; Dizdaroglu, M.; Wallace, S.S. The mouse ortholog of NEIL3 is a functional DNA glycosylase in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 4925–4930. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Liu, M.; Fleming, A.M.; Burrows, C.J.; Wallace, S.S. Neil3 and NEIL1 DNA glycosylases remove oxidative damages from quadruplex DNA and exhibit preferences for lesions in the telomeric sequence context. J. Biol. Chem. 2013, 288, 27263–27272. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Fleming, A.M.; Averill, A.M.; Burrows, C.J.; Wallace, S.S. The NEIL glycosylases remove oxidized guanine lesions from telomeric and promoter quadruplex DNA structures. Nucleic Acids Res. 2015, 43, 4039–4054. [Google Scholar] [CrossRef]
- Martin, P.R.; Couve, S.; Zutterling, C.; Albelazi, M.S.; Groisman, R.; Matkarimov, B.T.; Parsons, J.L.; Elder, R.H.; Saparbaev, M.K. The Human DNA glycosylases NEIL1 and NEIL3 Excise Psoralen-Induced DNA-DNA Cross-Links in a Four-Stranded DNA Structure. Sci. Rep. 2017, 7, 17438. [Google Scholar] [CrossRef]
- Semlow, D.R.; Zhang, J.; Budzowska, M.; Drohat, A.C.; Walter, J.C. Replication-Dependent Unhooking of DNA Interstrand Cross-Links by the NEIL3 Glycosylase. Cell 2016, 167, 498–511.e414. [Google Scholar] [CrossRef]
- Li, N.; Wang, J.; Wallace, S.S.; Chen, J.; Zhou, J.; D’Andrea, A.D. Cooperation of the NEIL3 and Fanconi anemia/BRCA pathways in interstrand crosslink repair. Nucleic Acids Res. 2020, 48, 3014–3028. [Google Scholar] [CrossRef]
- Carter, R.J.; Parsons, J.L. Base Excision Repair, a Pathway Regulated by Posttranslational Modifications. Mol. Cell. Biol. 2016, 36, 1426–1437. [Google Scholar] [CrossRef]
- Limpose, K.L.; Corbett, A.H.; Doetsch, P.W. BERing the burden of damage: Pathway crosstalk and posttranslational modification of base excision repair proteins regulate DNA damage management. DNA Repair (Amst) 2017, 56, 51–64. [Google Scholar] [CrossRef]
- Edmonds, M.J.; Parsons, J.L. Regulation of base excision repair proteins by ubiquitylation. Exp. Cell Res. 2014, 329, 132–138. [Google Scholar] [CrossRef]
- Su, S.; Zhang, Y.; Liu, P. Roles of Ubiquitination and SUMOylation in DNA Damage Response. Curr. Issues Mol. Biol. 2020, 35, 59–84. [Google Scholar] [CrossRef]
- Clague, M.J.; Barsukov, I.; Coulson, J.M.; Liu, H.; Rigden, D.J.; Urbe, S. Deubiquitylases from genes to organism. Physiol. Rev. 2013, 93, 1289–1315. [Google Scholar] [CrossRef]
- Parsons, J.L.; Dianova, I.I.; Khoronenkova, S.V.; Edelmann, M.J.; Kessler, B.M.; Dianov, G.L. USP47 is a deubiquitylating enzyme that regulates base excision repair by controlling steady-state levels of DNA polymerase beta. Mol. Cell 2011, 41, 609–615. [Google Scholar] [CrossRef]
- Parsons, J.L.; Tait, P.S.; Finch, D.; Dianova, I.I.; Allinson, S.L.; Dianov, G.L. CHIP-mediated degradation and DNA damage-dependent stabilization regulate base excision repair proteins. Mol. Cell 2008, 29, 477–487. [Google Scholar] [CrossRef]
- Parsons, J.L.; Tait, P.S.; Finch, D.; Dianova, I.I.; Edelmann, M.J.; Khoronenkova, S.V.; Kessler, B.M.; Sharma, R.A.; McKenna, W.G.; Dianov, G.L. Ubiquitin ligase ARF-BP1/Mule modulates base excision repair. EMBO J. 2009, 28, 3207–3215. [Google Scholar] [CrossRef]
- Edmonds, M.J.; Carter, R.J.; Nickson, C.M.; Williams, S.C.; Parsons, J.L. Ubiquitylation-dependent regulation of NEIL1 by Mule and TRIM26 is required for the cellular DNA damage response. Nucleic Acids Res. 2017, 45, 726–738. [Google Scholar] [CrossRef]
- Williams, S.C.; Parsons, J.L. NTH1 Is a New Target for Ubiquitylation-Dependent Regulation by TRIM26 Required for the Cellular Response to Oxidative Stress. Mol. Cell. Biol. 2018, 38, e00616-17. [Google Scholar] [CrossRef]
- Mohammadi, A.; Pour Abbasi, M.S.; Khorrami, S.; Khodamoradi, S.; Mohammadi Goldar, Z.; Ebrahimzadeh, F. The TRIM proteins in cancer: From expression to emerging regulatory mechanisms. Clin. Transl. Oncol. 2022, 24, 460–470. [Google Scholar] [CrossRef]
- Hatakeyama, S. TRIM Family Proteins: Roles in Autophagy, Immunity, and Carcinogenesis. Trends Biochem. Sci. 2017, 42, 297–311. [Google Scholar] [CrossRef]
- Wang, Y.; He, D.; Yang, L.; Wen, B.; Dai, J.; Zhang, Q.; Kang, J.; He, W.; Ding, Q.; He, D. TRIM26 functions as a novel tumor suppressor of hepatocellular carcinoma and its downregulation contributes to worse prognosis. Biochem. Biophys. Res. Commun. 2015, 463, 458–465. [Google Scholar] [CrossRef]
- Wang, K.; Chai, L.; Qiu, Z.; Zhang, Y.; Gao, H.; Zhang, X. Overexpression of TRIM26 suppresses the proliferation, metastasis, and glycolysis in papillary thyroid carcinoma cells. J. Cell Physiol. 2019, 234, 19019–19027. [Google Scholar] [CrossRef]
- Xie, X.; Li, H.; Pan, J.; Han, X. Knockdown of TRIM26 inhibits the proliferation, migration and invasion of bladder cancer cells through the Akt/GSK3beta/beta-catenin pathway. Chem. Biol. Interact. 2021, 337, 109366. [Google Scholar] [CrossRef]
- Wang, P.; Zhao, W.; Zhao, K.; Zhang, L.; Gao, C. TRIM26 negatively regulates interferon-beta production and antiviral response through polyubiquitination and degradation of nuclear IRF3. PLoS Pathog. 2015, 11, e1004726. [Google Scholar] [CrossRef]
- Zhao, J.; Cai, B.; Shao, Z.; Zhang, L.; Zheng, Y.; Ma, C.; Yi, F.; Liu, B.; Gao, C. TRIM26 positively regulates the inflammatory immune response through K11-linked ubiquitination of TAB1. Cell Death Differ 2021, 28, 3077–3091. [Google Scholar] [CrossRef]
- Li, X.; Yuan, J.; Song, C.; Lei, Y.; Xu, J.; Zhang, G.; Wang, W.; Song, G. Deubiquitinase USP39 and E3 ligase TRIM26 balance the level of ZEB1 ubiquitination and thereby determine the progression of hepatocellular carcinoma. Cell Death Differ 2021, 28, 2315–2332. [Google Scholar] [CrossRef]
- Hans, F.; Senarisoy, M.; Bhaskar Naidu, C.; Timmins, J. Focus on DNA Glycosylases-A Set of Tightly Regulated Enzymes with a High Potential as Anticancer Drug Targets. Int. J. Mol. Sci. 2020, 21, 9226. [Google Scholar] [CrossRef]
- Kumar, A.; Pant, M.C.; Singh, H.S.; Khandelwal, S. Reduced expression of DNA repair genes (XRCC1, XPD, and OGG1) in squamous cell carcinoma of head and neck in North India. Tumour Biol. 2012, 33, 111–119. [Google Scholar] [CrossRef]
- Paz-Elizur, T.; Ben-Yosef, R.; Elinger, D.; Vexler, A.; Krupsky, M.; Berrebi, A.; Shani, A.; Schechtman, E.; Freedman, L.; Livneh, Z. Reduced repair of the oxidative 8-oxoguanine DNA damage and risk of head and neck cancer. Cancer Res. 2006, 66, 11683–11689. [Google Scholar] [CrossRef] [PubMed]
- Goto, M.; Shinmura, K.; Igarashi, H.; Kobayashi, M.; Konno, H.; Yamada, H.; Iwaizumi, M.; Kageyama, S.; Tsuneyoshi, T.; Tsugane, S.; et al. Altered expression of the human base excision repair gene NTH1 in gastric cancer. Carcinogenesis 2009, 30, 1345–1352. [Google Scholar] [CrossRef] [PubMed]
- Tran, O.T.; Tadesse, S.; Chu, C.; Kidane, D. Overexpression of NEIL3 associated with altered genome and poor survival in selected types of human cancer. Tumour Biol. 2020, 42, 1010428320918404. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Zhang, G.; Ma, J.; Wu, H.; Xiong, J.; Huang, X.; Tian, Y.; Deng, T.; Han, X.; Sun, X.; et al. Upregulation of Nei-Like DNA Glycosylase 3 Predicts Poor Prognosis in Hepatocellular Carcinoma. J. Oncol. 2021, 2021, 1301671. [Google Scholar] [CrossRef] [PubMed]
- Canugovi, C.; Misiak, M.; Scheibye-Knudsen, M.; Croteau, D.L.; Mattson, M.P.; Bohr, V.A. Loss of NEIL1 causes defects in olfactory function in mice. Neurobiol. Aging 2015, 36, 1007–1012. [Google Scholar] [CrossRef] [PubMed]
- Canugovi, C.; Yoon, J.S.; Feldman, N.H.; Croteau, D.L.; Mattson, M.P.; Bohr, V.A. Endonuclease VIII-like 1 (NEIL1) promotes short-term spatial memory retention and protects from ischemic stroke-induced brain dysfunction and death in mice. Proc. Natl. Acad. Sci. USA 2012, 109, 14948–14953. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Nejad, M.I.; Varela, J.G.; Price, N.E.; Wang, Y.; Gates, K.S. A role for the base excision repair enzyme NEIL3 in replication-dependent repair of interstrand DNA cross-links derived from psoralen and abasic sites. DNA Repair (Amst) 2017, 52, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Fleming, A.M.; Ding, Y.; Burrows, C.J. Oxidative DNA damage is epigenetic by regulating gene transcription via base excision repair. Proc. Natl. Acad. Sci. USA 2017, 114, 2604–2609. [Google Scholar] [CrossRef]
- Allgayer, J.; Kitsera, N.; Bartelt, S.; Epe, B.; Khobta, A. Widespread transcriptional gene inactivation initiated by a repair intermediate of 8-oxoguanine. Nucleic Acids Res. 2016, 44, 7267–7280. [Google Scholar] [CrossRef]
- Pan, L.; Zhu, B.; Hao, W.; Zeng, X.; Vlahopoulos, S.A.; Hazra, T.K.; Hegde, M.L.; Radak, Z.; Bacsi, A.; Brasier, A.R.; et al. Oxidized Guanine Base Lesions Function in 8-Oxoguanine DNA Glycosylase-1-mediated Epigenetic Regulation of Nuclear Factor kappaB-driven Gene Expression. J. Biol. Chem. 2016, 291, 25553–25566. [Google Scholar] [CrossRef]
- Wang, R.; Hao, W.; Pan, L.; Boldogh, I.; Ba, X. The roles of base excision repair enzyme OGG1 in gene expression. Cell. Mol. Life Sci. 2018, 75, 3741–3750. [Google Scholar] [CrossRef] [PubMed]
- Fleming, A.M.; Burrows, C.J. Oxidative stress-mediated epigenetic regulation by G-quadruplexes. NAR Cancer 2021, 3, zcab038. [Google Scholar] [CrossRef]
- Bhakat, K.K.; Mokkapati, S.K.; Boldogh, I.; Hazra, T.K.; Mitra, S. Acetylation of human 8-oxoguanine-DNA glycosylase by p300 and its role in 8-oxoguanine repair in vivo. Mol. Cell. Biol. 2006, 26, 1654–1665. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Imam, S.Z.; Hashiguchi, K.; de Souza-Pinto, N.C.; Bohr, V.A. Phosphorylation of human oxoguanine DNA glycosylase (alpha-OGG1) modulates its function. Nucleic Acids Res. 2005, 33, 3271–3282. [Google Scholar] [CrossRef] [PubMed]
- Prakash, A.; Cao, V.B.; Doublie, S. Phosphorylation Sites Identified in the NEIL1 DNA Glycosylase Are Potential Targets for the JNK1 Kinase. PLoS ONE 2016, 11, e0157860. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Yang, C.; Hegde, M.L.; Hegde, P.M.; Mitra, J.; Pandey, A.; Dutta, A.; Datarwala, A.T.; Bhakat, K.K.; Mitra, S. Acetylation of oxidized base repair-initiating NEIL1 DNA glycosylase required for chromatin-bound repair complex formation in the human genome increases cellular resistance to oxidative stress. DNA Repair (Amst) 2018, 66–67, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.R.; Parsons, J.L. The E3 Ubiquitin Ligase NEDD4L Targets OGG1 for Ubiquitylation and Modulates the Cellular DNA Damage Response. Front. Cell Dev. Biol. 2020, 8, 607060. [Google Scholar] [CrossRef]
- Braselmann, H.; Michna, A.; Hess, J.; Unger, K. CFAssay: Statistical analysis of the colony formation assay. Radiat Oncol. 2015, 10, 223. [Google Scholar] [CrossRef] [PubMed]
- Nickson, C.M.; Moori, P.; Carter, R.J.; Rubbi, C.P.; Parsons, J.L. Misregulation of DNA damage repair pathways in HPV-positive head and neck squamous cell carcinoma contributes to cellular radiosensitivity. Oncotarget 2017, 8, 29963–29975. [Google Scholar] [CrossRef] [PubMed]
- Nickson, C.M.; Parsons, J.L. Monitoring regulation of DNA repair activities of cultured cells in-gel using the comet assay. Front. Genet. 2014, 5, 232. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Konis, S.M.R.; Hughes, J.R.; Parsons, J.L. TRIM26 Maintains Cell Survival in Response to Oxidative Stress through Regulating DNA Glycosylase Stability. Int. J. Mol. Sci. 2022, 23, 11613. https://doi.org/10.3390/ijms231911613
Konis SMR, Hughes JR, Parsons JL. TRIM26 Maintains Cell Survival in Response to Oxidative Stress through Regulating DNA Glycosylase Stability. International Journal of Molecular Sciences. 2022; 23(19):11613. https://doi.org/10.3390/ijms231911613
Chicago/Turabian StyleKonis, Sifaddin M. R., Jonathan R. Hughes, and Jason L. Parsons. 2022. "TRIM26 Maintains Cell Survival in Response to Oxidative Stress through Regulating DNA Glycosylase Stability" International Journal of Molecular Sciences 23, no. 19: 11613. https://doi.org/10.3390/ijms231911613
APA StyleKonis, S. M. R., Hughes, J. R., & Parsons, J. L. (2022). TRIM26 Maintains Cell Survival in Response to Oxidative Stress through Regulating DNA Glycosylase Stability. International Journal of Molecular Sciences, 23(19), 11613. https://doi.org/10.3390/ijms231911613