
Characterization and Comparative Analysis of Chloroplast Genomes in Five Uncaria Species Endemic to China

Abstract
1. Introduction
2. Results
2.1. Morphological Comparison
2.2. Chloroplast Genome Sequencing and Assembly
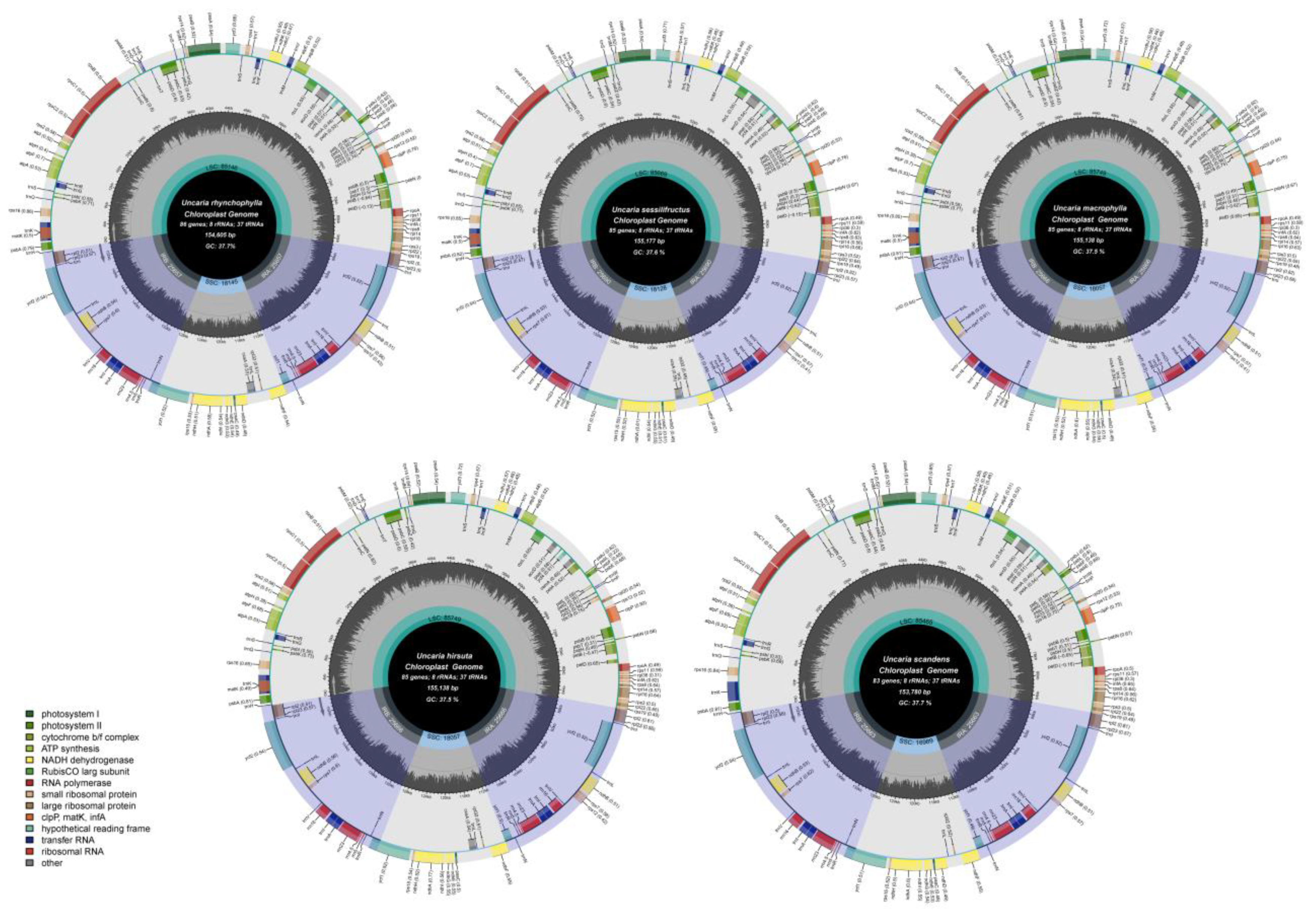
2.3. Chloroplast Genome Structure and Characteristics Analyses
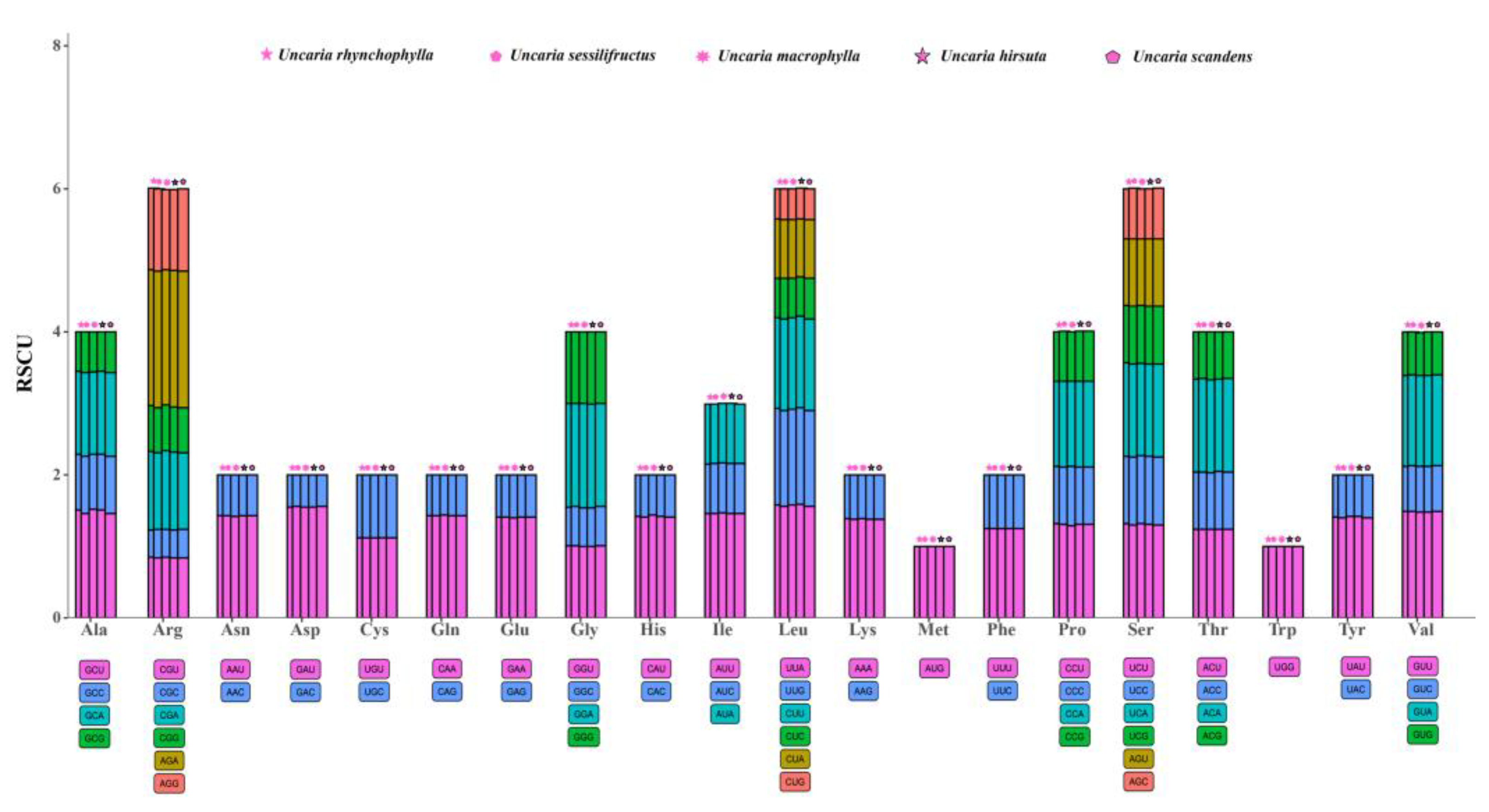
2.4. Codon Usage Analyses
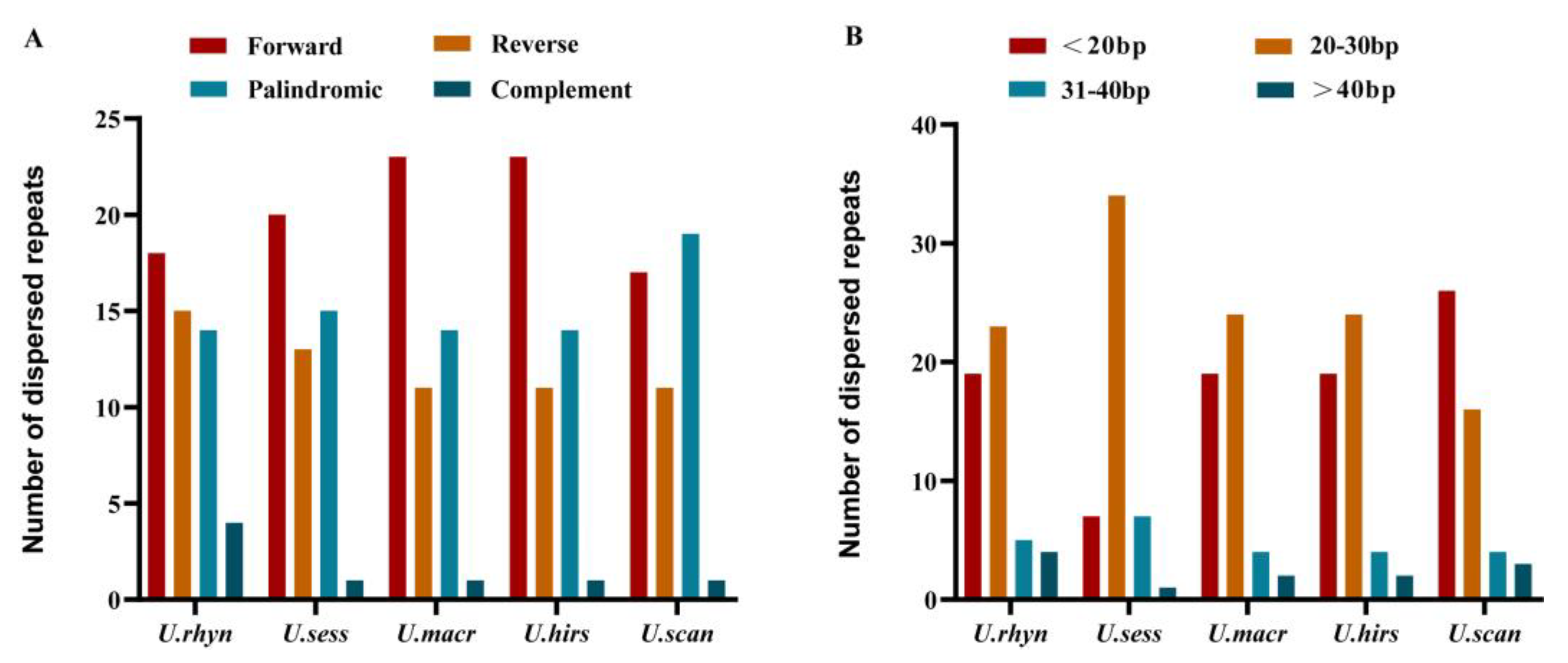
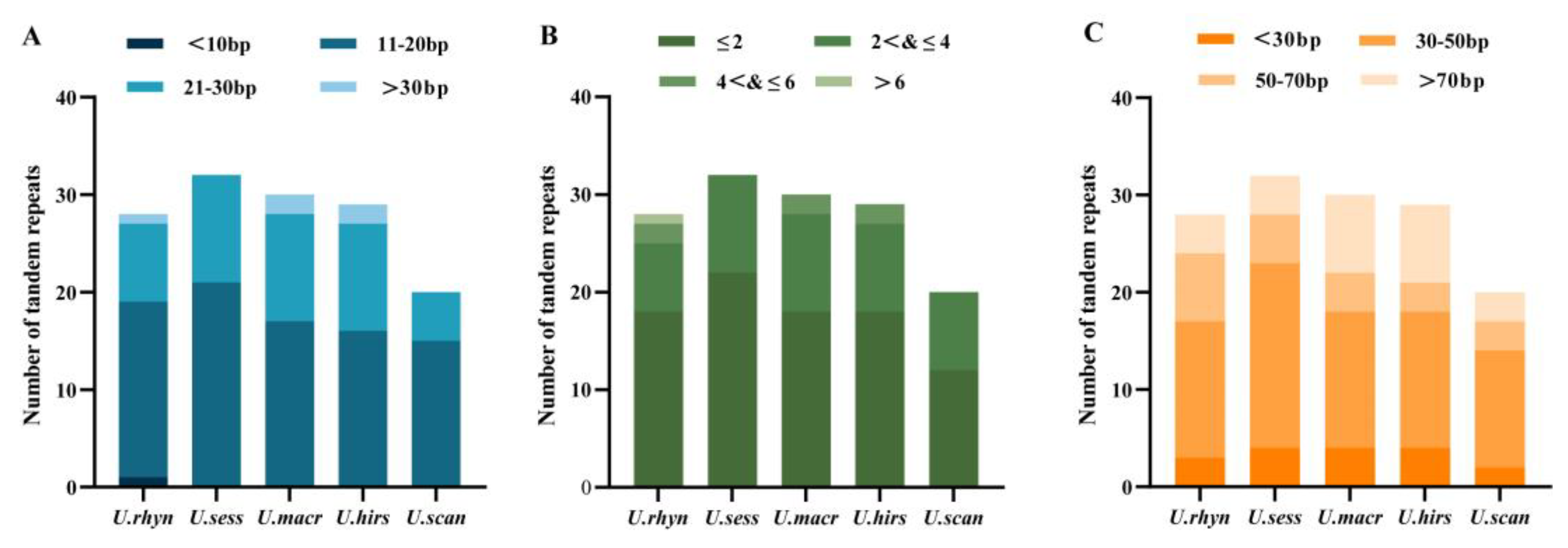
2.5. Repeat Sequence Analysis
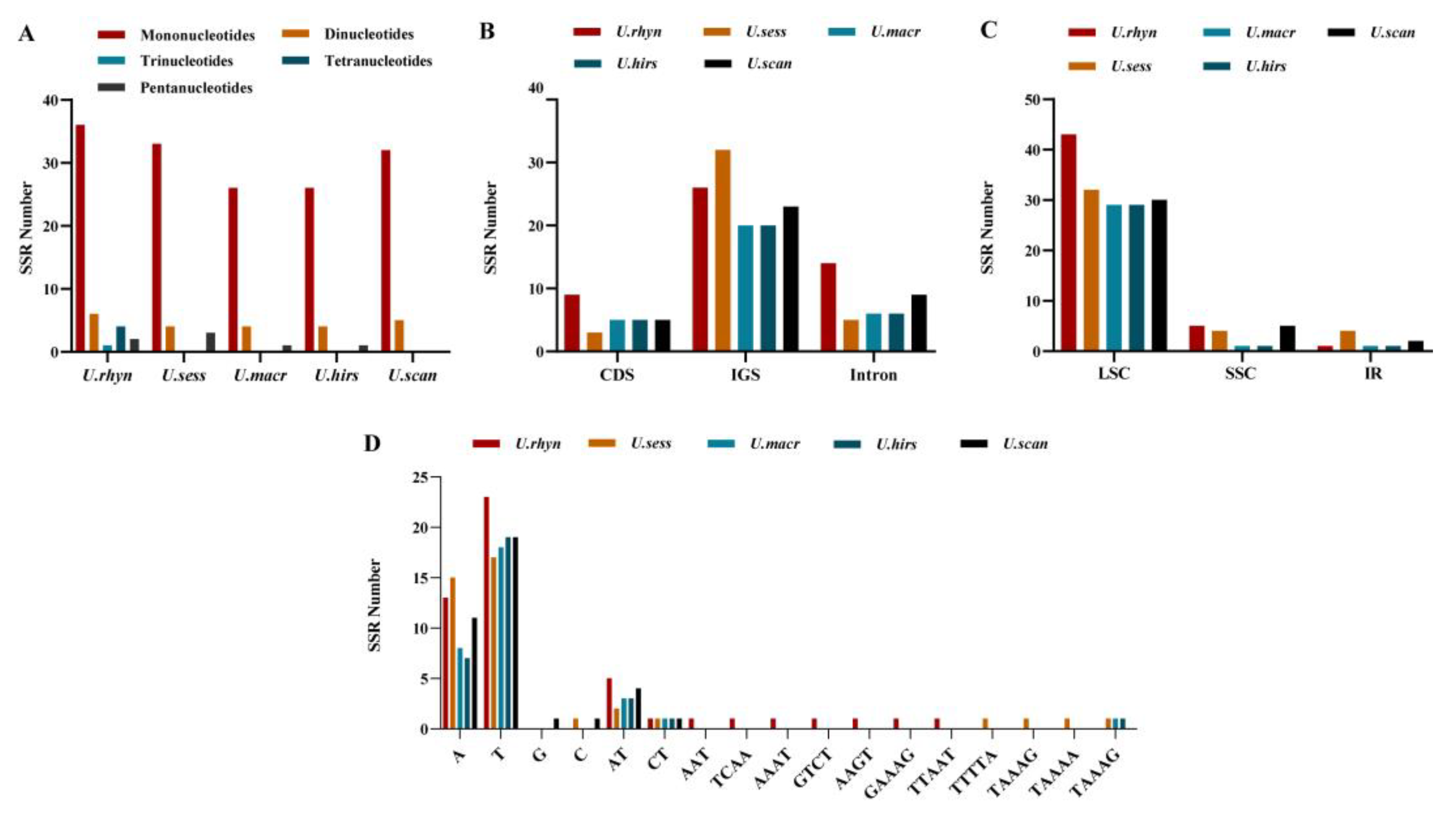
2.6. SSR Analysis
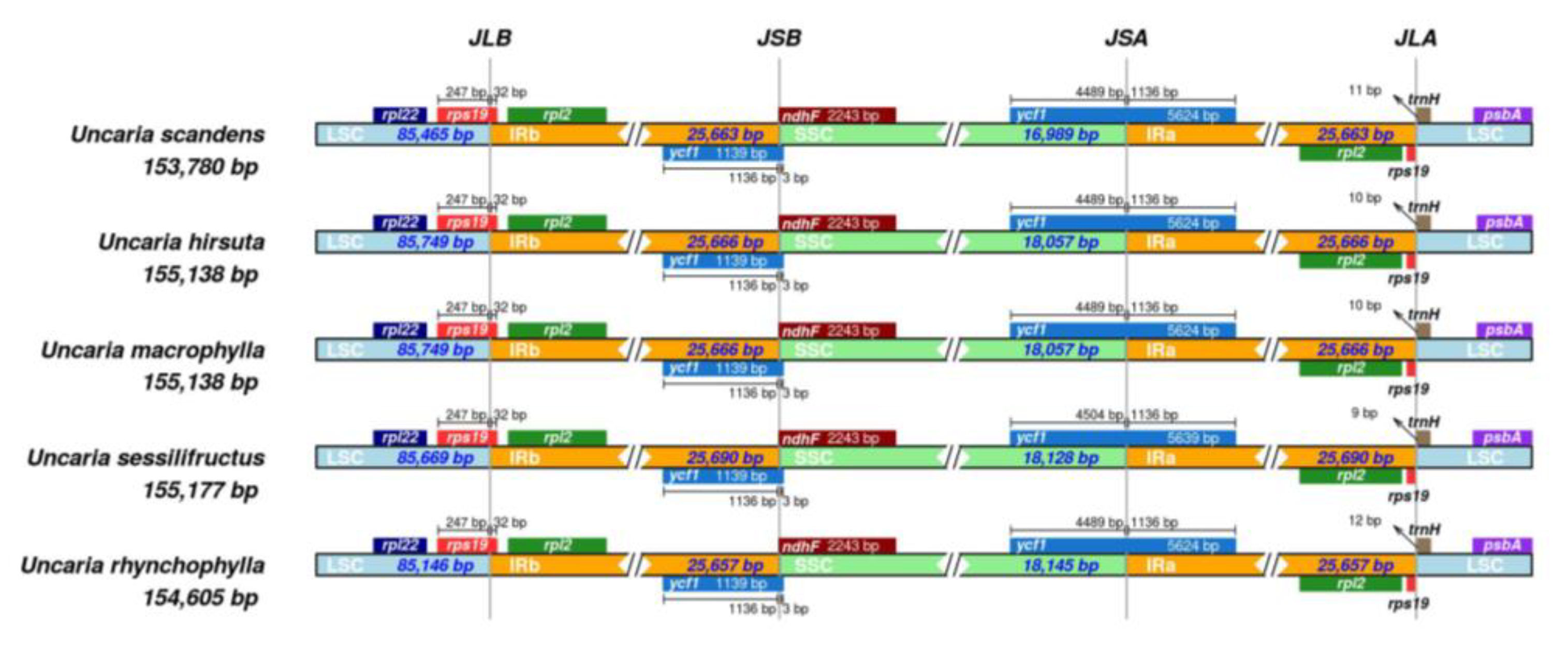
2.7. Contraction and Expansion of Inverted Repeats
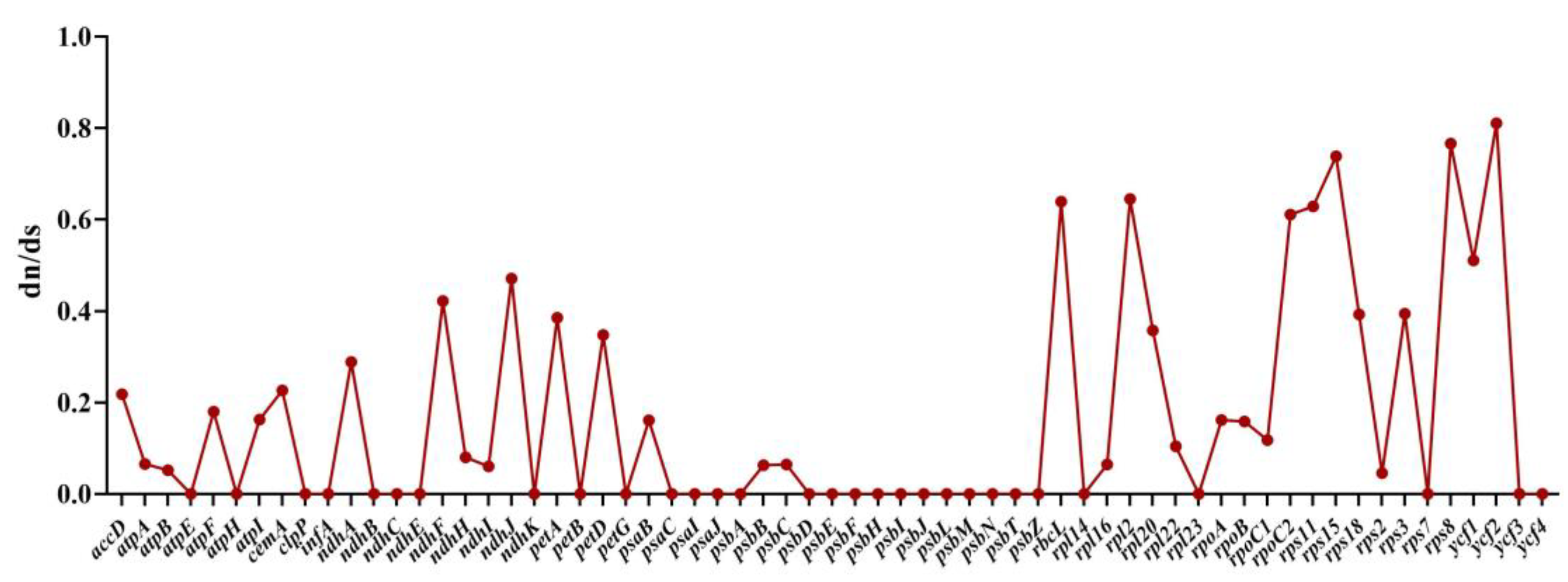
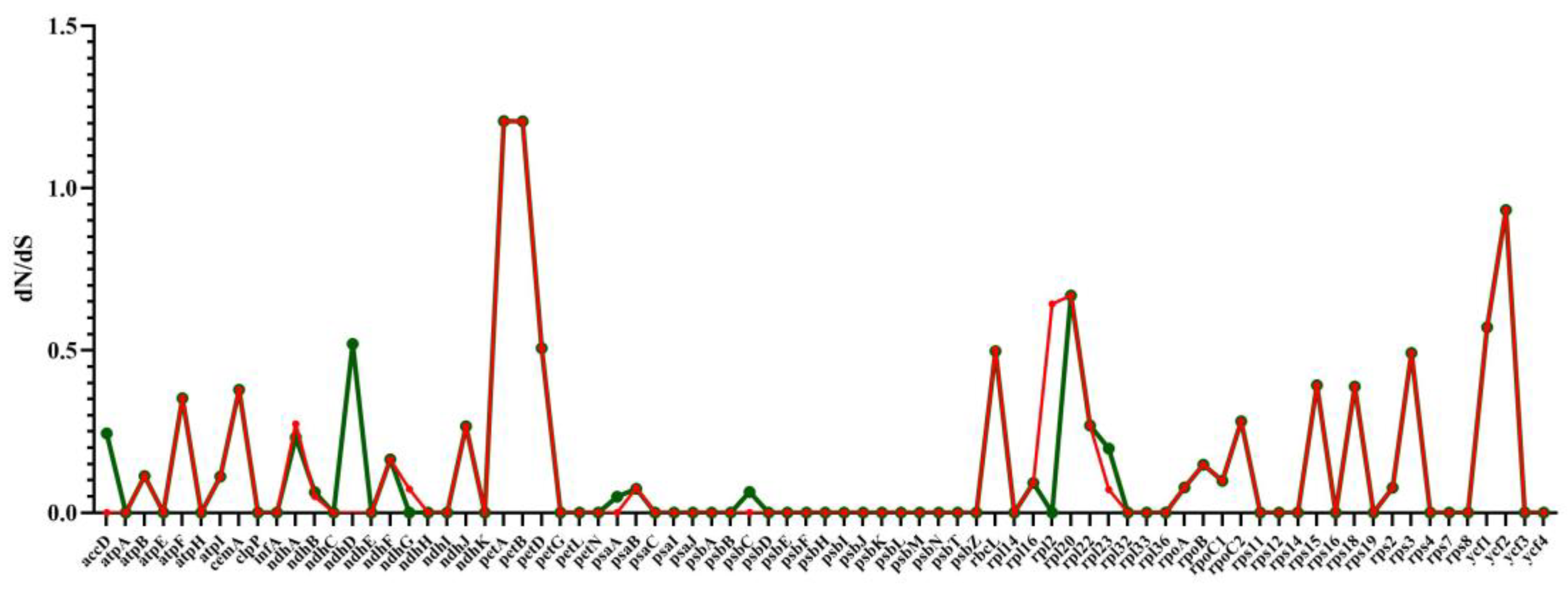
2.8. RNA Editing Site Analysis and Selective Pressure Analyses
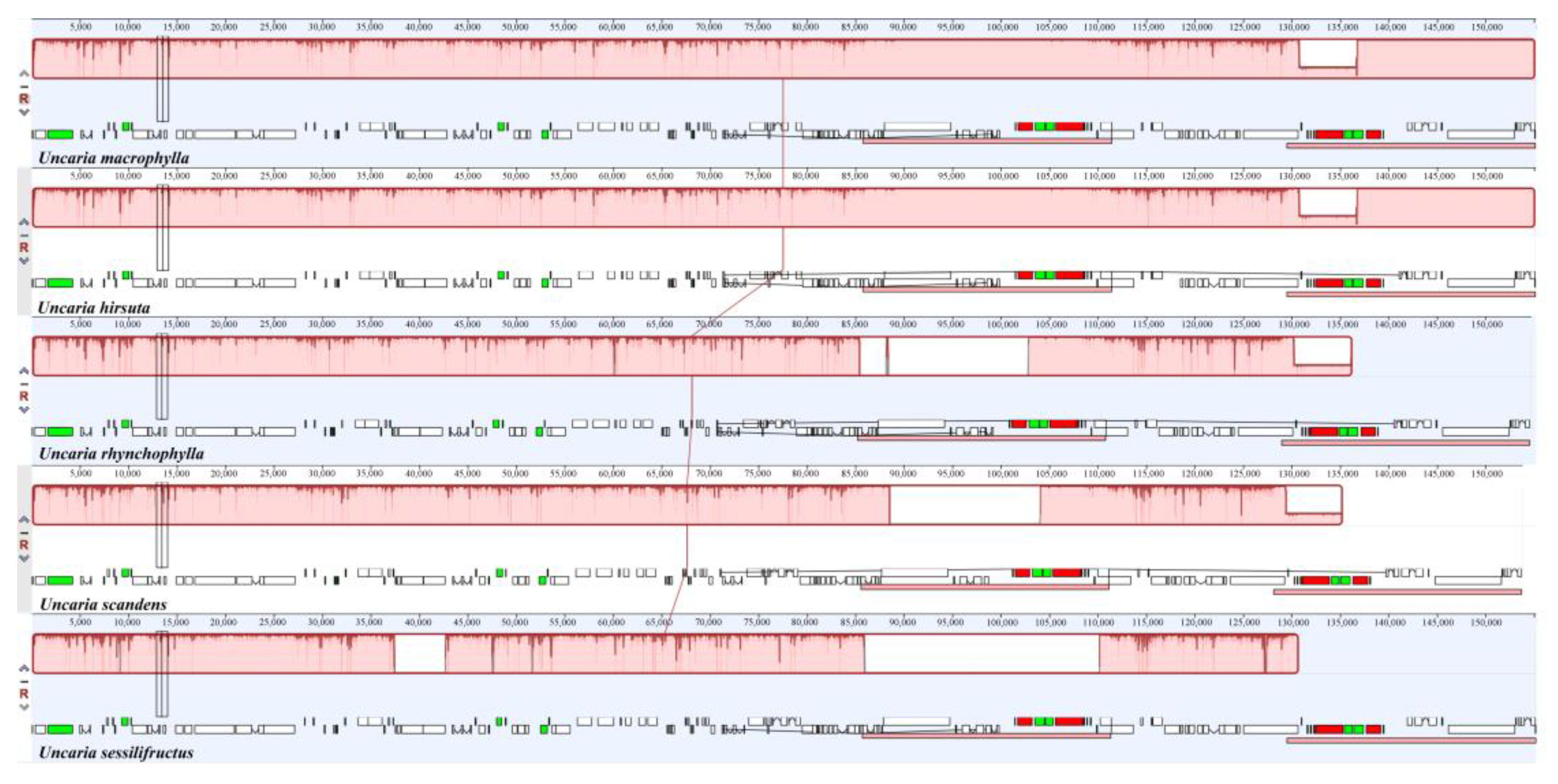
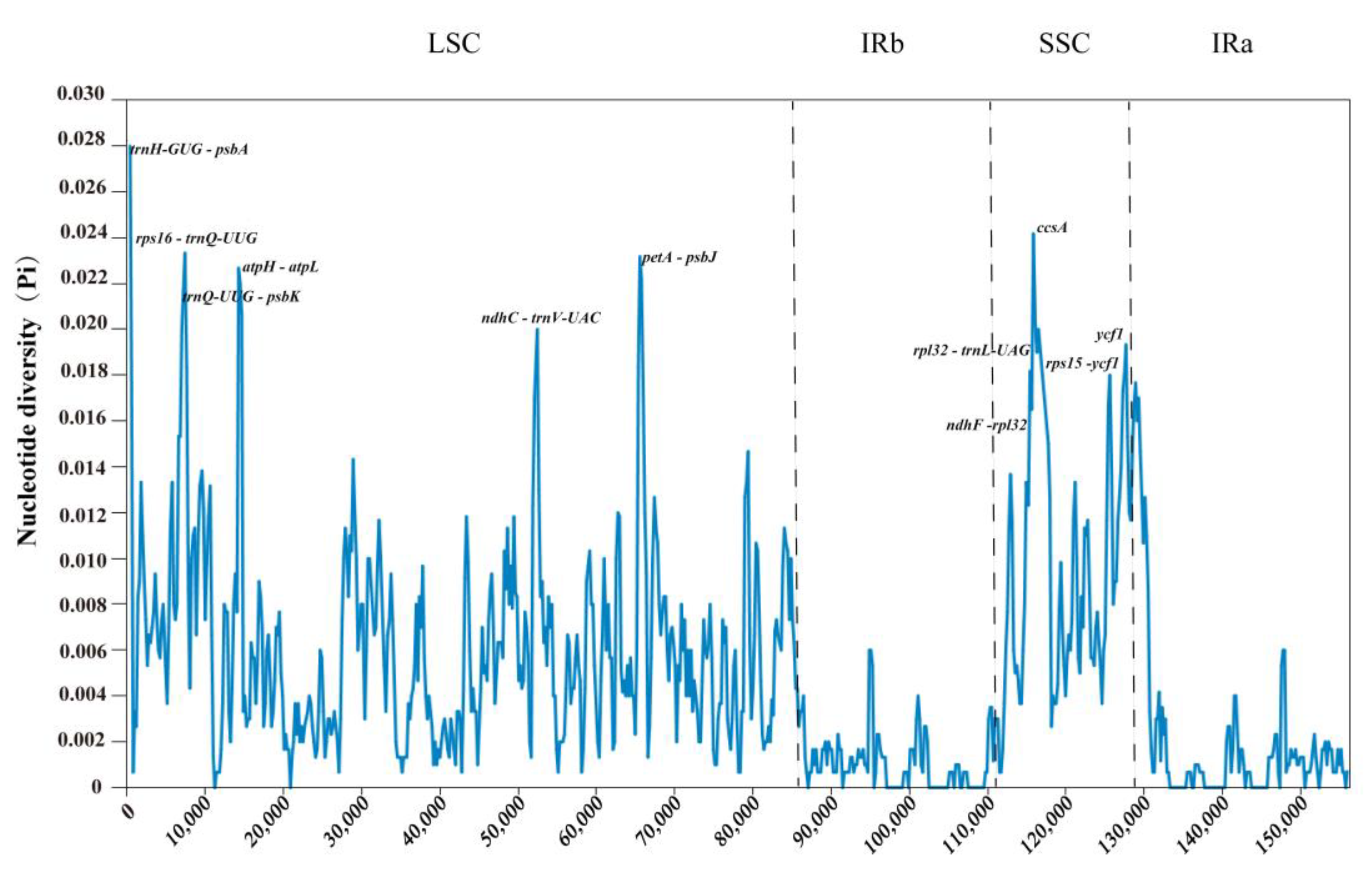
2.9. Sequence Divergence Analysis
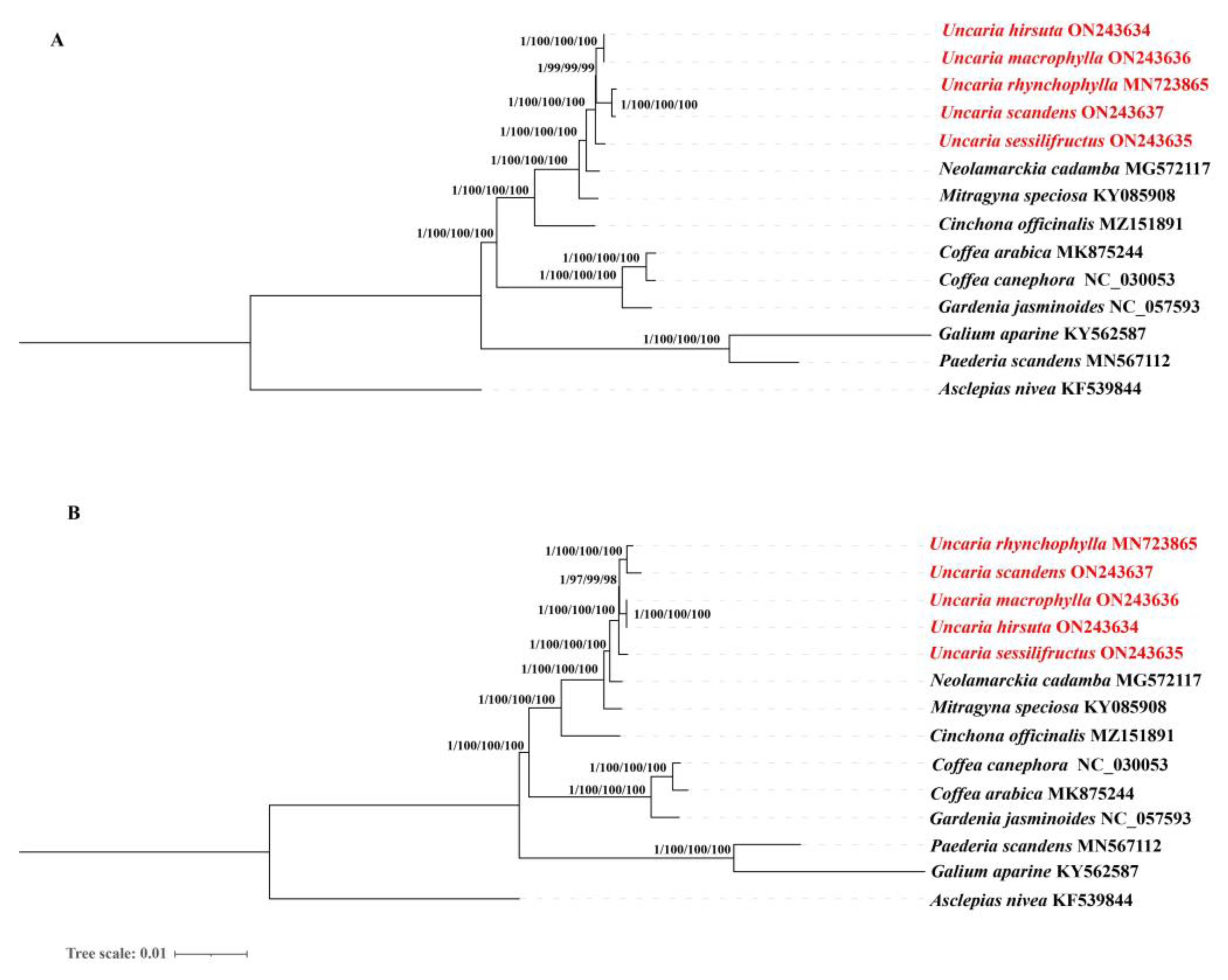
2.10. Phylogenetic Analysis
3. Discussion
4. Materials and Methods
4.1. Phenotype Measurement
4.2. Chloroplast DNA Extraction, Sequencing, Assembly and Annotation
4.3. Codon Usage Analyses
4.4. IR/SC Boundary Region
4.5. Characterization of Repeat Sequences and SSRs
4.6. Predict RNA Editing Sites and Selective Pressure Analyses
4.7. Sequence Divergence Analysis
4.8. Phylogenetic Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, M.; Chen, M.M.; Zhang, X.M.; Chen, S.N.; Liang, Z.S. The complete chloroplast genome sequence of traditional Chinese medicine Uncaria macrophylla (Rubiaceae). Mitochondrial DNA Part B 2022, 7, 694–695. [Google Scholar] [CrossRef] [PubMed]
- Laus, G. Advances in Chemistry and Bioactivity of the Genus Uncaria. Phytother. Res. 2004, 18, 259–274. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.F.; Liu, L.M.; Liu, Y.Y.; Liu, J.; Yan, L.; Pan, C.S.; Wang, M.X.; Wang, C.S.; Fan, J.Y.; Gao, Y.S.; et al. Inhibitory effect of rhynchophylline on contraction of cerebral arterioles to endothelin 1: Role of rho kinase—ScienceDirect. J. Ethnopharmacol. 2014, 155, 147–153. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, L.; Hu, L.J.; Liu, W.Y.; Feng, F.; Qu, W. Two new ortho benzoquinones from Uncaria rhynchophylla. Chin. J. Nat. Med. 2016, 14, 235. [Google Scholar] [CrossRef]
- Guo, Q.; Si, X.L.; Shi, Y.T.; Yang, H.S.; Liu, X.Y.; Liang, H.; Tu, P.F.; Zhang, Q.Y. Glucoconjugated Monoterpene Indole Alkaloids from Uncaria rhynchophylla. J. Nat. Prod. 2019, 82, 3288–3301. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Jiang, F.; Li, Y.L.; Jiang, Y.H.; Yang, W.Q.; Sheng, J.; Xu, W.J.; Zhu, Q.J. Rhynchophylla total alkaloid rescues autophagy, decreases oxidative stress and improves endothelial vasodilation in spontaneous hypertensive rats. Acta Pharmacol. Sin. 2018, 39, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, I.; Terabayashi, S.; Kubo, M.; Higuchi, M.; Komatsu, Y.; Okada, M.; Taki, K.; Kamei, J. Effect on locomotion of indole alkaloids from the hooks of Uncaria plants. Phytomedicine 1999, 6, 163–168. [Google Scholar] [CrossRef]
- Hu, S.Q.; Mak, S.H.; Zuo, X.L.; Li, H.T.; Wang, Y.Q.; Han, Y.F. Neuroprotection Against MPP+-Induced Cytotoxicity Through the Activation of PI3-K/Akt/GSK3β/MEF2D Signaling Pathway by Rhynchophylline, the Major Tetracyclic Oxindole Alkaloid Isolated From Uncaria rhynchophylla. Front. Pharmacol. 2018, 9, 768. [Google Scholar] [CrossRef]
- Hanwool, L.; Seung, B.; Jong, L.; Chulwon, K.; Jeong Hyeon, K.; Seok Geun, L.; Arunachalam, C.; Sulaiman, A.; Yang, W.; Jae Young, U. Isorhynchophylline, a Potent Plant Alkaloid, Induces Apoptotic and Anti-Metastatic Effects in Human Hepatocellular Carcinoma Cells through the Modulation of Diverse Cell Signaling Cascades. Int. J. Mol. Sci. 2017, 18, 1095. [Google Scholar]
- Wicke, S.; Schneeweiss, G.M.; De Pamphilis, C.W.; Kai, F.M.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef]
- Ruhlman, T.A.; Jansen, R.K. Plastid Genomes of Flowering Plants: Essential Principles. Methods Mol. Biol. 2021, 2317, 3–47. [Google Scholar] [PubMed]
- Yang, Z.; Wang, G.; Ma, Q.; Ma, W.; Liang, L.; Zhao, T. The complete chloroplast genomes of three Betulaceae species: Implications for molecular phylogeny and historical biogeography. PeerJ 2019, 7, e6320. [Google Scholar] [CrossRef] [PubMed]
- Ruhfel, B.; Gitzendanner, M.A.; Soltis, P.S.; Soltis, D.E.; Burleigh, J.G. From algae to angiosperms–inferring the phylogeny of green plants (Viridiplantae) from 360 plastid genomes. BMC Evol. Biol. 2014, 14, 23. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.L.; Zhou, J.G.; Cui, Y.X.; Wang, Y.; Duan, B.Z.; Yao, H. Identification of Ligularia Herbs Using the Complete Chloroplast Genome as a Super-Barcode. Front. Pharmacol. 2018, 9, 695. [Google Scholar] [CrossRef]
- Li, L.; Hu, Y.; He, M.; Zhang, B.; Hong, Y. Comparative chloroplast genomes: Insights into the evolution of the chloroplast genome of Camellia sinensis and the phylogeny of Camellia. BMC Genom. 2020, 22, 138. [Google Scholar] [CrossRef]
- Ning, C.U.I.; Bao-Sheng, L.I.A.O.; Liang, C.L.; Shi-Feng, L.I.; Zhang, H.; Jiang, X.U.; Xi-Wen, L.I.; Chen, S.L. Complete chloroplast genome of Salvia plebeia:organization, specific barcode and phylogenetic analysis. Chin. J. Nat. Med. 2020, 18, 563–572. [Google Scholar]
- Zhou, J.H.; Zhang, J.; Chen, H.T.; Ma, L.N.; Liu, Y.S. Analysis of synonymous codon usage in foot-and-mouth disease virus. Vet. Res. Commun. 2010, 34, 393–404. [Google Scholar] [CrossRef]
- Nie, X.; Lv, S.; Zhang, Y.; Du, X.; Wang, L.; Biradar, S.S.; Tan, X.; Wan, F.; Song, W.; Sergios, O.K. Complete Chloroplast Genome Sequence of a Major Invasive Species, Crofton Weed (Ageratina adenophora). PLoS ONE 2012, 7, e36869. [Google Scholar] [CrossRef]
- Yi, X.; Lei, G.; Wang, B.; Su, Y.J.; Wang, T. The Complete Chloroplast Genome Sequence of Cephalotaxus oliveri (Cephalotaxaceae): Evolutionary Comparison of Cephalotaxus Chloroplast DNAs and Insights into the Loss of Inverted Repeat Copies in Gymnosperms. Genome Biol. Evol. Dev. 2013, 5, 688–698. [Google Scholar] [CrossRef]
- Yang, Q.; Fu, G.F.; Wu, Z.Q.; Li, L.; Zhao, J.L.; Li, Q.J. Chloroplast Genome Evolution in Four Montane Zingiberaceae Taxa in China. Front. Plant Sci. 2021, 12, 774482. [Google Scholar] [CrossRef]
- Hu, Y.; Xing, W.; Song, H.; Zhu, H.; Liu, G.; Hu, Z. Evolutionary Analysis of Unicellular Species in Chlamydomonadales Through Chloroplast Genome Comparison With the Colonial Volvocine Algae. Front. Microbiol. 2019, 10, 1351. [Google Scholar] [CrossRef] [PubMed]
- Ollivier, M.; Rispe, C. Evolutionary Dynamics in the Aphid Genome: Search for Genes Under Positive Selection and Detection of Gene Family Expansions. In Evolutionary Biology–Concepts, Molecular and Morphological Evolution; Springer: Berlin/Heidelberg, Germany, 2010; pp. 133–142. [Google Scholar]
- Wei, X.P.; Li, H.J.; Che, P.; Guo, H.J.; Zhang, B.G.; Liu, H.T.; Qi, Y.D. Comparing chloroplast genomes of traditional Chinese herbs Schisandra sphenanthera and S. chinensis. Chin. Herb. Med. 2020, 12, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Du, L.; Ao, L.; Chen, J.; Li, W.; Hu, W.; Zhang, W.; Kyunghee, K.; Sang-Choon, L.; Yang, T.J. The Complete Chloroplast Genome Sequences of Five Epimedium Species: Lights into Phylogenetic and Taxonomic Analyses. Front. Plant Sci. 2016, 7, 306. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J.; Bell, C.D.; Soltis, P.S.; Soltis, D.E. Using plastid genome-scale data to resolve enigmatic relationships among basal angiosperms. Proc. Natl. Acad. Sci. USA 2007, 104, 19363–19368. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, J.W.; Yang, Y.; Li, X.N. Structural and Comparative Analysis of the Complete Chloroplast Genome of a Mangrove Plant: Scyphiphora hydrophyllacea Gaertn. f. and Related Rubiaceae Species. Forests 2019, 10, 1000. [Google Scholar] [CrossRef]
- Gao, L.; Su, Y.J.; Wang, T. Plastid genome sequencing, comparative genomics, and phylogenomics:Current status and prospects. J. Syst. Evol. 2010, 48, 77–93. [Google Scholar] [CrossRef]
- Oyebanji, O.; Zhang, R.; Chen, S.Y.; Yi, T.S. New Insights Into the Plastome Evolution of the Millettioid Phaseoloid Clade (Papilionoideae, Leguminosae). Front. Plant Sci. 2021, 12, 652483. [Google Scholar] [CrossRef]
- Frailey, D.C.; Chaluvadi, S.R.; Vaughn, J.N.; Coatney, C.G.; Bennetzen, J.L. Gene loss and genome rearrangement in the plastids of five Hemiparasites in the family Orobanchaceae. BMC Plant Biol. 2018, 18, 30. [Google Scholar] [CrossRef]
- Li, X.; Li, Y.F.; Zang, S.P.S.M.; El-Kassaby, Y.A.; Fang, Y.M. Evolutionary patterns of nucleotide substitution rates in plastid genomes of Quercus. Ecol. Evol. 2021, 11, 13401–13414. [Google Scholar] [CrossRef]
- Li, Y.; LÜ, G.H.; Zhang, X.N.; He, X.M. Chloroplast Genome Structure and Variation Analysis of Brassicaceae Species. Acta Bot. Boreali-Occident. Sin. 2017, 37, 1090–1101. [Google Scholar]
- Wang, W.; Yu, H.; Wang, J.; Lei, W.; Gao, J.; Qiu, X.; Wang, J. The Complete Chloroplast Genome Sequences of the Medicinal Plant Forsythia suspensa (Oleaceae). Int. J. Mol. Sci. 2017, 18, 2288. [Google Scholar] [CrossRef] [PubMed]
- Yi, D.K.; Lee, H.L.; Sun, B.Y.; Chung, M.Y.; Kim, K.J. The complete chloroplast DNA sequence of Eleutherococcus senticosus (Araliaceae); Comparative evolutionary analyses with other three asterids. Mol. Cells 2012, 33, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Lei, W.J.; Ni, D.P.; Wang, Y.J.; Shao, J.J.; Liu, C. Intraspecific and heteroplasmic variations, gene losses and inversions in the chloroplast genome of Astragalus membranaceus. Sci. Rep. 2016, 6, 21669. [Google Scholar] [CrossRef] [PubMed]
- Daniell, H.; Lin, C.S.; Yu, M.; Chang, W.J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef]
- Zhang, R.; Jiao, W.; Kai, H.; Ting, R.; Zeng, S.; Edward, B.; Liu, Z. Complete chloroplast genome sequence of Pedicularis cheilanthifolia, an alpine plant in China. Conserv. Genet. Resour. 2017, 9, 619–621. [Google Scholar] [CrossRef]
- Bock, R. Structure, function, and inheritance of plastid genomes. In Cell and Molecular Biology of Plastids; Springer: Berlin/Heidelberg, Germany, 2007; pp. 29–63. [Google Scholar]
- Ruhlman, T.A.; Jansen, R. The Plastid Genomes of Flowering Plants. Chloroplast Biotechnol. 2014, 1132, 3–38. [Google Scholar]
- Li, X.; Yang, J.B.; Wang, H.; Song, Y.; Yu, W.B. Plastid NDH pseudogenization and gene loss in a recently derived lineage from the largest hemiparasitic plant genus Pedicularis (Orobanchaceae). Plant Cell Physiol. 2021, 62, 971–984. [Google Scholar] [CrossRef]
- Hilu, K.W.; Borsch, T.; Müller, K.; Soltis, D.E.; Soltis, P.S.; Savolainen, V.; Chase, M.W.; Powell, M.P.; Alice, L.A.; Evans, R.; et al. Angiosperm phylogeny based on matK sequence information. Am. J. Bot. 2003, 90, 1758–1776. [Google Scholar] [CrossRef]
- Tamura, M.N.; Yamashita, J.; Fuse, S.; Haraguchi, M. Molecular phylogeny of monocotyledons inferred from combined analysis of plastid matK and rbcL gene sequences. J. Plant Res. 2004, 117, 109–120. [Google Scholar] [CrossRef]
- Graham, S.W.; Lam, V.K.Y.; Merckx, V.S.F.T. Plastomes on the edge: The evolutionary breakdown of mycoheterotroph plastid genomes. New Phytol. 2017, 214, 48–55. [Google Scholar] [CrossRef]
- Saldaña, C.L.; Grados, P.R.; Galarza, J.C.C.; Feijoo, S.; Abad, J.C.G.; Vásquez, H.V.; Maicelo, J.L.; Jhoncon, J.H.; Arbizu, C.I. Unlocking the Complete Chloroplast Genome of a Native Tree Species from the Amazon Basin, Capirona (Calycophyllum spruceanum, Rubiaceae), and Its Comparative Analysis with Other Ixoroideae Species. Genes 2022, 13, 113. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Li, Q.; Gao, J.; Qu, M.; Ding, P. The complete chloroplast genome sequence of the medicinal plant Morinda officinalis (Rubiaceae), an endemic to China. Mitochondrial DNA Part B 2016, 27, 4324–4325. [Google Scholar] [CrossRef] [PubMed]
- Duan, R.Y.; Huang, M.Y.; Yang, L.M.; Liu, Z.W. Characterization of the complete chloroplast genome of Emmenopterys henryi (Gentianales: Rubiaceae), an endangered relict tree species endemic to China. Conserv. Genet. Resour. 2017, 9, 459–461. [Google Scholar] [CrossRef]
- Mardanov, A.V.; Ravin, N.V.; Kuznetsov, B.B.; Samigullin, T.H.; Antonov, A.S.; Kolganova, T.V.; Skyabin, K.G. Complete Sequence of the Duckweed (Lemna minor) Chloroplast Genome: Structural Organization and Phylogenetic Relationships to Other Angiosperms. J. Mol. Evol. 2008, 66, 555–564. [Google Scholar] [CrossRef]
- Gao, L.; Yi, X.; Yang, Y.X.; Su, Y.J.; Wang, T. Complete chloroplast genome sequence of a tree fern Alsophila spinulosa: Insights into evolutionary changes in fern chloroplast genomes. BMC Evol. Biol. 2009, 9, 130. [Google Scholar] [CrossRef]
- Wanga, V.O.; Dong, X.; Oulo, M.A.; Mkala, E.M.; Wang, Q.F. Complete Chloroplast Genomes of Acanthochlamys bracteata (China) and Xerophyta (Africa) (Velloziaceae): Comparative Genomics and Phylogenomic Placement. Front. Plant Sci. 2021, 12, 1135. [Google Scholar] [CrossRef]
- Ren, J.; Tian, J.; Jiang, H.; Zhu, X.; Mutie, F.M.; Wanga, V.O.; Ding, S.; Yang, J.; Dong, X.; Chen, L.; et al. Comparative and Phylogenetic Analysis Based on the Chloroplast Genome of Coleanthus subtilis (Tratt.) Seidel, a Protected Rare Species of Monotypic Genus. Front. Plant Sci. 2022, 13, 828467. [Google Scholar] [CrossRef]
- Guo, S.; Liao, X.J.; Chen, S.Y.; Liao, B.S.; Guo, Y.M.; Cheng, R.Y.; Xiao, S.M.; Hu, H.Y.; Chen, J.; Pei, J.; et al. A Comparative Analysis of the Chloroplast Genomes of Four Polygonum Medicinal Plants. Front. Genet. 2022, 13, 764534. [Google Scholar] [CrossRef]
- Howe, C.J.; Barbrook, A.C.; Koumandou, V.L.; Nisbet, R.; Symington, H.A.; Wightman, T.F. Evolution of the chloroplast genome. Philos. Trans. R. Soc. B Biol. Sci. 2003, 358, 99–106. [Google Scholar] [CrossRef]
- Khrustalev, V.V.; Barkovsky, E.V. Mutational pressure is a cause of inter- and intragenomic differences in GC-content of simplex and varicello viruses. Comput. Biol. Chem. 2009, 33, 295–302. [Google Scholar] [CrossRef]
- Ding, S.X.; Dong, X.; Yang, J.X.; Guo, C.; Cao, B.B.; Guo, Y.; Hu, G.W. Complete Chloroplast Genome of Clethra fargesii Franch. an Original Sympetalous Plant from Central China: Comparative Analysis, Adaptive Evolution, and Phylogenetic Relationships. Forests 2021, 12, 441. [Google Scholar] [CrossRef]
- Kim, K.J.; Lee, H.L. Complete chloroplast genome sequences from Korean ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res. Int. J. Rapid Publ. Rep. Genes Genomes 2004, 11, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, C.; Miao, H.; Xiong, S. Insights from the Complete Chloroplast Genome into the Evolution of Sesamum indicum L. PLoS ONE 2013, 8, e80508. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Guo, J.; Cha, J.; Chae, M.; She, C.; Barral, J.M.; Sachs, M.S.; Liu, Y. Non-optimal codon usage affects expression, structure and function of clock protein FRQ. Nature 2013, 495, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Lyu, X.L.; Liu, Y. Non-optimal codon usage is critical for protein structure and function of the master general amino acid control regulator CPC-1. mBio 2020, 11, e2605–e2620. [Google Scholar] [CrossRef] [PubMed]
- Jian, H.Y.; Zhang, Y.H.; Yan, H.J.; Qiu, X.Q.; Wang, Q.G.; Li, S.B.; Zhang, S.D. The Complete Chloroplast Genome of a Key Ancestor of Modern Roses, Rosa chinensis var. spontanea, and a Comparison with Congeneric Species. Molecules 2018, 23, 389. [Google Scholar] [CrossRef] [PubMed]
- Somaratne, Y.; Guan, D.L.; Wang, W.Q.; Zhao, L.; Xu, S.Q. The Complete Chloroplast Genomes of Two Lespedeza Species: Insights into Codon Usage Bias, RNA Editing Sites, and Phylogenetic Relationships in Desmodieae (Fabaceae: Papilionoideae). Plants 2020, 9, 51. [Google Scholar] [CrossRef]
- Kuang, D.Y.; Wu, H.; Wang, Y.L.; Gao, L.M.; Lu, L. Complete chloroplast genome sequence of Magnolia kwangsiensis (Magnoliaceae): Implication for DNA barcoding and population. Genome 2011, 54, 663–673. [Google Scholar] [CrossRef]
- Dong, F.; Lin, Z.; Lin, J.; Ming, R.; Zhang, W. Chloroplast Genome of Rambutan and Comparative Analyses in Sapindaceae. Plants 2021, 10, 283. [Google Scholar] [CrossRef]
- Yu, X.; Zuo, L.; Lu, D.; Lu, B.; Yang, M.S.; Wang, J. Comparative analysis of chloroplast genomes of five Robinia species: Genome comparative and evolution analysis. Gene 2018, 689, 141–151. [Google Scholar] [CrossRef]
- Wang, M.; Liu, H.; Ge, L.; Xing, G.; Wang, M.; Weining, S.; Nie, X. Identification and Analysis of RNA Editing Sites in the Chloroplast Transcripts of Aegilops tauschii L. Genes 2017, 8, 13. [Google Scholar] [CrossRef] [PubMed]
- Josphat, S.; Li, Z.Z.; Andrew, G.; Liao, Y.Y. The Complete Chloroplast Genome Sequence of Tree of Heaven (Ailanthus altissima (Mill.) (Sapindales: Simaroubaceae), an Important Pantropical Tree. Int. J. Mol. Sci. 2018, 19, 929. [Google Scholar]
- Ibrar, A.; Biggs, P.J.; Matthews, P.J.; Collins, L.J.; Hendy, M.D.; Lockhart, P. Mutational Dynamics of Aroid Chloroplast Genomes. Genome Biol. Evol. Dev. 2012, 4, 1316–1323. [Google Scholar]
- Jungeun, L.; Kang, Y.; Chul, S.S.; Hyun, P.; Hyoungseok, L.; Szabolcs, S. Combined Analysis of the Chloroplast Genome and Transcriptome of the Antarctic Vascular Plant Deschampsia antarctica Desv. PLoS ONE 2014, 9, e92501. [Google Scholar]
- Liu, Q.; Li, X.Y.; Li, M.Z.; Xu, W.K.; Heslop, H.; John, S. Comparative chloroplast genome analyses of Avena: Insights into evolutionary dynamics and phylogeny. BMC Plant Biol. 2020, 20, 406. [Google Scholar] [CrossRef]
- Abdullah; Mehmood, F.; Shahzadi, I.; Ali, Z.; Islam, M.; Naeem, M.; Mirza, B.; Lockhart, P.J.; Ahmed, I.; Waheed, M.T. Correlations among oligonucleotide repeats, nucleotide substitutions, and insertion–deletion mutations in chloroplast genomes of plant family Malvaceae. J. Syst. Evol. Dev. 2020, 59, 388–402. [Google Scholar] [CrossRef]
- Liu, H.Y.; Yu, Y.; Deng, Y.Q.; Li, J.; Huang, Z.X.; Zhou, S.D. The Chloroplast Genome of Lilium henrici: Genome Structure and Comparative Analysis. Molecules 2018, 23, 1276. [Google Scholar] [CrossRef]
- Bejaoui, F.; Salas, J.J.; Nouairi, I.; Smaoui, A.; Abdelly, C.; Force, E.M.; Youssef, N.B. Changes in chloroplast lipid contents and chloroplast ultrastructure in Sulla carnosa and Sulla coronaria leaves under salt stress. J. Plant Physiol. 2016, 198, 32–38. [Google Scholar] [CrossRef]
- Dong, W.; Liu, H.; Xu, C.; Zuo, Y.; Chen, Z.; Zhou, S. A chloroplast genomic strategy for designing taxon specific DNA mini-barcodes: A case study on ginsengs. BMC Genet. 2014, 15, 138. [Google Scholar] [CrossRef]
- Jim, P.; Powell, W.; Hollingsworth, P.M. Chloroplast microsatellites: New tools for studies in plant ecology and evolution. Trends Ecol. Evol. 2001, 16, 142–147. [Google Scholar]
- Varshney, R.K.; Sigmund, R.; Börner, A.; Korzun, V.; Stein, N.; Sorrells, M.E.; Langridge, P.; Graner, A. Interspecific transferability and comparative mapping of barley EST-SSR markers in wheat, rye and rice. Plant Sci. 2005, 168, 195–202. [Google Scholar] [CrossRef]
- Rivas, J.D.L.; Lozano, J.; Ortiz, A.R. Comparative Analysis of Chloroplast Genomes: Functional Annotation, Genome-Based Phylogeny, and Deduced Evolutionary Patterns. Genome Res. 2002, 12, 567–583. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.M.; Wang, R.H.; Sha, H.K.; Liu, M.Z.; Tong, J.Q.; He, Q.L. The complete chloroplast genome sequence of Spiraea × vanhouttei (Briot) Zabel (Rosaceae). Mitochondrial DNA Part B 2022, 7, 505–506. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.H.; Chen, M.M.; Wu, Y.Z.; Wang, R.R.; Xiao, X.F.; Qi, Z.C.; Yan, X.L. The complete chloroplast genome sequence of wild Japanese pepper Tubocapsicum anomalum Makino (Solanaceae). Mitochondrial DNA Part B 2021, 6, 2322–2323. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Huang, Y.B.; Zhang, Z.C.; Xu, X.Y.; Wang, R.H.; Xua, L.; Qi, Z.C.; Wei, Y.K. The complete chloroplast genome of endangered Zhangjiajie sage Salvia daiguii Y. K. Wei & Y. B. Huang (Lamiaceae). Mitochondrial DNA Part B 2020, 5, 3833–3834. [Google Scholar] [PubMed]
- Qi, Z.C.; Shen, C.; Han, Y.W.; Shen, W.; Yang, M.; Liu, J.L.; Liang, Z.S.; Li, P.; Fu, C.X. Development of microsatellite loci in Mediterranean sarsaparilla (Smilax aspera; Smilacaceae) using transcriptome data. Appl. Plant Sci. 2017, 5, 1700005. [Google Scholar] [CrossRef]
- Lu, Q.X.; Ren, L.J.; Wang, Y.N.; Xiao, Y.; Li, Y.D.; Ying, J.N.; Zhang, Y.; Wang, R.H.; Qi, Z.C.; You, Z.Y. Characterization of the complete chloroplast genome of Sparganium glomeratum (Typhaceae) from Jilin Province, China and phylogenetic analysis. Mitochondrial DNA Part B 2021, 6, 3253–3254. [Google Scholar] [CrossRef]
- Feng, J.Y.; Jin, X.J.; Zhang, S.L.; Yang, J.W.; Fei, S.P.; Huang, Y.S.; Liu, Y.; Qi, Z.C.; Li, P. Smilax weniae, a New Species of Smilacaceae from Limestone Areas Bordering Guizhou and Guangxi, China. Plants 2022, 11, 1032. [Google Scholar] [CrossRef]
- Jin, J.J.; Yu, W.B.; Yang, J.B.; Song, Y.; Li, D.Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef]
- Shi, L.; Chen, H.; Jiang, M.; Wang, L.; Wu, X.; Huang, L.; Liu, C. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nuclc Acids Res. 2019, 47, W65–W73. [Google Scholar] [CrossRef]
- Drummond, A.J.B. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar]
- Uddin, A.; Choudhury, M.N.; Chakraborty, S. Factors influencing codon usage of mitochondrial ND1 gene in pisces, aves and mammals. Mitochondrion 2017, 37, 17–26. [Google Scholar] [CrossRef]
- Yu, T.H.; Li, J.S.; Yang, Y.; Qi, L.; Chen, B.B.; Zhao, F.Q.; Bao, Q.Y.; Wu, J.Y. Codon usage patterns and adaptive evolution of marine unicellular cyanobacteria Synechococcus and Prochlorococcus. Mol. Phylogenetics Evol. 2012, 62, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Fei, L.; Gao, L.; Cheng, A.; Wang, M.; Han, Q. Analysis of Codon Usage Patterns of Tianfu Goose Interferon Alpha. Procedia Environ. Sci. 2011, 8, 730–736. [Google Scholar]
- Downie, S.R.; Jansen, R.K. A Comparative Analysis of Whole Plastid Genomes from the Apiales: Expansion and Contraction of the Inverted Repeat, Mitochondrial to Plastid Transfer of DNA, and Identification of Highly Divergent Noncoding Regions. Syst. Bot. 2015, 40, 336–351. [Google Scholar] [CrossRef]
- Lu, Q.; Ye, W.; Lu, R.; Xu, W.; Qiu, Y. Phylogenomic and Comparative Analyses of Complete Plastomes of Croomia and Stemona (Stemonaceae). Int. J. Mol. Sci. 2018, 19, 2383. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple Alignment of Conserved Genomic Sequence with Rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef]
- Gary, B. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Peng, X.X.; Xu, X.Y.; Wang, Y.M.; Hawke, D.H.; Yu, S.X.; Han, L.; Zhou, Z.C.; Mojumdar, K.; Jeong, K.J.; Labrie, M. A-to-I RNA Editing Contributes to Proteomic Diversity in Cancer. Cancer Cell 2018, 33, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Jiang, X.; Wang, T.; Deng, Z.; Zhang, A.; Zhang, X. Dynamic landscape of mitochondrial Cytidine-to-Uridine RNA editing in tobacco (Nicotiana tabacum) shows its tissue specificity. Plant Cell Tissue Organ Cult. 2022, 148, 363–376. [Google Scholar] [CrossRef]
- Brauer, N.L.; Alon, S.; Porath, H.T.; Elstein, B.; Unger, R.; Ziv, T.; Admon, A.; Levanon, E.Y.; Rosenthal, J.J.C.; Eisenberg, E. Trade-off between Transcriptome Plasticity and Genome Evolution in Cephalopods. Cell 2017, 169, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Hanson, M.R.; Sutton, C.; Luis, B. Plant organelle gene expression: Altered by RNA editing. Trends Plant Sci. 1996, 1, 57–64. [Google Scholar] [CrossRef]
- Mower, J.P. The PREP suite: Predictive RNA editors for plant mitochondrial genes, chloroplast genes and user-defined alignments. Nucleic Acids Res. 2009, 37, W253–W259. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 737–751. [Google Scholar] [CrossRef]
- Yang, Z.H. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Frazer, K.A.; Lior, P.; Alexander, P.; Rubin, E.M.; Inna, D. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Zhang, D.H.; Gao, F.L.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; Haeseler, A.V.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | U. sessilifructus | U. macrophylla | U. hirsuta | U. scandens |
|---|---|---|---|---|
| Clean reads | 18,093,602 | 18,325,401 | 14,436,561 | 13,659,533 |
| Clean bases | 2,300,585,478 | 2,759,589,867 | 2,174,504,818 | 2,031,770,495 |
| Clean data | 1.27 G | 2.06 G | 1.69 G | 1.68 G |
| Read length/bp | 150 | 150 | 150 | 150 |
| Q30(%) | 92.88% | 95.21% | 93.50% | 92.58% |
| Length/bp | 155,177 | 155,138 | 155,138 | 153,780 |
| GenBank | ON243635 | ON243636 | ON243634 | ON243637 |
| U. rhynchophylla | U. sessilifructus | U. macrophylla | U. hirsuta | U. scandens | |
|---|---|---|---|---|---|
| Length (bp) | |||||
| Total | 154,605 | 155,177 | 155,138 | 155,138 | 153,780 |
| LSC | 85,146(55.07%) | 85,669(55.21%) | 85,749(55.27%) | 85,749(55.27%) | 85,465(55.58%) |
| SSC | 18,145(11.74%) | 18,128(11.68%) | 18,057(11.64%) | 18,057(11.64%) | 16,989(11.05%) |
| IR | 25,657(16.60%) | 25,690(16.56%) | 25,666(16.54%) | 25,666(16.54%) | 25,663(16.69%) |
| CDS | 80,517(52.08%) | 80,027(51.57%) | 80,069(51.61%) | 77,390(49.88%) | 77,526(50.41%) |
| tRNA | 2789(1.80%) | 2789(1.80%) | 2789(1.80%) | 2793(1.80%) | 2717(1.77%) |
| rRNA | 9048(5.85%) | 9048(5.83%) | 9048(5.83%) | 9070(5.85%) | 9048(5.88%) |
| GC content (%) | |||||
| Overall | 37.7 | 37.6 | 37.5 | 37.5 | 37.7 |
| LSC | 35.6 | 35.5 | 35.4 | 35.5 | 35.5 |
| SSC | 31.7 | 31.5 | 31.5 | 31.7 | 31.7 |
| IR | 43.2 | 43.2 | 43.2 | 43.2 | 43.2 |
| CDS | 37.9 | 37.9 | 37.9 | 37.9 | 38.1 |
| tRNA | 53.3 | 53.5 | 53.5 | 53.5 | 53.4 |
| rRNA | 55.4 | 55.4 | 55.4 | 55.3 | 55.4 |
| Number of genes | |||||
| Overall | 131 | 130 | 130 | 130 | 128 |
| CDS | 86 | 85 | 85 | 85 | 83 |
| tRNA | 37 | 37 | 37 | 37 | 37 |
| rRNA | 8 | 8 | 8 | 8 | 8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, M.-M.; Zhang, M.; Liang, Z.-S.; He, Q.-L. Characterization and Comparative Analysis of Chloroplast Genomes in Five Uncaria Species Endemic to China. Int. J. Mol. Sci. 2022, 23, 11617. https://doi.org/10.3390/ijms231911617
Chen M-M, Zhang M, Liang Z-S, He Q-L. Characterization and Comparative Analysis of Chloroplast Genomes in Five Uncaria Species Endemic to China. International Journal of Molecular Sciences. 2022; 23(19):11617. https://doi.org/10.3390/ijms231911617
Chicago/Turabian StyleChen, Min-Min, Miao Zhang, Zong-Suo Liang, and Qiu-Ling He. 2022. "Characterization and Comparative Analysis of Chloroplast Genomes in Five Uncaria Species Endemic to China" International Journal of Molecular Sciences 23, no. 19: 11617. https://doi.org/10.3390/ijms231911617
APA StyleChen, M.-M., Zhang, M., Liang, Z.-S., & He, Q.-L. (2022). Characterization and Comparative Analysis of Chloroplast Genomes in Five Uncaria Species Endemic to China. International Journal of Molecular Sciences, 23(19), 11617. https://doi.org/10.3390/ijms231911617