
A Comprehensive Review on Beneficial Effects of Catechins on Secondary Mitochondrial Diseases


Abstract
1. Introduction
2. Attenuation of Secondary Mitochondrial Diseases by Catechins
2.1. The Neurodegenerative Diseases
2.1.1. Alzheimer’s Disease
2.1.2. Parkinson’s Disease (PD)
2.1.3. Huntington’s Disease (HD)
2.2. Metabolic Disorders
2.2.1. Diabetes Mellitus
2.2.2. Metabolic Complications
Diabetic Cardiomyopathy (DCM)
Diabetic Nephropathy (DN)
3. Mechanisms of Catechins on Secondary Mitochondrial Diseases
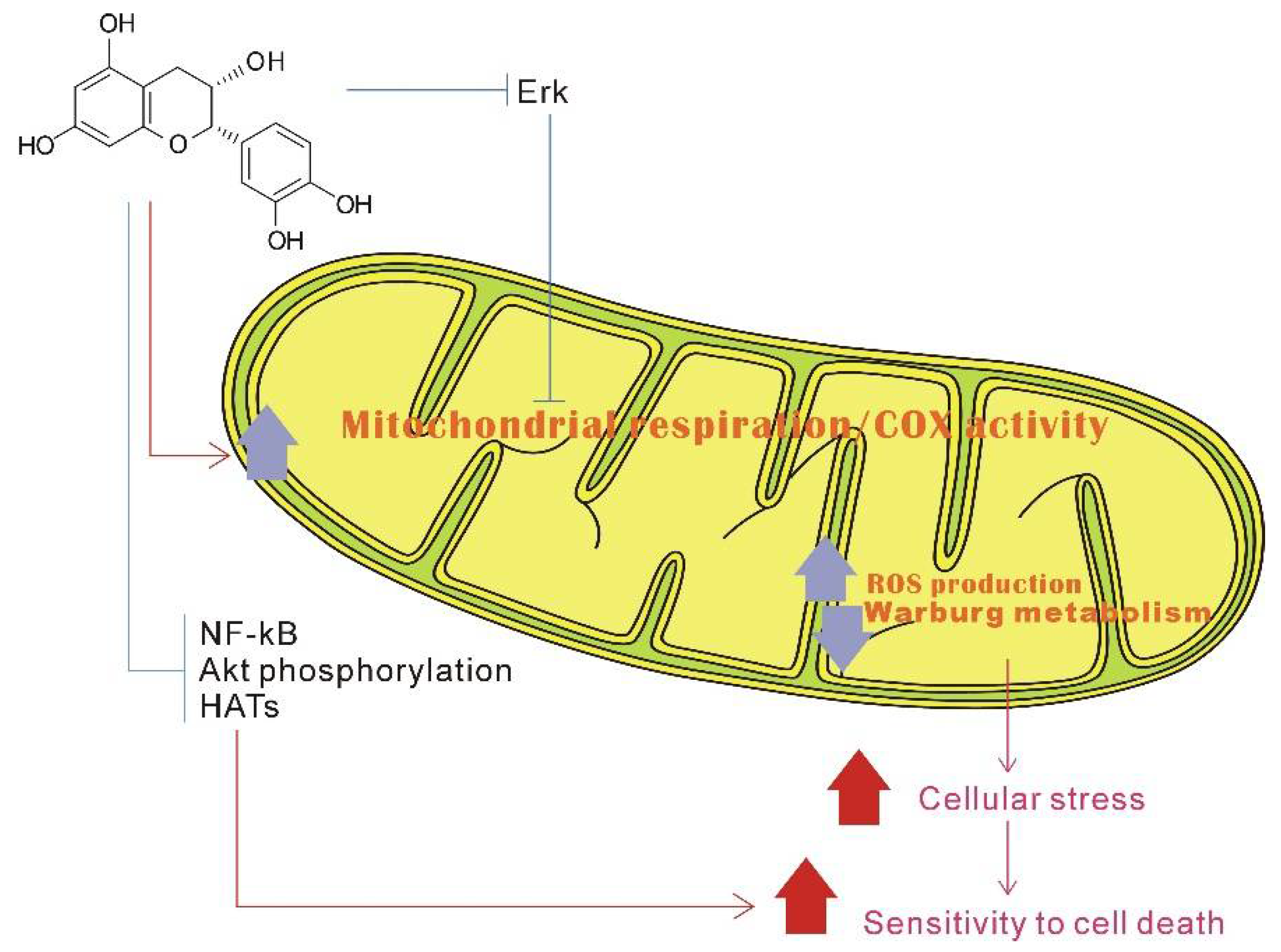
3.1. Modulate Cellular Homeostasis in the Mitochondria
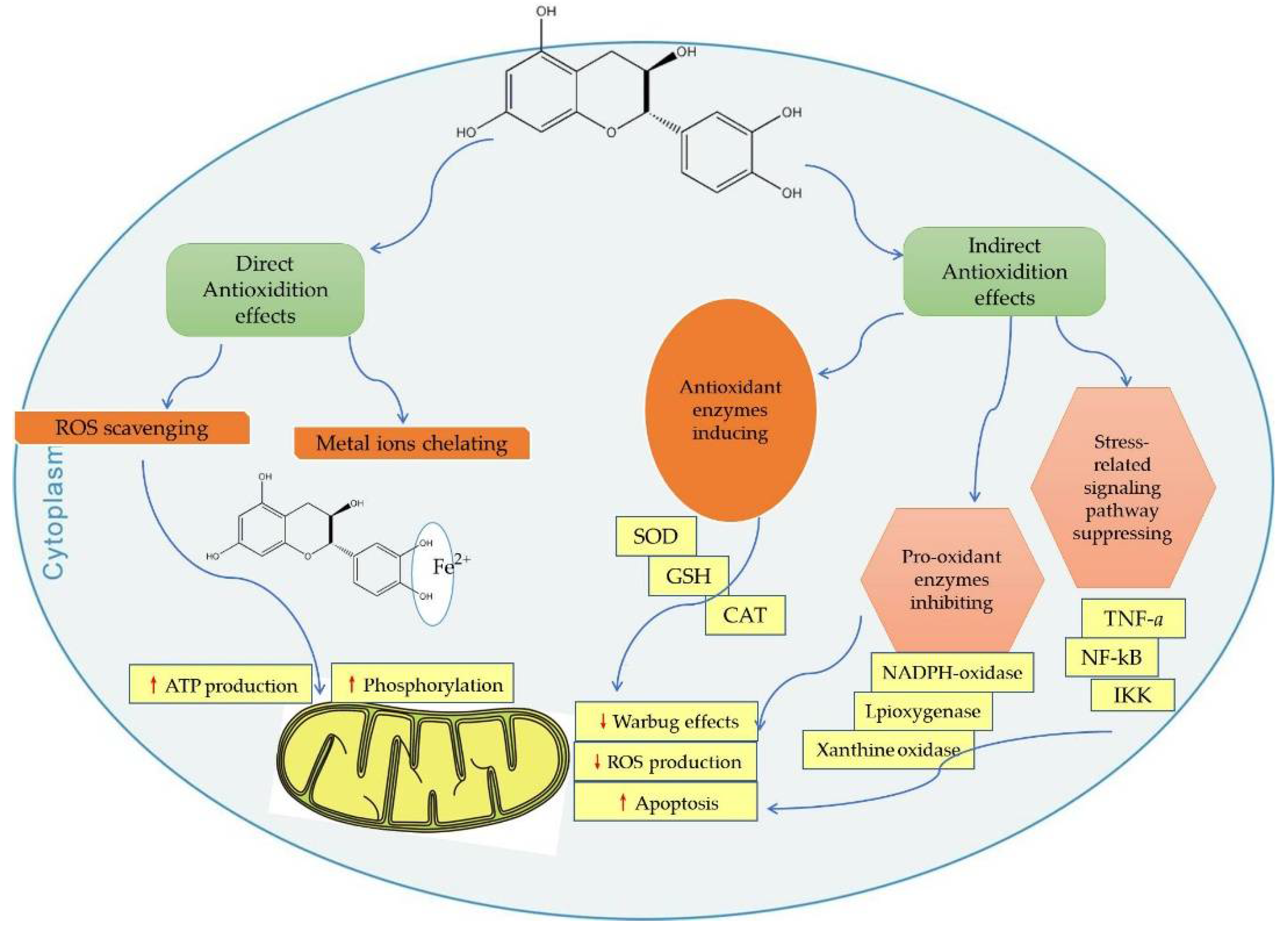
3.2. Act Directly as Radical Scavengers
3.3. Act Directly as Metal Chelator
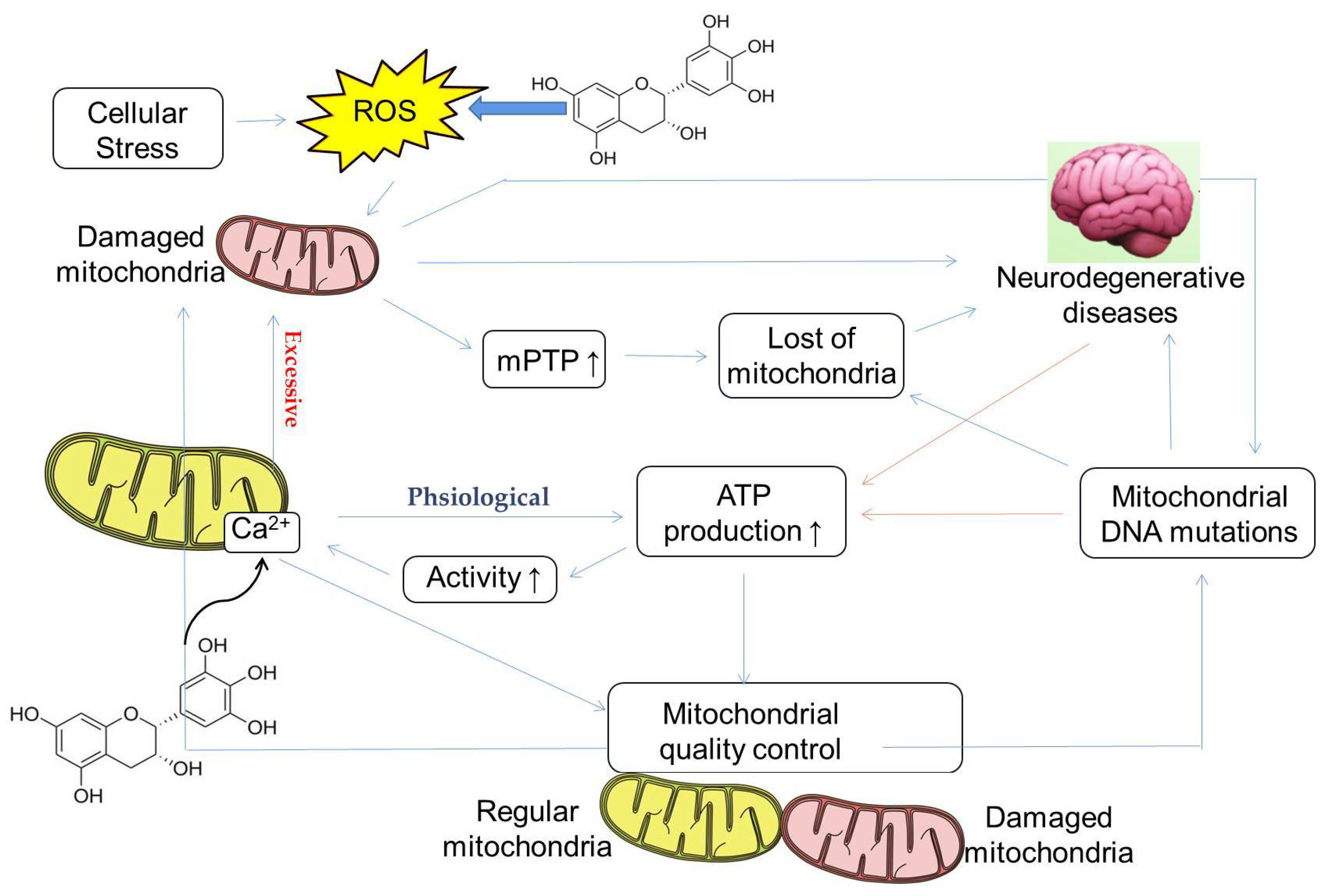
3.4. Protective Properties of mtDNA
3.5. Indirect Antioxidant—Upgrade Antioxidant Enzymes
4. Catechins and Their Regulation of the Metabolic Network Involved in Mitochondrial Function
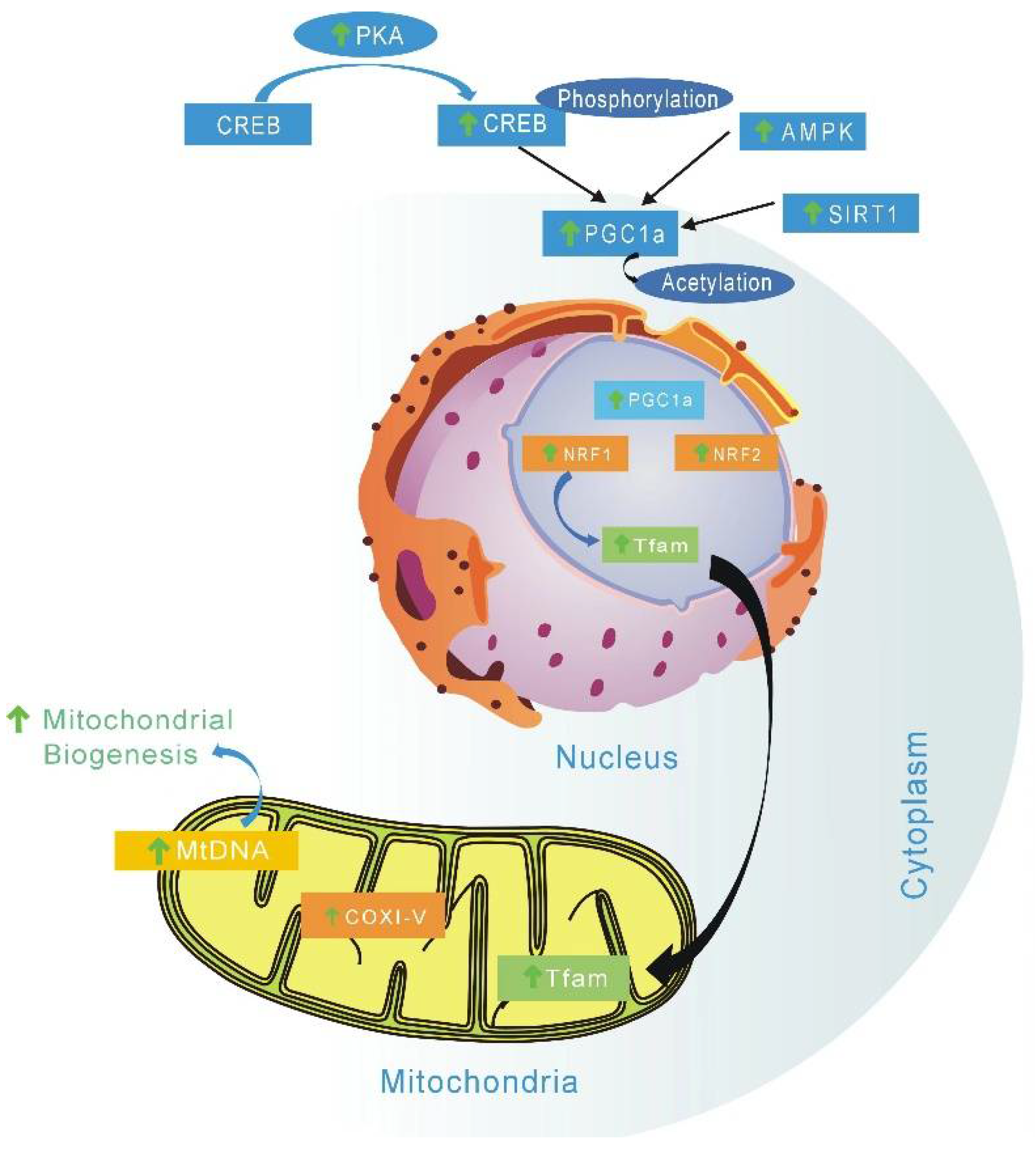
4.1. Regulate Mitochondrial Function and Biogenesis
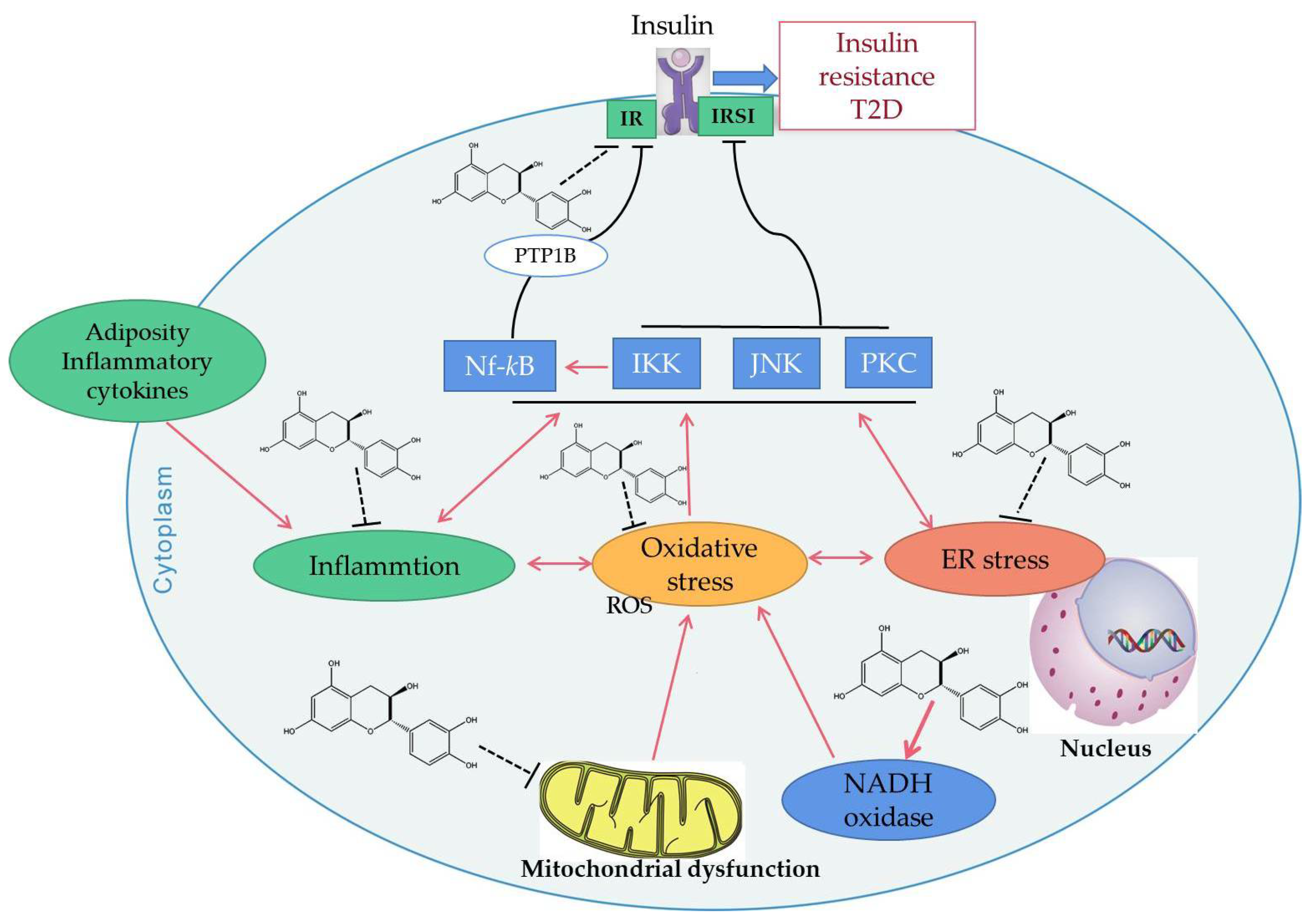
4.2. Improve Insulin Resistance
4.3. Regulation of Calcium Homeostasis
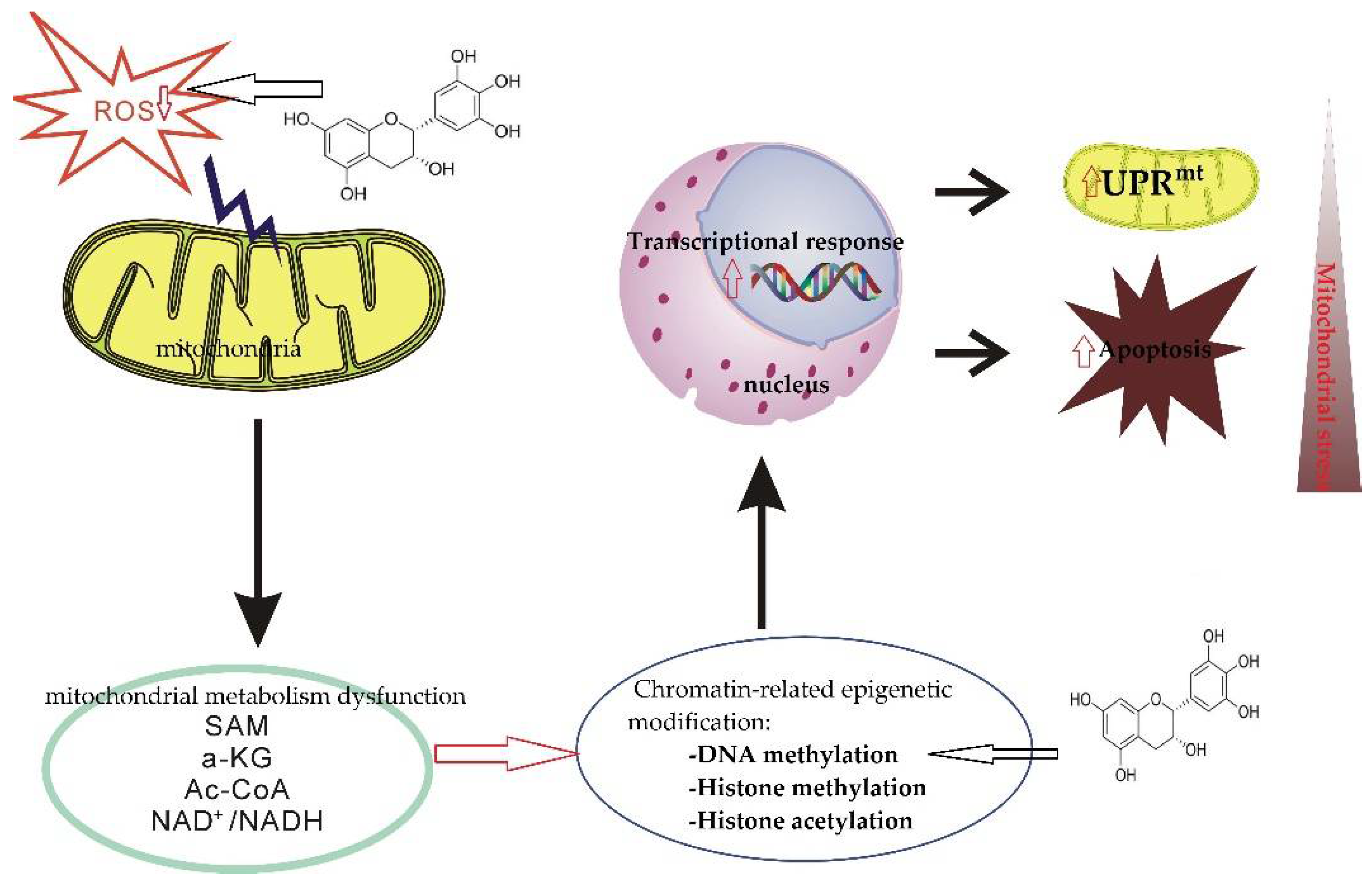
4.4. Regulate the Epigenetic Process
4.5. Regulate Energy Metabolism
5. Indirect Beneficial Effects of Catechins on Mitochondrial Disease
6. Discussion
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Valenti, D.; Vacca, R.A.; Moro, L.; Atlante, A. Mitochondria Can Cross Cell Boundaries: An Overview of the Biological Relevance, Pathophysiological Implications and Therapeutic Perspectives of Intercellular Mitochondrial Transfer. Int. J. Mol. Sci. 2021, 22, 8312. [Google Scholar] [CrossRef] [PubMed]
- El-Hattab, A.W.; Zarante, A.M.; Almannai, M.; Scaglia, F. Therapies for mitochondrial diseases and current clinical trials. Mol. Genet. Metab. 2017, 122, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Russell, O.M.; Gorman, G.S.; Lightowlers, R.N.; Turnbull, D.M. Mitochondrial Diseases: Hope for the Future. Cell 2020, 181, 168–188. [Google Scholar] [CrossRef]
- McKenzie, M.; Liolitsa, D.; Hanna, M.G. Mitochondrial Disease: Mutations and Mechanisms. Neurochem. Res. 2004, 29, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Chow, J.; Rahman, J.; Achermann, J.C.; Dattani, M.T.; Rahman, S. Mitochondrial disease and endocrine dysfunction. Nat. Rev. Endocrinol. 2017, 13, 92–104. [Google Scholar] [CrossRef]
- Poole, O.V.; Hanna, M.G.; Pitceathly, R.D. Mitochondrial disorders: Disease mechanisms and therapeutic approaches. Discov. Med. 2015, 20, 325–331. [Google Scholar]
- Friedrich, V.K.; Rubel, M.A.; Schurr, T.G. Mitochondrial genetic variation in human bioenergetics, adaptation, and adult disease. Am. J. Hum. Biol. 2022, 34, e23629. [Google Scholar] [CrossRef]
- Maassen, J.A.; Janssen, G.M.C.; Hart, L. Molecular mechanisms of mitochondrial diabetes (MIDD). Ann. Med. 2005, 37, 213–221. [Google Scholar] [CrossRef]
- Cao, J.; Wu, H.; Li, Z. Recent perspectives of pediatric mitochondrial diseases. Exp. Ther. Med. 2018, 16, 459. [Google Scholar] [CrossRef]
- Pearce, S.; Nezich, C.L.; Spinazzola, A. Mitochondrial diseases: Translation matters. Mol. Cell. Neurosci. 2013, 55, 1–12. [Google Scholar] [CrossRef]
- DiMauro, S.; Schon, E.A. Mitochondrial Respiratory-Chain Diseases. N. Engl. J. Med. 2003, 348, 2656–2668. [Google Scholar] [CrossRef] [PubMed]
- Niyazov, D.M.; Kahler, S.G.; Frye, R.E. Primary Mitochondrial Disease and Secondary Mitochondrial Dysfunction: Importance of Distinction for Diagnosis and Treatment. Mol. Syndr. 2016, 7, 122–137. [Google Scholar] [CrossRef] [PubMed]
- Orrenius, S.; Gogvadze, V.; Zhivotovsky, B. Mitochondrial Oxidative Stress: Implications for Cell Death. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 143–183. [Google Scholar] [CrossRef] [PubMed]
- Circu, M.L.; Moyer, M.P.; Harrison, L.; Aw, T.Y. Contribution of glutathione status to oxidant-induced mitochondrial DNA damage in colonic epithelial cells. Free Radic. Biol. Med. 2009, 47, 1190–1198. [Google Scholar] [CrossRef] [PubMed]
- Rachek, L.I.; Yuzefovych, L.V.; LeDoux, S.P.; Julie, N.L.; Wilson, G.L. Troglitazone, but not rosiglitazone, damages mitochondrial DNA and induces mitochondrial dysfunction and cell death in human hepatocytes. Toxicol. Appl. Pharmacol. 2009, 240, 348–354. [Google Scholar] [CrossRef]
- Ishisaka, A.; Kawabata, K.; Miki, S.; Shiba, Y.; Minekawa, S.; Nishikawa, T.; Mukai, R.; Terao, J.; Kawai, Y. Mitochondrial Dysfunction Leads to Deconjugation of Quercetin Glucuronides in Inflammatory Macrophages. PLoS ONE 2013, 8, e80843. [Google Scholar] [CrossRef]
- Bernatoniene, J.; Kopustinskiene, D.M. The Role of Catechins in Cellular Responses to Oxidative Stress. Molecules 2018, 23, 965. [Google Scholar] [CrossRef]
- Isemura, M. Catechin in Human Health and Disease. Molecules 2019, 24, 528. [Google Scholar] [CrossRef]
- Mika, M.; Kostogrys, R.B.; Franczyk-Żarów, M.; Wikiera, A.; Maślak, E. Anti-atherosclerotic activity of catechins depends on their stereoisomerism. Atherosclerosis 2015, 240, 125–130. [Google Scholar] [CrossRef]
- Chen, L.; Yang, X.; Jiao, H.; Zhao, B. Tea Catechins Protect against Lead-Induced ROS Formation, Mitochondrial Dysfunction, and Calcium Dysregulation in PC12 Cells. Chem. Res. Toxicol. 2003, 16, 1155–1161. [Google Scholar] [CrossRef]
- Yoon, Y.; Galloway, C.A.; Jhun, B.S.; Yu, T. Mitochondrial Dynamics in Diabetes. Antioxid. Redox Signal. 2011, 14, 439–457. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.-B.; Hall, A.R.; Hausenloy, D.J. Mitochondrial Dynamics in Cardiovascular Health and Disease. Antioxid. Redox Signal. 2013, 19, 400–414. [Google Scholar] [CrossRef] [PubMed]
- Boland, M.L.; Chourasia, A.H.; MacLeod, K.F. Mitochondrial Dysfunction in Cancer. Front. Oncol. 2013, 3, 292. [Google Scholar] [CrossRef] [PubMed]
- Zhan, M.; Brooks, C.; Liu, F.; Sun, L.; Dong, Z. Mitochondrial dynamics: Regulatory mechanisms and emerging role in renal pathophysiology. Kidney Int. 2013, 83, 568–581. [Google Scholar] [CrossRef]
- Chaturvedi, R.K.; Flint, B.M. Mitochondrial diseases of the brain. Free Radic. Biol. Med. 2013, 63, 1–29. [Google Scholar] [CrossRef]
- de Oliveira, M.R.; Nabavi, S.F.; Daglia, M.; Rastrelli, L.; Nabavi, S.M. Epigallocatechin gallate and mitochondria—A story of life and death. Pharmacol. Res. 2016, 104, 70–85. [Google Scholar] [CrossRef]
- Edeas, M.; Weissig, V. Targeting mitochondria: Strategies, innovations and challenges: The future of medicine will come through mitochondria. Mitochondrion 2013, 13, 389–390. [Google Scholar] [CrossRef]
- Swerdlow, R.H. Brain aging, Alzheimer’s disease, and mitochondria. Biochim. Biophys. Acta 2011, 1812, 1630–1639. [Google Scholar] [CrossRef]
- Macdonald, R.; Barnes, K.; Hastings, C.; Mortiboys, H. Mitochondrial abnormalities in Parkinson’s disease and Alzheimer’s disease: Can mitochondria be targeted therapeutically? Biochem. Soc. Trans. 2018, 46, 891–909. [Google Scholar] [CrossRef]
- Xie, C.; Zhuang, X.-X.; Niu, Z.; Ai, R.; Lautrup, S.; Zheng, S.; Jiang, Y.; Han, R.; Gupta, T.S.; Cao, S.; et al. Amelioration of Alzheimer’s disease pathology by mitophagy inducers identified via machine learning and a cross-species workflow. Nat. Biomed. Eng. 2022, 6, 76–93. [Google Scholar] [CrossRef]
- Dragicevic, N.; Smith, A.; Lin, X.; Yuan, F.; Copes, N.; Delic, V.; Tan, J.; Cao, C.; Shytle, R.D.; Bradshaw, P.C. Green tea epigallocatechin-3-gallate (EGCG) and other flavonoids reduce Alzheimer’s amyloid-induced mitochondrial dysfunction. J. Alzheimer’s Dis. 2011, 26, 507–521. [Google Scholar] [CrossRef] [PubMed]
- Pervin, M.; Unno, K.; Ohishi, T.; Tanabe, H.; Miyoshi, N.; Nakamura, Y. Beneficial Effects of Green Tea Catechins on Neurodegenerative Diseases. Molecules 2018, 23, 1297. [Google Scholar] [CrossRef] [PubMed]
- Fernando, W.; Somaratne, G.; Goozee, K.G.; Williams, S.; Singh, H.; Martins, R.N. Diabetes and Alzheimer’s Disease: Can Tea Phytochemicals Play a Role in Prevention? J. Alzheimer’s Dis. 2017, 59, 481–501. [Google Scholar] [CrossRef] [PubMed]
- Bieschke, J.; Russ, J.; Friedrich, R.P.; Ehrnhoefer, D.E.; Wobst, H.; Neugebauer, K.; Wanker, E.E. EGCG remodels mature alpha-synuclein and amyloid-beta fibrils and reduces cellular toxicity. Proc. Natl. Acad. Sci. USA 2010, 107, 7710–7715. [Google Scholar] [CrossRef]
- Ky, A.; Cth, B.; Mja, C. Multifaceted neuroprotective effects of (−)-epigallocatechin-3-gallate (EGCG) in Alzheimer’s disease: An overview of pre-clinical studies focused on β-amyloid peptide—ScienceDirect. Food Sci. Hum. Wellness 2022, 11, 11. [Google Scholar]
- Cerri, S.; Milanese, C.; Mastroberardino, P.G. Endocytic iron trafficking and mitochondria in Parkinson’s disease. Int. J. Biochem. Cell Biol. 2019, 110, 70–74. [Google Scholar] [CrossRef]
- Muñoz, Y.; Carrasco, C.M.; Campos, J.D.; Aguirre, P.; Núñez, M.T. Parkinson’s Disease: The Mitochondria-Iron Link. Park. Dis. 2016, 2016, 1–21. [Google Scholar] [CrossRef]
- Sherer, T.B.; Betarbet, R.; Greenamyre, J.T. Environment, mitochondria, and Parkinson’s disease. Neuroscientist 2002, 8, 192–197. [Google Scholar]
- Bose, A.; Beal, M.F. Mitochondrial dysfunction in Parkinson’s disease. J. Neurochem. 2016, 139 (Suppl. S1), 216–231. [Google Scholar] [CrossRef]
- Grünewald, A.; Kumar, K.R.; Sue, C.M. New insights into the complex role of mitochondria in Parkinson’s disease. Prog. Neurobiol. 2019, 177, 73–93. [Google Scholar] [CrossRef]
- Pogačnik, L.; Pirc, K.; Palmela, I.; Skrt, M.; Kim, K.S.; Brites, D.; Brito, M.A.; Ulrih, N.P.; Silva, R.F. Potential for brain accessibility and analysis of stability of selected flavonoids in relation to neuroprotection in vitro. Brain Res. 2016, 1651, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Mandel, S.A.; Avramovich-Tirosh, Y.; Reznichenko, L.; Zheng, H.; Weinreb, O.; Amit, T.; Youdim, M.B. Multifunctional Activities of Green Tea Catechins in Neuroprotection. Modulation of cell survival genes, iron-dependent oxidative stress and PKC signaling pathway. Neurosignals 2005, 14, 46–60. [Google Scholar] [CrossRef] [PubMed]
- Pantopoulos, K.; Porwal, S.K.; Tartakoff, A.; Devireddy, L. Mechanisms of Mammalian Iron Homeostasis. Biochemistry 2012, 51, 5705–5724. [Google Scholar] [CrossRef] [PubMed]
- Jomova, K.; Vondrakova, D.; Lawson, M.; Valko, M. Metals, oxidative stress and neurodegenerative disorders. Mol. Cell. Biochem. 2010, 345, 91–104. [Google Scholar] [CrossRef]
- Tanaka, K.; Miyake, Y.; Fukushima, W.; Sasaki, S.; Kiyohara, C.; Tsuboi, Y.; Yamada, T.; Oeda, T.; Miki, T.; Kawamura, N.; et al. Intake of Japanese and Chinese teas reduces risk of Parkinson’s disease. Park. Relat. Disord. 2011, 17, 446–450. [Google Scholar] [CrossRef]
- Qi, H.; Li, S. Dose-response meta-analysis on coffee, tea and caffeine consumption with risk of Parkinson’s disease. Geriatr. Gerontol. Int. 2014, 14, 430–439. [Google Scholar] [CrossRef]
- Hosseini, N.; Babakhani, B.; Hosseini, A.; Vahabi, Z.; Soltanzadeh, A. Non-genetic factors associated with the risk of Parkinson’s disease in Iranian patients. Funct. Neurol. 2013, 28, 107–113. [Google Scholar]
- Mandel, S.; Amit, T.; Reznichenko, L.; Weinreb, O.; Youdim, M.B.H. Green tea catechins as brain-permeable, natural iron chelators-antioxidants for the treatment of neurodegenerative disorders. Mol. Nutr. Food Res. 2006, 50, 229–234. [Google Scholar] [CrossRef]
- Mandel, S.; Weinreb, O.; Reznichenko, L.; Kalfon, L.; Amit, T. Green tea catechins as brain-permeable, non toxic iron chelators to "iron out iron" from the brain. J. Neural. Transm. Suppl. 2006, 71, 249–257. [Google Scholar]
- Grinberg, L.N.; Newmark, H.; Kitrossky, N.; Rahamim, E.; Chevion, M.; Rachmilewitz, E.A. Protective effects of tea polyphenols against oxidative damage to red blood cells. Biochem. Pharmacol. 1997, 54, 973–978. [Google Scholar] [CrossRef]
- Kumamoto, M.; Sonda, T.; Nagayama, K.; Tabata, M. Effects of pH and Metal Ions on Antioxidative Activities of Catechins. Biosci. Biotechnol. Biochem. 2001, 65, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Sharifi-Rad, M.; Anil Kumar, N.V.; Zucca, P.; Varoni, E.M.; Dini, L.; Panzarini, E.; Rajkovic, J.; Tsouh Fokou, P.V.; Azzini, E.; Peluso, I.; et al. Lifestyle, Oxidative Stress, and Antioxidants: Back and Forth in the Pathophysiology of Chronic Diseases. Front. Physiol. 2020, 11, 694. [Google Scholar] [CrossRef]
- Vinther-Jensen, T.; Nielsen, T.T.; Budtz-Jorgensen, E.; Larsen, I.U.; Hansen, M.M.; Hasholt, L.; Hjermind, L.E.; Nielsen, J.E.; Norremolle, A. Psychiatric and cognitive symptoms in Huntington’s disease are modified by polymorphisms in catecholamine regulating enzyme genes. Clin. Genet. 2016, 89, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Duennwald, M.L.; Shorter, J. Countering amyloid polymorphism and drug resistance with minimal drug cocktails. Prion 2010, 4, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Mandel, S.; Weinreb, O.; Amit, T.; Youdim, M.B. Cell signaling pathways in the neuroprotective actions of the green tea polyphenol (−)-epigallocatechin-3-gallate: Implications for neurodegenerative diseases. J. Neurochem. 2004, 88, 1555–1569. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.B.; Gutekunst, W.R.; Joyner, P.M.; Duan, W.; Li, Q.; Ross, C.A.; Williams, T.D.; Cichewicz, R.H. Bioactivity Profiling with Parallel Mass Spectrometry Reveals an Assemblage of Green Tea Metabolites Affording Protection against Human Huntingtin and α-Synuclein Toxicity. J. Agric. Food Chem. 2007, 55, 9450–9456. [Google Scholar] [CrossRef]
- Hudson, S.A.; Ecroyd, H.; Dehle, F.C.; Musgrave, I.F.; Carver, J.A. (−)-epigallocatechin-3-gallate (EGCG) maintains kappa-casein in its pre-fibrillar state without redirecting its aggregation pathway. J. Mol. Biol. 2009, 392, 689–700. [Google Scholar] [CrossRef]
- Itoh, K.; Nakamura, K.; Iijima, M.; Sesaki, H. Mitochondrial dynamics in neurodegeneration. Trends Cell Biol. 2013, 23, 64–71. [Google Scholar] [CrossRef]
- Kumar, P.; Kumar, A. Protective effects of epigallocatechin gallate following 3-nitropropionic acid-induced brain damage: Possible nitric oxide mechanisms. Psychopharmacology 2009, 207, 257–270. [Google Scholar] [CrossRef]
- He, J.; Xu, L.; Yang, L.; Sun, C. Anti-oxidative effects of catechins and theaflavins on glutamate-induced HT22 cell damage. RSC Adv. 2019, 9, 21418–21428. [Google Scholar] [CrossRef]
- Nath, S.; Bachani, M.; Harshavardhana, D.; Steiner, J.P. Catechins protect neurons against mitochondrial toxins and HIV proteins via activation of the BDNF pathway. J. Neurovirol. 2012, 18, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Levites, Y.; Amit, T.; Youdim, M.B.; Mandel, S. Involvement of Protein Kinase C Activation and Cell Survival/ Cell Cycle Genes in Green Tea Polyphenol (−)-Epigallocatechin 3-Gallate Neuroprotective Action. J. Biol. Chem. 2002, 277, 30574–30580. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.T.; Jung, C.H.; Lee, S.R.; Bae, J.H.; Baek, W.K.; Suh, M.H.; Park, J.; Park, C.W.; Suh, S.I. The green tea polyphenol (−)-epigallocatechin gallate attenuates beta-amyloid-induced neurotoxicity in cultured hippocampal neurons. Life Sci. 2001, 70, 603–614. [Google Scholar] [CrossRef]
- Levites, Y.; Amit, T.; Mandel, S.; Youdim, M.B. Neuroprotection and neurorescue against Abeta toxicity and PKC-dependent release of nonamyloidogenic soluble precursor protein by green tea polyphenol (−)-epigallocatechin-3-gallate. FASEB J. 2003, 17, 952–954. [Google Scholar] [CrossRef] [PubMed]
- Ban, J.Y.; Jeon, S.Y.; Bae, K.; Song, K.S.; Seong, Y.H. Catechin and epicatechin from Smilacis chinae rhizome protect cultured rat cortical neurons against amyloid beta protein (25-35)-induced neurotoxicity through inhibition of cytosolic calcium elevation. Life Sci. 2006, 79, 2251–2259. [Google Scholar] [CrossRef]
- Bai, L.; Li, X.; He, L.; Zheng, Y.; Lu, H.; Li, J.; Zhong, L.; Tong, R.; Jiang, Z.; Shi, J.; et al. Antidiabetic Potential of Flavonoids from Traditional Chinese Medicine: A Review. Am. J. Chin. Med. 2019, 47, 933–957. [Google Scholar] [CrossRef]
- American Diabetes Association Professional Practice, C. 3. Prevention or Delay of Type 2 Diabetes and Associated Comorbidities: Standards of Medical Care in Diabetes-2022. Diabetes Care 2022, 45 (Suppl. S1), S39–S45. [Google Scholar] [CrossRef]
- Fernyhough, P.; McGavock, J. Mechanisms of disease: Mitochondrial dysfunction in sensory neuropathy and other complications in diabetes. Handb. Clin. Neurol. 2014, 126, 353–377. [Google Scholar]
- Sack, M.N. Type 2 diabetes, mitochondrial biology and the heart. J. Mol. Cell. Cardiol. 2009, 46, 842–849. [Google Scholar] [CrossRef]
- Maassen, J.A.T.H.L.; Van Essen, E.; Heine, R.J.; Nijpels, G.; Jahangir, T.R.; Raap, A.K.; Janssen, G.M.; Lemkes, H.H. Mitochondrial diabetes: Molecular mechanisms and clinical presentation. Diabetes 2004, 53 (Suppl. S1), S103–S109. [Google Scholar] [CrossRef]
- Zhang, Z.; Ding, Y.; Dai, X.; Wang, J.; Li, Y. Epigallocatechin-3-gallate protects pro-inflammatory cytokine induced injuries in insulin-producing cells through the mitochondrial pathway. Eur. J. Pharmacol. 2011, 670, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Zhen, W.; Yuskavage, J.; Liu, D. Epigallocatechin gallate delays the onset of type 1 diabetes in spontaneous non-obese diabetic mice. Br. J. Nutr. 2011, 105, 1218–1225. [Google Scholar] [CrossRef]
- Hosoda, K.; Wang, M.-F.; Liao, M.-L.; Chuang, C.-K.; Iha, M.; Clevidence, B.; Yamamoto, S. Antihyperglycemic Effect of Oolong Tea in Type 2 Diabetes. Diabetes Care 2003, 26, 1714–1718. [Google Scholar] [CrossRef] [PubMed]
- Crespy, V.; Williamson, G. A Review of the Health Effects of Green Tea Catechins in In Vivo Animal Models. J. Nutr. 2004, 134, 3431S–3440S. [Google Scholar] [CrossRef] [PubMed]
- Wolfram, S.; Raederstorff, D.; Preller, M.; Wang, Y.; Teixeira, S.R.; Riegger, C.; Weber, P. Epigallocatechin Gallate Supplementation Alleviates Diabetes in Rodents. J. Nutr. 2006, 136, 2512–2518. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.A.; Silva, D.M.; De Morais, A.J.; Mota, J.F.; Botelho, P.B. Therapeutic potential of green tea on risk factors for type 2 diabetes in obese adults—A review. Obes. Rev. 2016, 17, 1316–1328. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Zhao, Y.; Suo, S.; Liu, Y.; Zhao, B. Green tea catechins ameliorate adipose insulin resistance by improving oxidative stress. Free Radic. Biol. Med. 2012, 52, 1648–1657. [Google Scholar] [CrossRef]
- Nagao, T.; Meguro, S.; Hase, T.; Otsuka, K.; Komikado, M.; Tokimitsu, I.; Yamamoto, T.; Yamamoto, K. A Catechin-rich Beverage Improves Obesity and Blood Glucose Control in Patients with Type 2 Diabetes. Obesity 2009, 17, 310–317. [Google Scholar] [CrossRef]
- Kunkel, G.H.; Kunkel, C.J.; Ozuna, H.; Miralda, I.; Tyagi, S.C. TFAM overexpression reduces pathological cardiac remodeling. Mol. Cell. Biochem. 2019, 454, 139–152. [Google Scholar] [CrossRef]
- Dobrin, J.S.; Lebeche, D. Diabetic cardiomyopathy: Signaling defects and therapeutic approaches. Expert Rev. Cardiovasc. Ther. 2010, 8, 373–391. [Google Scholar] [CrossRef][Green Version]
- Retnakaran, R.; Zinman, B. Type 1 diabetes, hyperglycaemia, and the heart. Lancet 2008, 371, 1790–1799. [Google Scholar] [CrossRef]
- Ni, R.; Cao, T.; Xiong, S.; Ma, J.; Fan, G.-C.; Lacefield, J.C.; Lu, Y.; Le Tissier, S.; Peng, T. Therapeutic inhibition of mitochondrial reactive oxygen species with mito-TEMPO reduces diabetic cardiomyopathy. Free Radic. Biol. Med. 2016, 90, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Al-Rasheed, N.M.; Al-Rasheed, N.M.; Hasan, I.H.; Al-Amin, M.A.; Al-Ajmi, H.N.; Mahmoud, A.M. Sitagliptin attenuates cardiomyopathy by modulating the JAK/STAT signaling pathway in experimental diabetic rats. Drug. Des. Devel. Ther. 2016, 10, 2095–2107. [Google Scholar] [PubMed]
- Barman, C.; Pandey, R.; Singh, J.; Sodhi, K. Molecular insights into diabetic cardiomyopathy. Int. J. Res. Med. Sci. 2015, 3, 1564–1570. [Google Scholar] [CrossRef][Green Version]
- Lee, W.-S.; Kim, J. Diabetic cardiomyopathy: Where we are and where we are going. Korean J. Intern. Med. 2017, 32, 404–421. [Google Scholar] [CrossRef]
- Yan, L.-J. Pathogenesis of Chronic Hyperglycemia: From Reductive Stress to Oxidative Stress. J. Diabetes Res. 2014, 2014, 1–11. [Google Scholar] [CrossRef]
- Yilmaz, S.; Canpolat, U.; Aydogdu, S.; Abboud, H.E. Diabetic Cardiomyopathy; Summary of 41 years. Korean Circ. J. 2015, 45, 266–272. [Google Scholar] [CrossRef]
- Bugger, H.; Abel, E.D. Mitochondria in the diabetic heart. Cardiovasc. Res. 2010, 88, 229–240. [Google Scholar] [CrossRef]
- Yu, T.; Robotham, J.L.; Yoon, Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc. Natl. Acad. Sci. USA 2006, 103, 2653–2658. [Google Scholar] [CrossRef]
- Othman, A.I.; El-Sawi, M.R.; El-Missiry, M.A.; Abukhalil, M.H. Epigallocatechin-3-gallate protects against diabetic cardiomyopathy through modulating the cardiometabolic risk factors, oxidative stress, inflammation, cell death and fibrosis in streptozotocin-nicotinamide-induced diabetic rats. Biomed. Pharmacother. 2017, 94, 362–373. [Google Scholar] [CrossRef]
- Yang, X.H.; Pan, Y.; Zhan, X.L.; Zhang, B.L.; Guo, L.L.; Jin, H.M. Epigallocatechin-3-gallate Attenuates Renal Damage by Suppressing Oxidative Stress in Diabetic db/db Mice. Oxidative Med. Cell. Longev. 2016, 2016, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Collins, Q.F.; Liu, H.-Y.; Pi, J.; Liu, Z.; Quon, M.J.; Cao, W. Epigallocatechin-3-gallate (EGCG), A Green Tea Polyphenol, Suppresses Hepatic Gluconeogenesis through 5′-AMP-activated Protein Kinase. J. Biol. Chem. 2007, 282, 30143–30149. [Google Scholar] [CrossRef]
- Waltner-Law, M.E.; Wang, X.L.; Law, B.K.; Hall, R.K.; Nawano, M.; Granner, D.K. Epigallocatechin Gallate, a Constituent of Green Tea, Represses Hepatic Glucose Production. J. Biol. Chem. 2002, 277, 34933–34940. [Google Scholar]
- Li, T.; Liu, J.; Zhang, X.; Ji, G. Antidiabetic activity of lipophilic (−)-epigallocatechin-3-gallate derivative under its role of α-glucosidase inhibition. Biomed. Pharmacother. 2007, 61, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Suzuki, M.; Satsu, H.; Arai, S.; Hara, Y.; Suzuki, K.; Miyamoto, Y.; Shimizu, M. Green Tea Polyphenols Inhibit the Sodium-Dependent Glucose Transporter of Intestinal Epithelial Cells by a Competitive Mechanism. J. Agric. Food Chem. 2000, 48, 5618–5623. [Google Scholar] [CrossRef] [PubMed]
- Koyama, Y.; Abe, K.; Sano, Y.; Ishizaki, Y.; Njelekela, M.; Shoji, Y.; Hara, Y.; Isemura, M. Effects of green tea on gene expression of hepatic gluconeogenic enzymes in vivo. Planta Med. 2004, 70, 1100–1102. [Google Scholar] [CrossRef]
- Nelson, R.H. Hyperlipidemia as a Risk Factor for Cardiovascular Disease. Prim. Care Clin. Off. Pr. 2012, 40, 195–211. [Google Scholar] [CrossRef]
- Ramesh, E.; Elanchezhian, R.; Sakthivel, M.; Jayakumar, T.; Senthil Kumar, R.S.; Geraldine, P.; Thomas, P.A. Epigallocatechin gallate improves serum lipid profile and erythrocyte and cardiac tissue antioxidant parameters in Wistar rats fed an atherogenic diet. Fundam. Clin. Pharmacol. 2008, 22, 275–284. [Google Scholar] [CrossRef]
- Zhong, W.; Huan, X.-D.; Cao, Q.; Yang, J. Cardioprotective effect of epigallocatechin-3-gallate against myocardial infarction in hypercholesterolemic rats. Exp. Ther. Med. 2015, 9, 405–410. [Google Scholar] [CrossRef]
- Raederstorff, D.G.; Schlachter, M.F.; Elste, V.; Weber, P. Effect of EGCG on lipid absorption and plasma lipid levels in rats. J. Nutr. Biochem. 2003, 14, 326–332. [Google Scholar] [CrossRef]
- Goto, T.; Saito, Y.; Morikawa, K.; Kanamaru, Y.; Nagaoka, S. Epigallocatechin gallate changes mRNA expression level of genes involved in cholesterol metabolism in hepatocytes. Br. J. Nutr. 2012, 107, 769–773. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, K.; Hirose, S.; Nagaoka, S.; Yanase, E. Interaction between Tea Polyphenols and Bile Acid Inhibits Micellar Cholesterol Solubility. J. Agric. Food Chem. 2016, 64, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Nan, C.; Chen, Y.; Tian, J.; Jean-Charles, P.-Y.; Getfield, C.; Wang, X.; Huang, X. Calcium desensitizer catechin reverses diastolic dysfunction in mice with restrictive cardiomyopathy. Arch. Biochem. Biophys. 2015, 573, 69–76. [Google Scholar] [CrossRef]
- Jaquenod De Giusti, C.; Palomeque, J.; Mattiazzi, A. Ca2+ mishandling and mitochondrial dysfunction: A converging road to prediabetic and diabetic cardiomyopathy. Pflug. Arch. 2022, 474, 33–61. [Google Scholar] [CrossRef] [PubMed]
- Quan, J.; Jia, Z.; Lv, T.; Zhang, L.; Liu, L.; Pan, B.; Zhu, J.; Gelb, I.J.; Huang, X.; Tian, J. Green tea extract catechin improves cardiac function in pediatric cardiomyopathy patients with diastolic dysfunction. J. Biomed. Sci. 2019, 26, 32. [Google Scholar] [CrossRef] [PubMed]
- Tadano, N.; Du, C.-K.; Yumoto, F.; Morimoto, S.; Ohta, M.; Xie, M.-F.; Nagata, K.; Zhan, D.-Y.; Lu, Q.-W.; Miwa, Y.; et al. Biological actions of green tea catechins on cardiac troponin C. J. Cereb. Blood Flow Metab. 2010, 161, 1034–1043. [Google Scholar] [CrossRef] [PubMed]
- Devika, P.T.; Stanely Mainzen Prince, P. (−)Epigallocatechin-gallate (EGCG) prevents mitochondrial damage in isoproterenol-induced cardiac toxicity in albino Wistar rats: A transmission electron microscopic and in vitro study. Pharmacol. Res. 2008, 57, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Liou, Y.; Hsu, G. (−)-Epigalocathine-3-gallate attenuates oxidative stress by modulation of redox signaling in H9c2 cultured rat cardiac myoblasts. FASEB J. 2011, 25, 1093.8. [Google Scholar] [CrossRef]
- Adikesavan, G.; Vinayagam, M.M.; Abdulrahman, L.A.; Chinnasamy, T. (−)-Epigallocatechin-gallate (EGCG) stabilize the mitochondrial enzymes and inhibits the apoptosis in cigarette smoke-induced myocardial dysfunction in rats. Mol. Biol. Rep. 2013, 40, 6533–6545. [Google Scholar] [CrossRef]
- Yu, N.H.; Pei, H.; Huang, Y.-P.; Li, Y.-F. (−)-Epigallocatechin-3-Gallate Inhibits Arsenic-Induced Inflammation and Apoptosis through Suppression of Oxidative Stress in Mice. Cell. Physiol. Biochem. 2017, 41, 1788–1800. [Google Scholar] [CrossRef]
- Al Hroob, A.M.; Abukhalil, M.H.; Hussein, O.E.; Mahmoud, A.M. Pathophysiological mechanisms of diabetic cardiomyopathy and the therapeutic potential of epigallocatechin-3-gallate. Biomed. Pharmacother. 2019, 109, 2155–2172. [Google Scholar] [CrossRef]
- Chennasamudram, S.P.; Kudugunti, S.; Boreddy, P.R.; Moridani, M.Y.; Vasylyeva, T.L. Renoprotective effects of (+)-catechin in streptozotocin-induced diabetic rat model. Nutr. Res. 2012, 32, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, R.E. Proximal Tubulopathy: Prime Mover and Key Therapeutic Target in Diabetic Kidney Disease. Diabetes 2017, 66, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Blaine, J. Proteinuria: Basic Mechanisms, Pathophysiology and Clinical Relevance || Pathophysiology of Diabetic Nephropathy; Springer: Berlin/Heidelberg, Germany, 2016; Chapter 4; pp. 41–65. [Google Scholar] [CrossRef]
- Haraguchi, R.; Kohara, Y.; Matsubayashi, K.; Kitazawa, R.; Kitazawa, S. New Insights into the Pathogenesis of Diabetic Nephropathy: Proximal Renal Tubules Are Primary Target of Oxidative Stress in Diabetic Kidney. Acta Histochem. Cytochem. 2020, 53, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Sun, L.; Li, D.; Lai, X.; Wen, S.; Chen, R.; Zhang, Z.; Li, Q.; Sun, S. Green tea peptides ameliorate diabetic nephropathy by inhibiting the TGF-beta/Smad signaling pathway in mice. Food Funct. 2022, 13, 3258–3270. [Google Scholar] [CrossRef]
- Renno, W.M.; Abdeen, S.; Alkhalaf, M.; Asfar, S. Effect of green tea on kidney tubules of diabetic rats. Br. J. Nutr. 2008, 100, 652–659. [Google Scholar] [CrossRef]
- Yokozawa, T.; Noh, J.S.; Park, C.H. Green Tea Polyphenols for the Protection against Renal Damage Caused by Oxidative Stress. Evid. -Based Complement. Altern. Med. 2012, 2012, 845917. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Wang, L.; Zhou, Q.; Yan, S.; Li, Z.; Sheng, J.; Zhang, W. (+)-Catechin ameliorates diabetic nephropathy by trapping methylglyoxal in type 2 diabetic mice. Mol. Nutr. Food Res. 2014, 58, 2249–2260. [Google Scholar] [CrossRef]
- Varatharajan, R.; Sattar, M.Z.; Chung, I.; Abdulla, M.A.; Kassim, N.M.; Abdullah, N.A. Antioxidant and pro-oxidant effects of oil palm (Elaeis guineensis) leaves extract in experimental diabetic nephropathy: A duration-dependent outcome. BMC Complement. Altern. Med. 2013, 13, 242. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Liu, X.; Zhang, H.; Song, Y.; Li, T.; Liu, X.; Liu, Y.; Guo, L.; Wang, F.; Yang, T.; et al. Epigallocatechin gallate upregulates NRF2 to prevent diabetic nephropathy via disabling KEAP1. Free Radic. Biol. Med. 2017, 108, 840–857. [Google Scholar] [CrossRef]
- Gerardo, Y.-E.F.; Andrade-Sierra, J.; Pazarin-Villasenor, L.; Santana-Arciniega, C.; De Jesus, T.-V.E.; Samuel, C.-I.J.; Angel, Z.-V.M.; Martin, P.-F.F. The Role of Dietary Antioxidants on Oxidative Stress in Diabetic Nephropathy. Iran. J. Kidney Dis. 2020, 14, 81–94. [Google Scholar]
- Bao, H.; Peng, A. The Green Tea Polyphenol(−)-epigallocatechin-3-gallate and its beneficial roles in chronic kidney disease. J. Transl. Intern. Med. 2016, 4, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Ladeira, L.C.M.; dos Santos, E.C.; Santos, T.A.; da Silva, J.; Lima, G.D.A.; Machado-Neves, M.; da Silva, R.C.; Freitas, M.B.; Maldonado, I. Green tea infusion prevents diabetic nephropathy aggravation in recent-onset type 1 diabetes regardless of glycemic control. J. Ethnopharmacol. 2021, 274, 114032. [Google Scholar] [CrossRef] [PubMed]
- Borges, C.M.; Papadimitriou, A.; Duarte, D.A.; Lopes de Faria, J.M.; Lopes de Faria, J.B. The use of green tea polyphenols for treating residual albuminuria in diabetic nephropathy: A double-blind randomised clinical trial. Sci. Rep. 2016, 6, 28282. [Google Scholar] [CrossRef]
- Hayashi, D.; Wang, L.; Ueda, S.; Yamanoue, M.; Ashida, H.; Shirai, Y. The mechanisms of ameliorating effect of a green tea polyphenol on diabetic nephropathy based on diacylglycerol kinase α. Sci. Rep. 2020, 10, 11790. [Google Scholar] [CrossRef]
- Raposo, D.; Morgado, C.; Pereira-Terra, P.; Tavares, I. Nociceptive spinal cord neurons of laminae I–III exhibit oxidative stress damage during diabetic neuropathy which is prevented by early antioxidant treatment with epigallocatechin-gallate (EGCG). Brain Res. Bull. 2015, 110, 68–75. [Google Scholar] [CrossRef]
- Tiwari, V.; Kuhad, A.; Chopral, K. Amelioration of functional, biochemical and molecular deficits by epigallocatechin gallate in experimental model of alcoholic neuropathy. Eur. J. Pain 2011, 15, 286–292. [Google Scholar] [CrossRef]
- Mohan, T.; Narasimhan, K.K.S.; Ravi, D.B.; Velusamy, P.; Chandrasekar, N.; Chakrapani, L.N.; Srinivasan, A.; Karthikeyan, P.; Kannan, P.; Tamilarasan, B.; et al. Role of Nrf2 dysfunction in the pathogenesis of diabetic nephropathy: Therapeutic prospect of epigallocatechin-3-gallate. Free Radic. Biol. Med. 2020, 160, 227–238. [Google Scholar] [CrossRef]
- Vasan, K.; Werner, M.; Chandel, N.S. Mitochondrial Metabolism as a Target for Cancer Therapy. Cell Metab. 2020, 32, 341–352. [Google Scholar] [CrossRef]
- Assuncao, M.; Andrade, J.P. Protective action of green tea catechins in neuronal mitochondria during aging. Front. Biosci. 2015, 20, 247–262. [Google Scholar]
- Oyewole, A.O.; Birch-Machin, M.A. Mitochondria-targeted antioxidants. FASEB J. 2015, 29, 4766–4771. [Google Scholar] [CrossRef]
- Srividhya, R.; Kalaiselvi, P. Neuroprotective potential of epigallo catechin-3-gallate in PC-12 cells. Neurochem. Res. 2013, 38, 486–493. [Google Scholar] [CrossRef]
- Rimessi, A.; Previati, M.; Nigro, F.; Wieckowski, M.R.; Pinton, P. Mitochondrial reactive oxygen species and inflammation: Molecular mechanisms, diseases and promising therapies. Int. J. Biochem. Cell Biol. 2016, 81, 281–293. [Google Scholar] [CrossRef]
- Taslimi, P.; Kocyigit, U.M.; Tüzün, B.; Kirici, M. Biological effects and molecular docking studies of Catechin 5-O-gallate: Antioxidant, anticholinergics, antiepileptic and antidiabetic potentials. J. Biomol. Struct. Dyn. 2020, 40, 2489–2497. [Google Scholar] [CrossRef]
- Pitceathly, R.D.; Rahman, S.; Hanna, M.G. Single deletions in mitochondrial DNA – Molecular mechanisms and disease phenotypes in clinical practice. Neuromuscul. Disord. 2012, 22, 577–586. [Google Scholar] [CrossRef]
- Ferrari, C.K. Functional foods, herbs and nutraceuticals: Towards biochemical mechanisms of healthy aging. Biogerontology 2004, 5, 275–289. [Google Scholar] [CrossRef]
- Iwai, K.; Iwamura, Y.; Yamashita, S.; Wadano, Y.; Mesaki, N. Effect of tea catechins on mitochondrial DNA 4977-bp deletions in human leucocytes. Mutat. Res. Mol. Mech. Mutagen. 2006, 595, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Srividhya, R.; Zarkovic, K.; Stroser, M.; Waeg, G.; Zarkovic, N.; Kalaiselvi, P. Mitochondrial alterations in aging rat brain: Effective role of (−)-epigallo catechin gallate. Int. J. Dev. Neurosci. 2009, 27, 223–231. [Google Scholar] [CrossRef]
- Zhong, J.; Tan, Y.; Lu, J.; Liu, J.; Xiao, X.; Zhu, P.; Chen, S.; Zheng, S.; Chen, Y.; Hu, Y.; et al. Therapeutic contribution of melatonin to the treatment of septic cardiomyopathy: A novel mechanism linking Ripk3-modified mitochondrial performance and endoplasmic reticulum function. Redox Biol. 2019, 26, 101287. [Google Scholar] [CrossRef]
- Wei, Y.; Corbalán-Campos, J.; Gurung, R.; Natarelli, L.; Zhu, M.; Exner, N.; Erhard, F.; Greulich, F.; Geißler, C.; Uhlenhaut, N.H.; et al. Dicer in Macrophages Prevents Atherosclerosis by Promoting Mitochondrial Oxidative Metabolism. Circulation 2018, 138, 2007–2020. [Google Scholar] [CrossRef]
- Jomova, K.; Valko, M. Advances in metal-induced oxidative stress and human disease. Toxicology 2011, 283, 65–87. [Google Scholar] [CrossRef]
- Dai, W.; Jiang, L. Dysregulated Mitochondrial Dynamics and Metabolism in Obesity, Diabetes, and Cancer. Front. Endocrinol. 2019, 10, 570. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.-T.; Zhang, W.-F.; Luo, P.; He, F.; Ge, X.-Y.; Zhang, Z.; Hu, C.-P. Epigallocatechin-3-gallate ameliorates hypoxia-induced pulmonary vascular remodeling by promoting mitofusin-2-mediated mitochondrial fusion. Eur. J. Pharmacol. 2017, 809, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. Free Radical Theory of Aging: An Update: Increasing the Functional Life Span. Ann. N. Y. Acad. Sci. 2006, 1067, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Yue, R.; Xia, X.; Jiang, J.; Yang, D.; Han, Y.; Chen, X.; Cai, Y.; Li, L.; Wang, W.E.; Zeng, C. Mitochondrial DNA Oxidative Damage Contributes to Cardiomyocyte Ischemia/Reperfusion-Injury in Rats: Cardioprotective Role of Lycopene. J. Cell. Physiol. 2015, 230, 2128–2141. [Google Scholar] [CrossRef]
- Pena-Bautista, C.; Baquero, M.; Vento, M.; Chafer-Pericas, C. Free radicals in Alzheimer’s disease: Lipid peroxidation biomarkers. Clin. Chim. Acta 2019, 491, 85–90. [Google Scholar] [CrossRef]
- Umeno, A.; Biju, V.; Yoshida, Y. In vivo ROS production and use of oxidative stress-derived biomarkers to detect the onset of diseases such as Alzheimer’s disease, Parkinson’s disease, and diabetes. Free Radic. Res. 2017, 5, 413–427. [Google Scholar] [CrossRef]
- Zhong, R.Z.; Xiao, W.J.; Zhou, D.W.; Tan, C.Y.; Tan, Z.L.; Han, X.F.; Zhou, C.S.; Tang, S.X. Effect of tea catechins on regulation of cell proliferation and antioxidant enzyme expression in H2 O2 -induced primary hepatocytes of goat in vitro. J. Anim. Physiol. Anim. Nutr. 2013, 97, 475–484. [Google Scholar] [CrossRef]
- Abib, R.T.; Peres, K.C.; Barbosa, A.M.; Peres, T.V.; Bernardes, A.; Zimmermann, L.M.; Quincozes-Santos, A.; Fiedler, H.D.; Leal, R.B.; Farina, M.; et al. Epigallocatechin-3-gallate protects rat brain mitochondria against cadmium-induced damage. Food Chem. Toxicol. 2011, 49, 2618–2623. [Google Scholar] [CrossRef]
- Xian, H.; Watari, K.; Sanchez-Lopez, E.; Offenberger, J.; Onyuru, J.; Sampath, H.; Ying, W.; Hoffman, H.M.; Shadel, G.S.; Karin, M. Oxidized DNA fragments exit mitochondria via mPTP- and VDAC-dependent channels to activate NLRP3 inflammasome and interferon signaling. Immunity 2022, 55, 1370–1385.e8. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, J.; Sun, X.; Shi, X.; Wang, L.; Huang, L.; Zhou, W. Evaluation of the neuroprotective effect of EGCG: A potential mechanism of mitochondrial dysfunction and mitochondrial dynamics after subarachnoid hemorrhage. Food Funct. 2018, 9, 6349–6359. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Xu, F.; Si, S.; Zhao, X.; Bi, S.; Cen, Y. Mitochondrial DNA-Induced Inflammatory Responses and Lung Injury in Thermal Injury Rat Model: Protective Effect of Epigallocatechin Gallate. J. Burn. Care Res. 2017, 38, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.-Y.; Gu, J.; Fan, J.-X.; Zhang, H.-W.; Xu, F.; Liang, H.-M.; Fan, K.-J.; Xiao, Z.-H.; Zhang, E.-Y.; Hu, J. Epigallocatechin gallate attenuates mitochondrial DNA-induced inflammatory damage in the development of ventilator-induced lung injury. Phytomedicine 2018, 48, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Charles, A.-L.; Meyer, A.; Dal-Ros, S.; Auger, C.; Keller, N.; Ramamoorthy, T.G.; Zoll, J.; Metzger, D.; Schini-Kerth, V.; Geny, B. Polyphenols prevent ageing-related impairment in skeletal muscle mitochondrial function through decreased reactive oxygen species production. Exp. Physiol. 2013, 98, 536–545. [Google Scholar] [CrossRef]
- Li, P.; Liu, A.; Xiong, W.; Lin, H.; Xiao, W.; Huang, J.; Zhang, S.; Liu, Z. Catechins enhance skeletal muscle performance. Crit. Rev. Food Sci. Nutr. 2020, 60, 515–528. [Google Scholar] [CrossRef]
- Picca, A.; Lezza, A.M. Regulation of mitochondrial biogenesis through TFAM-mitochondrial DNA interactions: Useful insights from aging and calorie restriction studies. Mitochondrion 2015, 25, 67–75. [Google Scholar] [CrossRef]
- Perez-De-Arce, K.; Foncea, R.; Leighton, F. Reactive oxygen species mediates homocysteine-induced mitochondrial biogenesis in human endothelial cells: Modulation by antioxidants. Biochem. Biophys. Res. Commun. 2005, 338, 1103–1109. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y.; Zhu, Q.; Li, T.; Lu, H.; Wei, N.; Huang, Y.; Shi, R.; Ma, X.; Wang, X.; et al. Anti-skin-aging effect of epigallocatechin gallate by regulating epidermal growth factor receptor pathway on aging mouse model induced by d -Galactose. Mech. Ageing Dev. 2017, 164, 1–7. [Google Scholar] [CrossRef]
- Chakrabarty, S.; Kabekkodu, S.P.; Singh, R.P.; Thangaraj, K.; Singh, K.K.; Satyamoorthy, K. Mitochondria in health and disease. Mitochondrion 2018, 43, 25–29. [Google Scholar] [CrossRef]
- Choi, Y.S.; Kim, S.; Kyu Lee, H.; Lee, K.-U.; Pak, Y.K. In vitro methylation of nuclear respiratory factor-1 binding site suppresses the promoter activity of mitochondrial transcription factor A. Biochem. Biophys. Res. Commun. 2004, 314, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Irrcher, I.; Adhihetty, P.J.; Joseph, A.-M.; Ljubicic, V.; Hood, D.A. Regulation of Mitochondrial Biogenesis in Muscle by Endurance Exercise. Sports Med. 2003, 33, 783–793. [Google Scholar] [CrossRef] [PubMed]
- Ha, T.; Kim, M.K.; Park, K.-S.; Jung, W.; Choo, H.; Chong, Y. Structural Modification of (−)-Epigallocatechin Gallate (EGCG) Shows Significant Enhancement in Mitochondrial Biogenesis. J. Agric. Food Chem. 2018, 66, 3850–3859. [Google Scholar] [CrossRef] [PubMed]
- Jäger, S.; Handschin, C.; St.-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef]
- Rothenberg, D.O.; Zhou, C.; Zhang, L. A Review on the Weight-Loss Effects of Oxidized Tea Polyphenols. Molecules 2018, 23, 1176. [Google Scholar] [CrossRef]
- Pal, S.; Porwal, K.; Rajak, S.; Sinha, R.A.; Chattopadhyay, N. Selective dietary polyphenols induce differentiation of human osteoblasts by adiponectin receptor 1-mediated reprogramming of mitochondrial energy metabolism. Biomed. Pharmacother. 2020, 127, 110207. [Google Scholar] [CrossRef]
- Dugar, S.; Villarreal, F.; Hollinger, F.H.; Mahajan, D.; Ramirez-Sanchez, I.; Moreno-Ulloa, A.; Ceballos, G.; Schreiner, G. 11-beta-hydroxysterols as possible endogenous stimulators of mitochondrial biogenesis as inferred from epicatechin molecular mimicry. Pharmacol. Res. 2020, 151, 104540. [Google Scholar] [CrossRef]
- Moreno-Ulloa, A.; Nájera-García, N.; Hernández, M.; Ramírez-Sánchez, I.; Taub, P.R.; Su, Y.; Beltrán-Partida, E.; Ceballos, G.; Dugar, S.; Schreiner, G.; et al. A pilot study on clinical pharmacokinetics and preclinical pharmacodynamics of (+)-epicatechin on cardiometabolic endpoints. Food Funct. 2018, 9, 307–319. [Google Scholar] [CrossRef]
- Lilja, S.; Oldenburg, J.; Pointner, A.; Dewald, L.; Lerch, M.; Hippe, B.; Switzeny, O.; Haslberger, A. Epigallocatechin Gallate Effectively Affects Senescence and Anti-SASP via SIRT3 in 3T3-L1 Preadipocytes in Comparison with Other Bioactive Substances. Oxidative Med. Cell. Longev. 2020, 2020, 1–13. [Google Scholar] [CrossRef]
- Bourebaba, N.; Kornicka-Garbowska, K.; Marycz, K.; Bourebaba, L.; Kowalczuk, A. Laurus nobilis ethanolic extract attenuates hyperglycemia and hyperinsulinemia-induced insulin resistance in HepG2 cell line through the reduction of oxidative stress and improvement of mitochondrial biogenesis—Possible implication in pharmacotherapy. Mitochondrion 2021, 59, 190–213. [Google Scholar] [CrossRef]
- Valenti, D.; de Bari, L.; de Rasmo, D.; Signorile, A.; Henrion-Caude, A.; Contestabile, A.; Vacca, R.A. The polyphenols resveratrol and epigallocatechin-3-gallate restore the severe impairment of mitochondria in hippocampal progenitor cells from a Down syndrome mouse model. Biochim. Biophys. Acta 2016, 1862, 1093–1104. [Google Scholar] [CrossRef] [PubMed]
- Korish, A.A.; Arafah, M.M. Catechin combined with vitamins C and E ameliorates insulin resistance (IR) and atherosclerotic changes in aged rats with chronic renal failure (CRF). Arch. Gerontol. Geriatr. 2008, 46, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Abolhassani, N.; Leon, J.; Sheng, Z.; Oka, S.; Hamasaki, H.; Iwaki, T.; Nakabeppu, Y. Molecular pathophysiology of impaired glucose metabolism, mitochondrial dysfunction, and oxidative DNA damage in Alzheimer’s disease brain. Mech. Ageing Dev. 2017, 161, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Santamarina, A.B.; Carvalho-Silva, M.; Gomes, L.M.; Okuda, M.H.; Santana, A.A.; Streck, E.L.; Seelaender, M.; do Nascimento, C.M.O.; Ribeiro, E.B.; Lira, F.S.; et al. Decaffeinated green tea extract rich in epigallocatechin-3-gallate prevents fatty liver disease by increased activities of mitochondrial respiratory chain complexes in diet-induced obesity mice. J. Nutr. Biochem. 2015, 26, 1348–1356. [Google Scholar] [CrossRef] [PubMed]
- Gleichmann, M.; Mattson, M.P. Neuronal Calcium Homeostasis and Dysregulation. Antioxid. Redox Signal. 2011, 14, 1261–1273. [Google Scholar] [CrossRef]
- Boyman, L.; Karbowski, M.; Lederer, W.J. Regulation of Mitochondrial ATP Production: Ca(2+) Signaling and Quality Control. Trends Mol. Med. 2020, 26, 21–39. [Google Scholar] [CrossRef]
- Wang, J.-H.; Cheng, J.; Li, C.-R.; Ye, M.; Ma, Z.; Cai, F. Modulation of Ca(2)(+) Signals by Epigallocatechin-3-gallate(EGCG) in Cultured Rat Hippocampal Neurons. Int. J. Mol. Sci. 2011, 12, 742–754. [Google Scholar] [CrossRef]
- Kim, H.J.; Yum, K.S.; Sung, J.H.; Rhie, D.J.; Kim, M.J.; Min, D.S.; Hahn, S.J.; Kim, M.S.; Jo, Y.H.; Yoon, S.H. Epigallocatechin-3-gallate increases intracellular [Ca2+] in U87 cells mainly by influx of extracellular Ca2+ and partly by release of intracellular stores. Naunyn. Schmiedebergs. Arch. Pharmacol. 2004, 369, 260–267. [Google Scholar]
- Marchetti, C.; Gavazzo, P.; Burlando, B. Epigallocatechin-3-gallate mobilizes intracellular Ca2+ in prostate cancer cells through combined Ca2+ entry and Ca2+-induced Ca2+ release. Life Sci. 2020, 258, 118232. [Google Scholar] [CrossRef]
- Rinaldi, D.E.; Ontiveros, M.Q.; Saffioti, N.A.; Vigil, M.A.; Mangialavori, I.C.; Rossi, R.C.; Rossi, J.P.; Espelt, M.V.; Ferreira-Gomes, M.S. Epigallocatechin 3-gallate inhibits the plasma membrane Ca2+-ATPase: Effects on calcium homeostasis. Heliyon 2021, 7, e06337. [Google Scholar] [CrossRef]
- Peleg, S.; Feller, C.; Ladurner, A.G.; Imhof, A. The Metabolic Impact on Histone Acetylation and Transcription in Ageing. Trends Biochem. Sci. 2016, 41, 700–711. [Google Scholar] [CrossRef] [PubMed]
- Bai, M.; Yang, L.; Liao, H.; Liang, X.; Xie, B.; Xiong, J.; Tao, X.; Chen, X.; Cheng, Y.; Chen, X.; et al. Metformin sensitizes endometrial cancer cells to chemotherapy through IDH1-induced Nrf2 expression via an epigenetic mechanism. Oncogene 2018, 37, 5666–5681. [Google Scholar] [CrossRef] [PubMed]
- Nandakumar, V.; Vaid, M.; Katiyar, S.K. (−)-Epigallocatechin-3-gallate reactivates silenced tumor suppressor genes, Cip1/p21 and p16INK4a, by reducing DNA methylation and increasing histones acetylation in human skin cancer cells. Carcinogenesis 2011, 32, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Yiannakopoulou, E.C. Targeting DNA Methylation with Green Tea Catechins. Pharmacology 2015, 95, 111–116. [Google Scholar] [CrossRef]
- Weng, Y.-P.; Hung, P.-F.; Ku, W.-Y.; Chang, C.-Y.; Wu, B.-H.; Wu, M.-H.; Yao, J.-Y.; Yang, J.-R.; Lee, C.-H. The inhibitory activity of gallic acid against DNA methylation: Application of gallic acid on epigenetic therapy of human cancers. Oncotarget 2018, 9, 361–374. [Google Scholar] [CrossRef]
- Lee, W.J.; Shim, J.-Y.; Zhu, B.T. Mechanisms for the Inhibition of DNA Methyltransferases by Tea Catechins and Bioflavonoids. Mol. Pharmacol. 2005, 68, 1018–1030. [Google Scholar] [CrossRef]
- Sur, S.; Panda, C.K. Molecular aspects of cancer chemopreventive and therapeutic efficacies of tea and tea polyphenols. Nutrition 2017, 43-44, 8–15. [Google Scholar] [CrossRef]
- Yang, C.S.; Zhang, J.; Zhang, L.; Huang, J.; Wang, Y. Mechanisms of body weight reduction and metabolic syndrome alleviation by tea. Mol. Nutr. Food Res. 2016, 60, 160–174. [Google Scholar] [CrossRef]
- Yang, C.S.; Hong, J. Prevention of Chronic Diseases by Tea: Possible Mechanisms and Human Relevance. Annu. Rev. Nutr. 2013, 33, 161–181. [Google Scholar] [CrossRef]
- Huang, J.; Wang, Y.; Xie, Z.; Zhou, Y.; Zhang, Y.; Wan, X. The anti-obesity effects of green tea in human intervention and basic molecular studies. Eur. J. Clin. Nutr. 2014, 68, 1075–1087. [Google Scholar] [CrossRef]
- Long, Y.C.; Zierath, J.R. AMP-activated protein kinase signaling in metabolic regulation. J. Clin. Invest. 2006, 116, 1776–1783. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. AMPK: Positive and negative regulation, and its role in whole-body energy homeostasis. Curr. Opin. Cell Biol. 2015, 33, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Bezzina, R.; Hinch, E.; Lewandowski, P.A.; Cameron-Smith, D.; Mathai, M.L.; Jois, M.; Sinclair, A.J.; Begg, D.P.; Wark, J.D.; et al. Green tea, black tea, and epigallocatechin modify body composition, improve glucose tolerance, and differentially alter metabolic gene expression in rats fed a high-fat diet. Nutr. Res. 2009, 29, 784–793. [Google Scholar] [CrossRef]
- Lambert, J.D.; Sang, S.; Hong, J.; Kwon, S.-J.; Lee, M.-J.; Ho, C.-T.; Yang, C.S. Peracetylation as a Means of Enhancing in Vitro Bioactivity and Bioavailability of Epigallocatechin-3-Gallate. Drug Metab. Dispos. 2006, 34, 2111–2116. [Google Scholar] [CrossRef]
- Liu, C.; Jin, Y.; Fan, Z. The Mechanism of Warburg Effect-Induced Chemoresistance in Cancer. Front. Oncol. 2021, 11. [Google Scholar] [CrossRef]
- Samec, M.; Liskova, A.; Koklesova, L.; Samuel, S.M.; Zhai, K.; Buhrmann, C.; Varghese, E.; Abotaleb, M.; Qaradakhi, T.; Zulli, A.; et al. Flavonoids against the Warburg phenotype—concepts of predictive, preventive and personalised medicine to cut the Gordian knot of cancer cell metabolism. EPMA J. 2020, 11, 377–398. [Google Scholar] [CrossRef]
- Han, J.H.; Kim, M.; Kim, H.J.; Jang, S.B.; Bae, S.J.; Lee, I.K.; Ryu, D.; Ha, K.T. Targeting Lactate Dehydrogenase A with Catechin Resensitizes SNU620/5FU Gastric Cancer Cells to 5-Fluorouracil. Int. J. Mol. Sci. 2021, 22, 10. [Google Scholar] [CrossRef]
- Chen, S.; Nishi, M.; Morine, Y.; Shimada, M.; Tokunaga, T.; Kashihara, H.; Takasu, C.; Yamada, S.; Wada, Y. Epigallocatechin-3-gallate hinders metabolic coupling to suppress colorectal cancer malignancy through targeting aerobic glycolysis in cancer-associated fibroblasts. Int. J. Oncol. 2022, 60, 1–13. [Google Scholar] [CrossRef]
- Shay, J.; Elbaz, H.A.; Lee, I.; Zielske, S.P.; Malek, M.H.; Huttemann, M. Molecular Mechanisms and Therapeutic Effects of (−)-Epicatechin and Other Polyphenols in Cancer, Inflammation, Diabetes, and Neurodegeneration. Oxid. Med. Cell. Longev. 2015, 2015, 181260. [Google Scholar] [CrossRef]
- Khojaste, E.; Ahmadizadeh, C. Catechin Metabolites along with Curcumin Inhibit Proliferation and Induce Apoptosis in Cervical Cancer Cells by Regulating VEGF Expression In-Vitro. Nutr. Cancer 2022, 74, 1048–1057. [Google Scholar] [CrossRef] [PubMed]
- Kopustinskiene, D.M.; Bernatoniene, J.; Jakstas, V.; Morkuniene, R. Chapter 22—The effects of catechins on the cardiac mitochondria. In Mitochondrial Physiology and Vegetal Molecules; de Oliveira, M.R., Ed.; Academic Press: London, UK, 2021; pp. 471–487. [Google Scholar]
- Khiewkamrop, P.; Phunsomboon, P.; Richert, L.; Pekthong, D.; Srisawang, P. Epistructured catechins, EGCG and EC facilitate apoptosis induction through targeting de novo lipogenesis pathway in HepG2 cells. Cancer Cell Int. 2018, 18, 46. [Google Scholar] [CrossRef] [PubMed]
- Nakazato, T.; Ito, K.; Miyakawa, Y.; Kinjo, K.; Yamada, T.; Hozumi, N.; Ikeda, Y.; Kizaki, M. Catechin, a green tea component, rapidly induces apoptosis of myeloid leukemic cells via modulation of reactive oxygen species production in vitro and inhibits tumor growth in vivo. Haematologica 2005, 90, 317–325. [Google Scholar] [PubMed]
- Philips, B.J.; Coyle, C.H.; Morrisroe, S.N.; Chancellor, M.B.; Yoshimura, N. Induction of apoptosis in human bladder cancer cells by green tea catechins. Biomed. Res. 2009, 30, 207–215. [Google Scholar] [CrossRef]
- Han, D.H.; Jeong, J.H.; Kim, J.H. Anti-proliferative and apoptosis induction activity of green tea polyphenols on human promyelocytic leukemia HL-60 cells. Anticancer Res. 2009, 29, 1417–1421. [Google Scholar]
- Ong, S.-B.; Gustafsson, A.B. New roles for mitochondria in cell death in the reperfused myocardium. Cardiovasc. Res. 2012, 94, 190–196. [Google Scholar] [CrossRef]
- Giorgio, V.; Guo, L.; Bassot, C.; Petronilli, V.; Bernardi, P. Calcium and regulation of the mitochondrial permeability transition. Cell Calcium 2018, 70, 56–63. [Google Scholar] [CrossRef]
- Choi, Y.-J.; Jeong, Y.-J.; Lee, Y.-J.; Kwon, H.-M.; Kang, Y.-H. (−)Epigallocatechin Gallate and Quercetin Enhance Survival Signaling in Response to Oxidant-Induced Human Endothelial Apoptosis. J. Nutr. 2005, 135, 707–713. [Google Scholar] [CrossRef]
- Choi, J.-S.; Choi, Y.-J.; Shin, S.-Y.; Li, J.; Kang, S.-W.; Bae, J.-Y.; Kim, D.S.; Ji, G.-E.; Kang, J.-S.; Kang, Y.-H. Dietary Flavonoids Differentially Reduce Oxidized LDL-Induced Apoptosis in Human Endothelial Cells: Role of MAPK- and JAK/STAT-Signaling. J. Nutr. 2008, 138, 983–990. [Google Scholar] [CrossRef]
- Huang, J.; Tang, X.; Liang, X.; Wen, Q.; Zhang, S.; Xuan, F.; Jian, J.; Lin, X.; Huang, R. The Effects of 17-Methoxyl-7-Hydroxy-Benzene-Furanchalcone on the Pressure Overload-Induced Progression of Cardiac Hypertrophy to Cardiac Failure. PLoS ONE 2014, 9, e91834. [Google Scholar] [CrossRef]
- Zhang, C.; Liao, P.; Liang, R.; Zheng, X.; Jian, J. Epigallocatechin gallate prevents mitochondrial impairment and cell apoptosis by regulating miR-30a/p53 axis. Phytomedicine 2019, 61, 152845. [Google Scholar] [CrossRef] [PubMed]
- Townsend, P.A.; Scarabelli, T.M.; Pasini, E.; Gitti, G.; Menegazzi, M.; Suzuki, H.; Knight, R.A.; Latchman, D.S.; Stephanou, A. Epigallocatechin-3-gallate inhibits STAT-1 activation and protects cardiac myocytes from ischemia/reperfusion-induced apoptosis. FASEB J. 2004, 18, 1621–1623. [Google Scholar] [CrossRef]
- Xuan, F.; Jian, J. Epigallocatechin gallate exerts protective effects against myocardial ischemia/reperfusion injury through the PI3K/Akt pathway-mediated inhibition of apoptosis and the restoration of the autophagic flux. Int. J. Mol. Med. 2016, 38, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Piao, C.S.; Kim, D.S.; Ha, K.C.; Kim, H.R.; Chae, H.J.; Chae, S.W. The Protective Effect of Epigallocatechin-3 Gallate on Ischemia/Reperfusion Injury in Isolated Rat Hearts: An ex vivo Approach. Korean J. Physiol. Pharmacol. 2011, 15, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Mohan, T.; Velusamy, P.; Chakrapani, L.N.; Srinivasan, A.K.; Singh, A.; Johnson, T.; Periandavan, K. Impact of EGCG Supplementation on the Progression of Diabetic Nephropathy in Rats: An Insight into Fibrosis and Apoptosis. J. Agric. Food Chem. 2017, 65, 8028–8036. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wu, M.; Lu, F.; Luo, N.; He, Z.P.; Yang, H. Involvement of alpha7 nAChR signaling cascade in epigallocatechin gallate suppression of beta-amyloid-induced apoptotic cortical neuronal insults. Mol. Neurobiol. 2014, 49, 66–77. [Google Scholar] [CrossRef]
- Payne, A.; Nahashon, S.; Taka, E.; Adinew, G.M.; Soliman, K.F.A. Epigallocatechin-3-Gallate (EGCG): New Therapeutic Perspectives for Neuroprotection, Aging, and Neuroinflammation for the Modern Age. Biomolecules 2022, 12, 371. [Google Scholar] [CrossRef]
- Chu, K.O.; Wang, C.C.R.; Chu, C.Y.; Chan, K.P.; Rogers, M.S.; Choy, K.W.; Pang, C.P. Pharmacokinetic studies of green tea catechins in maternal plasma and fetuses in rats. J. Pharm. Sci. 2006, 95, 1372–1381. [Google Scholar] [CrossRef]
- Huo, Y.; Zhang, Q.; Li, Q.; Geng, B.; Bi, K. Development of a UFLC-MS/MS method for the simultaneous determination of seven tea catechins in rat plasma and its application to a pharmacokinetic study after administration of green tea extract. J. Pharm. Biomed. Anal. 2016, 125, 229–235. [Google Scholar] [CrossRef]
- Mustata, G.T.; Rosca, M.; Biemel, K.M.; Reihl, O.; Smith, M.A.; Viswanathan, A.; Strauch, C.Y.D.; Tang, J.; Kern, T.S.; Lederer, M.O.; et al. Paradoxical effects of green tea (Camellia sinensis) and antioxidant vitamins in diabetic rats: Improved retinopathy and renal mitochondrial defects but deterioration of collagen matrix glycoxidation and cross-linking. Diabetes 2005, 54, 517–526. [Google Scholar] [CrossRef]
- Hirai, M.; Hotta, Y.; Ishikawa, N.; Wakida, Y.; Fukuzawa, Y.; Isobe, F.; Nakano, A.; Chiba, T.; Kawamura, N. Protective effects of EGCg or GCg, a green tea catechin epimer, against postischemic myocardial dysfunction in guinea-pig hearts. Life Sci. 2007, 80, 1020–1032. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Xu, L.; Yang, L.; Wang, X. Epigallocatechin Gallate Is the Most Effective Catechin Against Antioxidant Stress via Hydrogen Peroxide and Radical Scavenging Activity. Med Sci. Monit. 2018, 24, 8198–8206. [Google Scholar] [CrossRef] [PubMed]
- Eslami, A.C.; Pasanphan, W.; Wagner, B.A.; Buettner, G.R. Free radicals produced by the oxidation of gallic acid: An electron paramagnetic resonance study. Chem. Central J. 2010, 4, 15. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Pukala, T.L.; Musgrave, I.F.; Williams, D.M.; Dehle, F.C.; Carver, J.A. Gallic acid is the major component of grape seed extract that inhibits amyloid fibril formation. Bioorg. Med. Chem. Lett. 2013, 23, 6336–6340. [Google Scholar] [CrossRef]
- Wang, H.; Fowler, M.I.; Messenger, D.J.; Ordaz-Ortiz, J.J.; Gu, X.; Shi, S.; Terry, L.A.; Berry, M.J.; Lian, G.; Wang, S. Inhibition of the intestinal postprandial glucose transport by gallic acid and gallic acid derivatives. Food Funct. 2021, 12, 5399–5406. [Google Scholar] [CrossRef]
- Bashar, S.M.; Elhadidy, M.G.; Mostafa, A.F.; Hamed, B.; Helmy, S.; Abd-Elmoniem, H.A. Hepatoprotective effect of gallic acid against type 2-induced diabetic liver injury in male rats through modulation of fetuin-A and GLP-1 with involvement of ERK1/2/NF-κB and Wnt1/beta-catenin signaling pathways. Gen. Physiol. Biophys. 2021, 40, 221–234. [Google Scholar] [CrossRef]
- Sohrabi, F.; Dianat, M.; Badavi, M.; Radan, M.; Mard, S.A. Gallic acid suppresses inflammation and oxidative stress through modulating Nrf2-HO-1-NF-κB signaling pathways in elastase-induced emphysema in rats. Environ. Sci. Pollut. Res. Int. 2021, 28, 56822–56834. [Google Scholar] [CrossRef]
- Niittykoski, M.; Haapalinna, A.; Sirviö, J. Selegiline reduces N-methyl-D-aspartic acid induced perturbation of neurotransmission but it leaves NMDA receptor dependent long-term potentiation intact in the hippocampus. J. Neural Transm. 2003, 110, 1225–1240. [Google Scholar] [CrossRef]
- Oi, Y.; Hou, I.-C.; Fujita, H.; Yazawa, K. Antiobesity Effects of Chinese Black Tea (Pu-erh Tea) Extract and Gallic Acid. Phytotherapy Res. 2012, 26, 475–481. [Google Scholar] [CrossRef]
- Zhang, D.Y.; Shao, W.F.; Liu, Z.H.; Liu, Y.L.; Huang, Y.W. Study of gallic acid in pu-erh tea on the peroxisome proliferators activated receptors function. Acta Nutr. Sin. 2009, 31, 47–50. [Google Scholar]
- Chao, J.; Lau, W.K.; Huie, M.J.; Ho, Y.-S.; Yu, M.-S.; Lai, C.S.; Wang, M.; Yuen, W.-H.; Lam, W.H.; Chan, T.H.; et al. A pro-drug of the green tea polyphenol (−)-epigallocatechin-3-gallate (EGCG) prevents differentiated SH-SY5Y cells from toxicity induced by 6-hydroxydopamine. Neurosci. Lett. 2010, 469, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.C.; Xu, H.; Man, C.W.; Zhang, T.; Chu, K.O.; Chu, C.Y.; Cheng, J.T.Y.; Li, G.; He, Y.X.; Qin, L.; et al. Prodrug of green tea epigallocatechin-3-gallate (Pro-EGCG) as a potent anti-angiogenesis agent for endometriosis in mice. Angiogenesis 2013, 16, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Chiou, Y.-S.; Sang, S.; Cheng, K.-H.; Ho, C.-T.; Wang, Y.-J.; Pan, M.-H. Peracetylated (−)-epigallocatechin-3-gallate (AcEGCG) potently prevents skin carcinogenesis by suppressing the PKD1-dependent signaling pathway in CD34 + skin stem cells and skin tumors. Carcinogenesis 2013, 34, 1315–1322. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Man, G.C.W.; Chan, T.H.; Kwong, J.; Wang, C.C. A prodrug of green tea polyphenol (–)-epigallocatechin-3-gallate (Pro-EGCG) serves as a novel angiogenesis inhibitor in endometrial cancer. Cancer Lett. 2018, 412, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Duangjan, C.; Curran, S.P. Oolonghomobisflavans from Camellia sinensis increase Caenorhabditis elegans lifespan and healthspan. GeroScience 2022, 44, 533–545. [Google Scholar] [CrossRef]
- Pervin, M.; Unno, K.; Takagaki, A.; Isemura, M.; Nakamura, Y. Function of Green Tea Catechins in the Brain: Epigallocatechin Gallate and its Metabolites. Int. J. Mol. Sci. 2019, 20, 3630. [Google Scholar] [CrossRef]
- Fujihara, T.; Nakagawa-Izumi, A.; Ozawa, T.; Numata, O. High-Molecular-Weight Polyphenols from Oolong Tea and Black Tea: Purification, Some Properties, and Role in Increasing Mitochondrial Membrane Potential. Biosci. Biotechnol. Biochem. 2007, 71, 711–719. [Google Scholar] [CrossRef]
- Kikuchi, A.; Shiba, K.; Ozawa, T.; Nakano, K.; Inaba, K.; Numata, O. Black Tea High-Molecular-Weight Polyphenol Increases the Motility of Sea Urchin Sperm by Activating Mitochondrial Respiration. Biosci. Biotechnol. Biochem. 2012, 76, 2321–2324. [Google Scholar] [CrossRef][Green Version]
- Eguchi, T.; Kumagai, C.; Fujihara, T.; Takemasa, T.; Ozawa, T.; Numata, O. Black Tea High-Molecular-Weight Polyphenol Stimulates Exercise Training-Induced Improvement of Endurance Capacity in Mouse via the Link between AMPK and GLUT4. PLoS ONE 2013, 8, e69480. [Google Scholar] [CrossRef]
- Aoki, Y.; Ozawa, T.; Takemasa, T.; Numata, O. Black Tea High-Molecular-Weight Polyphenol-Rich Fraction Promotes Hypertrophy during Functional Overload in Mice. Molecules 2017, 22, 548. [Google Scholar] [CrossRef]
- Aoki, Y.; Ozawa, T.; Numata, O.; Takemasa, T. High-Molecular-Weight Polyphenol-Rich Fraction of Black Tea Does Not Prevent Atrophy by Unloading, But Promotes Soleus Muscle Mass Recovery from Atrophy in Mice. Nutrients 2019, 11, 2131. [Google Scholar] [CrossRef] [PubMed]
- Ehrnhoefer, D.E.; Duennwald, M.; Markovic, P.; Wacker, J.L.; Engemann, S.; Roark, M.; Legleiter, J.; Marsh, J.L.; Thompson, L.M.; Lindquist, S.; et al. Green tea (−)-epigallocatechin-gallate modulates early events in huntingtin misfolding and reduces toxicity in Huntington’s disease models. Hum. Mol. Genet. 2006, 15, 2743–2751. [Google Scholar] [CrossRef] [PubMed]
- Xiang, C.; Xiao, X.; Jiang, B.; Zhou, M.; Zhang, Y.; Li, H.; Hu, Z. Epigallocatechin-3-gallate protects from high glucose induced podocyte apoptosis via suppressing endoplasmic reticulum stress. Mol. Med. Rep. 2017, 16, 6142–6147. [Google Scholar] [CrossRef] [PubMed]
- Koonyosying, P.; Kongkarnka, S.; Uthaipibull, C.; Svasti, S.; Fucharoen, S.; Srichairatanakool, S. Green tea extract modulates oxidative tissue injury in beta-thalassemic mice by chelation of redox iron and inhibition of lipid peroxidation. Biomed. Pharmacother. 2018, 108, 1694–1702. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experimental Objects | Drugs | Duration and Treatment Schedule | Measurement Indicators | Type of Effect | References |
|---|---|---|---|---|---|
| Transgenic drosophila melanogaster expressing Htt93Q in all neurons | EGCG | Fed with the compound of sugar supplemented with 500 mm EGCG | The number of rhabdomeres per ommatidium, motor function | EGCG reduces photoreceptor degeneration and improves the abnormal motor ability in HD transgenic flies | [243] |
| Male Wistar rats | EGCG | 3-NP, L-arginine, andL-NAME and administered intraperitoneally to animals. EGCG was suspended in 0.05% w/v sodium carboxy-methyl-cellulose solution and administered by oral route in a constant volume of 0.5 mL/100 g of body weight | Lesion volume | EGCG treatment significantly reversed 3-NP-induced rat striatal degeneration as compared to 3-NP-treated group. | [59] |
| NB SH-SY5Y cells | EGCG, 6-OHDA | EGCG was added 15 min before insult with 6-OHDA for a subsequent 24 or 48 h, respectively. The inhibitor of PKC, GF 109203X, was added 30 min before treatment with EGCG. | Neuronal cell injury was evaluated by a colorimetric assay for mitochondrial function using the MTT test | Pretreatment for 15 min with EGCG conferred significant protection against 6-OHDA neurotoxicity | [62] |
| Hippocampal neuron from 18-day-old embryo Sprague–Dawley rats | βA (25–35) and EGCG | βA (25–100 μM) and/or EGCG (10 μM) was added to the culture medium. | Cell viability | EGCG effectively promoted the survival of βA (25–35)-treated neuronal cells. | [63] |
| Rat insulin-secreting RINm5F cells | EGCG from green tea | In a set of experiments, cells were pretreated with 0, 10, 20, 40 μM EGCG for 1 h prior to cytokine stimulation. | Cell viability, Insulin secretion | EGCG pretreatment prevented cells from this cytokine-induced death in RINm5F cells, with viability back to control level; EGCG can prevent the inhibitory effects of these cytokines on insulin release | [71] |
| Female NOD/LtJ mice | EGCG from green tea | Give either 0 or 0.05% (w/v) of EGCG in drinking-water | non-fasting blood glucose, the general clinical condition and mortality of mice | EGCG (0.05% in drinking water) significantly ameliorated hyperglycemia and delayed the onset of T1D in NOD mice | [72] |
| 20 free-living subjects who had type 2 diabetes and took hyperglycemic drugs as prescribed | Oolong tea | Subjects consumed oolong tea (1500 mL) or water for 30 days each in a randomized crossover design. Tea was not consumed for 14 days prior to treatments. | Plasma glucose | The plasma glucose and fructosamine concentrations of diabetes patients decreased significantly (p < 0.001 and 0.001, respectively) after drinking oolong tea, but did not change after drinking water. | [73] |
| db/db mice | Dietary EGCG | Mice consumed a modified AIN-93diet containing EGCG at concentrations of 2.5, 5.0, or 10.0 g/kg of diet (EGCG 0.25%, 0.5%, or 1% w:w, n¼9/group) | Blood glucose levels | A pronounced decrease of glucose levels was observed in food-deprived db/db mice treated with EGCG | [75] |
| Eight-week-old obese female KK-ay and C57BL/6J mice | GTCs | Mice were treated with GTCs for 4 weeks | Plasma glucose levels | GTCs feeding decreased the blood glucose content, random blood glucose content (RBG), fasting blood glucose content (FBG), and 2-h blood glucose content (2HBG) of KK-ay mice, and increased their glucose tolerance. | [77] |
| Twelve pediatric cardiomyopathy patients with diastolic dysfunction | Green tea extract catechins | Oral administration for 12 months | Heart rate and blood pressure, systolic and diastolic functions, isovolumetric relaxation time, LVESD, LVEDD, LVESV | an increase in left ventricle end diastolic volume and stroke volume were observed with echocardiography | [105] |
| cTnT transgenic mice | Catechins | Bind EGCG to cTn subunits | Force-pCa relationships in the skinned cardiac muscle fibers | EGCG reversed the increased myofilament Ca2+ sensitivity of mutant mice, improved the diastolic dysfunction of the hearts of these mice, and increased their cardiac output. | [106] |
| Male Wistar rats | Streptozotocin (STZ), nicotinamide and (EGCG) | Oral EGCG treatment for rats induced experimental diabetes (2 mg/kg body wt) | Blood glucose, insulin, and glycosylated hemoglobin (HbA1c), serum lipid profile, the degree of cardiac apoptosis | EGCG had a positive effect against diabetes-induced cardiomyopathy by modulating the cardiometabolic risk factors, inflammation, oxidative stress, DNA damage, and cell death. | [90] |
| Male C57 BLKS/J genetic background (db/db) mice and their non-diabetic lean littermates (db/m; 6-week-old) and their kidneys | CE | Treated mice with CE for 16 weeks | Serum creatinine concentrations, urea levels, renal AGE levels, and morphometric changes | CE treatment for 16 weeks significantly lowered plasma creatinine and urea levels in diabetic db/db mice; CE showed notably protective effect on DN. | [119] |
| EA·hy926 cell line | CE | Cells were exposed to FBS-free medium containing CE (0, 250, 500, and 1000 nM) for 2 h followed by co-treatment with CE and high glucose (25 mM) for 6 or 24 h | Proinflammatory cytokines levels, IL-1β levels | CE dose-dependently abolished high glucose-induced IL-1β secretion | [119] |
| Male Sprague–Dawley rats | OPLE containing 1.1% (−) catechin gallate and 1.5% ferulic acid | The rats, after confirmation of diabetes induced by STZ, were treated with 1000 mg kg−1OPLE, which was dissolved in distilled water given daily for either 4 or 12 weeks by oral administration | Urinary protein concentration, Glomerular filtration rate (GFR), 8-OHdG levels in 2 h urine samples | Catechin gallate attenuated renal dysfunction (hyperfiltration, proteinuria) and suppressed increases in oxidative stress markers (8-OHdG, LPO) and the fibrotic cytokine, TGF-β1 | [120] |
| C57BL/6 wild type mice | sodium citrate or STZ, EGCG | The diabetic mice (induced by STZ) and age-matched controls were then treated daily by subcutaneously injected EGCG (100 mg/kg) or normal saline daily, for a total period of 24 weeks. | Blood glucose, urinary albumin and urinary creatinine, renal pathological changes | The diabetic mice had a marked accumulation of fibrosis in the kidney, expansion of the mesangial matrix, and enlargement of the glomerular area, effects of which were significantly ameliorated by EGCG | [121] |
| Mouse podocytes | EGCG | Cells were exposed to different conditions of reagents containing varying concentrations of glucose and EGCG for 24, 48 or 72 h. | Cell viability, injury, and apoptosis. | EGCG promotes podocyte proliferation and attenuates high glucose-induced podocyte injury, reducing podocyte apoptosis induced by high glucose | [244] |
| The mouse hippocampal neuronal cell line HT-22 | Eight tea catechin derivatives including EGCG | Cells were exposed to the indicated catechin derivative compounds for 3 h and H2O2 for 45 min | Cell viability, antioxidant properties | EGCG is the most effective polyphenol against H2O2-induced HT22 cell stress and exhibits a strong ability to reduce ROS production and radical scavenging | [223] |
| Wild type (WT) and inbred heterozygous β-globin knockout (BKO, muβ+/−) mice | Green tea extract (GTE) contained 24% EGCG | BKO mice were fed with a 0.2% (w/w) TMH-ferrocene supplemented diet (Fe diet) for 3 months and GTE (50 mg EGCG equivalent) for daily oral administration | Tissue iron concentration (TIC), tissue & plasma MDA (one of the lipid-peroxidation products) concentrations | GTE significantly reduced the plasma NTBI of iron-loaded BKO (p < 0.05) and diminished the increase of MDA | [245] |
| Young (3–4 months old; 15 ± 20 g) and aged (above 24 months; 420 ± 20 g) male albino rats of Wistar strain | EGCG | Rats were administered EGCG (2 mg/kg body weight/day) dissolved in saline through oral gavage for a period of 30 days | Superoxide dismutase (SOD), the activity of catalase, the level of ascorbic acid, estimation of lipid peroxidation (LPO) | EGCG supplementation resulted in the increment of the nonenzymic antioxidant status to an appreciable extent and improved the lipid peroxidation status to a considerable extent | [139] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, B.; Zhang, W.; Lin, C.; Zhang, L. A Comprehensive Review on Beneficial Effects of Catechins on Secondary Mitochondrial Diseases. Int. J. Mol. Sci. 2022, 23, 11569. https://doi.org/10.3390/ijms231911569
Chen B, Zhang W, Lin C, Zhang L. A Comprehensive Review on Beneficial Effects of Catechins on Secondary Mitochondrial Diseases. International Journal of Molecular Sciences. 2022; 23(19):11569. https://doi.org/10.3390/ijms231911569
Chicago/Turabian StyleChen, Baoyi, Wenting Zhang, Chuyuan Lin, and Lingyun Zhang. 2022. "A Comprehensive Review on Beneficial Effects of Catechins on Secondary Mitochondrial Diseases" International Journal of Molecular Sciences 23, no. 19: 11569. https://doi.org/10.3390/ijms231911569
APA StyleChen, B., Zhang, W., Lin, C., & Zhang, L. (2022). A Comprehensive Review on Beneficial Effects of Catechins on Secondary Mitochondrial Diseases. International Journal of Molecular Sciences, 23(19), 11569. https://doi.org/10.3390/ijms231911569