
Therapeutic Advances in Immunotherapies for Hematological Malignancies


Abstract
1. Introduction
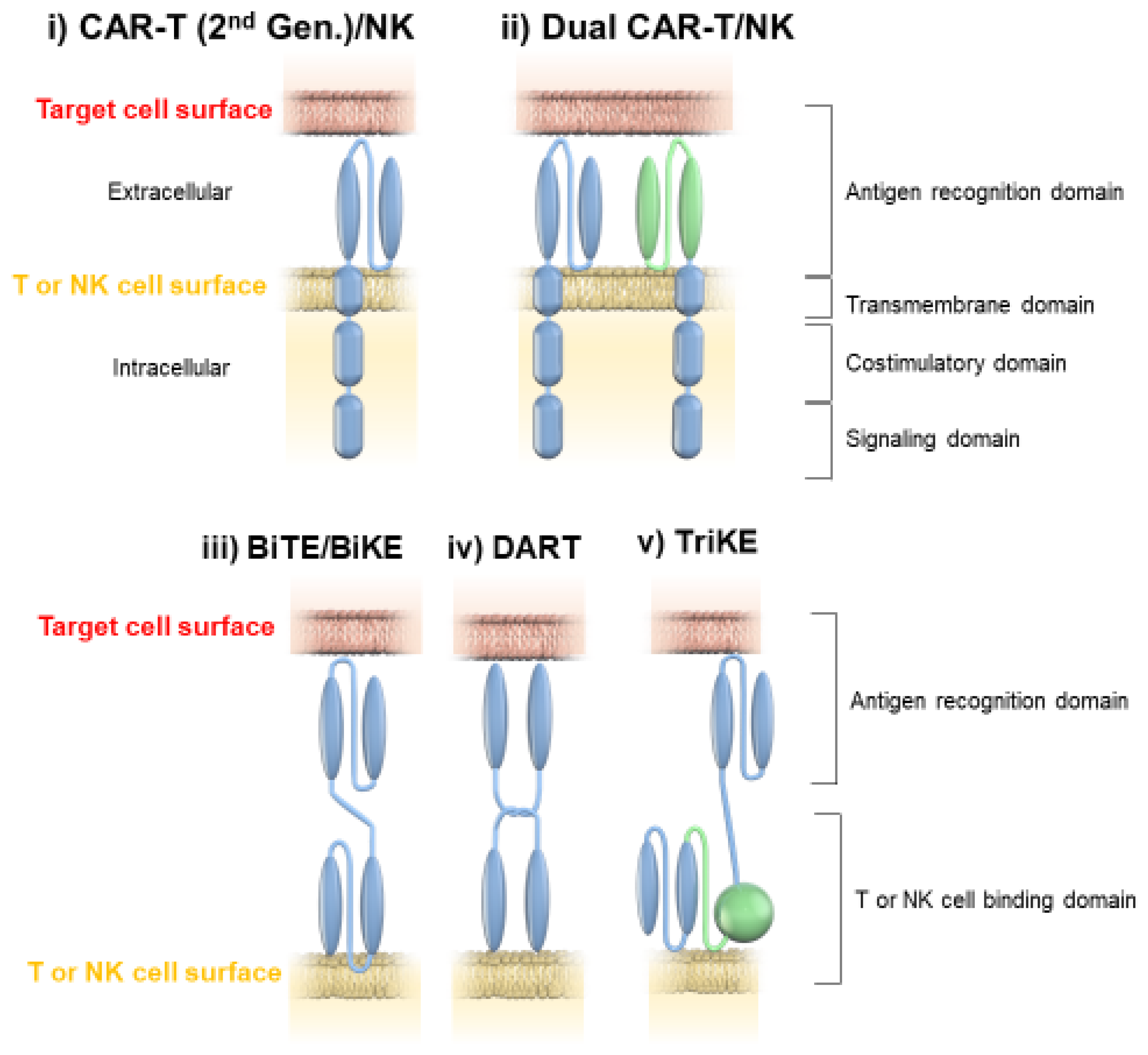
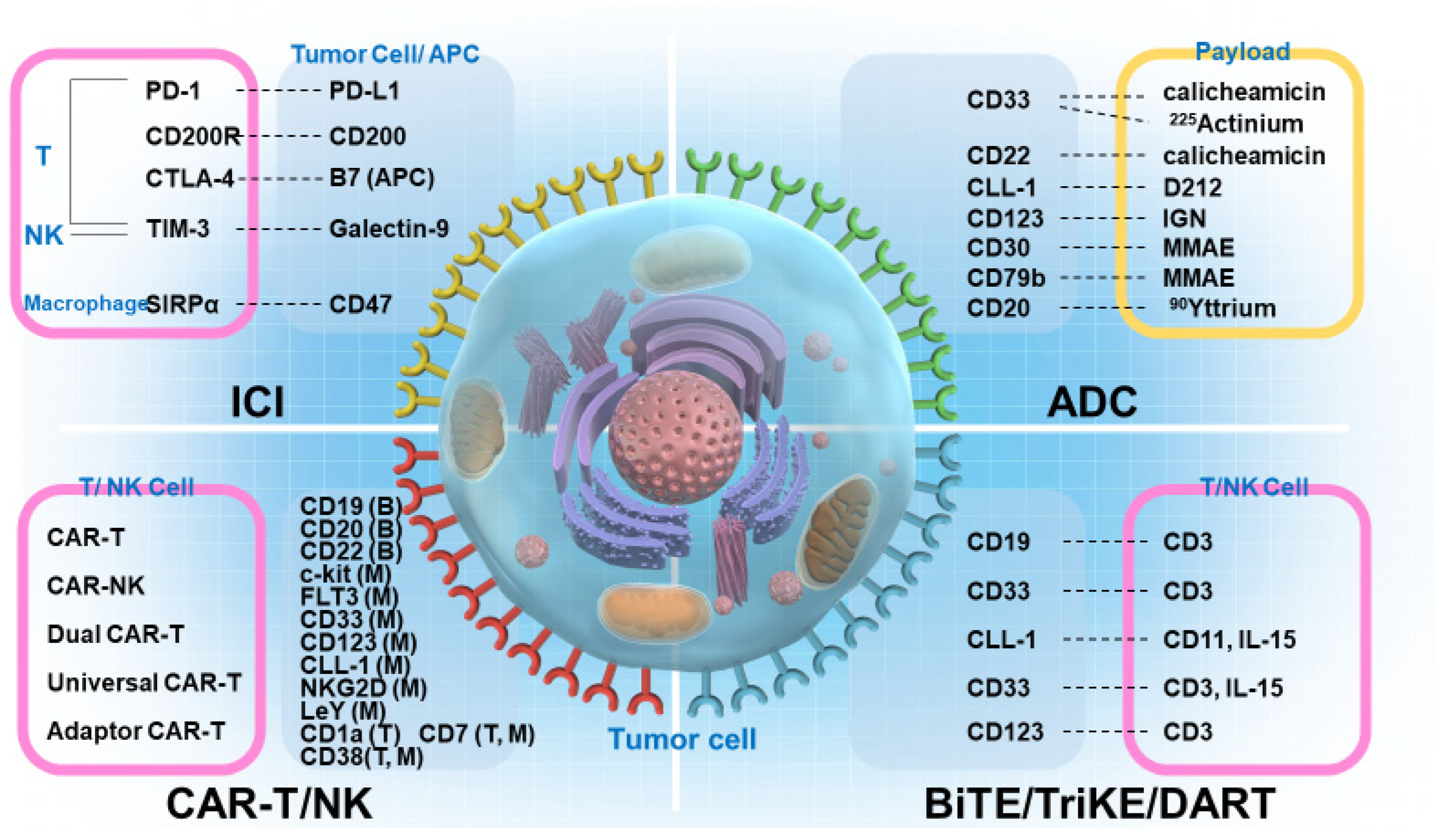
2. BiTEs, BiKEs, Checkpoint Inhibitory T-Cell–Engaging Antibodies, TriKEs, and DART
2.1. Development of BiTEs
2.2. Characteristics of BiTE, BiKE, TriKE, and DART
2.3. CD19/CD3 BiTEs
2.4. CD20/CD3 BiTEs
2.5. BiTE for Acute Myeloid Leukemia
3. CAR-T
3.1. Development of CAR-T
3.2. Dual-Targeted CAR-T Therapy
3.3. Universal CAR-T Cells
3.4. CAR-T Therapy for AML
3.5. CAR-T Therapy for T-Cell Acute Lymphoblastic Leukemia
3.6. Adaptor CAR-T Therapy
3.7. PD-1–Blocking CAR-T Therapy
4. ICIs
4.1. Representative ICI Molecules
4.2. PD-1 and CTLA-4
4.3. T-Cell Immunoglobulin and Mucin Domain-3

{kind=link}
{kind=link}
| Agents | Target | Author | Year | Phase | Objects | Cases | Survival Rate (%) | CR/CRi (%) | Median Survival (Months) | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| Pembrolizumab + decitabine | PD-1 | Lindblad et al. | 2018 | I/II | rrAML | 10 | 50 | 20 | 10 | [155] |
| Nivolumab + azaticidine | PD-1 | Daver et al. | 2018 | PhII | rrAML | 70 | 77 | 21 | 6.3 | [154] |
| Nivolumab + azaticidine +ipilimimab | 20 | NA | 36 | NR | ||||||
| Nivolumab + cytarabine + idarubicin | PD-1 | Assi et al. | 2018 | PhII | AML, hrMDS | 44 | NA | NA | 18.5 | [156] |
| MBG453 + decitabine | TIM-3 | Borate et al. | 2019 | PhIb | AML, hrMDS | 31 | 35 | 23 | 2.1-17.9 | [172] |
| Nivolumab + cytarabine + idarubicin | PD-1 | Ravandi et al. | 2019 | PhII | AML, hrMDS | 44 | 55 | 78 | 18.5 | [157] |
| Avelumab + decitabine | PD-L1 | Zheng et al. | 2021 | PhI | AML | 7 | NA | NA | 3.2 | [158] |
4.4. Lymphocyte Activation Gene 3
4.5. CD47
5. ADCs
6. Other Emerging Immunotherapies
7. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jabbour, E.; Faderl, S.; Sasaki, K.; Kadia, T.; Daver, N.; Pemmaraju, N.; Patel, K.; Khoury, J.D.; Bueso-Ramos, C.; Bohannan, Z.; et al. Phase 2 study of low-dose clofarabine plus cytarabine for patients with higher-risk myelodysplastic syndrome who have relapsed or are refractory to hypomethylating agents. Cancer 2017, 123, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Jabbour, E.; Short, N.J.; Montalban-Bravo, G.; Huang, X.; Bueso-Ramos, C.; Qiao, W.; Yang, H.; Zhao, C.; Kadia, T.; Borthakur, G.; et al. Randomized phase 2 study of low-dose decitabine vs low-dose azacitidine in lower-risk MDS and MDS/MPN. Blood 2017, 130, 1514–1522. [Google Scholar] [CrossRef] [PubMed]
- Morita, K.; Jain, N.; Kantarjian, H.; Takahashi, K.; Fang, H.; Konopleva, M.; El Hussein, S.; Wang, F.; Short, N.J.; Maiti, A.; et al. Outcome of T-cell acute lymphoblastic leukemia/lymphoma: Focus on near-ETP phenotype and differential impact of nelarabine. Am. J. Hematol. 2021, 96, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Kantarjian, H.; Sasaki, K.; Marx, K.; Jain, N.; Savoy, J.M.; DiPippo, A.; Jammal, N.; Bravo, G.M.; Kadia, T.; et al. Intrathecal prophylaxis with 12 versus 8 administrations reduces the incidence of central nervous system relapse in patients with newly diagnosed Philadelphia chromosome positive acute lymphoblastic leukemia. Am. J. Hematol. 2022. Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Jabbour, E.; Montalban-Bravo, G.; Darbaniyan, F.; Do, K.-A.; Class, C.; Short, N.J.; Kanagal-Shamana, R.; Kadia, T.; Borthakur, G.; et al. Low-Dose Decitabine versus Low-Dose Azacitidine in Lower-Risk MDS. NEJM Evid. 2022, 1, EVIDoa2200034. [Google Scholar] [CrossRef]
- Sasaki, K.; Jabbour, E.J.; Ravandi, F.; Short, N.J.; Thomas, D.A.; Garcia-Manero, G.; Daver, N.G.; Kadia, T.M.; Konopleva, M.Y.; Jain, N.; et al. Hyper-CVAD plus ponatinib versus hyper-CVAD plus dasatinib as frontline therapy for patients with Philadelphia chromosome-positive acute lymphoblastic leukemia: A propensity score analysis. Cancer 2016, 122, 3650–3656. [Google Scholar] [CrossRef]
- Sasaki, K.; Kantarjian, H.; Wierda, W.; Ravandi-Kashani, F.; Jorgensen, J.; Wang, S.A.; Khoury, J.; Daver, N.; Burger, J.; Di Nardo, C.D.; et al. Phase 2 study of hyper-CMAD with liposomal vincristine for patients with newly diagnosed acute lymphoblastic leukemia. Am. J. Hematol. 2020, 95, 734–739. [Google Scholar] [CrossRef]
- Sasaki, K.; Kantarjian, H.M.; Morita, K.; Short, N.J.; Konopleva, M.; Jain, N.; Ravandi, F.; Garcia-Manero, G.; Wang, S.; Khoury, J.D.; et al. Hyper-CVAD plus ofatumumab versus hyper-CVAD plus rituximab as frontline therapy in adults with Philadelphia chromosome-negative acute lymphoblastic leukemia: A propensity score analysis. Cancer 2021, 127, 3381–3389. [Google Scholar] [CrossRef]
- Bannon, S.A.; Routbort, M.J.; Montalban-Bravo, G.; Mehta, R.S.; Jelloul, F.Z.; Takahashi, K.; Daver, N.; Oran, B.; Pemmaraju, N.; Borthakur, G.; et al. Next-Generation Sequencing of DDX41 in Myeloid Neoplasms Leads to Increased Detection of Germline Alterations. Front. Oncol. 2020, 10, 582213. [Google Scholar] [CrossRef]
- Kanagal-Shamanna, R.; Montalban-Bravo, G.; Sasaki, K.; Darbaniyan, F.; Jabbour, E.; Bueso-Ramos, C.; Wei, Y.; Chien, K.; Kadia, T.; Ravandi, F.; et al. Only SF3B1 mutation involving K700E independently predicts overall survival in myelodysplastic syndromes. Cancer 2021, 127, 3552–3565. [Google Scholar] [CrossRef]
- Montalban-Bravo, G.; Kanagal-Shamanna, R.; Class, C.A.; Sasaki, K.; Ravandi, F.; Cortes, J.E.; Daver, N.; Takahashi, K.; Short, N.J.; DiNardo, C.D.; et al. Outcomes of acute myeloid leukemia with myelodysplasia related changes depend on diagnostic criteria and therapy. Am. J. Hematol. 2020, 95, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Montalban-Bravo, G.; Kanagal-Shamanna, R.; Sasaki, K.; Patel, K.; Ganan-Gomez, I.; Jabbour, E.; Kadia, T.; Ravandi, F.; DiNardo, C.; Borthakur, G.; et al. NPM1 mutations define a specific subgroup of MDS and MDS/MPN patients with favorable outcomes with intensive chemotherapy. Blood Adv. 2019, 3, 922–933. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Kanagal-Shamanna, R.; Montalban-Bravo, G.; Assi, R.; Jabbour, E.; Ravandi, F.; Kadia, T.; Pierce, S.; Takahashi, K.; Nogueras Gonzalez, G.; et al. Impact of the variant allele frequency of ASXL1, DNMT3A, JAK2, TET2, TP53, and NPM1 on the outcomes of patients with newly diagnosed acute myeloid leukemia. Cancer 2020, 126, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, Y.; Kantarjian, H.M.; Short, N.J.; Wang, F.; Furudate, K.; Uryu, H.; Garris, R.; Jain, N.; Sasaki, K.; Ravandi, F.; et al. Genetic correlates in patients with Philadelphia chromosome-positive acute lymphoblastic leukemia treated with Hyper-CVAD plus dasatinib or ponatinib. Leukemia 2022, 36, 1253–1260. [Google Scholar] [CrossRef]
- Chien, K.S.; Class, C.A.; Montalban-Bravo, G.; Wei, Y.; Sasaki, K.; Naqvi, K.; Ganan-Gomez, I.; Yang, H.; Soltysiak, K.A.; Kanagal-Shamanna, R.; et al. LILRB4 expression in chronic myelomonocytic leukemia and myelodysplastic syndrome based on response to hypomethylating agents. Leuk. Lymphoma 2020, 61, 1493–1499. [Google Scholar] [CrossRef]
- Montalban-Bravo, G.; Class, C.A.; Ganan-Gomez, I.; Kanagal-Shamanna, R.; Sasaki, K.; Richard-Carpentier, G.; Naqvi, K.; Wei, Y.; Yang, H.; Soltysiak, K.A.; et al. Transcriptomic analysis implicates necroptosis in disease progression and prognosis in myelodysplastic syndromes. Leukemia 2020, 34, 872–881. [Google Scholar] [CrossRef]
- Montalban-Bravo, G.; Kanagal-Shamanna, R.; Darbaniyan, F.; Siddiqui, M.T.; Sasaki, K.; Wei, Y.; Yang, H.; Chien, K.S.; Naqvi, K.; Jabbour, E.; et al. Clinical, genomic, and transcriptomic differences between myelodysplastic syndrome/myeloproliferative neoplasm with ring sideroblasts and thrombocytosis (MDS/MPN-RS-T) and myelodysplastic syndrome with ring sideroblasts (MDS-RS). Am. J. Hematol. 2021, 96, E246–E249. [Google Scholar] [CrossRef]
- Sakhdari, A.; Class, C.; Montalban-Bravo, G.; Sasaki, K.; Bueso-Ramos, C.E.; Patel, K.P.; Routbort, M.J.; Loghavi, S.; Ok, C.Y.; Quesada, A.; et al. Immunohistochemical loss of enhancer of Zeste Homolog 2 (EZH2) protein expression correlates with EZH2 alterations and portends a worse outcome in myelodysplastic syndromes. Mod. Pathol. 2022, 35, 1212–1219. [Google Scholar] [CrossRef]
- Short, N.J.; Kantarjian, H.; Kanagal-Shamanna, R.; Sasaki, K.; Ravandi, F.; Cortes, J.; Konopleva, M.; Issa, G.C.; Kornblau, S.M.; Garcia-Manero, G.; et al. Ultra-accurate Duplex Sequencing for the assessment of pretreatment ABL1 kinase domain mutations in Ph+ ALL. Blood Cancer J. 2020, 10, 61. [Google Scholar] [CrossRef]
- Yang, H.; Garcia-Manero, G.; Sasaki, K.; Montalban-Bravo, G.; Tang, Z.; Wei, Y.; Kadia, T.; Chien, K.; Rush, D.; Nguyen, H.; et al. High-resolution structural variant profiling of myelodysplastic syndromes by optical genome mapping uncovers cryptic aberrations of prognostic and therapeutic significance. Leukemia 2022, 36, 2306–2316. [Google Scholar] [CrossRef]
- Jabbour, E.; Short, N.J.; Jorgensen, J.L.; Yilmaz, M.; Ravandi, F.; Wang, S.A.; Thomas, D.A.; Khoury, J.; Champlin, R.E.; Khouri, I.; et al. Differential impact of minimal residual disease negativity according to the salvage status in patients with relapsed/refractory B-cell acute lymphoblastic leukemia. Cancer 2017, 123, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Short, N.J.; Kantarjian, H.M.; Sasaki, K.; Cortes, J.E.; Ravandi, F.; Thomas, D.A.; Garcia-Manero, G.; Khouri, I.; Kebriaei, P.; Champlin, R.E.; et al. Prognostic significance of day 14 bone marrow evaluation in adults with Philadelphia chromosome-negative acute lymphoblastic leukemia. Cancer 2016, 122, 3812–3820. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.; Kantarjian, H.; Wang, X.; Khoury, J.D.; Ravandi, F.; Jorgensen, J.; Short, N.J.; Loghavi, S.; Cortes, J.; Garcia-Manero, G.; et al. The early achievement of measurable residual disease negativity in the treatment of adults with Philadelphia-negative B-cell acute lymphoblastic leukemia is a strong predictor for survival. Am. J. Hematol. 2020, 95, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Ravandi, F.; Kadia, T.; DiNardo, C.; Borthakur, G.; Short, N.; Jain, N.; Daver, N.; Jabbour, E.; Garcia-Manero, G.; et al. Prediction of survival with intensive chemotherapy in acute myeloid leukemia. Am. J. Hematol. 2022, 97, 865–876. [Google Scholar] [CrossRef]
- Tefferi, A.; Gangat, N.; Al-Kali, A.; Alkhateeb, H.; Shah, M.; Patnaik, M.S.; Elliott, M.A.; Hogan, W.J.; Litzow, M.R.; Hook, C.C.; et al. A dynamic 3-factor survival model for acute myeloid leukemia that accounts for response to induction chemotherapy. Am. J. Hematol. 2022, 97, 1127–1134. [Google Scholar] [CrossRef]
- Dalle, I.A.; Paranal, R.; Zarka, J.; Paul, S.; Sasaki, K.; Li, W.; Ning, J.; Short, N.J.; Ohanian, M.; Cortes, J.E.; et al. Impact of luteinizing hormone suppression on hematopoietic recovery after intensive chemotherapy in patients with leukemia. Haematologica 2021, 106, 1097–1105. [Google Scholar] [CrossRef]
- Sasaki, K.; Jabbour, E.; Short, N.J.; Jain, N.; Ravandi, F.; Pui, C.H.; Kantarjian, H. Acute lymphoblastic leukemia: A population-based study of outcome in the United States based on the surveillance, epidemiology, and end results (SEER) database, 1980-2017. Am. J. Hematol. 2021, 96, 650–658. [Google Scholar] [CrossRef]
- Sasaki, K.; Ravandi, F.; Kadia, T.M.; DiNardo, C.D.; Short, N.J.; Borthakur, G.; Jabbour, E.; Kantarjian, H.M. De novo acute myeloid leukemia: A population-based study of outcome in the United States based on the Surveillance, Epidemiology, and End Results (SEER) database, 1980 to 2017. Cancer 2021, 127, 2049–2061. [Google Scholar] [CrossRef]
- Short, N.J.; Jabbour, E.; Naqvi, K.; Patel, A.; Ning, J.; Sasaki, K.; Nogueras-Gonzalez, G.M.; Bose, P.; Kornblau, S.M.; Takahashi, K.; et al. A phase II study of omacetaxine mepesuccinate for patients with higher-risk myelodysplastic syndrome and chronic myelomonocytic leukemia after failure of hypomethylating agents. Am. J. Hematol. 2019, 94, 74–79. [Google Scholar] [CrossRef]
- Morita, K.; Kantarjian, H.M.; Sasaki, K.; Issa, G.C.; Jain, N.; Konopleva, M.; Short, N.J.; Takahashi, K.; DiNardo, C.D.; Kadia, T.M.; et al. Outcome of patients with chronic myeloid leukemia in lymphoid blastic phase and Philadelphia chromosome-positive acute lymphoblastic leukemia treated with hyper-CVAD and dasatinib. Cancer 2021, 127, 2641–2647. [Google Scholar] [CrossRef]
- Sasaki, K.; Kantarjian, H.M.; Kadia, T.; Patel, K.; Loghavi, S.; Garcia-Manero, G.; Jabbour, E.J.; DiNardo, C.; Pemmaraju, N.; Daver, N.; et al. Sorafenib plus intensive chemotherapy improves survival in patients with newly diagnosed, FLT3-internal tandem duplication mutation-positive acute myeloid leukemia. Cancer 2019, 125, 3755–3766. [Google Scholar] [CrossRef] [PubMed]
- Short, N.J.; Kantarjian, H.; Ravandi, F.; Huang, X.; Xiao, L.; Garcia-Manero, G.; Plunkett, W.; Gandhi, V.; Sasaki, K.; Pemmaraju, N.; et al. A phase I/II randomized trial of clofarabine or fludarabine added to idarubicin and cytarabine for adults with relapsed or refractory acute myeloid leukemia. Leuk. Lymphoma 2018, 59, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Jabbour, E.; Short, N.J.; Ravandi, F.; Huang, X.; Xiao, L.; Garcia-Manero, G.; Plunkett, W.; Gandhi, V.; Sasaki, K.; Pemmaraju, N.; et al. A randomized phase 2 study of idarubicin and cytarabine with clofarabine or fludarabine in patients with newly diagnosed acute myeloid leukemia. Cancer 2017, 123, 4430–4439. [Google Scholar] [CrossRef]
- Sasaki, K.; Jabbour, E.; Cortes, J.; Kadia, T.; Garcia-Manero, G.; Borthakur, G.; Jain, P.; Pierce, S.; Daver, N.; Takahashi, K.; et al. Outcome of Patients with Therapy-Related Acute Myeloid Leukemia with or Without a History of Myelodysplasia. Clin. Lymphoma Myeloma Leuk. 2016, 16, 616–624. [Google Scholar] [CrossRef]
- Maiti, A.; Qiao, W.; Sasaki, K.; Ravandi, F.; Kadia, T.M.; Jabbour, E.J.; Daver, N.G.; Borthakur, G.; Garcia-Manero, G.; Pierce, S.A.; et al. Venetoclax with decitabine vs intensive chemotherapy in acute myeloid leukemia: A propensity score matched analysis stratified by risk of treatment-related mortality. Am. J. Hematol. 2021, 96, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Rausch, C.R.; DiNardo, C.D.; Maiti, A.; Jammal, N.J.; Kadia, T.M.; Marx, K.R.; Borthakur, G.; Savoy, J.M.; Pemmaraju, N.; DiPippo, A.J.; et al. Duration of cytopenias with concomitant venetoclax and azole antifungals in acute myeloid leukemia. Cancer 2021, 127, 2489–2499. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Lachowiez, C.A.; Takahashi, K.; Loghavi, S.; Xiao, L.; Kadia, T.; Daver, N.; Adeoti, M.; Short, N.J.; Sasaki, K.; et al. Venetoclax Combined With FLAG-IDA Induction and Consolidation in Newly Diagnosed and Relapsed or Refractory Acute Myeloid Leukemia. J. Clin. Oncol. 2021, 39, 2768–2778. [Google Scholar] [CrossRef]
- Venugopal, S.; Shoukier, M.; Konopleva, M.; Dinardo, C.D.; Ravandi, F.; Short, N.J.; Andreeff, M.; Borthakur, G.; Daver, N.; Pemmaraju, N.; et al. Outcomes in patients with newly diagnosed TP53-mutated acute myeloid leukemia with or without venetoclax-based therapy. Cancer 2021, 127, 3541–3551. [Google Scholar] [CrossRef]
- Reville, P.K.; Sasaki, K.; Kantarjian, H.M.; Daver, N.G.; Yilmaz, M.; Dinardo, C.D.; Short, N.J.; Borthakur, G.; Pemmaraju, N.; Mehta, R.S.; et al. Improved outcomes among newly diagnosed patients with FMS-like tyrosine kinase 3 internal tandem duplication mutated acute myeloid leukemia treated with contemporary therapy: Revisiting the European LeukemiaNet adverse risk classification. Am. J. Hematol. 2022, 97, 329–337. [Google Scholar] [CrossRef]
- Yilmaz, M.; Kantarjian, H.; Short, N.J.; Reville, P.; Konopleva, M.; Kadia, T.; DiNardo, C.; Borthakur, G.; Pemmaraju, N.; Maiti, A.; et al. Hypomethylating agent and venetoclax with FLT3 inhibitor “triplet” therapy in older/unfit patients with FLT3 mutated AML. Blood Cancer J. 2022, 12, 77. [Google Scholar] [CrossRef]
- Jabbour, E.; Sasaki, K.; Short, N.J.; Ravandi, F.; Huang, X.; Khoury, J.D.; Kanagal-Shamanna, R.; Jorgensen, J.; Khouri, I.F.; Kebriaei, P.; et al. Long-term follow-up of salvage therapy using a combination of inotuzumab ozogamicin and mini-hyper-CVD with or without blinatumomab in relapsed/refractory Philadelphia chromosome-negative acute lymphoblastic leukemia. Cancer 2021, 127, 2025–2038. [Google Scholar] [CrossRef] [PubMed]
- Short, N.J.; Macaron, W.; Konopleva, M.; Ravandi, F.; Jain, N.; Issa, G.C.; Kadia, T.; Sasaki, K.; Kebriaei, P.; Yilmaz, M.; et al. Dismal outcomes of patients with relapsed/refractory Philadelphia chromosome-negative B-cell acute lymphoblastic leukemia after failure of both inotuzumab ozogamicin and blinatumomab. Am. J. Hematol. 2022, 97, e201–e204. [Google Scholar] [CrossRef] [PubMed]
- Jabbour, E.; Short, N.J.; Ravandi, F.; Huang, X.; Daver, N.; DiNardo, C.D.; Konopleva, M.; Pemmaraju, N.; Wierda, W.; Garcia-Manero, G.; et al. Combination of hyper-CVAD with ponatinib as first-line therapy for patients with Philadelphia chromosome-positive acute lymphoblastic leukaemia: Long-term follow-up of a single-centre, phase 2 study. Lancet Haematol. 2018, 5, e618–e627. [Google Scholar] [CrossRef]
- Kantarjian, H.; Ravandi, F.; Short, N.J.; Huang, X.; Jain, N.; Sasaki, K.; Daver, N.; Pemmaraju, N.; Khoury, J.D.; Jorgensen, J.; et al. Inotuzumab ozogamicin in combination with low-intensity chemotherapy for older patients with Philadelphia chromosome-negative acute lymphoblastic leukaemia: A single-arm, phase 2 study. Lancet Oncol. 2018, 19, 240–248. [Google Scholar] [CrossRef]
- Jabbour, E.; Ravandi, F.; Kebriaei, P.; Huang, X.; Short, N.J.; Thomas, D.; Sasaki, K.; Rytting, M.; Jain, N.; Konopleva, M.; et al. Salvage Chemoimmunotherapy with Inotuzumab Ozogamicin Combined with Mini-Hyper-CVD for Patients with Relapsed or Refractory Philadelphia Chromosome-Negative Acute Lymphoblastic Leukemia: A Phase 2 Clinical Trial. JAMA Oncol. 2018, 4, 230–234. [Google Scholar] [CrossRef]
- Issa, G.C.; Kantarjian, H.M.; Yin, C.C.; Qiao, W.; Ravandi, F.; Thomas, D.; Short, N.J.; Sasaki, K.; Garcia-Manero, G.; Kadia, T.M.; et al. Prognostic impact of pretreatment cytogenetics in adult Philadelphia chromosome-negative acute lymphoblastic leukemia in the era of minimal residual disease. Cancer 2017, 123, 459–467. [Google Scholar] [CrossRef]
- Short, N.J.; Kantarjian, H.M.; Sasaki, K.; Ravandi, F.; Ko, H.; Cameron Yin, C.; Garcia-Manero, G.; Cortes, J.E.; Garris, R.; O’Brien, S.M.; et al. Poor outcomes associated with +der(22)t(9;22) and -9/9p in patients with Philadelphia chromosome-positive acute lymphoblastic leukemia receiving chemotherapy plus a tyrosine kinase inhibitor. Am. J. Hematol. 2017, 92, 238–243. [Google Scholar] [CrossRef]
- Kadia, T.M.; Reville, P.K.; Wang, X.; Rausch, C.R.; Borthakur, G.; Pemmaraju, N.; Daver, N.G.; DiNardo, C.D.; Sasaki, K.; Issa, G.C.; et al. Phase II Study of Venetoclax Added to Cladribine Plus Low-Dose Cytarabine Alternating with 5-Azacitidine in Older Patients with Newly Diagnosed Acute Myeloid Leukemia. J. Clin. Oncol. 2022, Jco2102823, Online ahead of print. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Venugopal, S.; Lachowiez, C.A.; Takahashi, K.; Loghavi, S.; Montalban-Bravo, G.; Wang, X.; Carraway, H.E.; Sekeres, M.A.; Sukkur, A.; et al. Targeted therapy with the mutant IDH2 inhibitor enasidenib for high-risk IDH2-mutant myelodysplastic syndrome. Blood Adv. 2022. Online ahead of print. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Lachowiez, C.A.; Takahashi, K.; Loghavi, S.; Kadia, T.; Daver, N.; Xiao, L.; Adeoti, M.; Short, N.J.; Sasaki, K.; et al. Venetoclax combined with FLAG-IDA induction and consolidation in newly diagnosed acute myeloid leukemia. Am. J. Hematol. 2022, 97, 1035–1043. [Google Scholar] [CrossRef]
- De Gast, G.C.; Haagen, I.A.; van Houten, A.A.; Klein, S.C.; Duits, A.J.; de Weger, R.A.; Vroom, T.M.; Clark, M.R.; Phillips, J.; van Dijk, A.J.; et al. CD8 T cell activation after intravenous administration of CD3 x CD19 bispecific antibody in patients with non-Hodgkin lymphoma. Cancer Immunol. Immunother. 1995, 40, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Loffler, A.; Kufer, P.; Lutterbuse, R.; Zettl, F.; Daniel, P.T.; Schwenkenbecher, J.M.; Riethmuller, G.; Dorken, B.; Bargou, R.C. A recombinant bispecific single-chain antibody, CD19 x CD3, induces rapid and high lymphoma-directed cytotoxicity by unstimulated T lymphocytes. Blood 2000, 95, 2098–2103. [Google Scholar] [CrossRef] [PubMed]
- Nagorsen, D.; Kufer, P.; Baeuerle, P.A.; Bargou, R. Blinatumomab: A historical perspective. Pharmacol. Ther. 2012, 136, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Merchant, A.M.; Zhu, Z.; Yuan, J.Q.; Goddard, A.; Adams, C.W.; Presta, L.G.; Carter, P. An efficient route to human bispecific IgG. Nat. Biotechnol. 1998, 16, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, W.; Regula, J.T.; Bahner, M.; Schanzer, J.; Croasdale, R.; Durr, H.; Gassner, C.; Georges, G.; Kettenberger, H.; Imhof-Jung, S.; et al. Immunoglobulin domain crossover as a generic approach for the production of bispecific IgG antibodies. Proc. Natl. Acad. Sci. USA 2011, 108, 11187–11192. [Google Scholar] [CrossRef] [PubMed]
- Wiernik, A.; Foley, B.; Zhang, B.; Verneris, M.R.; Warlick, E.; Gleason, M.K.; Ross, J.A.; Luo, X.; Weisdorf, D.J.; Walcheck, B.; et al. Targeting natural killer cells to acute myeloid leukemia in vitro with a CD16 x 33 bispecific killer cell engager and ADAM17 inhibition. Clin. Cancer Res. 2013, 19, 3844–3855. [Google Scholar] [CrossRef] [PubMed]
- Vallera, D.A.; Felices, M.; McElmurry, R.; McCullar, V.; Zhou, X.; Schmohl, J.U.; Zhang, B.; Lenvik, A.J.; Panoskaltsis-Mortari, A.; Verneris, M.R.; et al. IL15 Trispecific Killer Engagers (TriKE) Make Natural Killer Cells Specific to CD33+ Targets While Also Inducing Persistence, In Vivo Expansion, and Enhanced Function. Clin. Cancer Res. 2016, 22, 3440–3450. [Google Scholar] [CrossRef]
- Bock, A.M.; Nowakowski, G.S.; Wang, Y. Bispecific Antibodies for Non-Hodgkin Lymphoma Treatment. Curr. Treat. Options Oncol. 2022, 23, 155–170. [Google Scholar] [CrossRef]
- Kantarjian, H.; Stein, A.; Gokbuget, N.; Fielding, A.K.; Schuh, A.C.; Ribera, J.M.; Wei, A.; Dombret, H.; Foa, R.; Bassan, R.; et al. Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2017, 376, 836–847. [Google Scholar] [CrossRef]
- Gökbuget, N.; Dombret, H.; Bonifacio, M.; Reichle, A.; Graux, C.; Faul, C.; Diedrich, H.; Topp, M.S.; Brüggemann, M.; Horst, H.A.; et al. Blinatumomab for minimal residual disease in adults with B-cell precursor acute lymphoblastic leukemia. Blood 2018, 131, 1522–1531. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; Jain, N.; Garcia-Manero, G.; Welch, M.A.; Ravandi, F.; Wierda, W.G.; Jabbour, E.J. The cure of leukemia through the optimist’s prism. Cancer 2022, 128, 240–259. [Google Scholar] [CrossRef] [PubMed]
- Jabbour, E.; Sasaki, K.; Ravandi, F.; Huang, X.; Short, N.J.; Khouri, M.; Kebriaei, P.; Burger, J.; Khoury, J.; Jorgensen, J.; et al. Chemoimmunotherapy with inotuzumab ozogamicin combined with mini-hyper-CVD, with or without blinatumomab, is highly effective in patients with Philadelphia chromosome-negative acute lymphoblastic leukemia in first salvage. Cancer 2018, 124, 4044–4055. [Google Scholar] [CrossRef] [PubMed]
- Jabbour, E.J.; Sasaki, K.; Ravandi, F.; Short, N.J.; Garcia-Manero, G.; Daver, N.; Kadia, T.; Konopleva, M.; Jain, N.; Cortes, J.; et al. Inotuzumab ozogamicin in combination with low-intensity chemotherapy (mini-HCVD) with or without blinatumomab versus standard intensive chemotherapy (HCVAD) as frontline therapy for older patients with Philadelphia chromosome-negative acute lymphoblastic leukemia: A propensity score analysis. Cancer 2019, 125, 2579–2586. [Google Scholar] [CrossRef]
- Reusch, U.; Duell, J.; Ellwanger, K.; Herbrecht, C.; Knackmuss, S.H.; Fucek, I.; Eser, M.; McAleese, F.; Molkenthin, V.; Gall, F.L.; et al. A tetravalent bispecific TandAb (CD19/CD3), AFM11, efficiently recruits T cells for the potent lysis of CD19(+) tumor cells. MAbs 2015, 7, 584–604. [Google Scholar] [CrossRef]
- Bannerji, R.; Arnason, J.E.; Advani, R.H.; Brown, J.R.; Allan, J.N.; Ansell, S.M.; Barnes, J.A.; O’Brien, S.M.; Chavez, J.C.; Duell, J.; et al. Odronextamab, a human CD20xCD3 bispecific antibody in patients with CD20-positive B-cell malignancies (ELM-1): Results from the relapsed or refractory non-Hodgkin lymphoma cohort in a single-arm, multicentre, phase 1 trial. Lancet Haematol. 2022, 9, e327–e339. [Google Scholar] [CrossRef]
- Budde, L.E.; Assouline, S.; Sehn, L.H.; Schuster, S.J.; Yoon, S.S.; Yoon, D.H.; Matasar, M.J.; Bosch, F.; Kim, W.S.; Nastoupil, L.J.; et al. Single-Agent Mosunetuzumab Shows Durable Complete Responses in Patients with Relapsed or Refractory B-Cell Lymphomas: Phase I Dose-Escalation Study. J. Clin. Oncol. 2022, 40, 481–491. [Google Scholar] [CrossRef]
- Tavarozzi, R.; Manzato, E. The Role of Bispecific Antibodies in Non-Hodgkin’s Lymphoma: From Structure to Prospective Clinical Use. Antibodies 2022, 11, 16. [Google Scholar] [CrossRef]
- Morschhauser, F.; Bishton, M.; Eyre, T.A.; Bachy, E.; Cartron, G.; Ysebaert, L.; Bobillo, S.; Gutierrez, N.C.; Budde, L.E.; Fox, C.P.; et al. Mosunetuzumab in Combination with Lenalidomide Has a Manageable Safety Profile and Encouraging Activity in Patients with Relapsed/Refractory Follicular Lymphoma: Initial Results from a Phase Ib Study. Blood 2021, 138, 129. [Google Scholar] [CrossRef]
- Nastoupil, L.J.; Morschhauser, F.; Scholz, C.W.; Bishton, M.; Yoon, S.-S.; Giri, P.; Wei, M.C.; Knapp, A.; Li, C.-C.; Bottos, A.; et al. CELESTIMO: A phase III trial evaluating the efficacy and safety of mosunetuzumab plus lenalidomide versus rituximab plus lenalidomide in patients with relapsed or refractory follicular lymphoma who have received ≥ 1 line of systemic therapy. J. Clin. Oncol. 2022, 40, TPS7588. [Google Scholar] [CrossRef]
- Hutchings, M.; Mous, R.; Clausen, M.R.; Johnson, P.; Linton, K.M.; Chamuleau, M.E.D.; Lewis, D.J.; Sureda Balari, A.; Cunningham, D.; Oliveri, R.S.; et al. Dose escalation of subcutaneous epcoritamab in patients with relapsed or refractory B-cell non-Hodgkin lymphoma: An open-label, phase 1/2 study. Lancet 2021, 398, 1157–1169. [Google Scholar] [CrossRef]
- Hutchings, M.; Morschhauser, F.; Iacoboni, G.; Carlo-Stella, C.; Offner, F.C.; Sureda, A.; Salles, G.; Martinez-Lopez, J.; Crump, M.; Thomas, D.N.; et al. Glofitamab, a Novel, Bivalent CD20-Targeting T-Cell-Engaging Bispecific Antibody, Induces Durable Complete Remissions in Relapsed or Refractory B-Cell Lymphoma: A Phase I Trial. J. Clin. Oncol. 2021, 39, 1959–1970. [Google Scholar] [CrossRef] [PubMed]
- Durben, M.; Schmiedel, D.; Hofmann, M.; Vogt, F.; Nubling, T.; Pyz, E.; Buhring, H.J.; Rammensee, H.G.; Salih, H.R.; Grosse-Hovest, L.; et al. Characterization of a bispecific FLT3 X CD3 antibody in an improved, recombinant format for the treatment of leukemia. Mol. Ther. 2015, 23, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Jitschin, R.; Saul, D.; Braun, M.; Tohumeken, S.; Völkl, S.; Kischel, R.; Lutteropp, M.; Dos Santos, C.; Mackensen, A.; Mougiakakos, D. CD33/CD3-bispecific T-cell engaging (BiTE®) antibody construct targets monocytic AML myeloid-derived suppressor cells. J. Immunother. Cancer 2018, 6, 116. [Google Scholar] [CrossRef]
- Ravandi, F.; Walter, R.B.; Subklewe, M.; Buecklein, V.; Jongen-Lavrencic, M.; Paschka, P.; Ossenkoppele, G.J.; Kantarjian, H.M.; Hindoyan, A.; Agarwal, S.K.; et al. Updated results from phase I dose-escalation study of AMG 330, a bispecific T-cell engager molecule, in patients with relapsed/refractory acute myeloid leukemia (R/R AML). J. Clin. Oncol. 2020, 38, 7508. [Google Scholar] [CrossRef]
- Roboz, G.J.; Chun, P.; Guenot, J.; Gojo, I.; Oehler, V.G.; Long, M.; Altman, J.K.; Cortes, J.E.; Westervelt, P. Phase 1 First-in-Human Trial of AMV564, a Bivalent Bispecific (2:2) CD33/CD3 T-Cell Engager, in Patients with Relapsed/Refractory Acute Myeloid Leukemia (AML). Blood 2019, 134, 834. [Google Scholar] [CrossRef]
- Herrmann, M.C.K.; Deiser, K.; Brauchle, B.; Marcinek, A.; Ogrinc Wagner, A.; Rataj, F.; Mocikat, R.; Metzeler, K.H.; Spiekermann, K.; Kobold, S.; et al. Bifunctional PD-1 X aCD3 X aCD33 fusion protein reverses adaptive immune escape in acute myeloid leukemia. Blood 2018, 132, 2484–2494. [Google Scholar] [CrossRef] [PubMed]
- Ehninger, A.; Kramer, M.; Rollig, C.; Thiede, C.; Bornhauser, M.; von Bonin, M.; Wermke, M.; Feldmann, A.; Bachmann, M.; Ehninger, G.; et al. Distribution and levels of cell surface expression of CD33 and CD123 in acute myeloid leukemia. Blood Cancer J. 2014, 4, e218. [Google Scholar] [CrossRef]
- Jordan, C.T.; Upchurch, D.; Szilvassy, S.J.; Guzman, M.L.; Howard, D.S.; Pettigrew, A.L.; Meyerrose, T.; Rossi, R.; Grimes, B.; Rizzieri, D.A.; et al. The interleukin-3 receptor alpha chain is a unique marker for human acute myelogenous leukemia stem cells. Leukemia 2000, 14, 1777–1784. [Google Scholar] [CrossRef]
- Testa, U.; Riccioni, R.; Militi, S.; Coccia, E.; Stellacci, E.; Samoggia, P.; Latagliata, R.; Mariani, G.; Rossini, A.; Battistini, A.; et al. Elevated expression of IL-3Ralpha in acute myelogenous leukemia is associated with enhanced blast proliferation, increased cellularity, and poor prognosis. Blood 2002, 100, 2980–2988. [Google Scholar] [CrossRef]
- Ravandi, F.; Bashey, A.; Stock, W.; Foran, J.M.; Mawad, R.; Egan, D.; Blum, W.; Yang, A.; Pastore, A.; Johnson, C.; et al. Complete Responses in Relapsed/Refractory Acute Myeloid Leukemia (AML) Patients on a Weekly Dosing Schedule of Vibecotamab (XmAb14045), a CD123 x CD3 T Cell-Engaging Bispecific Antibody; Initial Results of a Phase 1 Study. Blood 2020, 136, 4–5. [Google Scholar] [CrossRef]
- Uy, G.L.; Aldoss, I.; Foster, M.C.; Sayre, P.H.; Wieduwilt, M.J.; Advani, A.S.; Godwin, J.E.; Arellano, M.L.; Sweet, K.L.; Emadi, A.; et al. Flotetuzumab as salvage immunotherapy. Blood 2021, 137, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.; Doucette, K.; Norsworthy, K. Recent drug approvals for acute myeloid leukemia. J. Hematol. Oncol. 2019, 12, 100. [Google Scholar] [CrossRef] [PubMed]
- Leong, S.R.; Sukumaran, S.; Hristopoulos, M.; Totpal, K.; Stainton, S.; Lu, E.; Wong, A.; Tam, L.; Newman, R.; Vuillemenot, B.R.; et al. An anti-CD3/anti-CLL-1 bispecific antibody for the treatment of acute myeloid leukemia. Blood 2017, 129, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Arvindam, U.S.; van Hauten, P.M.M.; Schirm, D.; Schaap, N.; Hobo, W.; Blazar, B.R.; Vallera, D.A.; Dolstra, H.; Felices, M.; Miller, J.S. A trispecific killer engager molecule against CLEC12A effectively induces NK-cell mediated killing of AML cells. Leukemia 2021, 35, 1586–1596. [Google Scholar] [CrossRef]
- Stirewalt, D.L.; Radich, J.P. The role of FLT3 in haematopoietic malignancies. Nat. Rev. Cancer 2003, 3, 650–665. [Google Scholar] [CrossRef]
- Brauchle, B.; Goldstein, R.L.; Karbowski, C.M.; Henn, A.; Li, C.M.; Bücklein, V.L.; Krupka, C.; Boyle, M.C.; Koppikar, P.; Haubner, S.; et al. Characterization of a Novel FLT3 BiTE Molecule for the Treatment of Acute Myeloid Leukemia. Mol. Cancer Ther. 2020, 19, 1875–1888. [Google Scholar] [CrossRef]
- Moore, P.A.; Zhang, W.; Rainey, G.J.; Burke, S.; Li, H.; Huang, L.; Gorlatov, S.; Veri, M.C.; Aggarwal, S.; Yang, Y.; et al. Application of dual affinity retargeting molecules to achieve optimal redirected T-cell killing of B-cell lymphoma. Blood 2011, 117, 4542–4551. [Google Scholar] [CrossRef]
- Liu, L.; Lam, C.K.; Long, V.; Widjaja, L.; Yang, Y.; Li, H.; Jin, L.; Burke, S.; Gorlatov, S.; Brown, J.; et al. MGD011, A CD19 x CD3 Dual-Affinity Retargeting Bi-specific Molecule Incorporating Extended Circulating Half-life for the Treatment of B-Cell Malignancies. Clin. Cancer Res. 2017, 23, 1506–1518. [Google Scholar] [CrossRef]
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 720–724. [Google Scholar] [CrossRef]
- Moritz, D.; Wels, W.; Mattern, J.; Groner, B. Cytotoxic T lymphocytes with a grafted recognition specificity for ERBB2-expressing tumor cells. Proc. Natl. Acad. Sci. USA 1994, 91, 4318–4322. [Google Scholar] [CrossRef]
- Wang, M.; Munoz, J.; Goy, A.; Locke, F.L.; Jacobson, C.A.; Hill, B.T.; Timmerman, J.M.; Holmes, H.; Jaglowski, S.; Flinn, I.W.; et al. KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. N. Engl. J. Med. 2020, 382, 1331–1342. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Abramson, J.S.; Palomba, M.L.; Gordon, L.I.; Lunning, M.A.; Wang, M.; Arnason, J.; Mehta, A.; Purev, E.; Maloney, D.G.; Andreadis, C.; et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): A multicentre seamless design study. Lancet 2020, 396, 839–852. [Google Scholar] [CrossRef] [PubMed]
- Locke, F.L.; Ghobadi, A.; Jacobson, C.A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.O.; Lin, Y.; Braunschweig, I.; Hill, B.T.; Timmerman, J.M.; et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): A single-arm, multicentre, phase 1-2 trial. Lancet Oncol. 2019, 20, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef]
- Nie, Y.; Lu, W.; Chen, D.; Tu, H.; Guo, Z.; Zhou, X.; Li, M.; Tu, S.; Li, Y. Mechanisms underlying CD19-positive ALL relapse after anti-CD19 CAR T cell therapy and associated strategies. Biomark. Res. 2020, 8, 18. [Google Scholar] [CrossRef]
- Nahas, G.R.; Komanduri, K.V.; Pereira, D.; Goodman, M.; Jimenez, A.M.; Beitinjaneh, A.; Wang, T.P.; Lekakis, L.J. Incidence and risk factors associated with a syndrome of persistent cytopenias after CAR-T cell therapy (PCTT). Leuk. Lymphoma 2020, 61, 940–943. [Google Scholar] [CrossRef]
- Vadakekolathu, J.; Lai, C.; Reeder, S.; Church, S.E.; Hood, T.; Lourdusamy, A.; Rettig, M.P.; Aldoss, I.; Advani, A.S.; Godwin, J.; et al. TP53 abnormalities correlate with immune infiltration and associate with response to flotetuzumab immunotherapy in AML. Blood Adv. 2020, 4, 5011–5024. [Google Scholar] [CrossRef]
- Gödel, P.; Sieg, N.; Heger, J.M.; Kutsch, N.; Herling, C.; Bärmann, B.N.; Scheid, C.; Borchmann, P.; Holtick, U. Hematologic Rescue of CAR T-cell-mediated Prolonged Pancytopenia Using Autologous Peripheral Blood Hematopoietic Stem Cells in a Lymphoma Patient. Hemasphere 2021, 5, e545. [Google Scholar] [CrossRef]
- Strati, P.; Varma, A.; Adkins, S.; Nastoupil, L.J.; Westin, J.; Hagemeister, F.B.; Fowler, N.H.; Lee, H.J.; Fayad, L.E.; Samaniego, F.; et al. Hematopoietic recovery and immune reconstitution after axicabtagene ciloleucel in patients with large B-cell lymphoma. Haematologica 2021, 106, 2667–2672. [Google Scholar] [CrossRef]
- Ma, S.; Li, X.; Wang, X.; Cheng, L.; Li, Z.; Zhang, C.; Ye, Z.; Qian, Q. Current Progress in CAR-T Cell Therapy for Solid Tumors. Int. J. Biol. Sci. 2019, 15, 2548–2560. [Google Scholar] [CrossRef]
- Orlando, E.J.; Han, X.; Tribouley, C.; Wood, P.A.; Leary, R.J.; Riester, M.; Levine, J.E.; Qayed, M.; Grupp, S.A.; Boyer, M.; et al. Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat. Med. 2018, 24, 1504–1506. [Google Scholar] [CrossRef]
- Ruella, M.; Xu, J.; Barrett, D.M.; Fraietta, J.A.; Reich, T.J.; Ambrose, D.E.; Klichinsky, M.; Shestova, O.; Patel, P.R.; Kulikovskaya, I.; et al. Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic B cell. Nat. Med. 2018, 24, 1499–1503. [Google Scholar] [CrossRef]
- Cordoba, S.; Onuoha, S.; Thomas, S.; Pignataro, D.S.; Hough, R.; Ghorashian, S.; Vora, A.; Bonney, D.; Veys, P.; Rao, K.; et al. CAR T cells with dual targeting of CD19 and CD22 in pediatric and young adult patients with relapsed or refractory B cell acute lymphoblastic leukemia: A phase 1 trial. Nat. Med. 2021, 27, 1797–1805. [Google Scholar] [CrossRef]
- Borchmann, P.; Jühling, A.; Gödel, P.; Balke-Want, H.; Schmid, C.; Ayuk, F.A.; Holtkamp, S.; Preussner, L.; Zadoyan, G.; Hanssens, L.; et al. Phase I Trial of MB-CART2019.1, a Novel CD20 and CD19 Targeting Tandem Chimeric Antigen Receptor, in Patients with Relapsed or Refractory B-Cell Non-Hodgkin Lymphoma. Blood 2020, 136, 48. [Google Scholar] [CrossRef]
- Zah, E.; Lin, M.Y.; Silva-Benedict, A.; Jensen, M.C.; Chen, Y.Y. T Cells Expressing CD19/CD20 Bispecific Chimeric Antigen Receptors Prevent Antigen Escape by Malignant B Cells. Cancer Immunol. Res. 2016, 4, 498–508. [Google Scholar] [CrossRef]
- Schneider, D.; Xiong, Y.; Wu, D.; Nlle, V.; Schmitz, S.; Haso, W.; Kaiser, A.; Dropulic, B.; Orentas, R.J. A tandem CD19/CD20 CAR lentiviral vector drives on-target and off-target antigen modulation in leukemia cell lines. J. Immunother Cancer 2017, 5, 42. [Google Scholar] [CrossRef]
- Hu, Y.; Zhou, Y.; Zhang, M.; Ge, W.; Li, Y.; Yang, L.; Wei, G.; Han, L.; Wang, H.; Yu, S.; et al. CRISPR/Cas9-Engineered Universal CD19/CD22 Dual-Targeted CAR-T Cell Therapy for Relapsed/Refractory B-cell Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2021, 27, 2764–2772. [Google Scholar] [CrossRef]
- Jain, N.; Roboz, G.J.; Konopleva, M.; Liu, H.; Jabbour, E.; Poirot, C.; Schiffer-Manniou, C.; Gouble, A.; Haider, A.; Zernovak, O.; et al. Preliminary Results of Balli-01: A Phase I Study of UCART22 (allogeneic engineered T-cells expressing anti-CD22 Chimeric Antigen Receptor) in Adult Patients with Relapsed or Refractory (R/R) CD22+ B-Cell Acute Lymphoblastic Leukemia (B-ALL). Blood 2020, 136, 7–8. [Google Scholar] [CrossRef]
- Jain, N.; Roboz, G.J.; Konopleva, M.; Liu, H.; Schiller, G.J.; Jabbour, E.J.; Whitfield, D.; Haider, A.; Zernovak, O.; Frattini, M.G.; et al. Preliminary Results from the Flu/Cy/Alemtuzumab Arm of the Phase I BALLI-01 Trial of UCART22, an Anti-CD22 Allogeneic CAR-T Cell Product, in Adult Patients with Relapsed or Refractory (R/R) CD22+ B-Cell Acute Lymphoblastic Leukemia (B-ALL). Blood 2021, 138, 1746. [Google Scholar] [CrossRef]
- Perez, C.; Gruber, I.; Arber, C. Off-the-Shelf Allogeneic T Cell Therapies for Cancer: Opportunities and Challenges Using Naturally Occurring "Universal" Donor T Cells. Front. Immunol. 2020, 11, 583716. [Google Scholar] [CrossRef]
- Hofmann, S.; Schubert, M.L.; Wang, L.; He, B.; Neuber, B.; Dreger, P.; Muller-Tidow, C.; Schmitt, M. Chimeric Antigen Receptor (CAR) T Cell Therapy in Acute Myeloid Leukemia (AML). J. Clin. Med. 2019, 8, 200. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, P.; Li, Z.; He, Y.; Gan, W.; Jiang, H. Anti-CLL1 Chimeric Antigen Receptor T-Cell Therapy in Children with Relapsed/Refractory Acute Myeloid Leukemia. Clin. Cancer Res. 2021, 27, 3549–3555. [Google Scholar] [CrossRef]
- Jin, X.; Zhang, M.; Sun, R.; Lyu, H.; Xiao, X.; Zhang, X.; Li, F.; Xie, D.; Xiong, X.; Wang, J.; et al. First-in-human phase I study of CLL-1 CAR-T cells in adults with relapsed/refractory acute myeloid leukemia. J. Hematol. Oncol. 2022, 15, 88. [Google Scholar] [CrossRef]
- Lanza, F.; Castagnari, B.; Rigolin, G.; Moretti, S.; Latorraca, A.; Ferrari, L.; Bardi, A.; Castoldi, G. Flow cytometry measurement of GM-CSF receptors in acute leukemic blasts, and normal hemopoietic cells. Leukemia 1997, 11, 1700–1710. [Google Scholar] [CrossRef]
- Graf, M.; Hecht, K.; Reif, S.; Pelka-Fleischer, R.; Pfister, K.; Schmetzer, H. Expression and prognostic value of hemopoietic cytokine receptors in acute myeloid leukemia (AML): Implications for future therapeutical strategies. Eur. J. Haematol. 2004, 72, 89–106. [Google Scholar] [CrossRef]
- Nakazawa, Y.; Matsuda, K.; Kurata, T.; Sueki, A.; Tanaka, M.; Sakashita, K.; Imai, C.; Wilson, M.H.; Koike, K. Anti-proliferative effects of T cells expressing a ligand-based chimeric antigen receptor against CD116 on CD34(+) cells of juvenile myelomonocytic leukemia. J. Hematol. Oncol. 2016, 9, 27. [Google Scholar] [CrossRef]
- Hasegawa, A.; Saito, S.; Narimatsu, S.; Nakano, S.; Nagai, M.; Ohnota, H.; Inada, Y.; Morokawa, H.; Nakashima, I.; Morita, D.; et al. Mutated GM-CSF-based CAR-T cells targeting CD116/CD131 complexes exhibit enhanced anti-tumor effects against acute myeloid leukaemia. Clin. Transl. Immunol. 2021, 10, e1282. [Google Scholar] [CrossRef]
- Jetani, H.; Garcia-Cadenas, I.; Nerreter, T.; Thomas, S.; Rydzek, J.; Meijide, J.B.; Bonig, H.; Herr, W.; Sierra, J.; Einsele, H.; et al. CAR T-cells targeting FLT3 have potent activity against FLT3(-)ITD(+) AML and act synergistically with the FLT3-inhibitor crenolanib. Leukemia 2018, 32, 1168–1179. [Google Scholar] [CrossRef]
- Valverde, L.R.; Matutes, E.; Farahat, N.; Heffernan, A.; Owusu-Ankomah, K.; Morilla, R.; Catovsky, D. C-kit receptor (CD117) expression in acute leukemia. Ann. Hematol. 1996, 72, 11–15. [Google Scholar] [CrossRef]
- Wells, S.J.; Bray, R.A.; Stempora, L.L.; Farhi, D.C. CD117/CD34 expression in leukemic blasts. Am. J. Clin. Pathol. 1996, 106, 192–195. [Google Scholar] [CrossRef]
- Shima, T.; Miyamoto, T.; Kikushige, Y.; Yuda, J.; Tochigi, T.; Yoshimoto, G.; Kato, K.; Takenaka, K.; Iwasaki, H.; Mizuno, S.; et al. The ordered acquisition of Class II and Class I mutations directs formation of human t(8;21) acute myelogenous leukemia stem cell. Exp. Hematol. 2014, 42, 955–965.e5. [Google Scholar] [CrossRef]
- Myburgh, R.; Kiefer, J.D.; Russkamp, N.F.; Magnani, C.F.; Nunez, N.; Simonis, A.; Pfister, S.; Wilk, C.M.; McHugh, D.; Friemel, J.; et al. Anti-human CD117 CAR T-cells efficiently eliminate healthy and malignant CD117-expressing hematopoietic cells. Leukemia 2020, 34, 2688–2703. [Google Scholar] [CrossRef]
- Budde, L.; Song, J.Y.; Kim, Y.; Blanchard, S.; Wagner, J.; Stein, A.S.; Weng, L.; Del Real, M.; Hernandez, R.; Marcucci, E.; et al. Remissions of Acute Myeloid Leukemia and Blastic Plasmacytoid Dendritic Cell Neoplasm Following Treatment with CD123-Specific CAR T Cells: A First-in-Human Clinical Trial. Blood 2017, 130, 811. [Google Scholar] [CrossRef]
- Sugita, M.; Galetto, R.; Zong, H.; Ewing-Crystal, N.; Trujillo-Alonso, V.; Mencia-Trinchant, N.; Yip, W.; Filipe, S.; Lebuhotel, C.; Gouble, A.; et al. Allogeneic TCRαβ deficient CAR T-cells targeting CD123 in acute myeloid leukemia. Nat. Commun. 2022, 13, 2227. [Google Scholar] [CrossRef]
- Wang, Q.S.; Wang, Y.; Lv, H.Y.; Han, Q.W.; Fan, H.; Guo, B.; Wang, L.L.; Han, W.D. Treatment of CD33-directed chimeric antigen receptor-modified T cells in one patient with relapsed and refractory acute myeloid leukemia. Mol. Ther. 2015, 23, 184–191. [Google Scholar] [CrossRef]
- Tambaro, F.P.; Singh, H.; Jones, E.; Rytting, M.; Mahadeo, K.M.; Thompson, P.; Daver, N.; DiNardo, C.; Kadia, T.; Garcia-Manero, G.; et al. Autologous CD33-CAR-T cells for treatment of relapsed/refractory acute myelogenous leukemia. Leukemia 2021, 35, 3282–3286. [Google Scholar] [CrossRef]
- Tang, X.; Yang, L.; Li, Z.; Nalin, A.P.; Dai, H.; Xu, T.; Yin, J.; You, F.; Zhu, M.; Shen, W.; et al. Erratum: First-in-man clinical trial of CAR NK-92 cells: Safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am. J. Cancer Res. 2018, 8, 1899. [Google Scholar]
- Li, K.X.; Wu, H.Y.; Pan, W.Y.; Guo, M.Q.; Qiu, D.Z.; He, Y.J.; Li, Y.H.; Yang, D.H.; Huang, Y.X. A novel approach for relapsed/refractory FLT3(mut+) acute myeloid leukaemia: Synergistic effect of the combination of bispecific FLT3scFv/NKG2D-CAR T cells and gilteritinib. Mol. Cancer 2022, 21, 66. [Google Scholar] [CrossRef]
- Driouk, L.; Gicobi, J.K.; Kamihara, Y.; Rutherford, K.; Dranoff, G.; Ritz, J.; Baumeister, S.H.C. Chimeric Antigen Receptor T Cells Targeting NKG2D-Ligands Show Robust Efficacy Against Acute Myeloid Leukemia and T-Cell Acute Lymphoblastic Leukemia. Front. Immunol. 2020, 11, 580328. [Google Scholar] [CrossRef]
- Baumeister, S.H.; Murad, J.; Werner, L.; Daley, H.; Trebeden-Negre, H.; Gicobi, J.K.; Schmucker, A.; Reder, J.; Sentman, C.L.; Gilham, D.E.; et al. Phase I Trial of Autologous CAR T Cells Targeting NKG2D Ligands in Patients with AML/MDS and Multiple Myeloma. Cancer Immunol. Res. 2019, 7, 100–112. [Google Scholar] [CrossRef]
- Sallman, D.A.; Kerre, T.; Poire, X.; Havelange, V.; Lewalle, P.; Davila, M.L.; Wang, E.S.; Dekker, D.; Snykers, S.; Sotiropoulou, P.A.; et al. Remissions in Relapse/Refractory Acute Myeloid Leukemia Patients Following Treatment with NKG2D CAR-T Therapy without a Prior Preconditioning Chemotherapy. Blood 2018, 132, 902. [Google Scholar] [CrossRef]
- Gomes-Silva, D.; Srinivasan, M.; Sharma, S.; Lee, C.M.; Wagner, D.L.; Davis, T.H.; Rouce, R.H.; Bao, G.; Brenner, M.K.; Mamonkin, M. CD7-edited T cells expressing a CD7-specific CAR for the therapy of T-cell malignancies. Blood 2017, 130, 285–296. [Google Scholar] [CrossRef]
- Zhang, M.; Yang, L.; Fu, X.; Zhang, L.; Meng, H.; Li, L.; Li, X.; Wang, X.; Sun, Z.; Yu, H.; et al. First-in-human clinical trial of the autologous CD7-CART for relapsed/refractory ACUTE lymphoblastic leukemia/lymphoma. J. Clin. Oncol. 2020, 38, 3026. [Google Scholar] [CrossRef]
- Lu, P.; Liu, Y.; Yang, J.; Zhang, X.; Yang, X.; Wang, H.; Wang, L.; Wang, Q.; Jin, D.; Li, J.; et al. Naturally selected CD7 CAR-T therapy without genetic manipulations for T-ALL/LBL: First-in-human phase 1 clinical trial. Blood 2022, 140, 321–334. [Google Scholar] [CrossRef]
- Wang, X.; Li, S.; Gao, L.; Yuan, Z.; Wu, K.; Liu, L. Clinical safety and efficacy study of TruUCAR™ GC027: The first-in-human, universal CAR-T therapy for adult relapsed/refractory T-cell acute lymphoblastic leukemia (r/r T-ALL). Cancer Res. 2020, 80, CT052. [Google Scholar] [CrossRef]
- Sanchez-Martinez, D.; Baroni, M.L.; Gutierrez-Aguera, F.; Roca-Ho, H.; Blanch-Lombarte, O.; Gonzalez-Garcia, S.; Torrebadell, M.; Junca, J.; Ramirez-Orellana, M.; Velasco-Hernandez, T.; et al. Fratricide-resistant CD1a-specific CAR T cells for the treatment of cortical T-cell acute lymphoblastic leukemia. Blood 2019, 133, 2291–2304. [Google Scholar] [CrossRef]
- Seitz, C.M.; Mittelstaet, J.; Atar, D.; Hau, J.; Reiter, S.; Illi, C.; Kieble, V.; Engert, F.; Drees, B.; Bender, G.; et al. Novel adapter CAR-T cell technology for precisely controllable multiplex cancer targeting. Oncoimmunology 2021, 10, 2003532. [Google Scholar] [CrossRef]
- Rafiq, S.; Yeku, O.O.; Jackson, H.J.; Purdon, T.J.; van Leeuwen, D.G.; Drakes, D.J.; Song, M.; Miele, M.M.; Li, Z.; Wang, P.; et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat. Biotechnol. 2018, 36, 847–856. [Google Scholar] [CrossRef]
- Chong, E.A.; Alanio, C.; Svoboda, J.; Nasta, S.D.; Landsburg, D.J.; Lacey, S.F.; Ruella, M.; Bhattacharyya, S.; Wherry, E.J.; Schuster, S.J. Pembrolizumab for B-cell lymphomas relapsing after or refractory to CD19-directed CAR T-cell therapy. Blood 2022, 139, 1026–1038. [Google Scholar] [CrossRef] [PubMed]
- Tabata, R.; Chi, S.; Yuda, J.; Minami, Y. Emerging Immunotherapy for Acute Myeloid Leukemia. Int. J. Mol. Sci. 2021, 22, 1944. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Grasso, C.S.; Hugo, W.; Sandoval, S.; Torrejon, D.Y.; et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov. 2017, 7, 188–201. [Google Scholar] [CrossRef]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef] [PubMed]
- Berger, K.N.; Pu, J.J. PD-1 pathway and its clinical application: A 20year journey after discovery of the complete human PD-1 gene. Gene 2018, 638, 20–25. [Google Scholar] [CrossRef]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef]
- Blank, C.; Brown, I.; Peterson, A.C.; Spiotto, M.; Iwai, Y.; Honjo, T.; Gajewski, T.F. PD-L1/B7H-1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer Res. 2004, 64, 1140–1145. [Google Scholar] [CrossRef]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef]
- Haroun, F.; Solola, S.A.; Nassereddine, S.; Tabbara, I. PD-1 signaling and inhibition in AML and MDS. Ann. Hematol. 2017, 96, 1441–1448. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Bueso-Ramos, C.; DiNardo, C.; Estecio, M.R.; Davanlou, M.; Geng, Q.R.; Fang, Z.; Nguyen, M.; Pierce, S.; Wei, Y.; et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia 2014, 28, 1280–1288. [Google Scholar] [CrossRef] [PubMed]
- Goltz, D.; Gevensleben, H.; Grünen, S.; Dietrich, J.; Kristiansen, G.; Landsberg, J.; Dietrich, D. PD-L1 (CD274) promoter methylation predicts survival in patients with acute myeloid leukemia. Leukemia 2017, 31, 738–743. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.; Garcia-Manero, G.; Basu, S.; Boddu, P.C.; Alfayez, M.; Cortes, J.E.; Konopleva, M.; Ravandi-Kashani, F.; Jabbour, E.; Kadia, T.; et al. Efficacy, Safety, and Biomarkers of Response to Azacitidine and Nivolumab in Relapsed/Refractory Acute Myeloid Leukemia: A Nonrandomized, Open-Label, Phase II Study. Cancer Discov. 2019, 9, 370–383. [Google Scholar] [CrossRef] [PubMed]
- Goswami, M.; Gui, G.; Dillon, L.W.; Lindblad, K.E.; Thompson, J.; Valdez, J.; Kim, D.Y.; Ghannam, J.Y.; Oetjen, K.A.; Destefano, C.B.; et al. Pembrolizumab and decitabine for refractory or relapsed acute myeloid leukemia. J. Immunother. Cancer 2022, 10, e003392. [Google Scholar] [CrossRef]
- Daver, N.; Basu, S.; Garcia-Manero, G.; Abbas, H.A.; Konopleva, M.; Kadia, T.M.; DiNardo, C.D.; Borthakur, G.; Alotaibi, A.S.; Pemmaraju, N.; et al. Azacitidine (AZA) with Nivolumab (Nivo), and AZA with Nivo + Ipilimumab (Ipi) in Relapsed/Refractory (R/R) Acute Myeloid Leukemia: Clinical and Immune Biomarkers of Response. Blood 2020, 136, 43–45. [Google Scholar] [CrossRef]
- Ravandi, F.; Assi, R.; Daver, N.; Benton, C.B.; Kadia, T.; Thompson, P.A.; Borthakur, G.; Alvarado, Y.; Jabbour, E.J.; Konopleva, M.; et al. Idarubicin, cytarabine, and nivolumab in patients with newly diagnosed acute myeloid leukaemia or high-risk myelodysplastic syndrome: A single-arm, phase 2 study. Lancet Haematol. 2019, 6, e480–e488. [Google Scholar] [CrossRef]
- Zheng, H.; Mineishi, S.; Claxton, D.; Zhu, J.; Zhao, C.; Jia, B.; Ehmann, W.C.; Rybka, W.B.; Naik, S.; Songdej, N.; et al. A phase I clinical trial of avelumab in combination with decitabine as first line treatment of unfit patients with acute myeloid leukemia. Am. J. Hematol. 2021, 96, E46–E50. [Google Scholar] [CrossRef]
- Teft, W.A.; Kirchhof, M.G.; Madrenas, J. A molecular perspective of CTLA-4 function. Annu. Rev. Immunol. 2006, 24, 65–97. [Google Scholar] [CrossRef]
- Linsley, P.S.; Greene, J.L.; Brady, W.; Bajorath, J.; Ledbetter, J.A.; Peach, R. Human B7-1 (CD80) and B7-2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors. Immunity 1994, 1, 793–801. [Google Scholar] [CrossRef]
- Ramzi, M.; Iravani Saadi, M.; Yaghobi, R.; Arandi, N. Dysregulated Expression of CD28 and CTLA-4 Molecules in Patients with Acute Myeloid Leukemia and Possible Association with Development of Graft versus Host Disease after Hematopoietic Stem Cell Transplantation. Int. J. Organ Transplant. Med. 2019, 10, 84–90. [Google Scholar] [PubMed]
- Liao, D.; Wang, M.; Liao, Y.; Li, J.; Niu, T. A Review of Efficacy and Safety of Checkpoint Inhibitor for the Treatment of Acute Myeloid Leukemia. Front. Pharmacol. 2019, 10, 609. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.C.; Anderson, D.E.; Bregoli, L.; Hastings, W.D.; Kassam, N.; Lei, C.; Chandwaskar, R.; Karman, J.; Su, E.W.; Hirashima, M.; et al. Promotion of tissue inflammation by the immune receptor Tim-3 expressed on innate immune cells. Science 2007, 318, 1141–1143. [Google Scholar] [CrossRef]
- DeKruyff, R.H.; Bu, X.; Ballesteros, A.; Santiago, C.; Chim, Y.L.; Lee, H.H.; Karisola, P.; Pichavant, M.; Kaplan, G.G.; Umetsu, D.T.; et al. T cell/transmembrane, Ig, and mucin-3 allelic variants differentially recognize phosphatidylserine and mediate phagocytosis of apoptotic cells. J. Immunol. 2010, 184, 1918–1930. [Google Scholar] [CrossRef] [PubMed]
- Gleason, M.K.; Lenvik, T.R.; McCullar, V.; Felices, M.; O’Brien, M.S.; Cooley, S.A.; Verneris, M.R.; Cichocki, F.; Holman, C.J.; Panoskaltsis-Mortari, A.; et al. Tim-3 is an inducible human natural killer cell receptor that enhances interferon gamma production in response to galectin-9. Blood 2012, 119, 3064–3072. [Google Scholar] [CrossRef]
- Monney, L.; Sabatos, C.A.; Gaglia, J.L.; Ryu, A.; Waldner, H.; Chernova, T.; Manning, S.; Greenfield, E.A.; Coyle, A.J.; Sobel, R.A.; et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature 2002, 415, 536–541. [Google Scholar] [CrossRef] [PubMed]
- Ndhlovu, L.C.; Lopez-Vergès, S.; Barbour, J.D.; Jones, R.B.; Jha, A.R.; Long, B.R.; Schoeffler, E.C.; Fujita, T.; Nixon, D.F.; Lanier, L.L. Tim-3 marks human natural killer cell maturation and suppresses cell-mediated cytotoxicity. Blood 2012, 119, 3734–3743. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, J.; Wang, M.; Zhang, L.; Yu, L. One Stone, Two Birds: The Roles of Tim-3 in Acute Myeloid Leukemia. Front. Immunol. 2021, 12, 618710. [Google Scholar] [CrossRef]
- Li, C.; Chen, X.; Yu, X.; Zhu, Y.; Ma, C.; Xia, R.; Ma, J.; Gu, C.; Ye, L.; Wu, D. Tim-3 is highly expressed in T cells in acute myeloid leukemia and associated with clinicopathological prognostic stratification. Int. J. Clin. Exp. Pathol. 2014, 7, 6880–6888. [Google Scholar]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef]
- Kikushige, Y. TIM-3 in normal and malignant hematopoiesis: Structure, function, and signaling pathways. Cancer Sci. 2021, 112, 3419–3426. [Google Scholar] [CrossRef] [PubMed]
- Brunner, A.; Borate, U.; Esteve, J.; Porkka, K.; Knapper, S.; Vey, N.; Scholl, S.; Wermke, M.; Janssen, J.; Traer, E.; et al. AML-190: Anti-TIM-3 Antibody MBG453 in Combination with Hypomethylating Agents (HMAs) in Patients with High-Risk Myelodysplastic Syndrome (HR-MDS) and Acute Myeloid Leukemia: A Phase 1 Study. Clin. Lymphoma Myeloma Leuk. 2020, 20, S188–S189. [Google Scholar] [CrossRef]
- Curigliano, G.; Gelderblom, H.; Mach, N.; Doi, T.; Tai, D.; Forde, P.M.; Sarantopoulos, J.; Bedard, P.L.; Lin, C.C.; Hodi, F.S.; et al. Phase I/Ib Clinical Trial of Sabatolimab, an Anti-TIM-3 Antibody, Alone and in Combination with Spartalizumab, an Anti-PD-1 Antibody, in Advanced Solid Tumors. Clin. Cancer Res. 2021, 27, 3620–3629. [Google Scholar] [CrossRef] [PubMed]
- Baixeras, E.; Huard, B.; Miossec, C.; Jitsukawa, S.; Martin, M.; Hercend, T.; Auffray, C.; Triebel, F.; Piatier-Tonneau, D. Characterization of the lymphocyte activation gene 3-encoded protein. A new ligand for human leukocyte antigen class II antigens. J. Exp. Med. 1992, 176, 327–337. [Google Scholar] [CrossRef]
- Maruhashi, T.; Sugiura, D.; Okazaki, I.M.; Okazaki, T. LAG-3: From molecular functions to clinical applications. J. Immunother. Cancer 2020, 8, e001014. [Google Scholar] [CrossRef]
- Maruhashi, T.; Sugiura, D.; Okazaki, I.M.; Shimizu, K.; Maeda, T.K.; Ikubo, J.; Yoshikawa, H.; Maenaka, K.; Ishimaru, N.; Kosako, H.; et al. Binding of LAG-3 to stable peptide-MHC class II limits T cell function and suppresses autoimmunity and anti-cancer immunity. Immunity 2022, 55, 912–924.e918. [Google Scholar] [CrossRef]
- Yang, Z.Z.; Kim, H.J.; Villasboas, J.C.; Chen, Y.P.; Price-Troska, T.; Jalali, S.; Wilson, M.; Novak, A.J.; Ansell, S.M. Expression of LAG-3 defines exhaustion of intratumoral PD-1(+) T cells and correlates with poor outcome in follicular lymphoma. Oncotarget 2017, 8, 61425–61439. [Google Scholar] [CrossRef]
- Williams, P.; Basu, S.; Garcia-Manero, G.; Hourigan, C.S.; Oetjen, K.A.; Cortes, J.E.; Ravandi, F.; Jabbour, E.J.; Al-Hamal, Z.; Konopleva, M.; et al. The distribution of T-cell subsets and the expression of immune checkpoint receptors and ligands in patients with newly diagnosed and relapsed acute myeloid leukemia. Cancer 2019, 125, 1470–1481. [Google Scholar] [CrossRef]
- Nagasaki, J.; Togashi, Y.; Sugawara, T.; Itami, M.; Yamauchi, N.; Yuda, J.; Sugano, M.; Ohara, Y.; Minami, Y.; Nakamae, H.; et al. The critical role of CD4+ T cells in PD-1 blockade against MHC-II-expressing tumors such as classic Hodgkin lymphoma. Blood Adv. 2020, 4, 4069–4082. [Google Scholar] [CrossRef]
- Garcia-Marquez, M.A.; Thelen, M.; Reinke, S.; Keller, D.; Wennhold, K.; Lehmann, J.; Veldman, J.; Borchmann, S.; Rosenwald, A.; Sasse, S.; et al. Reverted exhaustion phenotype of circulating lymphocytes as immune correlate of anti-PD1 first-line treatment in Hodgkin lymphoma. Leukemia 2022, 36, 760–771. [Google Scholar] [CrossRef]
- Askeland, F.B.; Rasmussen, A.-M.; Schjesvold, F. Relapse from MRD Negativity as Indication for Treatment in Multiple Myeloma—The Remnant Study. Blood 2020, 136, 21–22. [Google Scholar] [CrossRef]
- Barclay, A.N.; Brown, M.H. The SIRP family of receptors and immune regulation. Nat. Rev. Immunol. 2006, 6, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Chao, M.P.; Alizadeh, A.A.; Tang, C.; Jan, M.; Weissman-Tsukamoto, R.; Zhao, F.; Park, C.Y.; Weissman, I.L.; Majeti, R. Therapeutic antibody targeting of CD47 eliminates human acute lymphoblastic leukemia. Cancer Res. 2011, 71, 1374–1384. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Jamieson, C.H.; Pang, W.W.; Park, C.Y.; Chao, M.P.; Majeti, R.; Traver, D.; van Rooijen, N.; Weissman, I.L. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell 2009, 138, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wang, F.; Guo, R.; Bian, Z.; Song, Y. Targeting macrophages in hematological malignancies: Recent advances and future directions. J. Hematol. Oncol. 2022, 15, 110. [Google Scholar] [CrossRef] [PubMed]
- Advani, R.; Flinn, I.; Popplewell, L.; Forero, A.; Bartlett, N.L.; Ghosh, N.; Kline, J.; Roschewski, M.; LaCasce, A.; Collins, G.P.; et al. CD47 Blockade by Hu5F9-G4 and Rituximab in Non-Hodgkin’s Lymphoma. N. Engl. J. Med. 2018, 379, 1711–1721. [Google Scholar] [CrossRef]
- Vyas, P.; Chao, M.; Takimoto, C.H.; Volkmer, J.-P.; Lin, M.; Van Elk, J.; Komrokji, R.S.; Garcia-Manero, G.; Daver, N.G.; Kambhampati, S.; et al. The First-in-Class Anti-CD47 Antibody Magrolimab (5F9) in Combination with Azacitidine Is Effective in MDS and AML Patients: Ongoing Phase 1b Results. Blood 2019, 134, 569. [Google Scholar] [CrossRef]
- Chen, S.H.; Dominik, P.K.; Stanfield, J.; Ding, S.; Yang, W.; Kurd, N.; Llewellyn, R.; Heyen, J.; Wang, C.; Melton, Z.; et al. Dual checkpoint blockade of CD47 and PD-L1 using an affinity-tuned bispecific antibody maximizes antitumor immunity. J. Immunother. Cancer 2021, 9, e003464. [Google Scholar] [CrossRef]
- Hills, R.K.; Castaigne, S.; Appelbaum, F.R.; Delaunay, J.; Petersdorf, S.; Othus, M.; Estey, E.H.; Dombret, H.; Chevret, S.; Ifrah, N.; et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: A meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014, 15, 986–996. [Google Scholar] [CrossRef]
- Amadori, S.; Suciu, S.; Selleslag, D.; Aversa, F.; Gaidano, G.; Musso, M.; Annino, L.; Venditti, A.; Voso, M.T.; Mazzone, C.; et al. Gemtuzumab Ozogamicin Versus Best Supportive Care in Older Patients with Newly Diagnosed Acute Myeloid Leukemia Unsuitable for Intensive Chemotherapy: Results of the Randomized Phase III EORTC-GIMEMA AML-19 Trial. J. Clin. Oncol. 2016, 34, 972–979. [Google Scholar] [CrossRef]
- Taksin, A.L.; Legrand, O.; Raffoux, E.; de Revel, T.; Thomas, X.; Contentin, N.; Bouabdallah, R.; Pautas, C.; Turlure, P.; Reman, O.; et al. High efficacy and safety profile of fractionated doses of Mylotarg as induction therapy in patients with relapsed acute myeloblastic leukemia: A prospective study of the alfa group. Leukemia 2007, 21, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Abedin, S.; Guru Murthy, G.S.G.; Hamadani, M.; Michaelis, L.C.; Runaas, L.; Carlson, K.-S.; Harrington, A.M.; Atallah, E.L. A Phase I Study of Lintuzumab Ac225 in Combination with CLAG-M Chemotherapy in Relapsed/Refractory AML. Blood 2020, 136, 9–10. [Google Scholar] [CrossRef]
- Raponi, S.; De Propris, M.S.; Intoppa, S.; Milani, M.L.; Vitale, A.; Elia, L.; Perbellini, O.; Pizzolo, G.; Foa, R.; Guarini, A. Flow cytometric study of potential target antigens (CD19, CD20, CD22, CD33) for antibody-based immunotherapy in acute lymphoblastic leukemia: Analysis of 552 cases. Leuk. Lymphoma 2011, 52, 1098–1107. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.M.; DeAngelo, D.J.; Stelljes, M.; Martinelli, G.; Liedtke, M.; Stock, W.; Gokbuget, N.; O’Brien, S.; Wang, K.; Wang, T.; et al. Inotuzumab Ozogamicin versus Standard Therapy for Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2016, 375, 740–753. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; DeAngelo, D.J.; Stelljes, M.; Liedtke, M.; Stock, W.; Gokbuget, N.; O’Brien, S.M.; Jabbour, E.; Wang, T.; Liang White, J.; et al. Inotuzumab ozogamicin versus standard of care in relapsed or refractory acute lymphoblastic leukemia: Final report and long-term survival follow-up from the randomized, phase 3 INO-VATE study. Cancer 2019, 125, 2474–2487. [Google Scholar] [CrossRef]
- Salvaris, R.; Fedele, P.L. Targeted Therapy in Acute Lymphoblastic Leukaemia. J. Pers. Med. 2021, 11, 715. [Google Scholar] [CrossRef]
- Bakker, A.B.; van den Oudenrijn, S.; Bakker, A.Q.; Feller, N.; van Meijer, M.; Bia, J.A.; Jongeneelen, M.A.; Visser, T.J.; Bijl, N.; Geuijen, C.A.; et al. C-type lectin-like molecule-1: A novel myeloid cell surface marker associated with acute myeloid leukemia. Cancer Res. 2004, 64, 8443–8450. [Google Scholar] [CrossRef]
- Zheng, B.; Yu, S.F.; Del Rosario, G.; Leong, S.R.; Lee, G.Y.; Vij, R.; Chiu, C.; Liang, W.C.; Wu, Y.; Chalouni, C.; et al. An Anti-CLL-1 Antibody-Drug Conjugate for the Treatment of Acute Myeloid Leukemia. Clin. Cancer Res. 2019, 25, 1358–1368. [Google Scholar] [CrossRef]
- Jiang, Y.P.; Liu, B.Y.; Zheng, Q.; Panuganti, S.; Chen, R.; Zhu, J.; Mishra, M.; Huang, J.; Dao-Pick, T.; Roy, S.; et al. CLT030, a leukemic stem cell-targeting CLL1 antibody-drug conjugate for treatment of acute myeloid leukemia. Blood Adv. 2018, 2, 1738–1749. [Google Scholar] [CrossRef]
- Gros, A.; Parkhurst, M.R.; Tran, E.; Pasetto, A.; Robbins, P.F.; Ilyas, S.; Prickett, T.D.; Gartner, J.J.; Crystal, J.S.; Roberts, I.M.; et al. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat. Med. 2016, 22, 433–438. [Google Scholar] [CrossRef]
- Roerden, M.; Nelde, A.; Walz, J.S. Neoantigens in Hematological Malignancies-Ultimate Targets for Immunotherapy? Front. Immunol. 2019, 10, 3004. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Leet, D.E.; Allesøe, R.L.; Oliveira, G.; Li, S.; Luoma, A.M.; Liu, J.; Forman, J.; Huang, T.; Iorgulescu, J.B.; et al. Personal neoantigen vaccines induce persistent memory T cell responses and epitope spreading in patients with melanoma. Nat. Med. 2021, 27, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Ushmorov, A.; Leithäuser, F.; Sakk, O.; Weinhaüsel, A.; Popov, S.W.; Möller, P.; Wirth, T. Epigenetic processes play a major role in B-cell-specific gene silencing in classical Hodgkin lymphoma. Blood 2006, 107, 2493–2500. [Google Scholar] [CrossRef]
- Kuhn, N.F.; Purdon, T.J.; van Leeuwen, D.G.; Lopez, A.V.; Curran, K.J.; Daniyan, A.F.; Brentjens, R.J. CD40 Ligand-Modified Chimeric Antigen Receptor T Cells Enhance Antitumor Function by Eliciting an Endogenous Antitumor Response. Cancer Cell 2019, 35, 473–488.e476. [Google Scholar] [CrossRef] [PubMed]
- Avanzi, M.P.; Yeku, O.; Li, X.; Wijewarnasuriya, D.P.; van Leeuwen, D.G.; Cheung, K.; Park, H.; Purdon, T.J.; Daniyan, A.F.; Spitzer, M.H.; et al. Engineered Tumor-Targeted T Cells Mediate Enhanced Anti-Tumor Efficacy Both Directly and through Activation of the Endogenous Immune System. Cell Rep. 2018, 23, 2130–2141. [Google Scholar] [CrossRef]
- Baryawno, N.; Przybylski, D.; Kowalczyk, M.S.; Kfoury, Y.; Severe, N.; Gustafsson, K.; Kokkaliaris, K.D.; Mercier, F.; Tabaka, M.; Hofree, M.; et al. A Cellular Taxonomy of the Bone Marrow Stroma in Homeostasis and Leukemia. Cell 2019, 177, 1915–1932.e1916. [Google Scholar] [CrossRef] [PubMed]
- Hirakawa, M.P.; Krishnakumar, R.; Timlin, J.A.; Carney, J.P.; Butler, K.S. Gene editing and CRISPR in the clinic: Current and future perspectives. Biosci. Rep. 2020, 40, BSR20200127. [Google Scholar] [CrossRef]
- Moeinafshar, A.; Hemmati, S.; Rezaei, N. Immunotherapy in AML: A brief review on emerging strategies. Clin. Transl. Oncol. 2021, 23, 2431–2447. [Google Scholar] [CrossRef]
- Fleming, V.; Hu, X.; Weber, R.; Nagibin, V.; Groth, C.; Altevogt, P.; Utikal, J.; Umansky, V. Targeting Myeloid-Derived Suppressor Cells to Bypass Tumor-Induced Immunosuppression. Front. Immunol. 2018, 9, 398. [Google Scholar] [CrossRef]
- Barrett, A.J. Acute myeloid leukaemia and the immune system: Implications for immunotherapy. Br. J. Haematol. 2020, 188, 147–158. [Google Scholar] [CrossRef]


| Characteristic | Odronextamab | Mosunetuzumab | Epcoritamab | Glofitamab |
|---|---|---|---|---|
| IgG | human IgG4 | human IgG1 | human IgG1 | human IgG like |
| Patients, n | 145 | 129 | 68 | 171 |
| Prior therapies, median | 3 | 4 | NA | 3 |
| Prior CAR-T therapy (%) | 29 | 11.6 | NA | 1.8 (2.9) |
| ORR (%) | 53 * (33 **) | 34.9 | 68 * (88 **) | 53.8* (65.7 **) |
| CR (%) | 100 * (27 **) | 19.4 | 45 * (38 **) | 36.8* (57.1 **) |
| CRS | ||||
| Any grade (%) | 28 | 27.4 | 59 | 50.3 * (71.4 **) |
| Grade > 3 (%) | 5.1 | 1 | 0 | 3.5 * (5.7 **) |
| NT | ||||
| Any grade (%) | 0 | NA | 6 | 43.3 * (31.4 **) |
| Grade > 3 (%) | 0 | 4.1 | 3 | NA |
| Clinical trial | NCT02290951 | NCT02500407 | NCT03625037 | NCT03075696 |
| Reference | [65] | [66] | [70] | [71] |
| Agents | Target | Author (Sposor) | Year (Estimated Completion Date) | Phase | Objects | Cases (Estimated Enrollment) | Survival Rate (%) | CR/CRi (%) | Median Survival (Months) | Clinical Trial | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|
| AMG330 | CD33/CD3 | Ravandi et al. | 2020 | I | rrAML | 42 | NA | 19 (8/42) | NA | NCT02520427 | [74] |
| Vixtimotamab (AMV564) | CD33/CD3 | Westervelt et al. | 2019 | I | rrAML | 35 | NA | 8.6 (3/35) | NA | NCT03144245 | [75] |
| Vibecotamab (XmAb14045) | CD123/CD3 | Ravandi et al. | 2020 | I | rrAML | 104 | NA | 14 (5/51) | NA | NCT02730312 | [80] |
| Flontetuzumab | CD123/CD3 | Uy et al. | 2021 | III | rrAML | 30 | 75 (6 m), 50 (12 m) | 26.7 | 10.2 | NCT02152956 | [81] |
| AMG427 | FLT3/CD3 | (Amgen) | (2022) | I | rrAML | (70) | NA | NA | NA | NCT03541369 | [86] |
| Agents | Target | Author (Sposor) | Year (Estimated Completion Date) | Phase | Objects | Cases (Estimated Enrollment) | Survival Rate (%) | CR/CRi (%) | Median Survival (Months) | Clinical Trial | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|
| CLL1 CAR-T | CLL-1 | Jin, X et al. | 2022 | I | rrAML | 10 | 60 | 70 (7/10) | 5.8 | - | [115] |
| CD123 CAR-T | CD123 | Budde et al. | 2017 | I | rrAML | 6 | NA | 50 (3/6) | NA | NCT02159495 | [125] |
| CD33CAR-T | CD33 | Tambaro et al. | 2021 | I | rrAML | 10 | 0 | NA | NA | NCT03126864 | [128] |
| NKG2D CAR-T | NKG2D | Baumeister et al. | 2019 | I | AML, MM | 12 | 75(3m), 42(6m) | NA | 4.7 | NCT02203825 | [132] |
| CYAD-01 | NKG2D | Sallman et al. | 2018 | I | AML, MDS, MM | 12 | NA | 42 | NA | NCT03018405 | [133] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nogami, A.; Sasaki, K. Therapeutic Advances in Immunotherapies for Hematological Malignancies. Int. J. Mol. Sci. 2022, 23, 11526. https://doi.org/10.3390/ijms231911526
Nogami A, Sasaki K. Therapeutic Advances in Immunotherapies for Hematological Malignancies. International Journal of Molecular Sciences. 2022; 23(19):11526. https://doi.org/10.3390/ijms231911526
Chicago/Turabian StyleNogami, Ayako, and Koji Sasaki. 2022. "Therapeutic Advances in Immunotherapies for Hematological Malignancies" International Journal of Molecular Sciences 23, no. 19: 11526. https://doi.org/10.3390/ijms231911526
APA StyleNogami, A., & Sasaki, K. (2022). Therapeutic Advances in Immunotherapies for Hematological Malignancies. International Journal of Molecular Sciences, 23(19), 11526. https://doi.org/10.3390/ijms231911526