
The Immuno-Oncology and Genomic Aspects of DNA-Hypomethylating Therapeutics in Acute Myeloid Leukemia


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Current Clinical Application of HMAs for AML
2.1. Combination of VEN and AZA in the Elderly
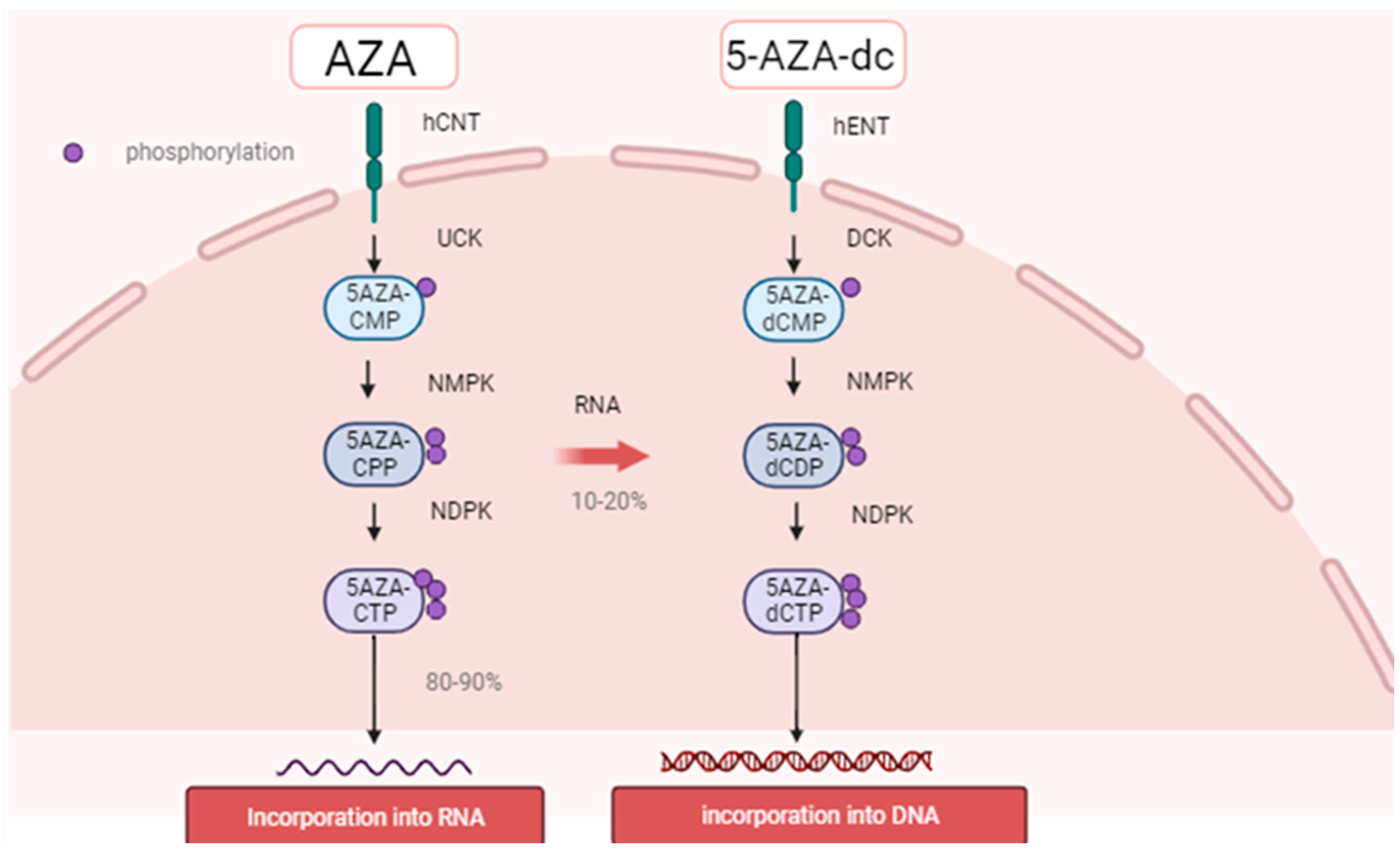
2.2. Mechanism of Action of HMAs
2.3. Pathophysiologic Features of Leukemic Stem Cells
2.3.1. Metabolic Changes in Leukemic Stem Cells
2.3.2. Amplification of Anti-Apoptotic Factors
2.4. VEN and Gilteritinib
2.5. Magrolimab Combination Therapy
2.6. AZA and Ivosidenib, Newly Therapy
3. Effect of HMAs on Tumor Immunity
3.1. HMA May Improve the Efficacy of CAR-T Cells
3.2. HMA-Induced Changes to the Immune System
3.3. The Effect of TP53 Mutation on Anti-Leukemic Immunity
4. Impact of Genetic Backgrounds on DNA-Hypomethylating Therapeutics
4.1. Aberrant Epigenetic Modification in Hematologic Malignancies
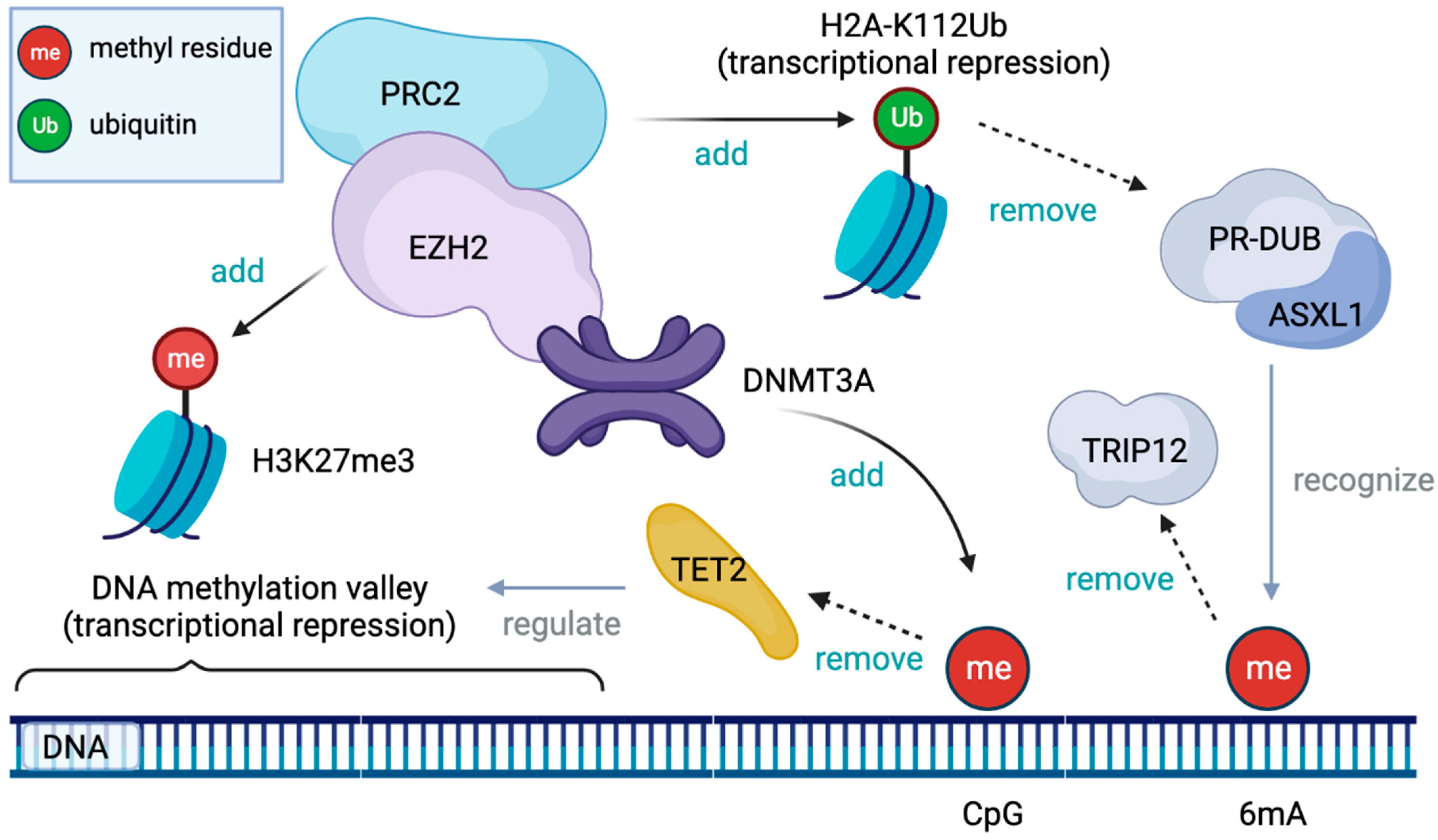
4.1.1. Physiologic Epigenetic Regulation of Hematopoiesis (Figure 2)

4.1.2. DNMT3A Mutation
4.1.3. TET2 Mutation
4.1.4. ASXL1 Mutation
4.1.5. EZH2 Mutation
4.2. Association between Anti-Leukemic Immunity and Epigenetic Dysregulation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Giles, F.J.; Borthakur, G.; Ravandi, F.; Faderl, S.; Verstovsek, S.; Thomas, D.; Wierda, W.; Ferrajoli, A.; Kornblau, S.; Pierce, S.; et al. The Haematopoietic Cell Transplantation Comorbidity Index Score Is Predictive of Early Death and Survival in Patients over 60 Years of Age Receiving Induction Therapy for Acute Myeloid Leukaemia. Br. J. Haematol. 2007, 136, 624–627. [Google Scholar] [CrossRef]
- Dombret, H.; Seymour, J.F.; Butrym, A.; Wierzbowska, A.; Selleslag, D.; Jang, J.H.; Kumar, R.; Cavenagh, J.; Schuh, A.C.; Candoni, A.; et al. International Phase 3 Study of Azacitidine vs. Conventional Care Regimens in Older Patients with Newly Diagnosed AML with >30% Blasts. Blood 2015, 126, 291–299. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Döhner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.H.; Strickland, S.A.; Hou, J.-Z.; Fiedler, W.; Lin, T.L.; Walter, R.B.; Enjeti, A.; Tiong, I.S.; Savona, M.; Lee, S.; et al. Venetoclax Combined with Low-Dose Cytarabine for Previously Untreated Patients with Acute Myeloid Leukemia: Results from a Phase Ib/II Study. J. Clin. Oncol. 2019, 37, 1277–1284. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, S.; Kadia, T.M.; Ravandi, F.; DiNardo, C.D.; Daver, N.; Borthakur, G.; Konopleva, M.; Garcia-Manero, G.; Montalban-Bravo, G.; Maiti, A.; et al. Impact of Frontline Treatment Approach in Patients with Secondary AML and Prior Hypomethylating Agent Exposure: A Retrospective Analysis of 562 Patients with Treated Secondary AML. Blood 2021, 138, 794. [Google Scholar] [CrossRef]
- Guan, Y.; Hogge, D.E. Proliferative Status of Primitive Hematopoietic Progenitors from Patients with Acute Myelogenous Leukemia (AML). Leukemia 2000, 14, 2135–2141. [Google Scholar] [CrossRef] [PubMed]
- Hope, K.J.; Jin, L.; Dick, J.E. Acute Myeloid Leukemia Originates from a Hierarchy of Leukemic Stem Cell Classes That Differ in Self-Renewal Capacity. Nat. Immunol. 2004, 5, 738–743. [Google Scholar] [CrossRef]
- Ishikawa, F.; Yoshida, S.; Saito, Y.; Hijikata, A.; Kitamura, H.; Tanaka, S.; Nakamura, R.; Tanaka, T.; Tomiyama, H.; Saito, N.; et al. Chemotherapy-Resistant Human AML Stem Cells Home to and Engraft within the Bone-Marrow Endosteal Region. Nat. Biotechnol. 2007, 25, 1315–1321. [Google Scholar] [CrossRef]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem Cells, Cancer, and Cancer Stem Cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef]
- Terpstra, W.; Ploemacher, R.E.; Prins, A.; van Lom, K.; Pouwels, K.; Wognum, A.W.; Wagemaker, G.; Löwenberg, B.; Wielenga, J.J. Fluorouracil Selectively Spares Acute Myeloid Leukemia Cells with Long-Term Growth Abilities in Immunodeficient Mice and in Culture. Blood 1996, 88, 1944–1950. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The Landscape of Somatic Copy-Number Alteration across Human Cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef]
- Denis, C.; Sopková-de Oliveira Santos, J.; Bureau, R.; Voisin-Chiret, A.S. Hot-Spots of Mcl-1 Protein. J. Med. Chem. 2020, 63, 928–943. [Google Scholar] [CrossRef] [PubMed]
- Grabow, S.; Delbridge, A.R.D.; Aubrey, B.J.; Vandenberg, C.J.; Strasser, A. Loss of a Single Mcl-1 Allele Inhibits MYC-Driven Lymphomagenesis by Sensitizing Pro-B Cells to Apoptosis. Cell Rep. 2016, 14, 2337–2347. [Google Scholar] [CrossRef] [PubMed]
- Glaser, S.P.; Lee, E.F.; Trounson, E.; Bouillet, P.; Wei, A.; Fairlie, W.D.; Izon, D.J.; Zuber, J.; Rappaport, A.R.; Herold, M.J.; et al. Anti-Apoptotic Mcl-1 Is Essential for the Development and Sustained Growth of Acute Myeloid Leukemia. Genes Dev. 2012, 26, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Pei, S.; Pollyea, D.A.; Gustafson, A.; Stevens, B.M.; Minhajuddin, M.; Fu, R.; Riemondy, K.A.; Gillen, A.E.; Sheridan, R.M.; Kim, J.; et al. Monocytic Subclones Confer Resistance to Venetoclax-Based Therapy in Patients with Acute Myeloid Leukemia. Cancer Discov. 2020, 10, 536–551. [Google Scholar] [CrossRef]
- Bolomsky, A.; Vogler, M.; Köse, M.C.; Heckman, C.A.; Ehx, G.; Ludwig, H.; Caers, J. MCL-1 Inhibitors, Fast-Lane Development of a New Class of Anti-Cancer Agents. J. Hematol. Oncol. 2020, 13, 173. [Google Scholar] [CrossRef]
- Jones, C.L.; Stevens, B.M.; D’Alessandro, A.; Reisz, J.A.; Culp-Hill, R.; Nemkov, T.; Pei, S.; Khan, N.; Adane, B.; Ye, H.; et al. Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell 2018, 34, 724–740.e4. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.; Perl, A.E.; Maly, J.; Levis, M.; Ritchie, E.; Litzow, M.R.; McCloskey, J.; Smith, C.C.; Schiller, G.J.; Bradley, T.; et al. Venetoclax in Combination with Gilteritinib Demonstrates Molecular Clearance of FLT3 Mutation in Relapsed/Refractory FLT3-Mutated Acute Myeloid Leukemia. Blood 2021, 138, 691. [Google Scholar] [CrossRef]
- Daver, N.; Konopleva, M.; Maiti, A.; Kadia, T.M.; DiNardo, C.D.; Loghavi, S.; Pemmaraju, N.; Jabbour, E.J.; Montalban-Bravo, G.; Tang, G.; et al. Phase I/II Study of Azacitidine (AZA) with Venetoclax (VEN) and Magrolimab (Magro) in Patients (Pts) with Newly Diagnosed Older/Unfit or High-Risk Acute Myeloid Leukemia (AML) and Relapsed/Refractory (R/R) AML. Blood 2021, 138, 371. [Google Scholar] [CrossRef]
- Montesinos, P.; Recher, C.; Vives, S.; Zarzycka, E.; Wang, J.; Bertani, G.; Heuser, M.; Calado, R.T.; Schuh, A.C.; Yeh, S.-P.; et al. Ivosidenib and Azacitidine in IDH1-Mutated Acute Myeloid Leukemia. N. Engl. J. Med. 2022, 386, 1519–1531. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, L.; Nomdedéu, J.F.; López, O.; Carnicer, M.J.; Bellido, M.; Aventín, A.; Brunet, S.; Sierra, J. Interleukin-3 Receptor Alpha Chain (CD123) Is Widely Expressed in Hematologic Malignancies. Haematologica 2001, 86, 1261–1269. [Google Scholar]
- El Khawanky, N.; Hughes, A.; Yu, W.; Myburgh, R.; Matschulla, T.; Taromi, S.; Aumann, K.; Clarson, J.; Vinnakota, J.M.; Shoumariyeh, K.; et al. Demethylating Therapy Increases Anti-CD123 CAR T Cell Cytotoxicity against Acute Myeloid Leukemia. Nat. Commun. 2021, 12, 6436. [Google Scholar] [CrossRef]
- Leick, M.B.; Silva, H.; Scarfò, I.; Larson, R.; Choi, B.D.; Bouffard, A.A.; Gallagher, K.; Schmidts, A.; Bailey, S.R.; Kann, M.C.; et al. Non-Cleavable Hinge Enhances Avidity and Expansion of CAR-T Cells for Acute Myeloid Leukemia. Cancer Cell 2022, 40, 494–508.e5. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.L.; Georgakilas, G.; Petrovic, J.; Kurachi, M.; Cai, S.; Harly, C.; Pear, W.S.; Bhandoola, A.; Wherry, E.J.; Vahedi, G. Lineage-Determining Transcription Factor TCF-1 Initiates the Epigenetic Identity of T Cell Development. Immunity 2018, 48, 243–257.e10. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Rivière, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef]
- Greenbaum, U.; Mahadeo, K.M.; Kebriaei, P.; Shpall, E.J.; Saini, N.Y. Chimeric Antigen Receptor T-Cells in B-Acute Lymphoblastic Leukemia: State of the Art and Future Directions. Front. Oncol. 2020, 10, 1594. [Google Scholar] [CrossRef]
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef]
- Wherry, E.J.; Ha, S.-J.; Kaech, S.M.; Haining, W.N.; Sarkar, S.; Kalia, V.; Subramaniam, S.; Blattman, J.N.; Barber, D.L.; Ahmed, R. Molecular Signature of CD8+ T Cell Exhaustion during Chronic Viral Infection. Immunity 2007, 27, 670–684. [Google Scholar] [CrossRef]
- Wherry, E.J.; Kurachi, M. Molecular and Cellular Insights into T Cell Exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef] [PubMed]
- Schietinger, A.; Philip, M.; Krisnawan, V.E.; Chiu, E.Y.; Delrow, J.J.; Basom, R.S.; Lauer, P.; Brockstedt, D.G.; Knoblaugh, S.E.; Hämmerling, G.J.; et al. Tumor-Specific T Cell Dysfunction Is a Dynamic Antigen-Driven Differentiation Program Initiated Early during Tumorigenesis. Immunity 2016, 45, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Schietinger, A.; Greenberg, P.D. Tolerance and Exhaustion: Defining Mechanisms of T Cell Dysfunction. Trends Immunol. 2014, 35, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tong, C.; Dai, H.; Wu, Z.; Han, X.; Guo, Y.; Chen, D.; Wei, J.; Ti, D.; Liu, Z.; et al. Low-Dose Decitabine Priming Endows CAR T Cells with Enhanced and Persistent Antitumour Potential via Epigenetic Reprogramming. Nat. Commun. 2021, 12, 409. [Google Scholar] [CrossRef]
- Chiappinelli, K.B.; Strissel, P.L.; Desrichard, A.; Li, H.; Henke, C.; Akman, B.; Hein, A.; Rote, N.S.; Cope, L.M.; Snyder, A.; et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via DsRNA Including Endogenous Retroviruses. Cell 2015, 162, 974–986. [Google Scholar] [CrossRef]
- Roulois, D.; Loo Yau, H.; Singhania, R.; Wang, Y.; Danesh, A.; Shen, S.Y.; Han, H.; Liang, G.; Jones, P.A.; Pugh, T.J.; et al. DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell 2015, 162, 961–973. [Google Scholar] [CrossRef]
- Legat, A.; Speiser, D.E.; Pircher, H.; Zehn, D.; Fuertes Marraco, S.A. Inhibitory Receptor Expression Depends More Dominantly on Differentiation and Activation than “Exhaustion” of Human CD8 T Cells. Front. Immunol. 2013, 4, 455. [Google Scholar] [CrossRef]
- Loo Yau, H.; Bell, E.; Ettayebi, I.; de Almeida, F.C.; Boukhaled, G.M.; Shen, S.Y.; Allard, D.; Morancho, B.; Marhon, S.A.; Ishak, C.A.; et al. DNA Hypomethylating Agents Increase Activation and Cytolytic Activity of CD8+ T Cells. Mol. Cell 2021, 81, 1469–1483.e8. [Google Scholar] [CrossRef]
- Lindsley, R.C.; Saber, W.; Mar, B.G.; Redd, R.; Wang, T.; Haagenson, M.D.; Grauman, P.V.; Hu, Z.-H.; Spellman, S.R.; Lee, S.J.; et al. Prognostic Mutations in Myelodysplastic Syndrome after Stem-Cell Transplantation. N. Engl. J. Med. 2017, 376, 536–547. [Google Scholar] [CrossRef] [PubMed]
- Yoshizato, T.; Nannya, Y.; Atsuta, Y.; Shiozawa, Y.; Iijima-Yamashita, Y.; Yoshida, K.; Shiraishi, Y.; Suzuki, H.; Nagata, Y.; Sato, Y.; et al. Genetic Abnormalities in Myelodysplasia and Secondary Acute Myeloid Leukemia: Impact on Outcome of Stem Cell Transplantation. Blood 2017, 129, 2347–2358. [Google Scholar] [CrossRef]
- Della Porta, M.G.; Gallì, A.; Bacigalupo, A.; Zibellini, S.; Bernardi, M.; Rizzo, E.; Allione, B.; van Lint, M.T.; Pioltelli, P.; Marenco, P.; et al. Clinical Effects of Driver Somatic Mutations on the Outcomes of Patients with Myelodysplastic Syndromes Treated with Allogeneic Hematopoietic Stem-Cell Transplantation. J. Clin. Oncol. 2016, 34, 3627–3637. [Google Scholar] [CrossRef] [PubMed]
- Montalban-Bravo, G.; Takahashi, K.; Garcia-Manero, G. Decitabine in TP53-Mutated AML. N. Engl. J. Med. 2017, 376, 796–797. [Google Scholar] [CrossRef] [PubMed]
- Sallman, D.A.; McLemore, A.F.; Aldrich, A.L.; Komrokji, R.S.; McGraw, K.L.; Dhawan, A.; Geyer, S.; Hou, H.-A.; Eksioglu, E.A.; Sullivan, A.; et al. TP53 Mutations in Myelodysplastic Syndromes and Secondary AML Confer an Immunosuppressive Phenotype. Blood 2020, 136, 2812–2823. [Google Scholar] [CrossRef]
- Suzuki, M.M.; Bird, A. DNA Methylation Landscapes: Provocative Insights from Epigenomics. Nat. Rev. Genet. 2008, 9, 465–476. [Google Scholar] [CrossRef]
- Weber, M.; Hellmann, I.; Stadler, M.B.; Ramos, L.; Pääbo, S.; Rebhan, M.; Schübeler, D. Distribution, Silencing Potential and Evolutionary Impact of Promoter DNA Methylation in the Human Genome. Nat. Genet. 2007, 39, 457–466. [Google Scholar] [CrossRef]
- Katrinli, S.; Maihofer, A.X.; Wani, A.H.; Pfeiffer, J.R.; Ketema, E.; Ratanatharathorn, A.; Baker, D.G.; Boks, M.P.; Geuze, E.; Kessler, R.C.; et al. Epigenome-Wide Meta-Analysis of PTSD Symptom Severity in Three Military Cohorts Implicates DNA Methylation Changes in Genes Involved in Immune System and Oxidative Stress. Mol. Psychiatry 2022, 27, 1720–1728. [Google Scholar] [CrossRef] [PubMed]
- Sammallahti, S.; Hidalgo, A.P.C.; Tuominen, S.; Malmberg, A.; Mulder, R.H.; Brunst, K.J.; Alemany, S.; McBride, N.S.; Yousefi, P.; Heiss, J.A.; et al. Maternal Anxiety during Pregnancy and Newborn Epigenome-Wide DNA Methylation. Mol. Psychiatry 2021, 26, 1832–1845. [Google Scholar] [CrossRef]
- Wang, X. Blood DNA Methylation and Type 2 Diabetes Mellitus: A Protocol for Systematic Review and Meta-Analysis; INPLASY—International Platform of Registered Systematic Review Protocols: New Castle, DE, USA, 2020. [Google Scholar]
- Jiang, Y.; Forno, E.; Han, Y.-Y.; Xu, Z.; Hu, D.; Boutaoui, N.; Eng, C.; Acosta-Pérez, E.; Huntsman, S.; Colón-Semidey, A.; et al. A Genome-Wide Study of DNA Methylation in White Blood Cells and Asthma in Latino Children and Youth. Epigenetics 2021, 16, 577–585. [Google Scholar] [CrossRef]
- Christiansen, C.; Castillo-Fernandez, J.E.; Domingo-Relloso, A.; Zhao, W.; El-Sayed Moustafa, J.S.; Tsai, P.-C.; Maddock, J.; Haack, K.; Cole, S.A.; Kardia, S.L.R.; et al. Novel DNA Methylation Signatures of Tobacco Smoking with Trans-Ethnic Effects. Clin. Epigenet. 2021, 13, 36. [Google Scholar] [CrossRef]
- Karabegovic, I. Epigenome-Wide Association Meta-Analysis of DNA Methylation with Coffee and Tea Consumption. Nat. Commun. 2021, 12, 2830. [Google Scholar] [CrossRef]
- Brown, K.D.; Robertson, K.D. DNMT1 Knockout Delivers a Strong Blow to Genome Stability and Cell Viability. Nat. Genet. 2007, 39, 289–290. [Google Scholar] [CrossRef] [PubMed]
- Holz-Schietinger, C.; Matje, D.M.; Harrison, M.F.; Reich, N.O. Oligomerization of DNMT3A Controls the Mechanism of de Novo DNA Methylation. J. Biol. Chem. 2011, 286, 41479–41488. [Google Scholar] [CrossRef] [PubMed]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA Methyltransferases Dnmt3a and Dnmt3b Are Essential for de Novo Methylation and Mammalian Development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef]
- Heyn, P.; Logan, C.V.; Fluteau, A.; Challis, R.C.; Auchynnikava, T.; Martin, C.-A.; Marsh, J.A.; Taglini, F.; Kilanowski, F.; Parry, D.A.; et al. Gain-of-Function DNMT3A Mutations Cause Microcephalic Dwarfism and Hypermethylation of Polycomb-Regulated Regions. Nat. Genet. 2019, 51, 96–105. [Google Scholar] [CrossRef]
- Putiri, E.L.; Robertson, K.D. Epigenetic Mechanisms and Genome Stability. Clin. Epigenet. 2011, 2, 299–314. [Google Scholar] [CrossRef]
- Xie, W.; Schultz, M.D.; Lister, R.; Hou, Z.; Rajagopal, N.; Ray, P.; Whitaker, J.W.; Tian, S.; Hawkins, R.D.; Leung, D.; et al. Epigenomic Analysis of Multilineage Differentiation of Human Embryonic Stem Cells. Cell 2013, 153, 1134–1148. [Google Scholar] [CrossRef]
- Jeong, M.; Sun, D.; Luo, M.; Huang, Y.; Challen, G.A.; Rodriguez, B.; Zhang, X.; Chavez, L.; Wang, H.; Hannah, R.; et al. Large Conserved Domains of Low DNA Methylation Maintained by Dnmt3a. Nat. Genet. 2014, 46, 17–23. [Google Scholar] [CrossRef]
- Li, Y.; Zheng, H.; Wang, Q.; Zhou, C.; Wei, L.; Liu, X.; Zhang, W.; Zhang, Y.; Du, Z.; Wang, X.; et al. Genome-Wide Analyses Reveal a Role of Polycomb in Promoting Hypomethylation of DNA Methylation Valleys. Genome Biol. 2018, 19, 18. [Google Scholar] [CrossRef] [PubMed]
- Boyer, L.A.; Plath, K.; Zeitlinger, J.; Brambrink, T.; Medeiros, L.A.; Lee, T.I.; Levine, S.S.; Wernig, M.; Tajonar, A.; Ray, M.K.; et al. Polycomb Complexes Repress Developmental Regulators in Murine Embryonic Stem Cells. Nature 2006, 441, 349–353. [Google Scholar] [CrossRef]
- Leeb, M.; Pasini, D.; Novatchkova, M.; Jaritz, M.; Helin, K.; Wutz, A. Polycomb Complexes Act Redundantly to Repress Genomic Repeats and Genes. Genes Dev. 2010, 24, 265–276. [Google Scholar] [CrossRef]
- Xie, H.; Xu, J.; Hsu, J.H.; Nguyen, M.; Fujiwara, Y.; Peng, C.; Orkin, S.H. Polycomb Repressive Complex 2 Regulates Normal Hematopoietic Stem Cell Function in a Developmental-Stage-Specific Manner. Cell Stem Cell 2014, 14, 68–80. [Google Scholar] [CrossRef]
- Kasinath, V.; Faini, M.; Poepsel, S.; Reif, D.; Feng, X.A.; Stjepanovic, G.; Aebersold, R.; Nogales, E. Structures of Human PRC2 with Its Cofactors AEBP2 and JARID2. Science 2018, 359, 940–944. [Google Scholar] [CrossRef]
- Jain, S.U.; Do, T.J.; Lund, P.J.; Rashoff, A.Q.; Diehl, K.L.; Cieslik, M.; Bajic, A.; Juretic, N.; Deshmukh, S.; Venneti, S.; et al. PFA Ependymoma-Associated Protein EZHIP Inhibits PRC2 Activity through a H3 K27M-like Mechanism. Nat. Commun. 2019, 10, 2146. [Google Scholar] [CrossRef]
- Viré, E.; Brenner, C.; Deplus, R.; Blanchon, L.; Fraga, M.; Didelot, C.; Morey, L.; Van Eynde, A.; Bernard, D.; Vanderwinden, J.-M.; et al. The Polycomb Group Protein EZH2 Directly Controls DNA Methylation. Nature 2006, 439, 871–874. [Google Scholar] [CrossRef]
- Hu, L.; Li, Z.; Cheng, J.; Rao, Q.; Gong, W.; Liu, M.; Shi, Y.G.; Zhu, J.; Wang, P.; Xu, Y. Crystal Structure of TET2-DNA Complex: Insight into TET-Mediated 5mC Oxidation. Cell 2013, 155, 1545–1555. [Google Scholar] [CrossRef] [PubMed]
- Agathocleous, M.; Meacham, C.E.; Burgess, R.J.; Piskounova, E.; Zhao, Z.; Crane, G.M.; Cowin, B.L.; Bruner, E.; Murphy, M.M.; Chen, W.; et al. Ascorbate Regulates Haematopoietic Stem Cell Function and Leukaemogenesis. Nature 2017, 549, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Hong, J.; Han, H.; Park, J.; Kim, D.; Park, H.; Ko, M.; Koh, Y.; Shin, D.-Y.; Yoon, S.-S. Decreased Vitamin C Uptake Mediated by SLC2A3 Promotes Leukaemia Progression and Impedes TET2 Restoration. Br. J. Cancer 2020, 122, 1445–1452. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.-S.; Kim, E.-J.; Park, U.-H.; Sin, H.-S.; Um, S.-J. Additional Sex Comb-like 1 (ASXL1), in Cooperation with SRC-1, Acts as a Ligand-Dependent Coactivator for Retinoic Acid Receptor. J. Biol. Chem. 2006, 281, 17588–17598. [Google Scholar] [CrossRef]
- Kolovos, P.; Nishimura, K.; Sankar, A.; Sidoli, S.; Cloos, P.A.; Helin, K.; Christensen, J. PR-DUB Maintains the Expression of Critical Genes through FOXK1/2- and ASXL1/2/3-Dependent Recruitment to Chromatin and H2AK119ub1 Deubiquitination. Genome Res. 2020, 30, 1119–1130. [Google Scholar] [CrossRef]
- Scheuermann, J.C.; de Ayala Alonso, A.G.; Oktaba, K.; Ly-Hartig, N.; McGinty, R.K.; Fraterman, S.; Wilm, M.; Muir, T.W.; Müller, J. Histone H2A Deubiquitinase Activity of the Polycomb Repressive Complex PR-DUB. Nature 2010, 465, 243–247. [Google Scholar] [CrossRef]
- de Napoles, M.; Mermoud, J.E.; Wakao, R.; Tang, Y.A.; Endoh, M.; Appanah, R.; Nesterova, T.B.; Silva, J.; Otte, A.P.; Vidal, M.; et al. Polycomb Group Proteins Ring1A/B Link Ubiquitylation of Histone H2A to Heritable Gene Silencing and X Inactivation. Dev. Cell 2004, 7, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Tamburri, S.; Lavarone, E.; Fernández-Pérez, D.; Conway, E.; Zanotti, M.; Manganaro, D.; Pasini, D. Histone H2AK119 Mono-Ubiquitination Is Essential for Polycomb-Mediated Transcriptional Repression. Mol. Cell 2020, 77, 840–856.e5. [Google Scholar] [CrossRef] [PubMed]
- Kweon, S.-M.; Chen, Y.; Moon, E.; Kvederaviciutė, K.; Klimasauskas, S.; Feldman, D.E. An Adversarial DNA N6-Methyladenine-Sensor Network Preserves Polycomb Silencing. Mol. Cell 2019, 74, 1138–1147.e6. [Google Scholar] [CrossRef] [PubMed]
- El Ghannam, D.; Taalab, M.M.; Ghazy, H.F.; Eneen, A.F. DNMT3A R882 Mutations in Patients with Cytogenetically Normal Acute Myeloid Leukemia and Myelodysplastic Syndrome. Blood Cells Mol. Dis. 2014, 53, 61–66. [Google Scholar] [CrossRef]
- Russler-Germain, D.A.; Spencer, D.H.; Young, M.A.; Lamprecht, T.L.; Miller, C.A.; Fulton, R.; Meyer, M.R.; Erdmann-Gilmore, P.; Townsend, R.R.; Wilson, R.K.; et al. The R882H DNMT3A Mutation Associated with AML Dominantly Inhibits Wild-Type DNMT3A by Blocking Its Ability to Form Active Tetramers. Cancer Cell 2014, 25, 442–454. [Google Scholar] [CrossRef]
- Hájková, H.; Marková, J.; Haškovec, C.; Sárová, I.; Fuchs, O.; Kostečka, A.; Cetkovský, P.; Michalová, K.; Schwarz, J. Decreased DNA Methylation in Acute Myeloid Leukemia Patients with DNMT3A Mutations and Prognostic Implications of DNA Methylation. Leuk. Res. 2012, 36, 1128–1133. [Google Scholar] [CrossRef]
- Qu, Y.; Lennartsson, A.; Gaidzik, V.I.; Deneberg, S.; Karimi, M.; Bengtzén, S.; Höglund, M.; Bullinger, L.; Döhner, K.; Lehmann, S. Differential Methylation in CN-AML Preferentially Targets Non-CGI Regions and Is Dictated by DNMT3A Mutational Status and Associated with Predominant Hypomethylation of HOX Genes. Epigenetics 2014, 9, 1108–1119. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, X.; Wang, X.Q.D.; Su, J.; Putluri, N.; Zhou, T.; Qu, Y.; Jeong, M.; Guzman, A.; Rosas, C.; et al. Dnmt3a Loss and Idh2 Neomorphic Mutations Mutually Potentiate Malignant Hematopoiesis. Blood 2020, 135, 845–856. [Google Scholar] [CrossRef]
- Traina, F.; Visconte, V.; Elson, P.; Tabarroki, A.; Jankowska, A.M.; Hasrouni, E.; Sugimoto, Y.; Szpurka, H.; Makishima, H.; O’Keefe, C.L.; et al. Impact of Molecular Mutations on Treatment Response to DNMT Inhibitors in Myelodysplasia and Related Neoplasms. Leukemia 2014, 28, 78–87. [Google Scholar] [CrossRef]
- Kuendgen, A.; Müller-Thomas, C.; Lauseker, M.; Haferlach, T.; Urbaniak, P.; Schroeder, T.; Brings, C.; Wulfert, M.; Meggendorfer, M.; Hildebrandt, B.; et al. Efficacy of Azacitidine Is Independent of Molecular and Clinical Characteristics—An Analysis of 128 Patients with Myelodysplastic Syndromes or Acute Myeloid Leukemia and a Review of the Literature. Oncotarget 2018, 9, 27882–27894. [Google Scholar] [CrossRef]
- Weissmann, S.; Alpermann, T.; Grossmann, V.; Kowarsch, A.; Nadarajah, N.; Eder, C.; Dicker, F.; Fasan, A.; Haferlach, C.; Haferlach, T.; et al. Landscape of TET2 Mutations in Acute Myeloid Leukemia. Leukemia 2012, 26, 934–942. [Google Scholar] [CrossRef]
- Langemeijer, S.M.C.; Kuiper, R.P.; Berends, M.; Knops, R.; Aslanyan, M.G.; Massop, M.; Stevens-Linders, E.; van Hoogen, P.; van Kessel, A.G.; Raymakers, R.A.P.; et al. Acquired Mutations in TET2 Are Common in Myelodysplastic Syndromes. Nat. Genet. 2009, 41, 838–842. [Google Scholar] [CrossRef] [PubMed]
- Moran-Crusio, K.; Reavie, L.; Shih, A.; Abdel-Wahab, O.; Ndiaye-Lobry, D.; Lobry, C.; Figueroa, M.E.; Vasanthakumar, A.; Patel, J.; Zhao, X.; et al. Tet2 Loss Leads to Increased Hematopoietic Stem Cell Self-Renewal and Myeloid Transformation. Cancer Cell 2011, 20, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Ko, M.; Huang, Y.; Jankowska, A.M.; Pape, U.J.; Tahiliani, M.; Bandukwala, H.S.; An, J.; Lamperti, E.D.; Koh, K.P.; Ganetzky, R.; et al. Impaired Hydroxylation of 5-Methylcytosine in Myeloid Cancers with Mutant TET2. Nature 2010, 468, 839–843. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 Mutations Result in a Hypermethylation Phenotype, Disrupt TET2 Function, and Impair Hematopoietic Differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef]
- Scourzic, L.; Mouly, E.; Bernard, O.A. TET Proteins and the Control of Cytosine Demethylation in Cancer. Genome Med. 2015, 7, 9. [Google Scholar] [CrossRef]
- Bejar, R.; Lord, A.; Stevenson, K.; Bar-Natan, M.; Pérez-Ladaga, A.; Zaneveld, J.; Wang, H.; Caughey, B.; Stojanov, P.; Getz, G.; et al. TET2 Mutations Predict Response to Hypomethylating Agents in Myelodysplastic Syndrome Patients. Blood 2014, 124, 2705–2712. [Google Scholar] [CrossRef]
- Itzykson, R.; Kosmider, O.; Cluzeau, T.; Mansat-De Mas, V.; Dreyfus, F.; Beyne-Rauzy, O.; Quesnel, B.; Vey, N.; Gelsi-Boyer, V.; Raynaud, S.; et al. Impact of TET2 Mutations on Response Rate to Azacitidine in Myelodysplastic Syndromes and Low Blast Count Acute Myeloid Leukemias. Leukemia 2011, 25, 1147–1152. [Google Scholar] [CrossRef]
- Shih, A.H.; Meydan, C.; Shank, K.; Garrett-Bakelman, F.E.; Ward, P.S.; Intlekofer, A.; Nazir, A.; Stein, E.; Knapp, K.; Glass, J.; et al. Combination Targeted Therapy to Disrupt Aberrant Oncogenic Signaling and Reverse Epigenetic Dysfunction in IDH2- and TET2-Mutant Acute Myeloid Leukemia. Cancer Discov. 2017, 7, 494–505. [Google Scholar] [CrossRef]
- Cedena, M.T.; Rapado, I.; Santos-Lozano, A.; Ayala, R.; Onecha, E.; Abaigar, M.; Such, E.; Ramos, F.; Cervera, J.; Díez-Campelo, M.; et al. Mutations in the DNA Methylation Pathway and Number of Driver Mutations Predict Response to Azacitidine in Myelodysplastic Syndromes. Oncotarget 2017, 8, 106948–106961. [Google Scholar] [CrossRef]
- Sasaki, K.; Kanagal-Shamanna, R.; Montalban-Bravo, G.; Assi, R.; Jabbour, E.; Ravandi, F.; Kadia, T.; Pierce, S.; Takahashi, K.; Nogueras Gonzalez, G.; et al. Impact of the Variant Allele Frequency of ASXL1, DNMT3A, JAK2, TET2, TP53, and NPM1 on the Outcomes of Patients with Newly Diagnosed Acute Myeloid Leukemia. Cancer 2020, 126, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, W.; Wang, M.; Li, Y.; Wang, X.; Yang, E.; Ming, J.; Quan, R.; Hu, X. Prognostic Value of ASXL1 Mutations in Patients with Primary Myelofibrosis and Its Relationship with Clinical Features: A Meta-Analysis. Ann. Hematol. 2021, 100, 465–479. [Google Scholar] [CrossRef]
- Yang, H.; Kurtenbach, S.; Guo, Y.; Lohse, I.; Durante, M.A.; Li, J.; Li, Z.; Al-Ali, H.; Li, L.; Chen, Z.; et al. Gain of Function of ASXL1 Truncating Protein in the Pathogenesis of Myeloid Malignancies. Blood 2018, 131, 328–341. [Google Scholar] [CrossRef]
- Bera, R.; Chiu, M.-C.; Huang, Y.-J.; Lin, T.-H.; Kuo, M.-C.; Shih, L.-Y. RUNX1 Mutations Promote Leukemogenesis of Myeloid Malignancies in ASXL1-Mutated Leukemia. J. Hematol. Oncol. 2019, 12, 104. [Google Scholar] [CrossRef]
- D’Altri, T.; Wilhelmson, A.S.; Schuster, M.B.; Wenzel, A.; Kalvisa, A.; Pundhir, S.; Hansen, A.M.; Porse, B.T. The ASXL1-G643W Variant Accelerates the Development of CEBPA Mutant Acute Myeloid Leukemia. Haematologica 2021, 106, 1000–1007. [Google Scholar] [CrossRef] [PubMed]
- Tobiasson, M.; McLornan, D.P.; Karimi, M.; Dimitriou, M.; Jansson, M.; Ben Azenkoud, A.; Jädersten, M.; Lindberg, G.; Abdulkadir, H.; Kulasekararaj, A.; et al. Mutations in Histone Modulators Are Associated with Prolonged Survival during Azacitidine Therapy. Oncotarget 2016, 7, 22103–22115. [Google Scholar] [CrossRef]
- Stasik, S.; Middeke, J.M.; Kramer, M.; Röllig, C.; Krämer, A.; Scholl, S.; Hochhaus, A.; Crysandt, M.; Brümmendorf, T.H.; Naumann, R.; et al. EZH2 Mutations and Impact on Clinical Outcome: An Analysis in 1604 Patients with Newly Diagnosed Acute Myeloid Leukemia. Haematologica 2020, 105, e228–e231. [Google Scholar] [CrossRef] [PubMed]
- Chase, A.; Cross, N.C.P. Aberrations of EZH2 in Cancer. Clin. Cancer Res. 2011, 17, 2613–2618. [Google Scholar] [CrossRef]
- Xu, F.; Liu, L.; Chang, C.-K.; He, Q.; Wu, L.-Y.; Zhang, Z.; Shi, W.-H.; Guo, J.; Zhu, Y.; Zhao, Y.-S.; et al. Genomic Loss of EZH2 Leads to Epigenetic Modifications and Overexpression of the HOX Gene Clusters in Myelodysplastic Syndrome. Oncotarget 2016, 7, 8119–8130. [Google Scholar] [CrossRef]
- Hasegawa, N.; Oshima, M.; Sashida, G.; Matsui, H.; Koide, S.; Saraya, A.; Wang, C.; Muto, T.; Takane, K.; Kaneda, A.; et al. Impact of Combinatorial Dysfunctions of Tet2 and Ezh2 on the Epigenome in the Pathogenesis of Myelodysplastic Syndrome. Leukemia 2017, 31, 861–871. [Google Scholar] [CrossRef]
- Saygin, C.; Hirsch, C.; Przychodzen, B.; Sekeres, M.A.; Hamilton, B.K.; Kalaycio, M.; Carraway, H.E.; Gerds, A.T.; Mukherjee, S.; Nazha, A.; et al. Mutations in DNMT3A, U2AF1, and EZH2 Identify Intermediate-Risk Acute Myeloid Leukemia Patients with Poor Outcome after CR1. Blood Cancer J 2018, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Li, X. The Role of Histone Methyltransferase EZH2 in Myelodysplastic Syndromes. Expert Rev. Hematol. 2012, 5, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Dufva, O.; Pölönen, P.; Brück, O.; Keränen, M.A.I.; Klievink, J.; Mehtonen, J.; Huuhtanen, J.; Kumar, A.; Malani, D.; Siitonen, S.; et al. Immunogenomic Landscape of Hematological Malignancies. Cancer Cell 2020, 38, 380–399.e13. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Urabe, A.; Chi, S.; Minami, Y. The Immuno-Oncology and Genomic Aspects of DNA-Hypomethylating Therapeutics in Acute Myeloid Leukemia. Int. J. Mol. Sci. 2023, 24, 3727. https://doi.org/10.3390/ijms24043727
Urabe A, Chi S, Minami Y. The Immuno-Oncology and Genomic Aspects of DNA-Hypomethylating Therapeutics in Acute Myeloid Leukemia. International Journal of Molecular Sciences. 2023; 24(4):3727. https://doi.org/10.3390/ijms24043727
Chicago/Turabian StyleUrabe, Akiko, SungGi Chi, and Yosuke Minami. 2023. "The Immuno-Oncology and Genomic Aspects of DNA-Hypomethylating Therapeutics in Acute Myeloid Leukemia" International Journal of Molecular Sciences 24, no. 4: 3727. https://doi.org/10.3390/ijms24043727
APA StyleUrabe, A., Chi, S., & Minami, Y. (2023). The Immuno-Oncology and Genomic Aspects of DNA-Hypomethylating Therapeutics in Acute Myeloid Leukemia. International Journal of Molecular Sciences, 24(4), 3727. https://doi.org/10.3390/ijms24043727