
ZnT2 Is Critical for TLR4-Mediated Cytokine Expression in Colonocytes and Modulates Mucosal Inflammation in Mice

Abstract
1. Introduction
2. Results
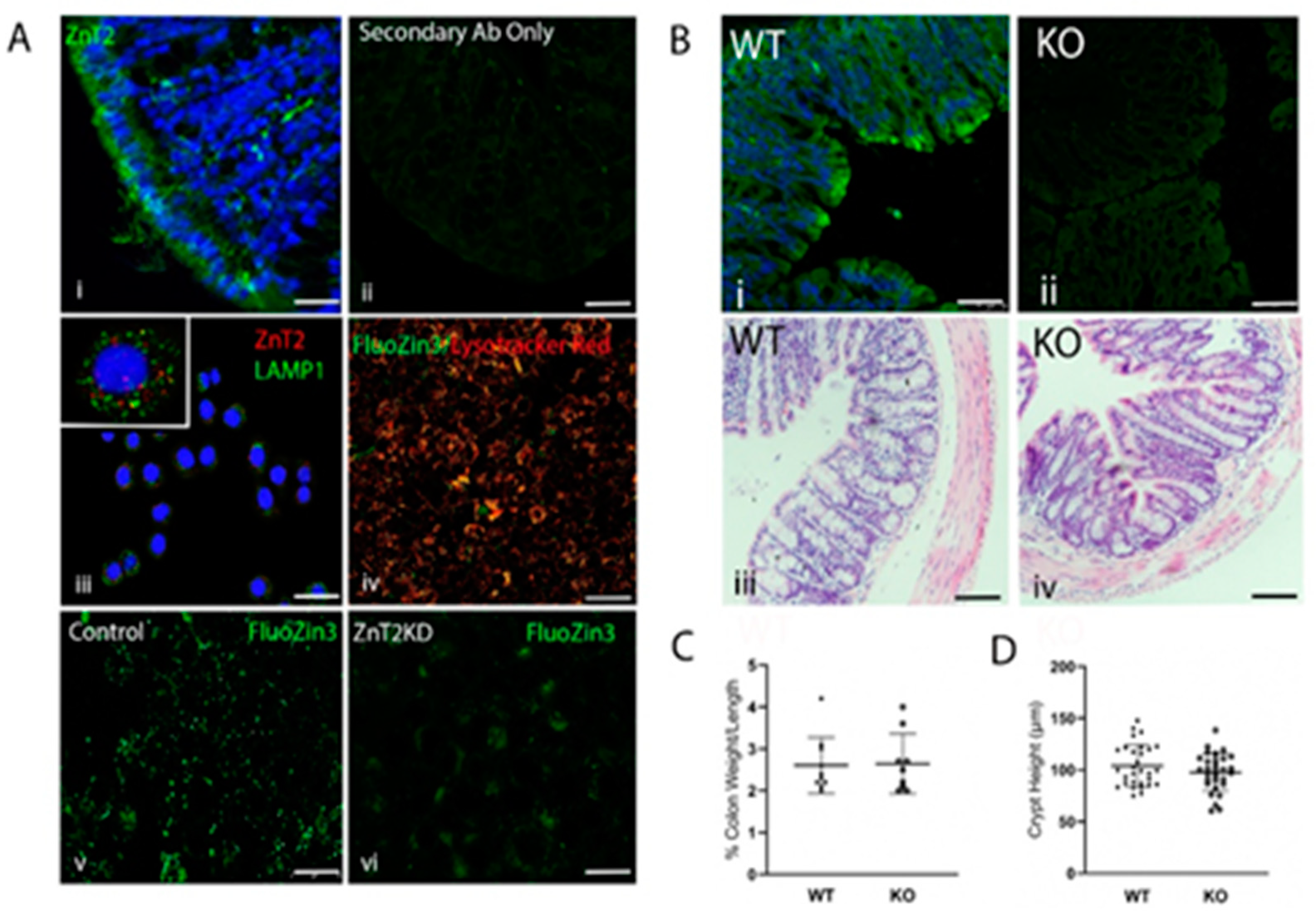
2.1. ZnT2 Is Expressed in the Colon and Localized to Acidic Vesicles in Colonocytes
2.2. Loss of ZnT2 Does Not Cause Colonic Inflammation or Pathology under Baseline Conditions
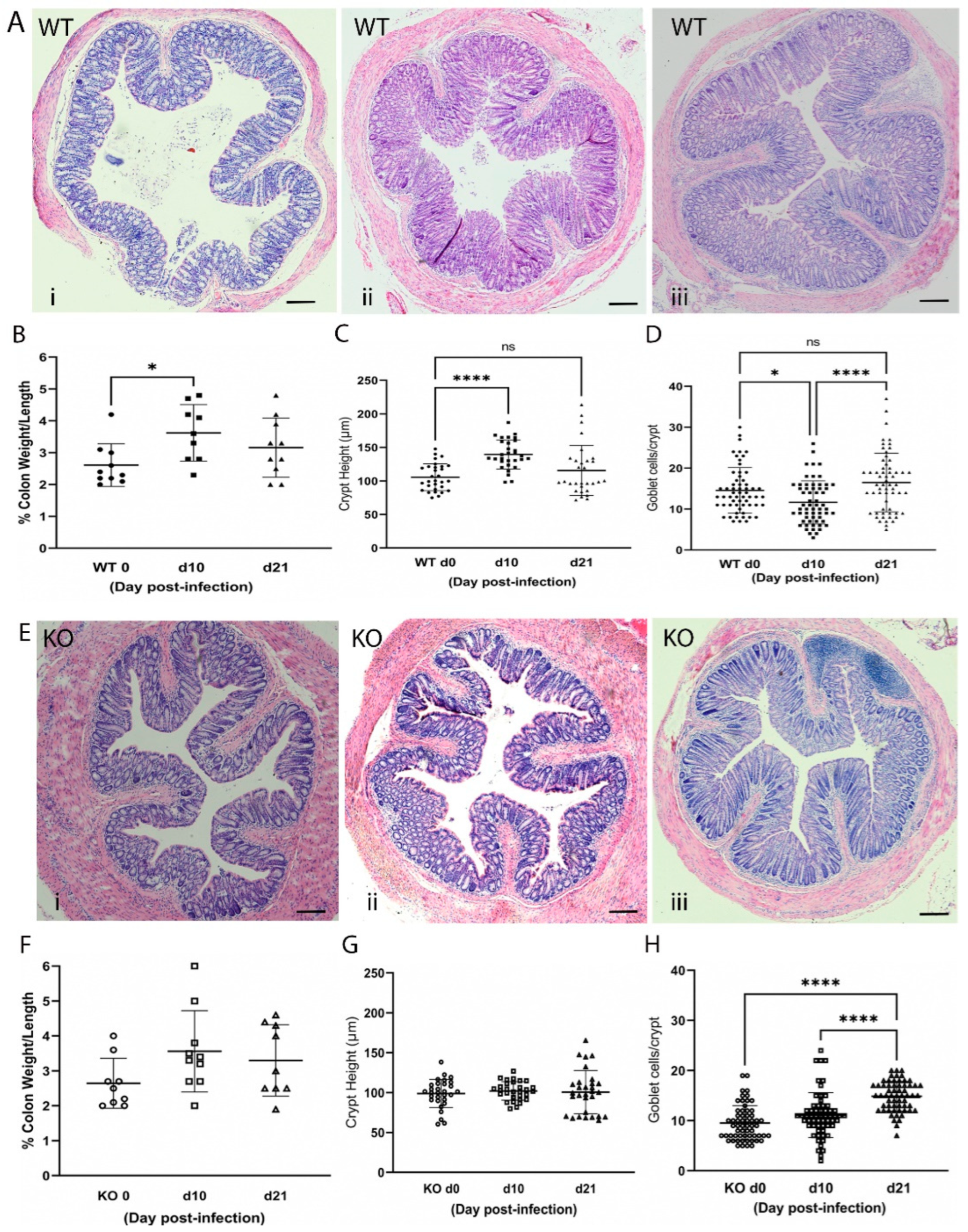
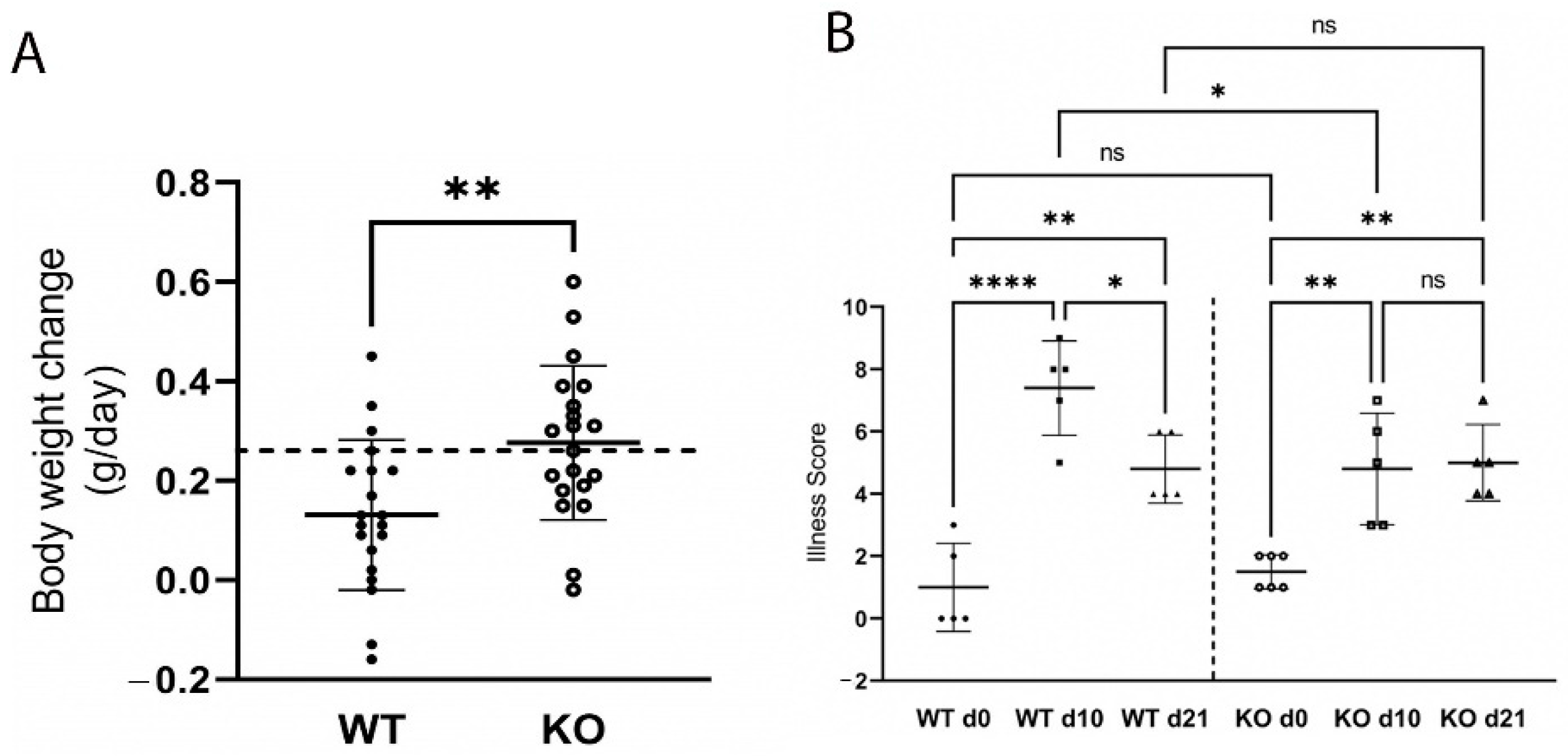
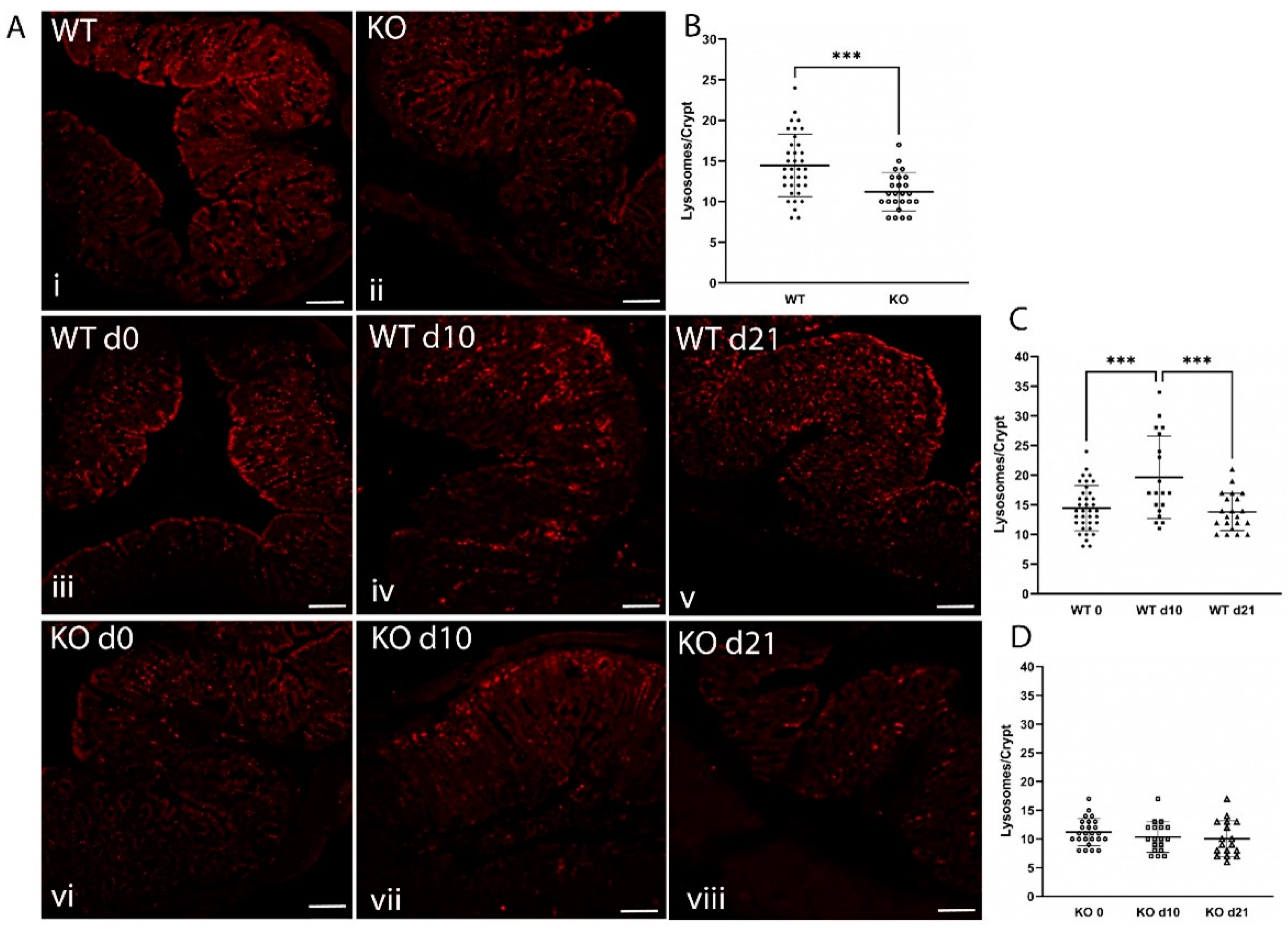
2.3. Loss of ZnT2 Impairs Response to Infectious Colitis
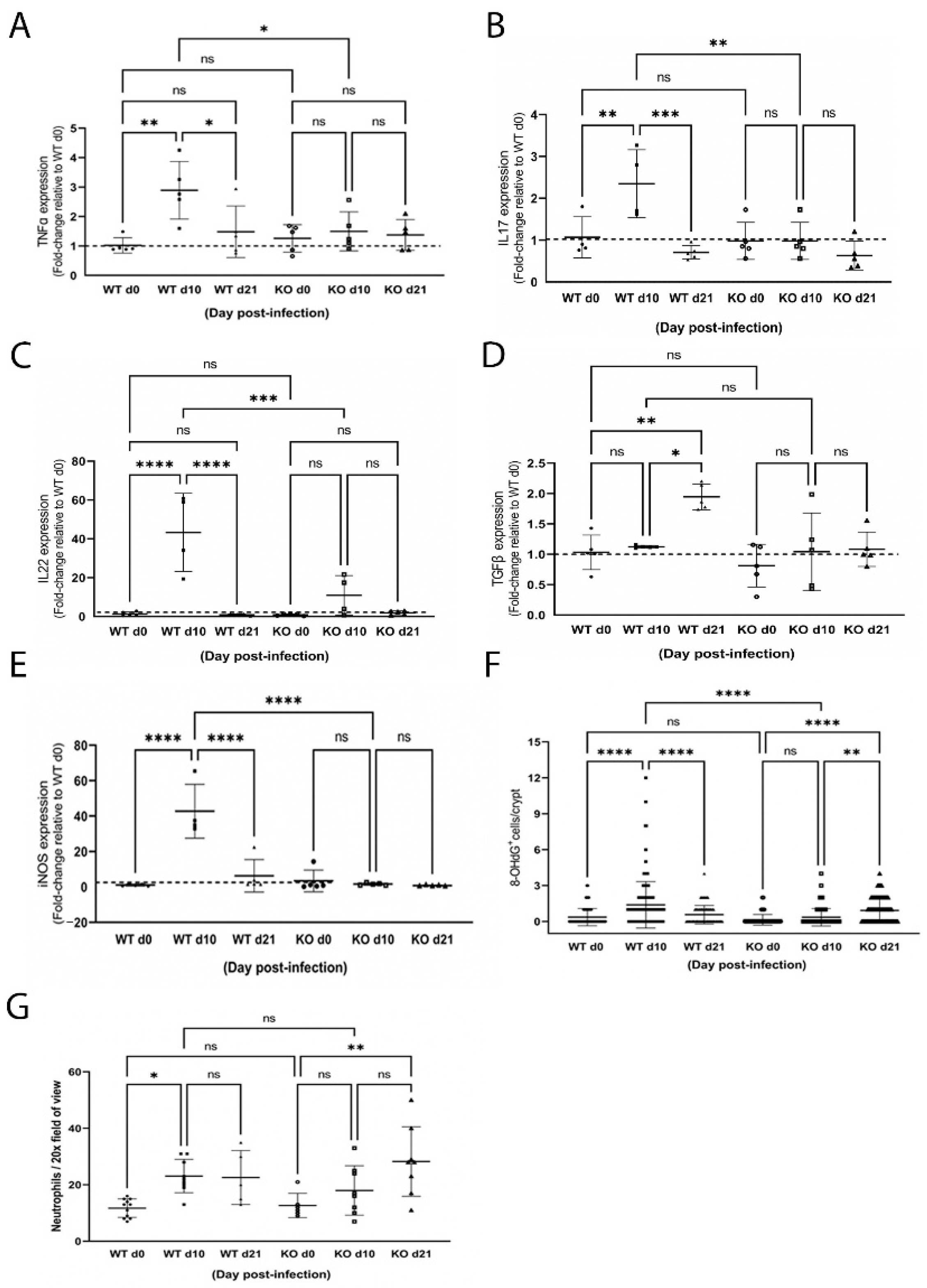
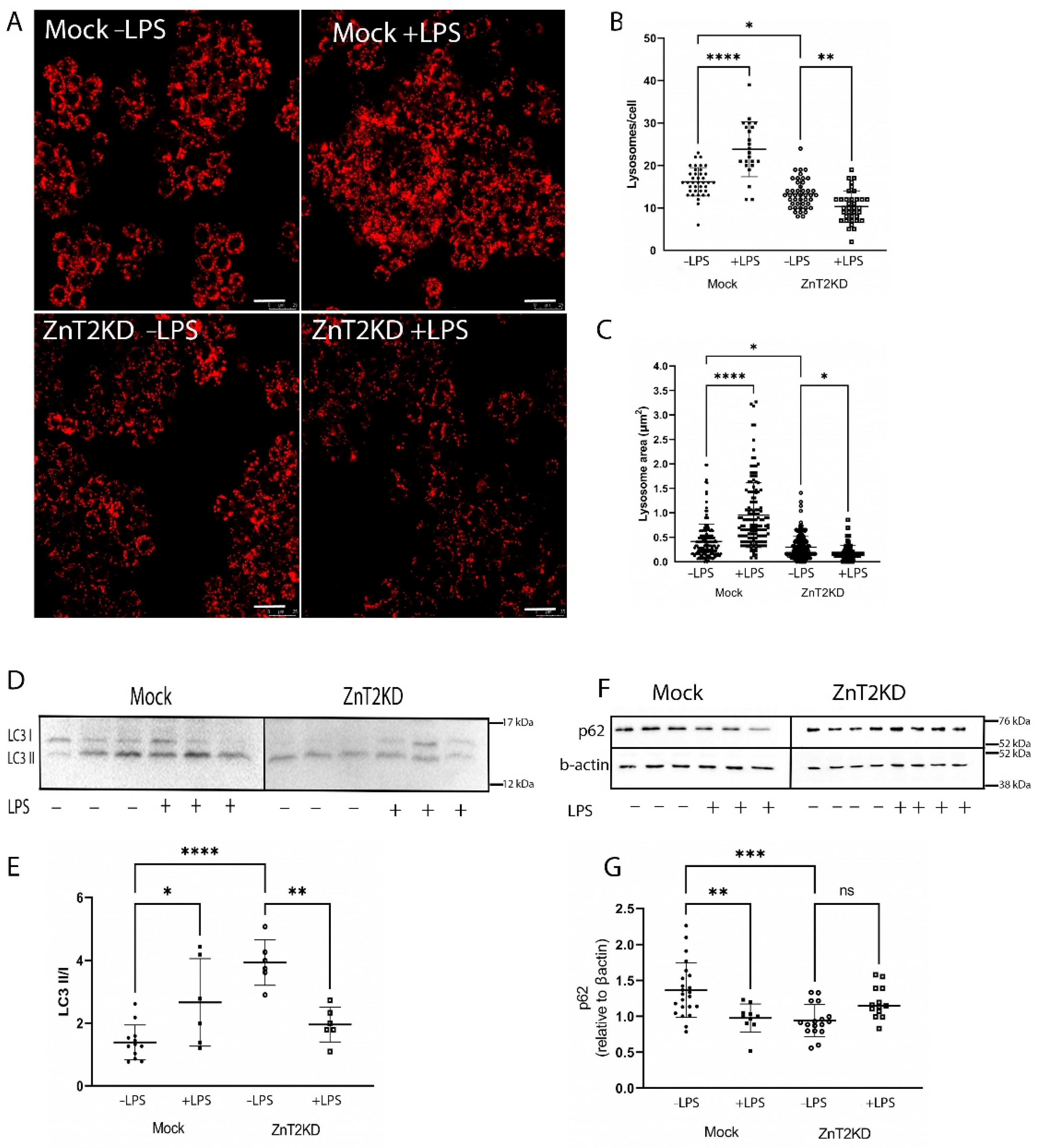
2.4. ZnT2 Is Important to Initiation of the Proinflammatory Response
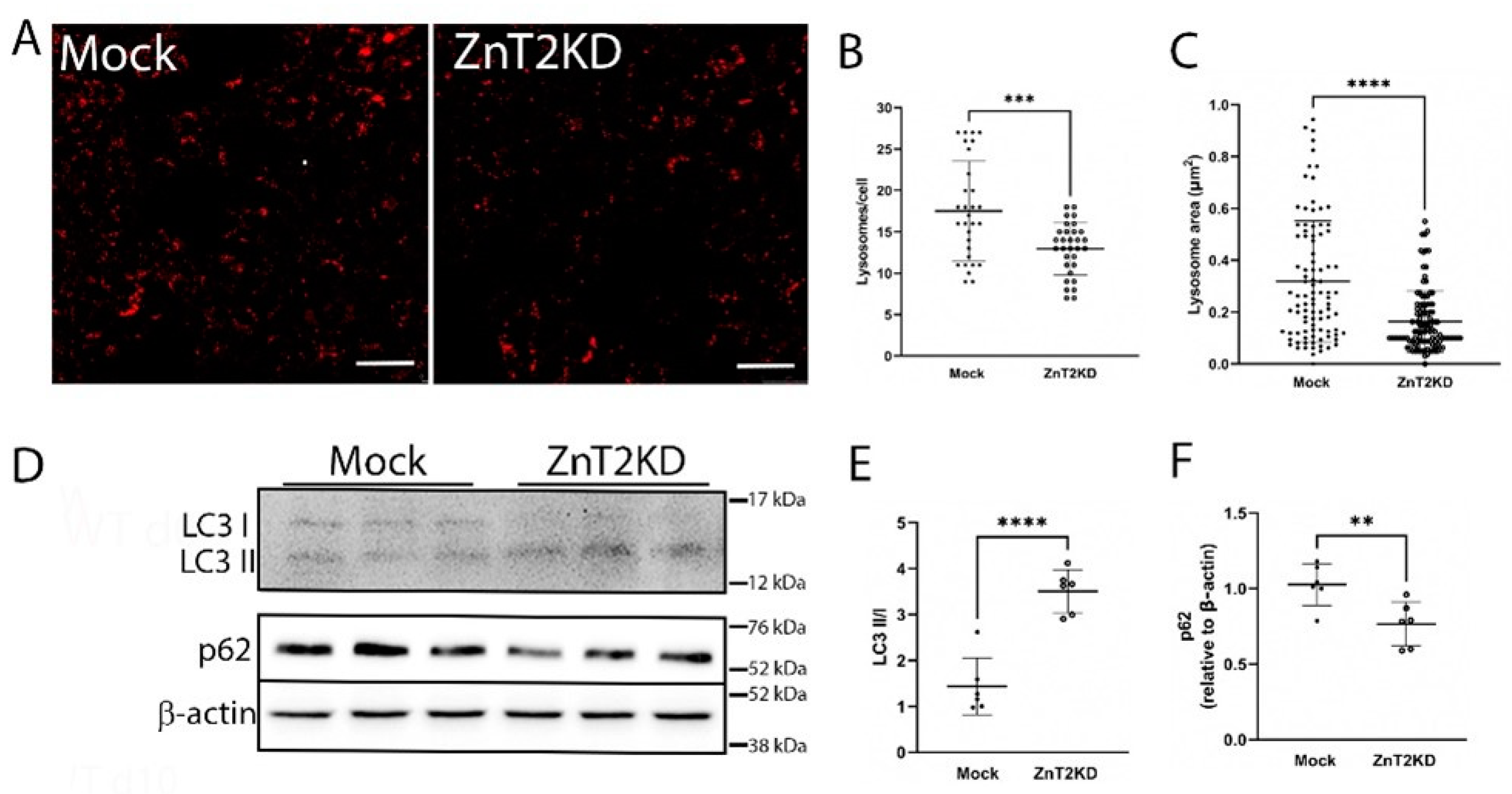
2.5. ZnT2 Is Important to Activation of Lysosome Biogenesis and Recovery
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Intestinal Infection Model
4.3. Histological Analysis
4.4. Illness Score
4.5. Cell Culture and Transfection
4.6. Immunofluorescent Imaging
4.6.1. Tissues
4.6.2. Cells
4.6.3. Lysosome Number
4.6.4. Intracellular Zn pools
4.7. Gene Expression
4.8. Immunoblotting
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Navaneethan, U.; Giannella, R.A. Infectious colitis. Curr. Opin. Gastroenterol. 2011, 27, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Nataro, J.P.; Kaper, J.B. Diarrheagenic escherichia coli. Clin. Microbiol. Rev. 1998, 11, 142–201. [Google Scholar] [CrossRef] [PubMed]
- Plastaras, L.; Vuitton, L.; Badet, N.; Koch, S.; Di Martino, V.; Delabrousse, E. Acute colitis: Differential diagnosis using multidetector ct. Clin. Radiol. 2015, 70, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Palmiter, R.D.; Cole, T.B.; Findley, S.D. ZnT-2, a mammalian protein that confers resistance to zinc by facilitating vesicular sequestration. EMBO J. 1996, 15, 1784–1791. [Google Scholar] [CrossRef]
- Rivera, O.C.; Geddes, D.T.; Barber-Zucker, S.; Zarivach, R.; Gagnon, A.; Soybel, D.I.; Kelleher, S.L. A common genetic variant in zinc transporter ZnT2 (thr288ser) is present in women with low milk volume and alters lysosome function and cell energetics. Am. J. Physiol. Cell. Physiol. 2020, 318, C1166–C1177. [Google Scholar] [CrossRef]
- Rivera, O.C.; Hennigar, S.R.; Kelleher, S.L. ZnT2 is critical for lysosome acidification and biogenesis during mammary gland involution. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 315, R323–R335. [Google Scholar] [CrossRef]
- Lee, S.; Hennigar, S.R.; Alam, S.; Nishida, K.; Kelleher, S.L. Essential role for zinc transporter 2 (ZnT2)-mediated zinc transport in mammary gland development and function during lactation. J. Biol. Chem. 2015, 290, 13064–13078. [Google Scholar] [CrossRef]
- Podany, A.B.; Wright, J.; Lamendella, R.; Soybel, D.I.; Kelleher, S.L. ZnT2-mediated zinc import into Paneth cell granules is necessary for coordinated secretion and paneth cell function in mice. Cell. Mol. Gastroenterol. Hepatol. 2016, 2, 369–383. [Google Scholar] [CrossRef]
- Pfaffl, M.W.; Windisch, W. Influence of zinc deficiency on the mrna expression of zinc transporters in adult rats. J. Trace. Elem. Med. Biol. 2003, 17, 97–106. [Google Scholar] [CrossRef]
- Golan, Y.; Lehvy, A.; Horev, G.; Assaraf, Y.G. High proportion of transient neonatal zinc deficiency causing alleles in the general population. J. Cell. Mol. Med. 2019, 23, 828–840. [Google Scholar] [CrossRef]
- Kelleher, S.L.; Alam, S.; Rivera, O.C.; Barber-Zucker, S.; Zarivach, R.; Wagatsuma, T.; Kambe, T.; Soybel, D.I.; Wright, J.; Lamendella, R. Loss-of-function SLC30A2 mutants are associated with gut dysbiosis and alterations in intestinal gene expression in preterm infants. Gut Microbes. 2022, 14, 2014739. [Google Scholar] [CrossRef] [PubMed]
- Hennigar, S.R.; Kelleher, S.L. Tnfα post-translationally targets ZnT2 to accumulate zinc in lysosomes. J. Cell. Physiol. 2015, 230, 2345–2350. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, C.L.; Hartland, E.L. Citrobacter rodentium infection model for the analysis of bacterial pathogenesis and mucosal immunology. Methods Mol. Biol. 2018, 1725, 77–89. [Google Scholar]
- Brady, O.A.; Martina, J.A.; Puertollano, R. Emerging roles for TFEB in the immune response and inflammation. Autophagy 2018, 14, 181–189. [Google Scholar] [CrossRef]
- Tang, X.; Zhu, Y. TLR4 signaling promotes immune escape of human colon cancer cells by inducing immunosuppressive cytokines and apoptosis resistance. Oncol. Res. 2012, 20, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Pietrzak, A.; Wierzbicki, M.; Wiktorska, M.; Brzezińska-Błaszczyk, E. Surface tlr2 and tlr4 expression on mature rat mast cells can be affected by some bacterial components and proinflammatory cytokines. Mediat. Inflamm. 2011, 2011, 427473. [Google Scholar] [CrossRef]
- Hu, L.; Cheng, S.; Li, Y.; Geng, S.; Ma, Y.; Han, X. Chitosan-zn chelate downregulates TLR4-nf-κb signal pathway of inflammatory response and cell death-associated proteins compared to inorganic zinc. Biol. Trace. Elem. Res. 2018, 184, 92–98. [Google Scholar] [CrossRef]
- Nardinocchi, L.; Pantisano, V.; Puca, R.; Porru, M.; Aiello, A.; Grasselli, A.; Leonetti, C.; Safran, M.; Rechavi, G.; Givol, D.; et al. Zinc downregulates HIF-1α and inhibits its activity in tumor cells in vitro and in vivo. PLoS ONE. 2010, 5, e15048. [Google Scholar] [CrossRef]
- Zhao, L.; Ma, R.; Zhang, L.; Yuan, X.; Wu, J.; He, L.; Liu, G.; Du, R. Inhibition of HIF-1a-mediated TLR4 activation decreases apoptosis and promotes angiogenesis of placental microvascular endothelial cells during severe pre-eclampsia pathogenesis. Placenta 2019, 83, 8–16. [Google Scholar] [CrossRef]
- Devisscher, L.; Hindryckx, P.; Olievier, K.; Peeters, H.; De Vos, M.; Laukens, D. Inverse correlation between metallothioneins and hypoxia-inducible factor 1 alpha in colonocytes and experimental colitis. Biochem. Biophys. Res. Commun. 2011, 416, 307–312. [Google Scholar] [CrossRef]
- Shi, Y.-J.; Hu, S.-J.; Zhao, Q.-Q.; Liu, X.-S.; Liu, C.; Wang, H. Toll-like receptor 4 (tlr4) deficiency aggravates dextran sulfate sodium (dss)-induced intestinal injury by down-regulating IL6, CCL2 and CSF3. Ann. Transl. Med. 2019, 7, 713. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Petris, M.J.; Peck, S.C. Separation of zinc-dependent and zinc-independent events during early LPS-stimulated TLR4 signaling in macrophage cells. FEBS Lett. 2014, 588, 2928–2935. [Google Scholar] [CrossRef] [PubMed]
- Haase, H.; Ober-Blöbaum, J.L.; Engelhardt, G.; Hebel, S.; Heit, A.; Heine, H.; Rink, L. Zinc signals are essential for lipopolysaccharide-induced signal transduction in monocytes. J. Immunol. 2008, 181, 6491–6502. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Tymoszuk, P.; Haschka, D.; Heeke, S.; Dichtl, S.; Petzer, V.; Seifert, M.; Hilbe, R.; Sopper, S.; Talasz, H.; et al. Salmonella utilizes zinc to subvert antimicrobial host defense of macrophages via modulation of nf-κb signaling. Infect. Immun. 2017, 85, e00418-17. [Google Scholar] [CrossRef] [PubMed]
- Bolte, L.A.; Vila, A.V.; Imhann, F.; Collij, V.; Gacesa, R.; Peters, V.; Wijmenga, C.; Kurilshikov, A.; Campmans-Kuijpers, M.J.E.; Fu, J.; et al. Long-term dietary patterns are associated with pro-inflammatory and anti-inflammatory features of the gut microbiome. Gut 2021, 70, 1287–1298. [Google Scholar] [CrossRef]
- Zhao, R.; Huang, F.; Shen, G.X. Dose-responses relationship in glucose lowering and gut dysbiosis to saskatoon berry powder supplementation in high fat-high sucrose diet-induced insulin resistant mice. Microorganisms 2021, 9, 1553. [Google Scholar] [CrossRef]
- Wang, Y.; Devkota, S.; Musch, M.W.; Jabri, B.; Nagler, C.; Antonopoulos, D.A.; Chervonsky, A.; Chang, E.B. Regional mucosa-associated microbiota determine physiological expression of TLR2 and TLR4 in murine colon. PLoS ONE 2010, 5, e13607. [Google Scholar] [CrossRef]
- Demirci, M.; Bahar Tokman, H.; Taner, Z.; Keskin, F.E.; Çağatay, P.; Ozturk Bakar, Y.; Özyazar, M.; Kiraz, N.; Kocazeybek, B.S. Bacteroidetes and firmicutes levels in gut microbiota and effects of hosts TLR2/TLR4 gene expression levels in adult type 1 diabetes patients in istanbul, Turkey. J. Diabetes Complicat. 2020, 34, 107449. [Google Scholar] [CrossRef]
- Cuesta, C.M.; Pascual, M.; Pérez-Moraga, R.; Rodríguez-Navarro, I.; García-García, F.; Ureña-Peralta, J.R.; Guerri, C. TLR4 deficiency affects the microbiome and reduces intestinal dysfunctions and inflammation in chronic alcohol-fed mice. Int. J. Mol. Sci. 2021, 22, 12830. [Google Scholar] [CrossRef]
- Sanjuan, M.A.; Dillon, C.P.; Tait, S.W.G.; Moshiach, S.; Dorsey, F.; Connell, S.; Komatsu, M.; Tanaka, K.; Cleveland, J.L.; Withoff, S.; et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 2007, 450, 1253–1257. [Google Scholar] [CrossRef]
- Xu, Y.; Jagannath, C.; Liu, X.D.; Sharafkhaneh, A.; Kolodziejska, K.E.; Eissa, N.T. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity 2007, 27, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.S.; Kehrl, J.H. Traf6 and a20 regulate lysine 63-linked ubiquitination of beclin-1 to control TLR4-induced autophagy. Sci. Signal. 2010, 3, ra42. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Klionsky, D.J. Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Dev. Cell. 2004, 6, 463–477. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Alam, S.; Hennigar, S.R.; Gallagher, C.; Soybel, D.I.; Kelleher, S.L. Exome sequencing of SLC30A2 identifies novel loss- and gain-of-function variants associated with breast cell dysfunction. J. Mammary Gland Biol. Neoplasia 2015, 20, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Nishida, K.; Hasegawa, A.; Yamasaki, S.; Uchida, R.; Ohashi, W.; Kurashima, Y.; Kunisawa, J.; Kimura, S.; Iwanaga, T.; Watarai, H.; et al. Mast cells play role in wound healing through the znt2/gpr39/il-6 axis. Sci. Rep. 2019, 9, 10842. [Google Scholar] [CrossRef]
- Bouladoux, N.; Harrison, O.J.; Belkaid, Y. The mouse model of infection with citrobacter rodentium. Curr. Protoc. Immunol. 2017, 119, 19.15.1–19.15.25. [Google Scholar] [CrossRef]
- Kang, E.; Zhou, G.; Yousefi, M.; Cayrol, R.; Xia, J.; Gruenheid, S. Loss of disease tolerance during citrobacter rodentium infection is associated with impaired epithelial differentiation and hyperactivation of t cell responses. Sci. Rep. 2018, 8, 847. [Google Scholar] [CrossRef]
- Seo, Y.A.; Lopez, V.; Kelleher, S.L. A histidine-rich motif mediates mitochondrial localization of znt2 to modulate mitochondrial function. Am. J. Physiol. Cell. Physiol. 2011, 300, C1479-89. [Google Scholar] [CrossRef]
- Dunn, K.W.; Kamocka, M.M.; McDonald, J.H. A practical guide to evaluating colocalization in biological microscopy. Am. J. Physiol Cell. Physiol. 2011, 300, C723–C742. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−δδct method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer | Sequence (5′ to 3′) |
|---|---|---|
| Mouse TNFA | Forward Reverse | TTGTCTACTCCCAGGTTCTCT GAGGTTGACTTTCTCCTGGTATG |
| Mouse TGFB1 | Forward Reverse | GGTGGTATACTGAGACACCTTG CCCAAGGAAAGGTAGGTGATAG |
| Mouse IL6 | Forward Reverse | GATAAGCTGGAGTCACAGAAGG TTGCCGAGTAGATCTCAAAGTG |
| Mouse IL17A | Forward Reverse | CCACGTTTCTCAGCAAACTTAC TGTGGAGGGCAGACAATTC |
| Mouse IL22 | Forward Reverse | CGACCAGAACATCCAGAAGAA GAGACATAAACAGCAGGTCCA |
| Mouse NOS2 | Forward Reverse | CCGCCGCTCTAATACTTA TTCATCAAGGAATTATACAGGAA |
| Mouse TLR4 | Forward Reverse | CCAGGTGAGCTGTAGCATTTA GAGCAAACAGCAGAGGAAGA |
| Mouse SLC30A2 | Forward Reverse | GGTTACAGATGCAAGAGGTAAGA TGGCCTGCAATGACAGATATAA |
| Mouse ACTB | Forward Reverse | AGGGAAATCGTGCGTGACAT GAACCGCTCGTTGCCAATAG |
| Human TLR4 | Forward Reverse | TCTAAGCCAGTGTCTCCATTTAC GAGGTCTGCCAACAAGGTATTA |
| Human CXCL8 | Forward Reverse | CTTGGCAGCCTTCCTGATTT GGGTGGAAAGGTTTGGAGTATG |
| Human TNFA | Forward Reverse | CTCTTCTGCCTGCTGCACTTTG ATGGGCTACAGGCTTGTCACTC |
| Human SLC30A2 | Forward Reverse | CCTGCGAGTGGTAGAACATT CTCAGCGGGTATAAAGCTAGTG |
| Human ACTB | Forward Reverse | CACCATTGGCAATGAGCGGTTC AGGTCTTTGCGGATGTCCACGT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McGourty, K.; Vijayakumar, R.; Wu, T.; Gagnon, A.; Kelleher, S.L. ZnT2 Is Critical for TLR4-Mediated Cytokine Expression in Colonocytes and Modulates Mucosal Inflammation in Mice. Int. J. Mol. Sci. 2022, 23, 11467. https://doi.org/10.3390/ijms231911467
McGourty K, Vijayakumar R, Wu T, Gagnon A, Kelleher SL. ZnT2 Is Critical for TLR4-Mediated Cytokine Expression in Colonocytes and Modulates Mucosal Inflammation in Mice. International Journal of Molecular Sciences. 2022; 23(19):11467. https://doi.org/10.3390/ijms231911467
Chicago/Turabian StyleMcGourty, Katherine, Ramya Vijayakumar, Tong Wu, Annie Gagnon, and Shannon L. Kelleher. 2022. "ZnT2 Is Critical for TLR4-Mediated Cytokine Expression in Colonocytes and Modulates Mucosal Inflammation in Mice" International Journal of Molecular Sciences 23, no. 19: 11467. https://doi.org/10.3390/ijms231911467
APA StyleMcGourty, K., Vijayakumar, R., Wu, T., Gagnon, A., & Kelleher, S. L. (2022). ZnT2 Is Critical for TLR4-Mediated Cytokine Expression in Colonocytes and Modulates Mucosal Inflammation in Mice. International Journal of Molecular Sciences, 23(19), 11467. https://doi.org/10.3390/ijms231911467