
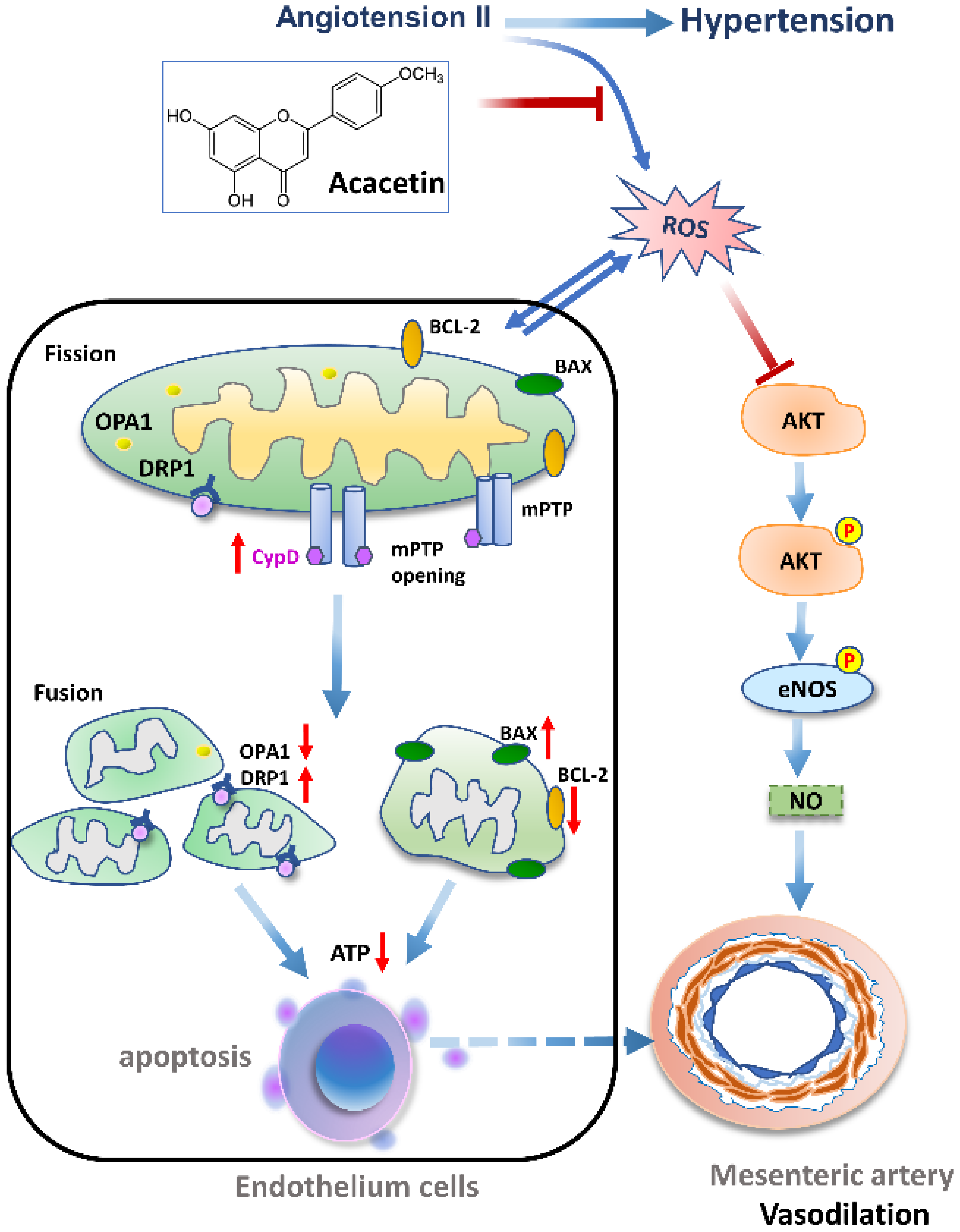
Restoration of Mitochondrial Function Is Essential in the Endothelium-Dependent Vasodilation Induced by Acacetin in Hypertensive Rats

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
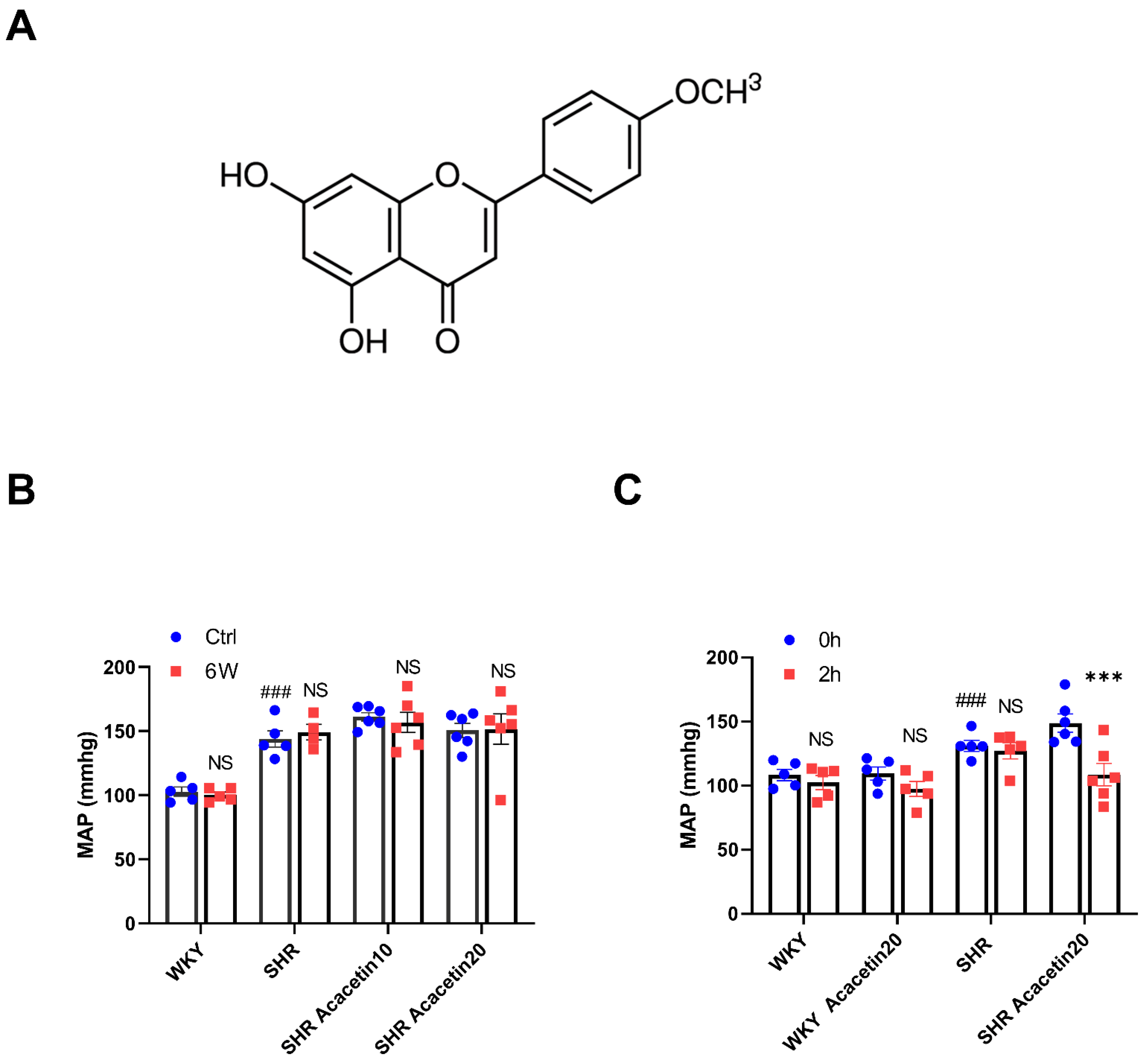
2.1. Acacetin Administered Intraperitoneally Reduced MAP in SHR
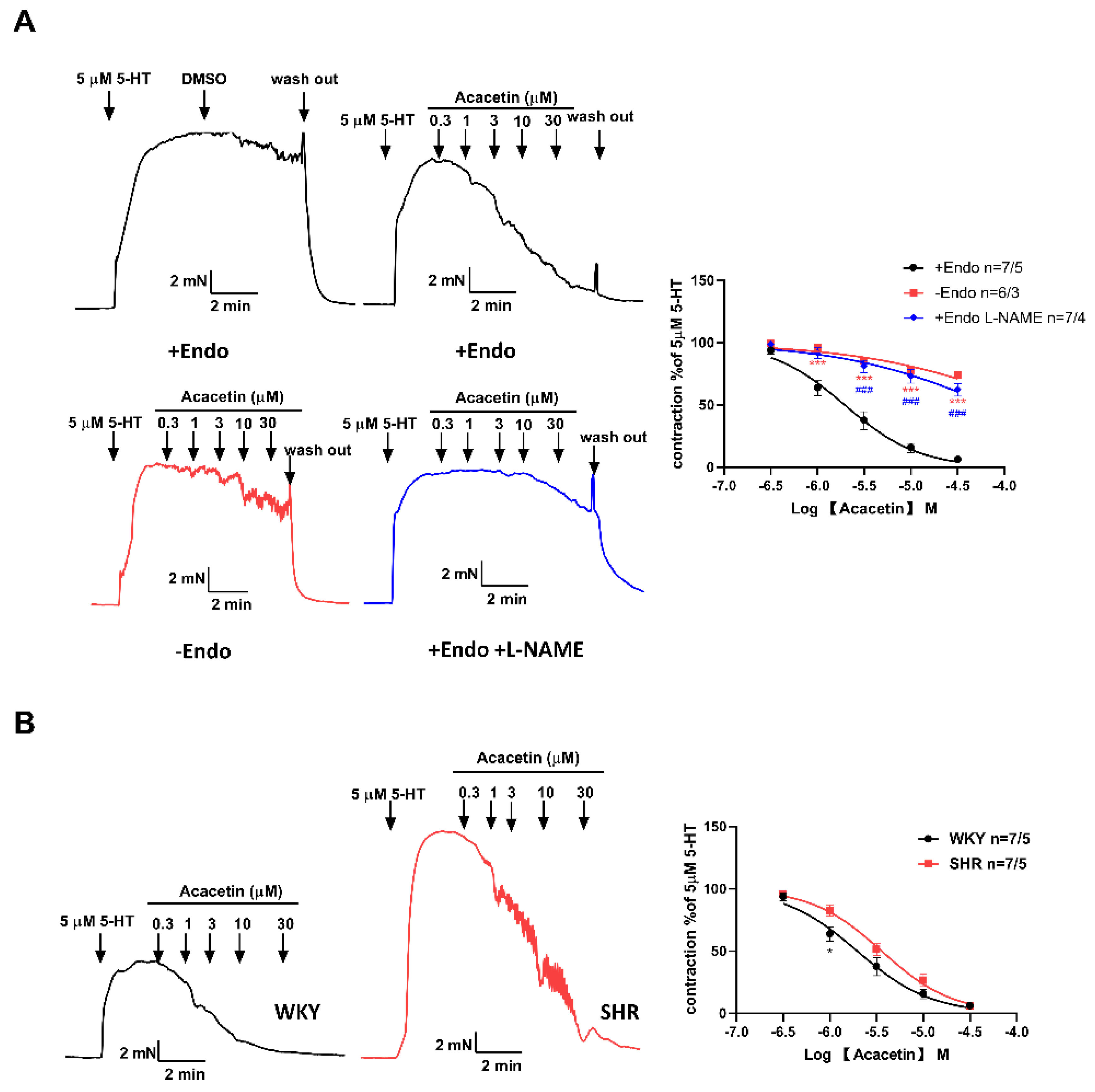
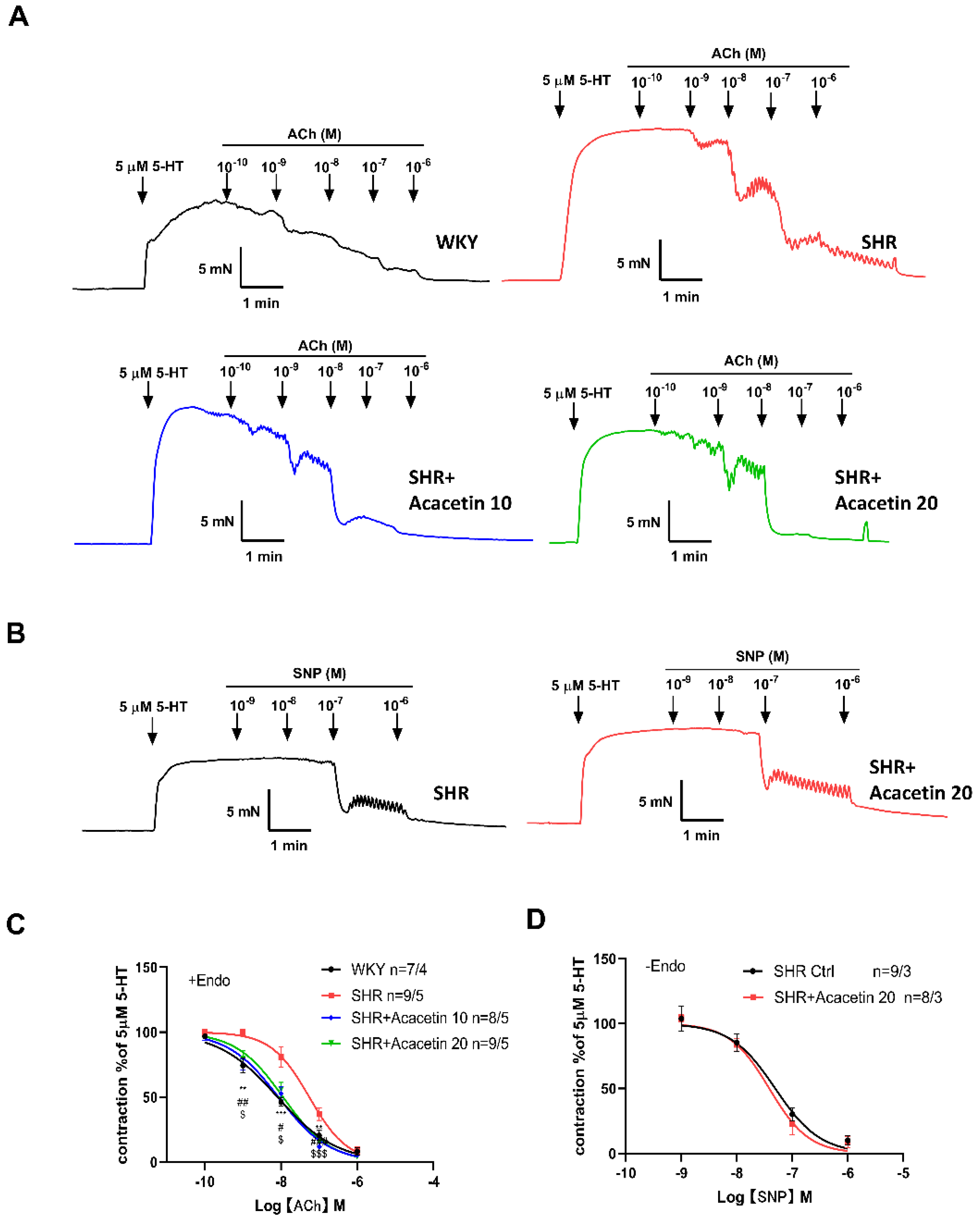
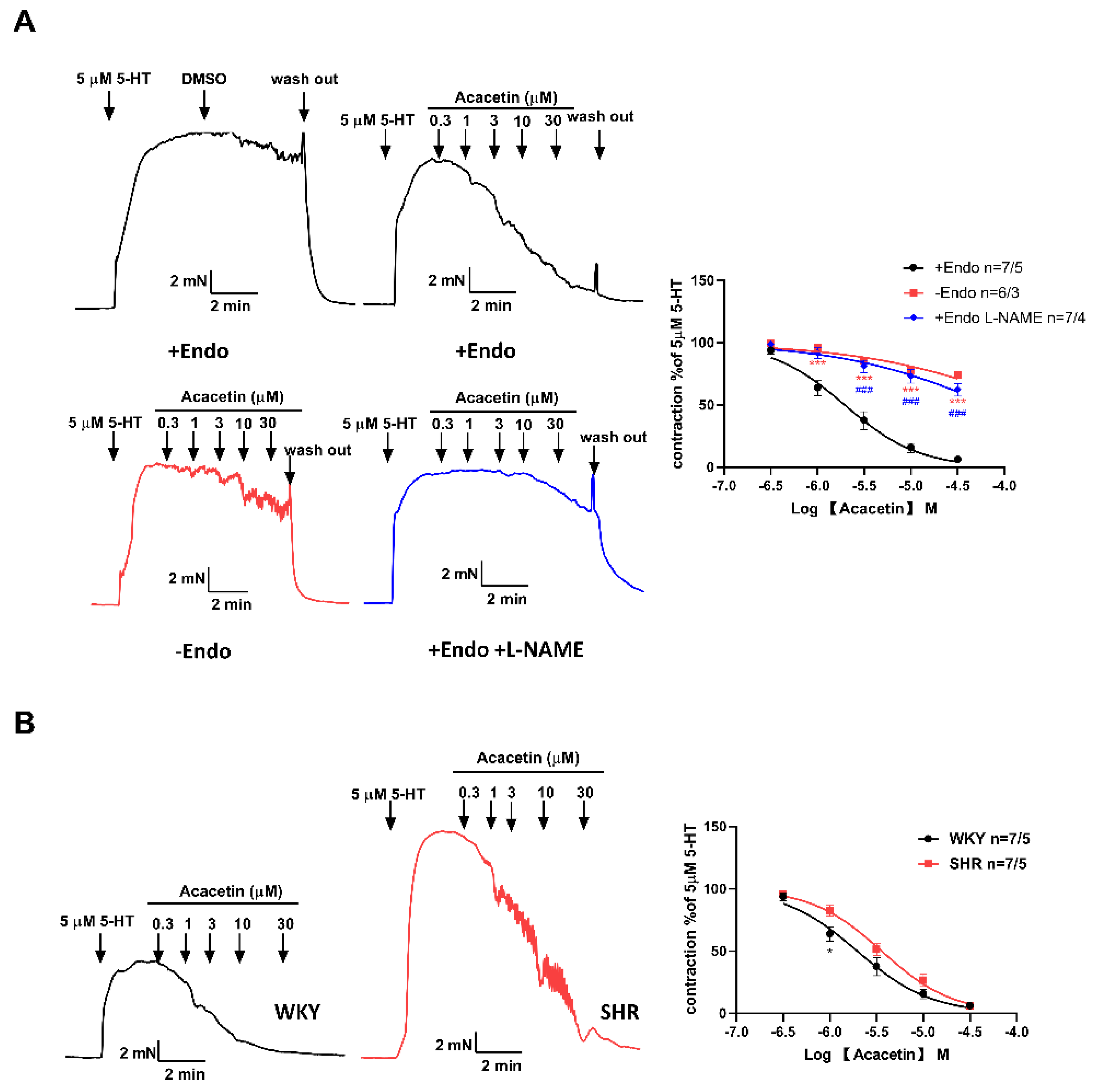
2.2. Acacetin Improved Endothelium-Dependent Vasodilation in SHR
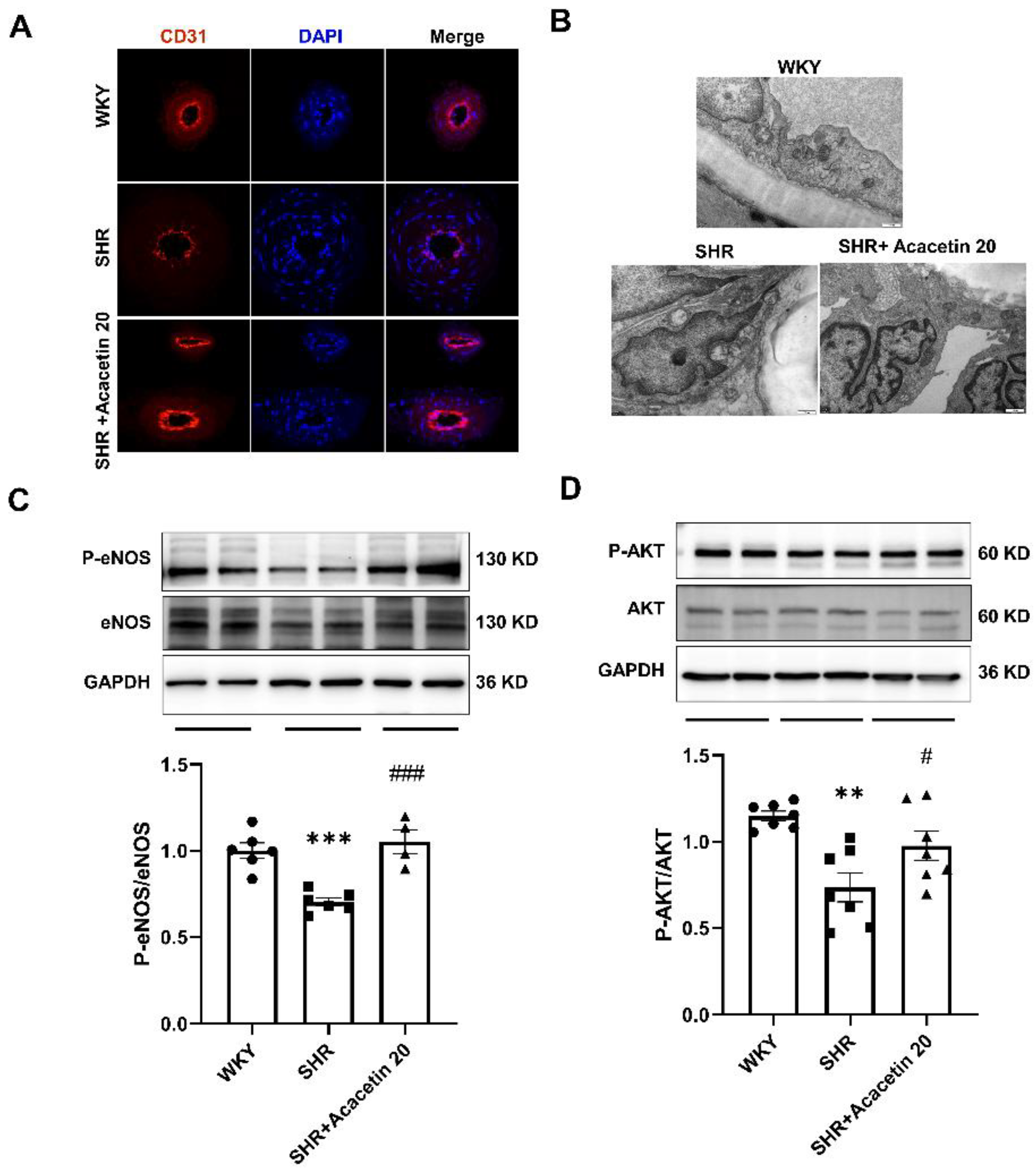
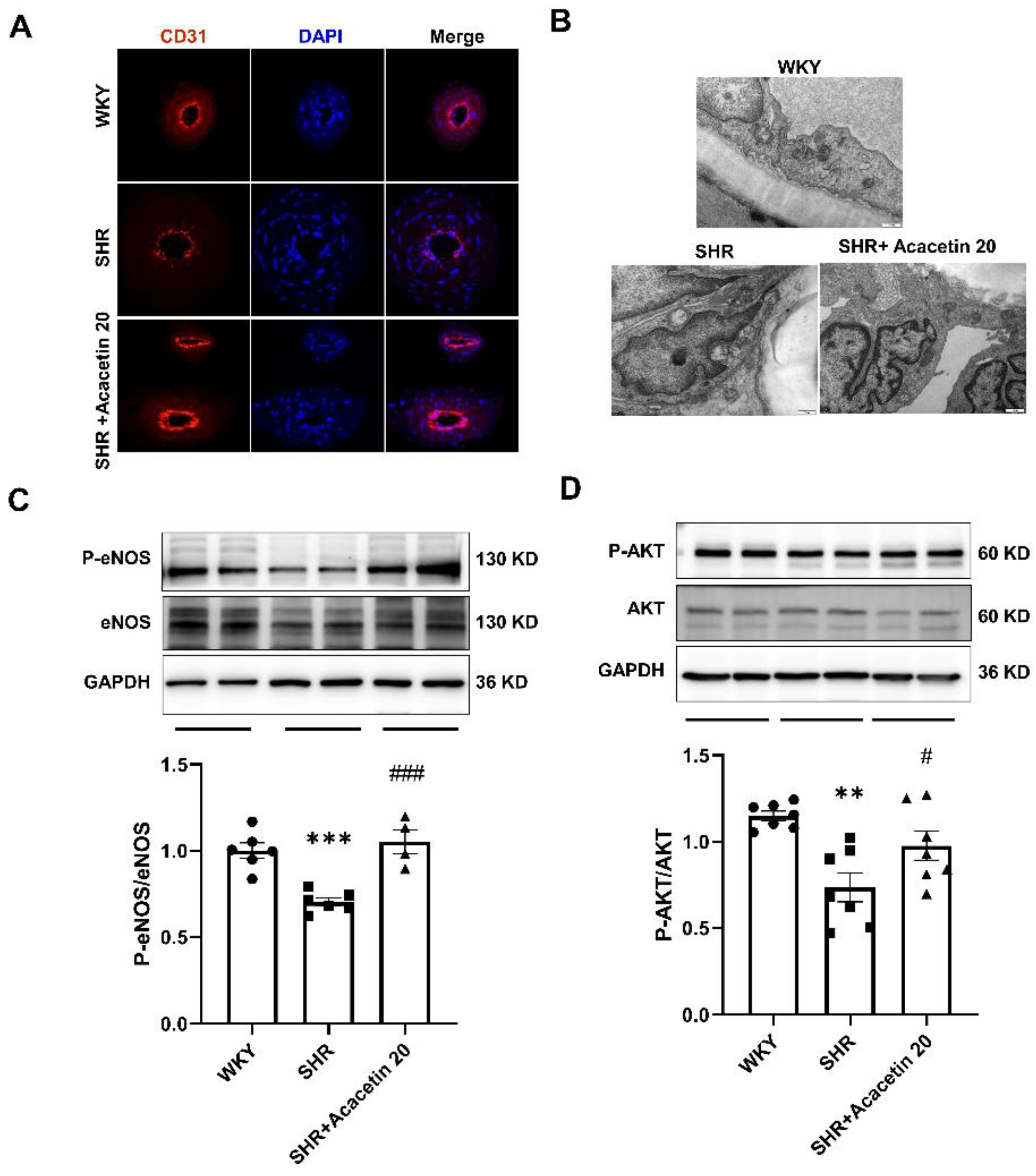
2.3. Acacetin Protected the Endothelium against Mitochondrial Injury in SHR through Activating the AKT/eNOS Pathway
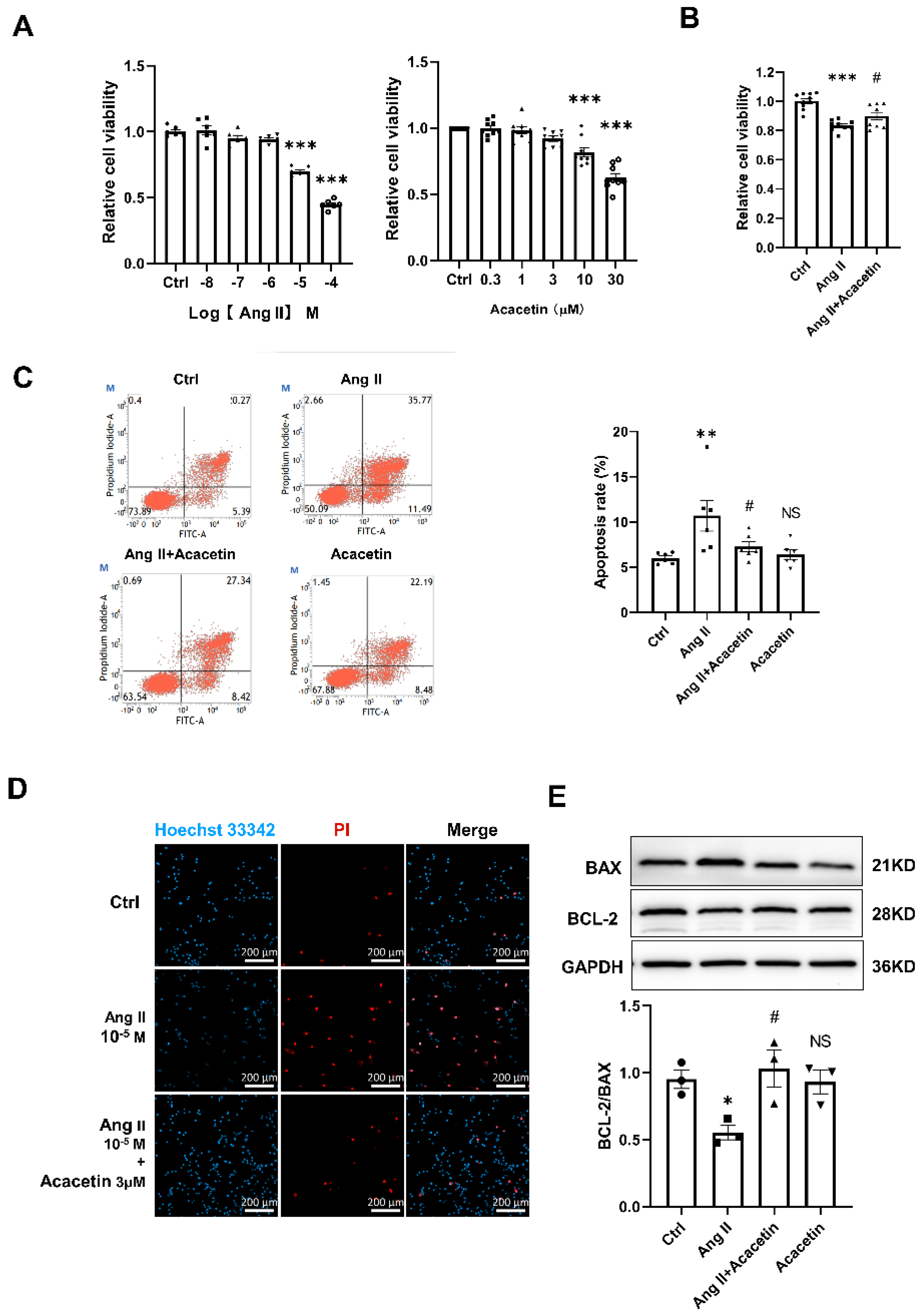
2.4. Acacetin Suppressed Ang II- Induced Apoptosis in HUVECs
2.5. Acacetin Protected against Mitochondria-Dependent Apoptosis Induced by Ang II via Regulating the Opening of mPTP and Mitochondrial Dynamics
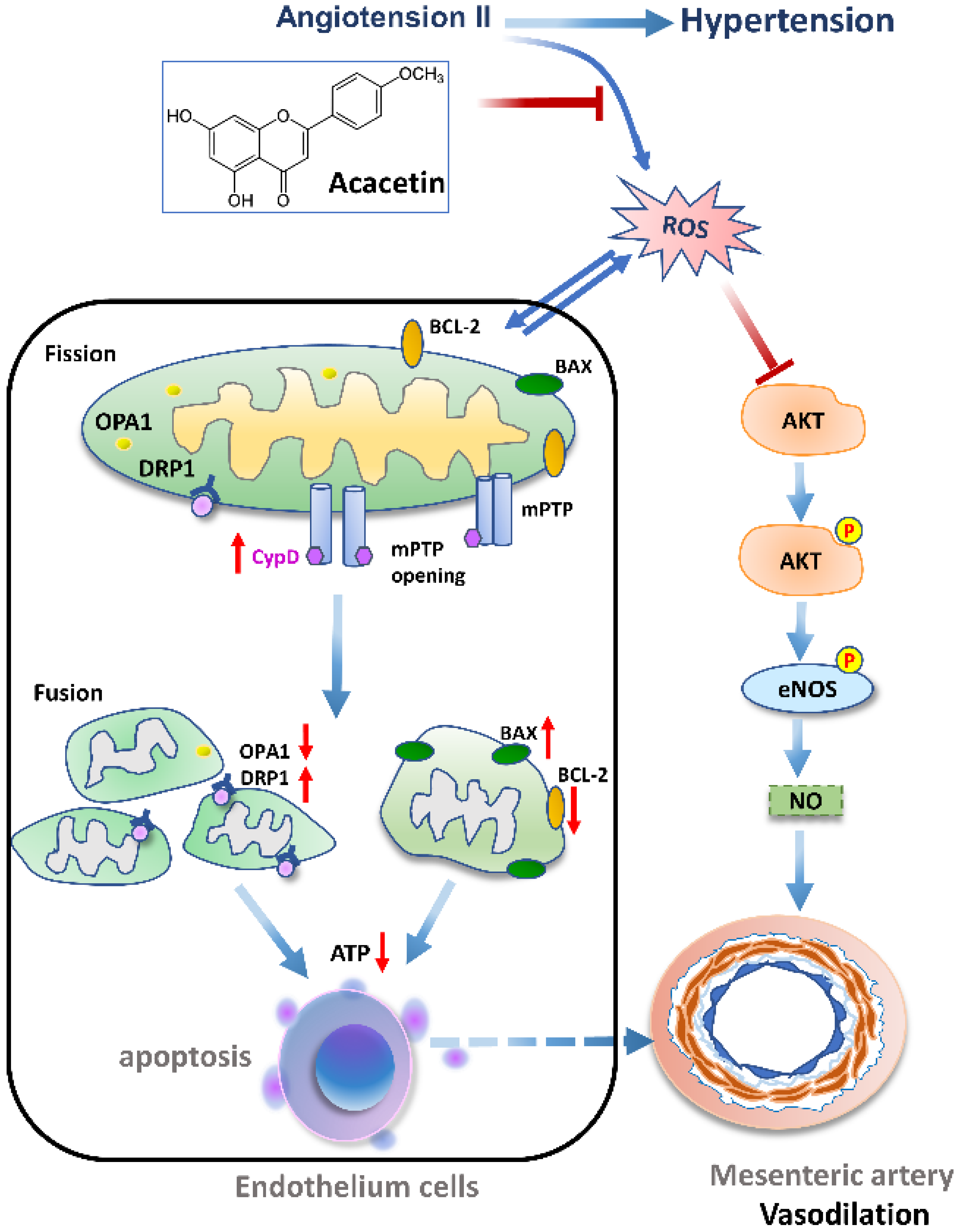
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Detection of Endothelium-Dependent and Independent Vasorelaxation
4.3. Immunofluorescence Staining and Transmission Electron Microscopy
4.4. Cell Culture
4.5. Immunoblotting
4.6. Cell Viability and Apoptosis Assays
4.7. Measurement of Intracellular ROS, ATP and mPTP Opening
4.8. Chemicals and Drugs
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gallo, G.; Volpe, M.; Savoia, C. Endothelial Dysfunction in Hypertension: Current Concepts and Clinical Implications. Front. Med. 2021, 8, 798958. [Google Scholar] [CrossRef] [PubMed]
- Luan, Y.; Ren, K.D.; Luan, Y.; Chen, X.; Yang, Y. Mitochondrial Dynamics: Pathogenesis and Therapeutic Targets of Vascular Diseases. Front. Cardiovasc. Med. 2021, 8, 770574. [Google Scholar] [CrossRef] [PubMed]
- Caja, S.; Enriquez, J.A. Mitochondria in endothelial cells: Sensors and integrators of environmental cues. Redox Biol. 2017, 12, 821–827. [Google Scholar] [CrossRef] [PubMed]
- Uchikado, Y.; Ikeda, Y.; Ohishi, M. Current Understanding of the Pivotal Role of Mitochondrial Dynamics in Cardiovascular Diseases and Senescence. Front. Cardiovasc. Med. 2022, 9, 905072. [Google Scholar] [CrossRef]
- Itani, H.A.; Dikalova, A.E.; McMaster, W.G.; Nazarewicz, R.R.; Bikineyeva, A.T.; Harrison, D.G.; Dikalov, S.I. Mitochondrial Cyclophilin D in Vascular Oxidative Stress and Hypertension. Hypertension 2016, 67, 1218–1227. [Google Scholar] [CrossRef]
- Duan, C.; Kuang, L.; Hong, C.; Xiang, X.; Liu, J.; Li, Q.; Peng, X.; Zhou, Y.; Wang, H.; Liu, L.; et al. Mitochondrial Drp1 recognizes and induces excessive mPTP opening after hypoxia through BAX-PiC and LRRK2-HK2. Cell Death Dis. 2021, 12, 1050. [Google Scholar] [CrossRef]
- Lahera, V.; Heras, N.D.L.; Farre, A.L.; Manucha, W.; Ferder, L. Role of Mitochondrial Dysfunction in Hypertension and Obesity. Curr. Hypertens. Rep. 2017, 19, 11. [Google Scholar] [CrossRef]
- Yang, D.; Liu, H.-Q.; Liu, F.-Y.; Guo, Z.; An, P.; Wang, M.-Y.; Yang, Z.; Fan, D.; Tang, Q.-Z. Mitochondria in Pathological Cardiac Hypertrophy Research and Therapy. Front. Cardiovasc. Med. 2021, 8, 822969. [Google Scholar] [CrossRef]
- Arillo, Z.C.; Castilla, C.Y.; Fernández, J.M.R.; Macías, R.Y.; Martínez, A.M. Pulmonary hypertension as a sign of onset of multiple mitochondrial dysfunction syndrome. An. Pediatr. 2021, 94, 185–187. [Google Scholar] [CrossRef]
- Singh, S.; Gupta, P.; Meena, A.; Luqman, S. A flavone with diverse therapeutic potential in cancer, inflammation, infections and other metabolic disorders. Food Chem. Toxicol. 2020, 145, 111708. [Google Scholar] [CrossRef]
- Song, F.; Mao, Y.-J.; Hu, Y.; Zhao, S.-S.; Wang, R.; Wu, W.-Y.; Li, G.-R.; Wang, Y.; Li, G. Acacetin attenuates diabetes-induced cardiomyopathy by inhibiting oxidative stress and energy metabolism via PPAR-alpha/AMPK pathway. Eur. J. Pharmacol. 2022, 922, 174916. [Google Scholar] [CrossRef]
- Cui, Y.-K.; Hong, Y.-X.; Wu, W.-Y.; Han, W.-M.; Wu, Y.; Wu, C.; Li, G.-R.; Wang, Y. Acacetin ameliorates cardiac hypertrophy by activating Sirt1/AMPK/PGC-1alpha pathway. Eur. J. Pharmacol. 2022, 920, 174858. [Google Scholar] [CrossRef]
- Wu, Y.; Song, F.; Li, Y.; Li, J.; Cui, Y.; Hong, Y.; Han, W.; Wu, W.; Lakhani, I.; Li, G.; et al. Acacetin exerts antioxidant potential against atherosclerosis through Nrf2 pathway in apoE(-/-) Mice. J. Cell Mol. Med. 2021, 25, 521–534. [Google Scholar] [CrossRef]
- Fusi, F.; Trezza, A.; Tramaglino, M.; Sgaragli, G.; Saponara, S.; Spiga, O. The beneficial health effects of flavonoids on the cardiovascular system: Focus on K(+) channels. Pharmacol. Res. 2020, 152, 104625. [Google Scholar] [CrossRef]
- Wei, Y.; Yuan, P.; Zhang, Q.; Fu, Y.; Hou, Y.; Gao, L.; Zheng, X.; Feng, W. Acacetin improves endothelial dysfunction and aortic fibrosis in insulin-resistant SHR rats by estrogen receptors. Mol. Biol. Rep. 2020, 47, 6899–6918. [Google Scholar] [CrossRef]
- Han, W.-M.; Chen, X.-C.; Li, G.-R.; Wang, Y. Acacetin Protects Against High Glucose-Induced Endothelial Cells Injury by Preserving Mitochondrial Function via Activating Sirt1/Sirt3/AMPK Signals. Front. Pharmacol. 2020, 11, 607796. [Google Scholar] [CrossRef]
- Bartáková, A.; Nováková, M. Secondary Metabolites of Plants as Modulators of Endothelium Functions. Int. J. Mol. Sci. 2021, 22, 2533. [Google Scholar] [CrossRef]
- Wu, Z.; Yao, H.; Xu, H.; Wang, Y.; Hu, W.; Lou, G.; Zhang, L.; Huang, C.; Jiang, C.; Zhou, S.; et al. Inhibition of eNOS by L-NAME resulting in rat hind limb developmental defects through PFKFB3 mediated angiogenetic pathway. Sci. Rep. 2020, 10, 16754. [Google Scholar] [CrossRef]
- Griendling, K.K.; Camargo, L.L.; Rios, F.J.; Alves-Lopes, R.; Montezano, A.C.; Touyz, R.M. Oxidative Stress and Hypertension. Circ. Res. 2021, 128, 993–1020. [Google Scholar] [CrossRef]
- Yu, Y.; Xu, L.-S.; Wu, Y.; Su, F.-F.; Zhou, X.-M.; Xu, C. The antihypertensive effect of MK on spontaneously hypertensive rats through the AMPK/Akt/eNOS/NO and ERK1/2/Cx43 signaling pathways. Hypertens. Res. 2021, 44, 781–790. [Google Scholar] [CrossRef]
- Guoliang, M.; Tang, X.; Xie, L.; Meng, G.; Ji, Y. Aliskiren improves endothelium-dependent relaxation of thoracic aorta by activating PI3K/Akt/eNOS signal pathway in SHR. Clin. Exp. Pharmacol. Physiol. 2016, 43, 450–458. [Google Scholar] [CrossRef]
- Xie, X.; Shu, R.; Yu, C.; Fu, Z.; Li, Z. Mammalian AKT, the Emerging Roles on Mitochondrial Function in Diseases. Aging Dis. 2022, 13, 157–174. [Google Scholar] [CrossRef]
- Islam, Z.; Kawaguchi, H.; Miura, N.; Miyoshi, N.; Yamazaki-Himeno, E.; Shiraishi, M.; Miyamoto, A.; Tanimoto, A. Hypertension alters the endothelial-dependent biphasic response of bradykinin in isolated Microminipig basilar artery. Microvasc. Res. 2017, 114, 52–57. [Google Scholar] [CrossRef]
- Biancardi, V.; Bomfim, G.F.; Reis, W.L.; Al-Gassimi, S.; Nunes, K.P. The interplay between Angiotensin II, TLR4 and hypertension. Pharmacol. Res. 2017, 120, 88–96. [Google Scholar] [CrossRef]
- Amanakis, G.; Murphy, E. Cyclophilin D: An Integrator of Mitochondrial Function. Front. Physiol. 2020, 11, 595. [Google Scholar] [CrossRef]
- Xiao, A.; Gan, X.; Chen, R.; Ren, Y.; Yu, H.; You, C. The cyclophilin D/Drp1 axis regulates mitochondrial fission contributing to oxidative stress-induced mitochondrial dysfunctions in SH-SY5Y cells. Biochem. Biophys. Res. Commun. 2017, 483, 765–771. [Google Scholar] [CrossRef]
- Beutner, G.; Alanzalon, R.E.; Porter, G.A., Jr. Cyclophilin D regulates the dynamic assembly of mitochondrial ATP synthase into synthasomes. Sci. Rep. 2017, 7, 14488. [Google Scholar] [CrossRef]
- Gilkerson, R.; De La Torre, P.; Vallier, S.S. Mitochondrial OMA1 and OPA1 as Gatekeepers of Organellar Structure/Function and Cellular Stress Response. Front. Cell Dev. Biol. 2021, 9, 626117. [Google Scholar] [CrossRef]
- Cicalese, S.M.; da Silva, J.F.; Priviero, F.; Webb, R.C.; Eguchi, S.; Tostes, R.C. Vascular Stress Signaling in Hypertension. Circ. Res. 2021, 128, 969–992. [Google Scholar] [CrossRef]
- Saenz-Medina, J.; Muñoz, M.; Rodriguez, C.; Sanchez, A.; Contreras, C.; Carballido-Rodríguez, J.; Prieto, D. Endothelial Dysfunction: An Intermediate Clinical Feature between Urolithiasis and Cardiovascular Diseases. Int. J. Mol. Sci. 2022, 23, 912. [Google Scholar] [CrossRef]
- Kumar, S.; Pandey, A.K. Chemistry and biological activities of flavonoids: An overview. Sci. World J. 2013, 2013, 162750. [Google Scholar] [CrossRef] [PubMed]
- Godo, S.; Shimokawa, H. Endothelial Functions. Arterioscler. Thromb. Vasc. Biol. 2017, 37, e108–e114. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Yan, J.; Li, W. Acacetin as a Potential Protective Compound against Cardiovascular Diseases. Evid. Based Complement. Altern. Med. 2022, 2022, 6265198. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Chen, R.-L.; Wang, Y.; Wu, W.-Y.; Li, G. Acacetin alleviates myocardial ischaemia/reperfusion injury by inhibiting oxidative stress and apoptosis via the Nrf-2/HO-1 pathway. Pharm. Biol. 2022, 60, 553–561. [Google Scholar] [CrossRef]
- Gonzalez, A.; Ravassa, S.; López, B.; Moreno, M.; Beaumont, J.; José, G.S.; Querejeta, R.; Bayés-Genís, A.; Díez, J. Myocardial Remodeling in Hypertension. Hypertension 2018, 72, 549–558. [Google Scholar] [CrossRef]
- Touyz, R.M.; Alves-Lopes, R.; Rios, F.J.; Camargo, L.L.; Anagnostopoulou, A.; Arner, A.; Montezano, A.C. Vascular smooth muscle contraction in hypertension. Cardiovasc. Res. 2018, 114, 529–539. [Google Scholar] [CrossRef]
- Claesson-Welsh, L.; Dejana, E.; McDonald, D.M. Permeability of the Endothelial Barrier: Identifying and Reconciling Controversies. Trends Mol. Med. 2021, 27, 314–331. [Google Scholar] [CrossRef]
- Liu, B.; Zhou, H.; Zhang, T.; Gao, X.; Tao, B.; Xing, H.; Zhuang, Z.; Dardik, A.; Kyriakides, T.R.; Goodwin, J.E. Loss of endothelial glucocorticoid receptor promotes angiogenesis via upregulation of Wnt/beta-catenin pathway. Angiogenesis 2021, 24, 631–645. [Google Scholar] [CrossRef]
- Flores-Flores, A.; Hernández-Abreu, O.; Rios, M.Y.; León-Rivera, I.; Aguilar-Guadarrama, B.; Castillo-España, P.; Perea-Arango, I.; Estrada-Soto, S. Vasorelaxant mode of action of dichloromethane-soluble extract from Agastache mexicana and its main bioactive compounds. Pharm. Biol. 2016, 54, 2807–2813. [Google Scholar] [CrossRef]
- Ambrosino, P.; Bachetti, T.; D’Anna, S.E.; Galloway, B.; Bianco, A.; D’Agnano, V.; Papa, A.; Motta, A.; Perrotta, F.; Maniscalco, M. Mechanisms and Clinical Implications of Endothelial Dysfunction in Arterial Hypertension. J. Cardiovasc. Dev. Dis. 2022, 9, 136. [Google Scholar] [CrossRef]
- Abate, M.; Festa, A.; Falco, M.; Lombardi, A.; Luce, A.; Grimaldi, A.; Zappavigna, S.; Sperlongano, P.; Irace, C.; Caraglia, M.; et al. Mitochondria as playmakers of apoptosis, autophagy and senescence. Semin. Cell Dev. Biol. 2020, 98, 139–153. [Google Scholar] [CrossRef]
- Bonora, M.; Morganti, C.; Morciano, G.; Giorgi, C.; Wieckowski, M.R.; Pinton, P. Comprehensive analysis of mitochondrial permeability transition pore activity in living cells using fluorescence-imaging-based techniques. Nat. Protoc. 2016, 11, 1067–1080. [Google Scholar] [CrossRef]
- Porter, J.G.A., Jr.; Beutner, G. Cyclophilin D, Somehow a Master Regulator of Mitochondrial Function. Biomolecules 2018, 8, 176. [Google Scholar] [CrossRef]
- Stockburger, C.; Eckert, S.; Eckert, G.P.; Friedland, K.; Müller, W.E. Mitochondrial Function, Dynamics, and Permeability Transition: A Complex Love Triangle as A Possible Target for the Treatment of Brain Aging and Alzheimer’s Disease. J. Alzheimers Dis. 2018, 64 (Suppl. S1), S455–S467. [Google Scholar] [CrossRef]
- Robert, P.; Nguyen, P.M.C.; Richard, A.; Grenier, C.; Chevrollier, A.; Munier, M.; Grimaud, L.; Proux, C.; Champin, T.; Lelièvre, E.; et al. Protective role of the mitochondrial fusion protein OPA1 in hypertension. FASEB J. 2021, 35, e21678. [Google Scholar] [CrossRef]
- Kleele, T.; Rey, T.; Winter, J.; Zaganelli, S.; Mahecic, D.; Lambert, H.P.; Ruberto, F.P.; Nemir, M.; Wai, T.; Pedrazzini, T.; et al. Distinct fission signatures predict mitochondrial degradation or biogenesis. Nature 2021, 593, 435–439. [Google Scholar] [CrossRef]
- Jin, J.-Y.; Wei, X.-X.; Zhi, X.-L.; Wang, X.-H.; Meng, D. Drp1-dependent mitochondrial fission in cardiovascular disease. Acta Pharmacol. Sin. 2021, 42, 655–664. [Google Scholar] [CrossRef]
- Cooper, H.A.; Cicalese, S.; Preston, K.J.; Kawai, T.; Okuno, K.; Choi, E.T.; Kasahara, S.; Uchida, H.A.; Otaka, N.; Scalia, R.; et al. Targeting mitochondrial fission as a potential therapeutic for abdominal aortic aneurysm. Cardiovasc. Res. 2021, 117, 971–982. [Google Scholar] [CrossRef]
- Deng, Y.; Li, S.; Chen, Z.; Wang, W.; Geng, B.; Cai, J. Mdivi-1, a mitochondrial fission inhibitor, reduces angiotensin-II- induced hypertension by mediating VSMC phenotypic switch. Biomed. Pharmacother. 2021, 140, 111689. [Google Scholar] [CrossRef]
- Huang, G.; Cong, Z.; Wang, X.; Yuan, Y.; Xu, R.; Lu, Z.; Wang, X.; Qi, J. Targeting HSP90 attenuates angiotensin II-induced adventitial remodelling via suppression of mitochondrial fission. Cardiovasc. Res. 2020, 116, 1071–1084. [Google Scholar] [CrossRef]
- Helfenberger, K.E.; Castillo, A.F.; Mele, P.G.; Fiore, A.; Herrera, L.; Finocchietto, P.; Podestá, E.J.; Poderoso, C. Angiotensin II stimulation promotes mitochondrial fusion as a novel mechanism involved in protein kinase compartmentalization and cholesterol transport in human adrenocortical cells. J. Steroid Biochem. Mol. Biol. 2019, 192, 105413. [Google Scholar] [CrossRef]
- Forrester, S.J.; Preston, K.J.; Cooper, H.A.; Boyer, M.J.; Escoto, K.M.; Poltronetti, A.J.; Elliott, K.J.; Kuroda, R.; Miyao, M.; Sesaki, H.; et al. Mitochondrial Fission Mediates Endothelial Inflammation. Hypertension 2020, 76, 267–276. [Google Scholar] [CrossRef]
- Liu, X.; Tan, H.; Liu, X.; Wu, Q. Correlation between the expression of Drp1 in vascular endothelial cells and inflammatory factors in hypertension rats. Exp. Ther. Med. 2018, 15, 3892–3898. [Google Scholar] [CrossRef]
- Hu, Q.; Zhang, H.; Cortés, N.G.; Wu, D.; Wang, P.; Zhang, J.; Mattison, J.A.; Smith, E.; Bettcher, L.F.; Wang, M.; et al. Increased Drp1 Acetylation by Lipid Overload Induces Cardiomyocyte Death and Heart Dysfunction. Circ. Res. 2020, 126, 456–470. [Google Scholar] [CrossRef]
- Han, H.; Tan, J.; Wang, R.; Wan, H.; He, Y.; Yan, X.; Guo, J.; Gao, Q.; Li, J.; Shang, S.; et al. PINK1 phosphorylates Drp1(S616) to regulate mitophagy-independent mitochondrial dynamics. EMBO Rep. 2020, 21, e48686. [Google Scholar] [CrossRef]
- Duan, C.; Kuang, L.; Xiang, X.; Zhang, J.; Zhu, Y.; Wu, Y.; Yan, Q.; Liu, L.; Li, T. Drp1 regulates mitochondrial dysfunction and dysregulated metabolism in ischemic injury via Clec16a-, BAX-, and GSH- pathways. Cell Death Dis. 2020, 11, 251. [Google Scholar] [CrossRef]
- Smolina, N.; Bruton, J.; Kostareva, A.; Sejersen, T. Assaying Mitochondrial Respiration as an Indicator of Cellular Metabolism and Fitness. Methods Mol. Biol. 2017, 1601, 79–87. [Google Scholar] [CrossRef]
- Basile, A.O.; Yahi, A.; Tatonetti, N.P. Artificial Intelligence for Drug Toxicity and Safety. Trends Pharmacol. Sci. 2019, 40, 624–635. [Google Scholar] [CrossRef]
- Lin, H.-T.; Shiou, Y.-L.; Jhuang, W.-J.; Lee, H.-C. Simultaneous Electrocardiography Recording and Invasive Blood Pressure Measurement in Rats. J. Vis. Exp. 2019, 143, e59115. [Google Scholar] [CrossRef]
- Zhou, Y.; Liu, X.; Zhang, X.; Wen, J.; Cheng, J.; Li, P.; Wang, N.; Zhou, X.; Xia, D.; Yang, Q.; et al. Decreased vasodilatory effect of Tanshinone A Sodium Sulfonate on mesenteric artery in hypertension. Eur. J. Pharmacol. 2019, 854, 365–371. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Dang, Q.; Li, Z.; Han, C.; Yang, Y.; Li, M.; Li, P. Restoration of Mitochondrial Function Is Essential in the Endothelium-Dependent Vasodilation Induced by Acacetin in Hypertensive Rats. Int. J. Mol. Sci. 2022, 23, 11350. https://doi.org/10.3390/ijms231911350
Li Y, Dang Q, Li Z, Han C, Yang Y, Li M, Li P. Restoration of Mitochondrial Function Is Essential in the Endothelium-Dependent Vasodilation Induced by Acacetin in Hypertensive Rats. International Journal of Molecular Sciences. 2022; 23(19):11350. https://doi.org/10.3390/ijms231911350
Chicago/Turabian StyleLi, Yuan, Qingya Dang, Zhiyi Li, Chuting Han, Yan Yang, Miaoling Li, and Pengyun Li. 2022. "Restoration of Mitochondrial Function Is Essential in the Endothelium-Dependent Vasodilation Induced by Acacetin in Hypertensive Rats" International Journal of Molecular Sciences 23, no. 19: 11350. https://doi.org/10.3390/ijms231911350
APA StyleLi, Y., Dang, Q., Li, Z., Han, C., Yang, Y., Li, M., & Li, P. (2022). Restoration of Mitochondrial Function Is Essential in the Endothelium-Dependent Vasodilation Induced by Acacetin in Hypertensive Rats. International Journal of Molecular Sciences, 23(19), 11350. https://doi.org/10.3390/ijms231911350