
Balanced Force Field ff03CMAP Improving the Dynamics Conformation Sampling of Phosphorylation Site


Abstract
1. Introduction
2. Material and Methods
2.1. Phosphorylated Protein Structural Propensity
2.2. Molecular Dynamics Simulation
2.3. Evaluation Metrics
3. Results
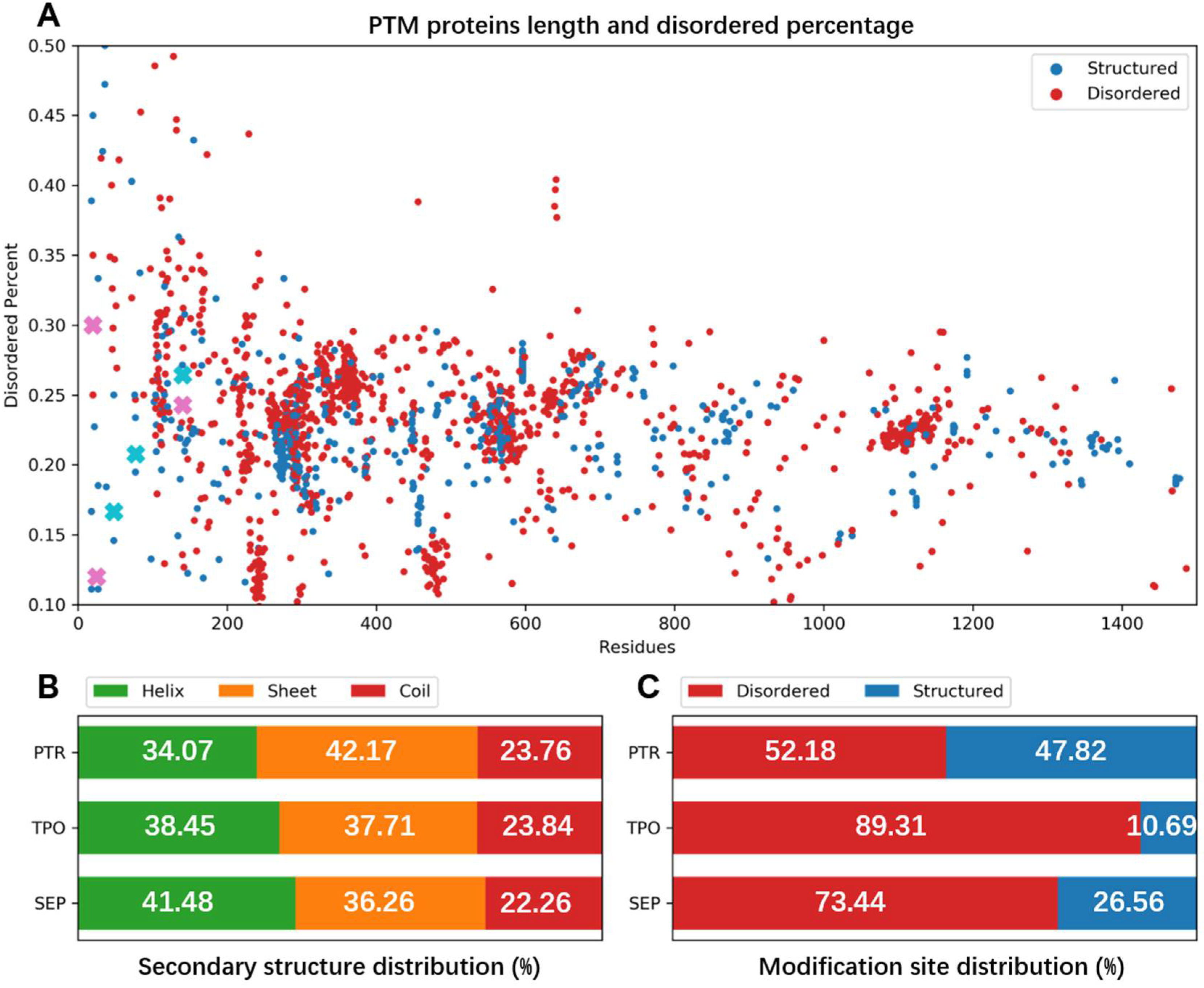
3.1. Structural Statistics of Phosphorylated Proteins in PDB Database
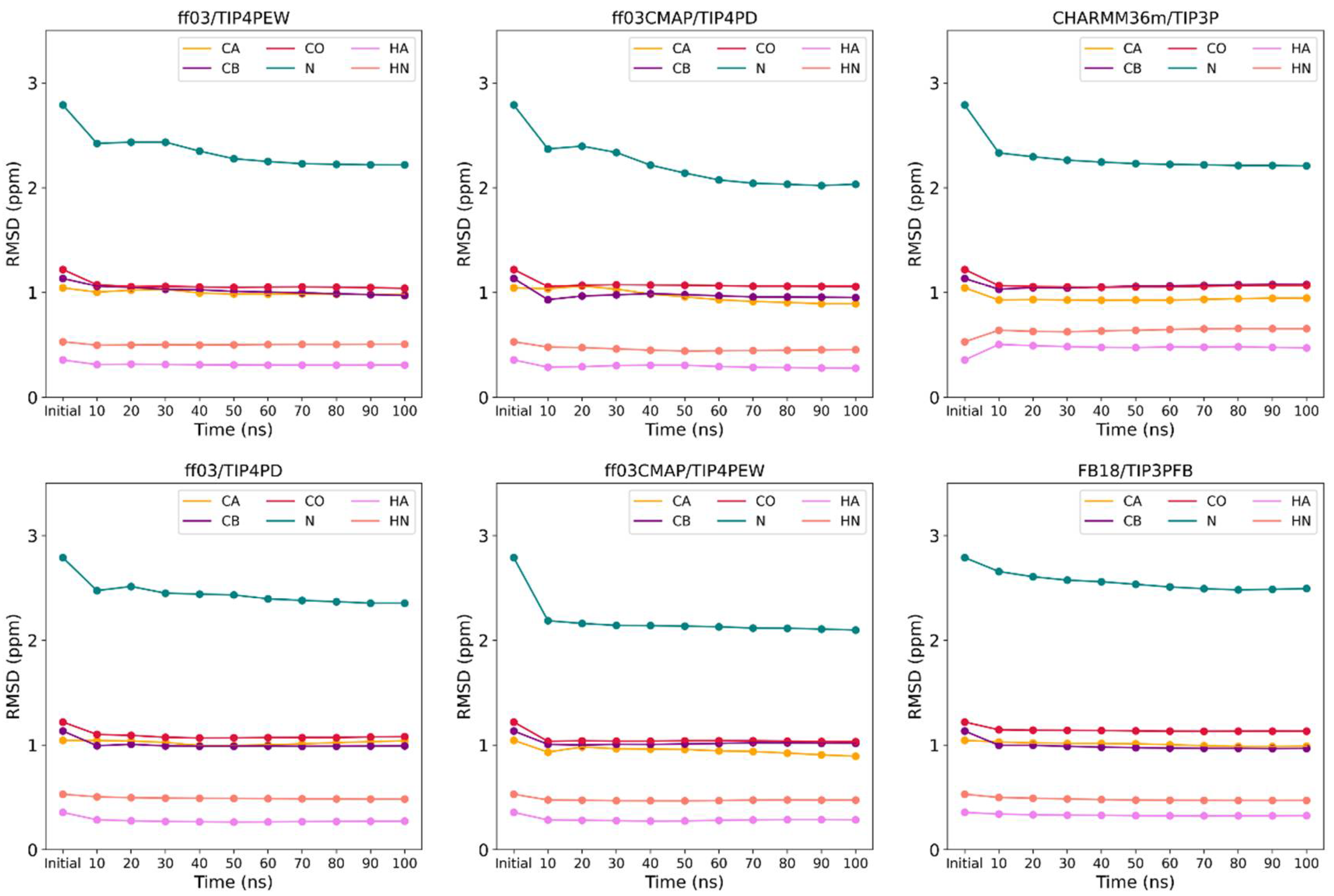
3.2. Convergence of Simulation System
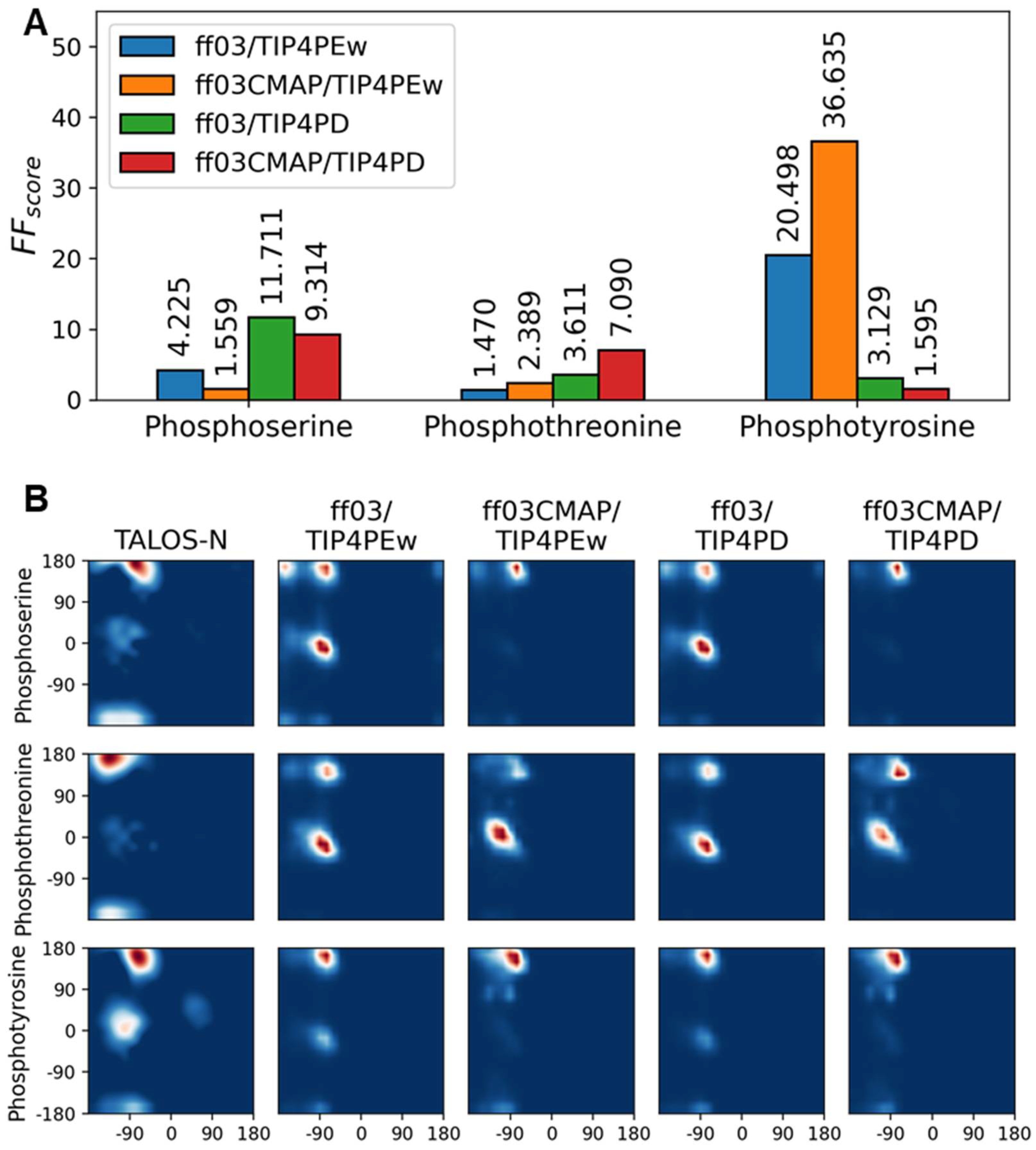
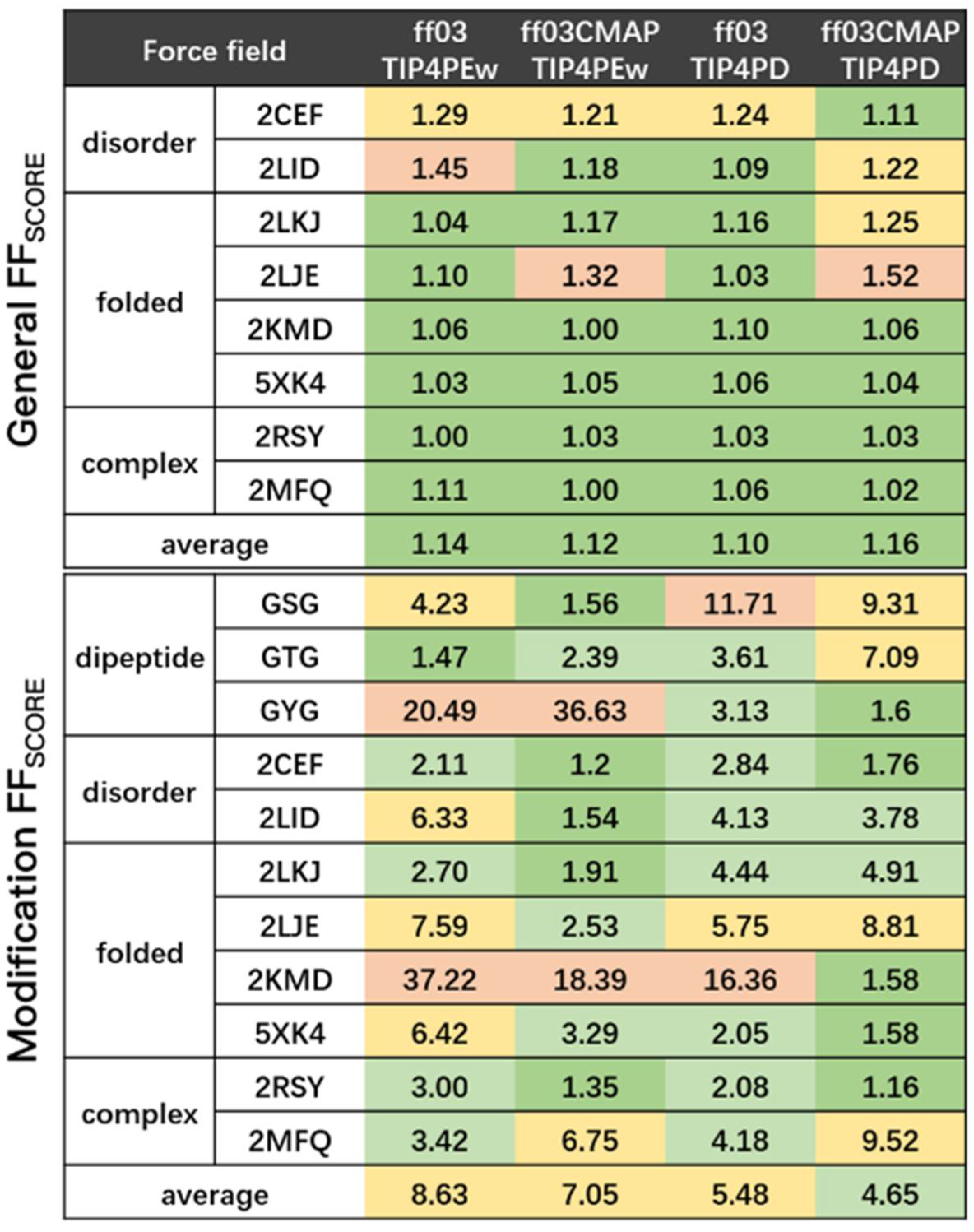
3.3. Phosphorylated Dipeptides
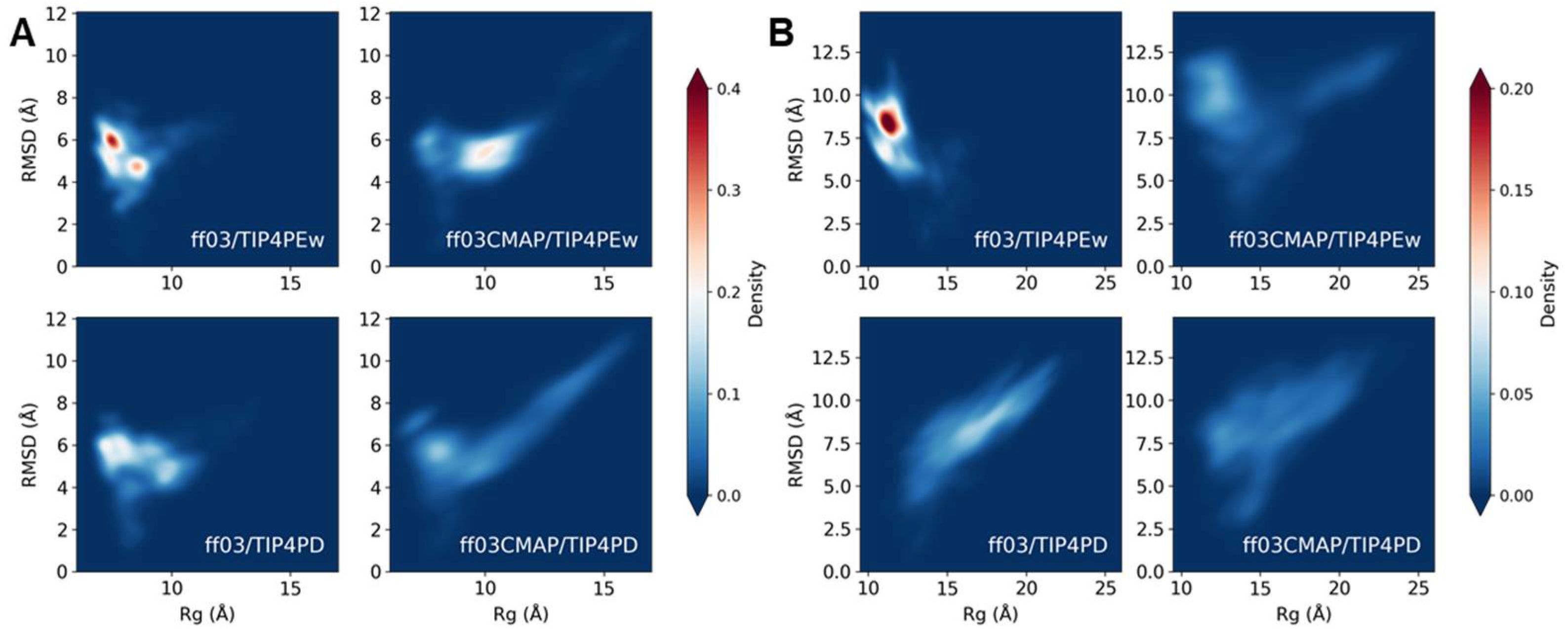
3.4. Phosphorylated Disordered Proteins
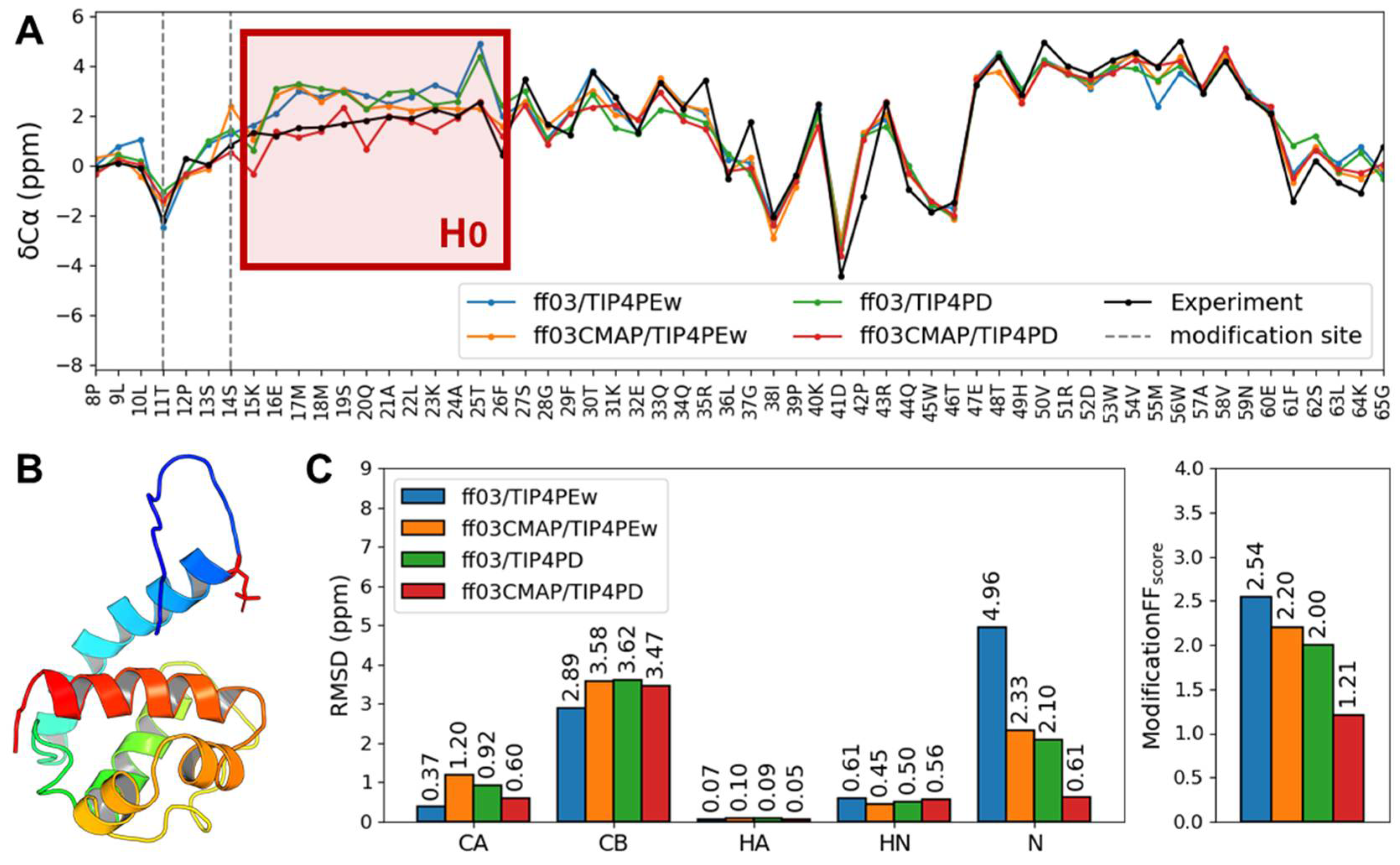
3.5. Phosphorylated Folded Proteins and Complexes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Khoury, G.A.; Baliban, R.C.; Floudas, C.A. Proteome-Wide Post-Translational Modification Statistics: Frequency Analysis and Curation of the Swiss-Prot Database. Sci. Rep. 2011, 1, 90. [Google Scholar] [CrossRef] [PubMed]
- Ardito, F.; Giuliani, M.; Perrone, D.; Troiano, G.; Muzio, L.L. The Crucial Role of Protein Phosphorylation in Cell Signaling and Its Use as Targeted Therapy (Review). Int. J. Mol. Med. 2017, 40, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Terman, J.R.; Kashina, A. Post-Translational Modification and Regulation of Actin. Curr. Opin. Cell Biol. 2013, 25, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Jenuwein, T.; Allis, C.D. Translating the Histone Code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef]
- Zecha, J.; Gabriel, W.; Spallek, R.; Chang, Y.-C.; Mergner, J.; Wilhelm, M.; Bassermann, F.; Kuster, B. Linking Post-Translational Modifications and Protein Turnover by Site-Resolved Protein Turnover Profiling. Nat. Commun. 2022, 13, 165. [Google Scholar] [CrossRef]
- Appella, E.; Anderson, C.W. Post-Translational Modifications and Activation of P53 by Genotoxic Stresses. Eur. J. Biochem. 2001, 268, 2764–2772. [Google Scholar] [CrossRef]
- Govin, J.; Caron, C.; Lestrat, C.; Rousseaux, S.; Khochbin, S. The Role of Histones in Chromatin Remodelling during Mammalian Spermiogenesis. Eur. J. Biochem. 2004, 271, 3459–3469. [Google Scholar] [CrossRef]
- Almeida, K.H.; Sobol, R.W. A Unified View of Base Excision Repair: Lesion-Dependent Protein Complexes Regulated by Post-Translational Modification. DNA Repair 2007, 6, 695–711. [Google Scholar] [CrossRef]
- Csizmok, V.; Forman-Kay, J.D. Complex Regulatory Mechanisms Mediated by the Interplay of Multiple Post-Translational Modifications. Curr. Opin. Struct. Biol. 2018, 48, 58–67. [Google Scholar] [CrossRef]
- Elbaum, M.B.; Zondlo, N.J. OGlcNAcylation and Phosphorylation Have Similar Structural Effects in α-Helices: Post-Translational Modifications as Inducible Start and Stop Signals in α-Helices, with Greater Structural Effects on Threonine Modification. Biochemistry 2014, 53, 2242–2260. [Google Scholar] [CrossRef]
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically Disordered Protein. J. Mol. Graph. Model. 2001, 19, 26–59. [Google Scholar] [CrossRef]
- Bah, A.; Forman-Kay, J.D. Modulation of Intrinsically Disordered Protein Function by Post-Translational Modifications. J. Biol. Chem. 2016, 291, 6696–6705. [Google Scholar] [CrossRef] [PubMed]
- Owen, I.; Shewmaker, F. The Role of Post-Translational Modifications in the Phase Transitions of Intrinsically Disordered Proteins. Int. J. Mol. Sci. 2019, 20, 5501. [Google Scholar] [CrossRef]
- Bah, A.; Vernon, R.M.; Siddiqui, Z.; Krzeminski, M.; Muhandiram, R.; Zhao, C.; Sonenberg, N.; Kay, L.E.; Forman-Kay, J.D. Folding of an Intrinsically Disordered Protein by Phosphorylation as a Regulatory Switch. Nature 2015, 519, 106–109. [Google Scholar] [CrossRef]
- Wu, J.; Li, D.; Liu, X.; Li, Q.; He, X.; Wei, J.; Li, M.; Rehman, A.U.; Xia, Y.; Wu, C.; et al. IDDB: A Comprehensive Resource Featuring Genes, Variants and Characteristics Associated with Infertility. Nucleic Acids Res. 2021, 49, D1218–D1224. [Google Scholar] [CrossRef]
- Slupsky, C.M.; Gentile, L.N.; Donaldson, L.W.; Mackereth, C.D.; Seidel, J.J.; Graves, B.J.; McIntosh, L.P. Structure of the Ets-1 Pointed Domain and Mitogen-Activated Protein Kinase Phosphorylation Site. Proc. Natl. Acad. Sci. USA 1998, 95, 12129–12134. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Jiang, F.; Wu, Y. Mechanism of Phosphorylation-Induced Folding of 4E-BP2 Revealed by Molecular Dynamics Simulations. J. Chem. Theory Comput. 2017, 13, 320–328. Available online: https://pubs.acs.org/doi/abs/10.1021/acs.jctc.6b00848?casa_token=C_nyHiGjT5kAAAAA:hbjhgAWkh2t36_rprkez0_2tUqifD0M5tOpTMqg0fMBfLYdIEt0iq2U5dvI5lji5LSyLcHtifeKX1Bi- (accessed on 16 April 2022). [CrossRef]
- Humphrey, S.J.; James, D.E.; Mann, M. Protein Phosphorylation: A Major Switch Mechanism for Metabolic Regulation. Trends Endocrinol. Metab. 2015, 26, 676–687. [Google Scholar] [CrossRef]
- Millar, A.H.; Heazlewood, J.L.; Giglione, C.; Holdsworth, M.J.; Bachmair, A.; Schulze, W.X. The Scope, Functions, and Dynamics of Posttranslational Protein Modifications. Annu. Rev. Plant Biol. 2019, 70, 119–151. [Google Scholar] [CrossRef]
- Smith, L.M.; Kelleher, N.L. Proteoform: A Single Term Describing Protein Complexity. Nat. Methods 2013, 10, 186–187. [Google Scholar] [CrossRef]
- Willems, P.; Horne, A.; Van Parys, T.; Goormachtig, S.; De Smet, I.; Botzki, A.; Van Breusegem, F.; Gevaert, K. The Plant PTM Viewer, a Central Resource for Exploring Plant Protein Modifications. Plant J. 2019, 99, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Zulawski, M.; Schulze, W.X. The Plant Kinome. In Plant Phosphoproteomics: Methods and Protocols; Methods in Molecular Biology; Schulze, W.X., Ed.; Springer: New York, NY, USA, 2015; pp. 1–23. ISBN 978-1-4939-2648-0. [Google Scholar]
- Homeyer, N.; Horn, A.H.; Lanig, H.; Sticht, H. AMBER Force-Field Parameters for Phosphorylated Amino Acids in Different Protonation States: Phosphoserine, Phosphothreonine, Phosphotyrosine, and Phosphohistidine. J. Mol. Modeling 2006, 12, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Khoury, G.A.; Thompson, J.P.; Smadbeck, J.; Kieslich, C.A.; Floudas, C.A. Forcefield_PTM: Ab Initio Charge and AMBER Forcefield Parameters for Frequently Occurring Post-Translational Modifications. J. Chem. Theory Comput. 2013, 9, 5653–5674. [Google Scholar] [CrossRef]
- Brooks, B.R.; Bruccoleri, R.E.; Olafson, B.D.; States, D.J.; Swaminathan, S.; Karplus, M. CHARMM: A Program for Macromolecular Energy, Minimization, and Dynamics Calculations. J. Comput. Chem. 1983, 4, 187–217. [Google Scholar] [CrossRef]
- Brooks, B.R.; Brooks, C.L., III; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The Biomolecular Simulation Program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]
- Margreitter, C.; Petrov, D.; Zagrovic, B. Vienna-PTM Web Server: A Toolkit for MD Simulations of Protein Post-Translational Modifications. Nucleic Acids Res. 2013, 41, W422–W426. [Google Scholar] [CrossRef] [PubMed]
- Petrov, D.; Margreitter, C.; Grandits, M.; Oostenbrink, C.; Zagrovic, B. A Systematic Framework for Molecular Dynamics Simulations of Protein Post-Translational Modifications. PLoS Comput. Biol. 2013, 9, e1003154. [Google Scholar] [CrossRef] [PubMed]
- Margreitter, C.; Reif, M.M.; Oostenbrink, C. Update on Phosphate and Charged Post-Translationally Modified Amino Acid Parameters in the GROMOS Force Field. J. Comput. Chem. 2017, 38, 714–720. [Google Scholar] [CrossRef]
- Vymetal, J.; Jurásková, V.; Vondrášek, J. AMBER and CHARMM Force Fields Inconsistently Portray the Microscopic Details of Phosphorylation. J. Chem. Theory Comput. 2018, 15, 665–679. [Google Scholar] [CrossRef]
- Rieloff, E.; Skepö, M. Phosphorylation of a Disordered Peptide—Structural Effects and Force Field Inconsistencies. J. Chem. Theory Comput. 2020, 16, 1924–1935. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved Side-Chain Torsion Potentials for the Amber Ff99SB Protein Force Field. Proteins Struct. Funct. Bioinform. 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A Point-Charge Force Field for Molecular Mechanics Simulations of Proteins Based on Condensed-Phase Quantum Mechanical Calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. Ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from Ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, H.; Yang, S.; Luo, R.; Chen, H.-F. Well-Balanced Force Field Ff03CMAP for Folded and Disordered Proteins. J. Chem. Theory Comput. 2019, 15, 6769–6780. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Kasavajhala, K.; Belfon, K.A.A.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q.; et al. Ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theory Comput. 2020, 16, 528–552. [Google Scholar] [CrossRef]
- Mackerell, A.D., Jr. Empirical Force Fields for Biological Macromolecules: Overview and Issues. J. Comput. Chem. 2004, 25, 1584–1604. [Google Scholar] [CrossRef]
- Song, D.; Luo, R.; Chen, H.-F. The IDP-Specific Force Field Ff14IDPSFF Improves the Conformer Sampling of Intrinsically Disordered Proteins. J. Chem. Inf. Model. 2017, 57, 1166–1178. [Google Scholar] [CrossRef]
- Chen, J.; Liu, H.; Cui, X.; Li, Z.; Chen, H.-F. RNA-Specific Force Field Optimization with CMAP and Reweighting. J. Chem. Inf. Model. 2022, 62, 372–385. [Google Scholar] [CrossRef]
- Song, D.; Liu, H.; Luo, R.; Chen, H.-F. Environment-Specific Force Field for Intrinsically Disordered and Ordered Proteins. J. Chem. Inf. Model. 2020, 60, 2257–2267. [Google Scholar] [CrossRef]
- Yang, S.; Liu, H.; Zhang, Y.; Lu, H.; Chen, H. Residue-Specific Force Field Improving the Sample of Intrinsically Disordered Proteins and Folded Proteins. J. Chem. Inf. Model. 2019, 59, 4793–4805. [Google Scholar] [CrossRef]
- Liu, H.; Song, D.; Lu, H.; Luo, R.; Chen, H.-F. Intrinsically Disordered Protein-Specific Force Field CHARMM36IDPSFF. Chem. Biol. Drug Des. 2018, 92, 1722–1735. [Google Scholar] [CrossRef]
- Ye, W.; Ji, D.; Wang, W.; Luo, R.; Chen, H.-F. Test and Evaluation of Ff99IDPs Force Field for Intrinsically Disordered Proteins. J. Chem. Inf. Model. 2015, 55, 1021–1029. [Google Scholar] [CrossRef]
- Wang, W.; Ye, W.; Jiang, C.; Luo, R.; Chen, H.-F. New Force Field on Modeling Intrinsically Disordered Proteins. Chem. Biol. Drug Des. 2014, 84, 253–269. [Google Scholar] [CrossRef]
- Horn, H.W.; Swope, W.C.; Pitera, J.W.; Madura, J.D.; Dick, T.J.; Hura, G.L.; Head-Gordon, T. Development of an Improved Four-Site Water Model for Biomolecular Simulations: TIP4P-Ew. J. Chem. Phys. 2004, 120, 9665–9678. [Google Scholar] [CrossRef]
- Piana, S.; Donchev, A.G.; Robustelli, P.; Shaw, D.E. Water Dispersion Interactions Strongly Influence Simulated Structural Properties of Disordered Protein States. J. Phys. Chem. B 2015, 119, 5113–5123. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of Protein Secondary Structure: Pattern Recognition of Hydrogen-Bonded and Geometrical Features. Biopolym. Orig. Res. Biomol. 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Touw, W.G.; Baakman, C.; Black, J.; Te Beek, T.A.; Krieger, E.; Joosten, R.P.; Vriend, G. A Series of PDB-Related Databanks for Everyday Needs. Nucleic Acids Res. 2015, 43, D364–D368. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef]
- Stoppelman, J.P.; Ng, T.T.; Nerenberg, P.S.; Wang, L.-P. Development and Validation of AMBER-FB15-Compatible Force Field Parameters for Phosphorylated Amino Acids. J. Phys. Chem. B 2021, 125, 11927–11942. [Google Scholar] [CrossRef]
- Bienkiewicz, E.A.; Lumb, K.J. Random-Coil Chemical Shifts of Phosphorylated Amino Acids. J. Biomol. NMR 1999, 15, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Sen, M.; Herzik, M., Jr.; Craft, J.W., Jr.; Creath, A.L.; Agrawal, S.; Ruf, W.; Legge, G.B. Spectroscopic Characterization of Successive Phosphorylation of the Tissue Factor Cytoplasmic Region. Open Spectrosc. J. 2009, 3, 58. [Google Scholar] [CrossRef] [PubMed]
- Havukainen, H.; Underhaug, J.; Wolschin, F.; Amdam, G.; Halskau, Ø. A Vitellogenin Polyserine Cleavage Site: Highly Disordered Conformation Protected from Proteolysis by Phosphorylation. J. Exp. Biol. 2012, 215, 1837–1846. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, L.; Meller, N.; Alder, N.; Byzova, T.; Vinogradova, O. Tyrosine Phosphorylation as a Conformational Switch a CASE Study of Integrin Β3 Cytoplasmic Tail. J. Biol. Chem. 2011, 286, 40943–40953. [Google Scholar] [CrossRef]
- Chua, G.-L.; Tang, X.-Y.; Amalraj, M.; Tan, S.-M.; Bhattacharjya, S. Structures and Interaction Analyses of Integrin AMβ2 Cytoplasmic Tails. J. Biol. Chem. 2011, 286, 43842–43854. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.L.; Kang, H.-S.; Lee, G.M.; Blaszczak, A.G.; Lau, D.K.; McIntosh, L.P.; Graves, B.J. Ras Signaling Requires Dynamic Properties of Ets1 for Phosphorylation-Enhanced Binding to Coactivator CBP. Proc. Natl. Acad. Sci. USA 2010, 107, 10026–10031. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Gong, Z.; Lu, Y.-B.; Liu, K.; Qin, L.-Y.; Ran, M.-L.; Zhang, C.-L.; Liu, Z.; Zhang, W.-P.; Tang, C. Ubiquitin S65 Phosphorylation Engenders a PH-Sensitive Conformational Switch. Proc. Natl. Acad. Sci. USA 2017, 114, 6770–6775. [Google Scholar] [CrossRef]
- Tanaka, H.; Akagi, K.; Oneyama, C.; Tanaka, M.; Sasaki, Y.; Kanou, T.; Lee, Y.-H.; Yokogawa, D.; Dobenecker, M.-W.; Nakagawa, A.; et al. Identification of a New Interaction Mode between the Src Homology 2 Domain of C-Terminal Src Kinase (Csk) and Csk-Binding Protein/Phosphoprotein Associated with Glycosphingolipid Microdomains. J. Biol. Chem. 2013, 288, 15240–15254. [Google Scholar] [CrossRef]
- Zeng, L.; Kuti, M.; Mujtaba, S.; Zhou, M.-M. Structural Insights into FRS2α PTB Domain Recognition by Neurotrophin Receptor TrkB. Proteins Struct. Funct. Bioinform. 2014, 82, 1534–1541. [Google Scholar] [CrossRef]
- Lee, T.-S.; Cerutti, D.S.; Mermelstein, D.; Lin, C.; LeGrand, S.; Giese, T.J.; Roitberg, A.; Case, D.A.; Walker, R.C.; York, D.M. GPU-Accelerated Molecular Dynamics and Free Energy Methods in Amber18: Performance Enhancements and New Features. J. Chem. Inf. Model. 2018, 58, 2043–2050. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N·log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Götz, A.W.; Poole, D.; Grand, S.L.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. Available online: https://pubs.acs.org/doi/abs/10.1021/ct400314y (accessed on 16 April 2022). [CrossRef] [PubMed]
- Rieloff, E.; Skepö, M. Molecular Dynamics Simulations of Phosphorylated Intrinsically Disordered Proteins: A Force Field Comparison. Int. J. Mol. Sci. 2021, 22, 10174. [Google Scholar] [CrossRef]
- Rieloff, E.; Skepö, M. The Effect of Multisite Phosphorylation on the Conformational Properties of Intrinsically Disordered Proteins. Int. J. Mol. Sci. 2021, 22, 11058. [Google Scholar] [CrossRef] [PubMed]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Shen, Y.; Bax, A. SPARTA+: A Modest Improvement in Empirical NMR Chemical Shift Prediction by Means of an Artificial Neural Network. J. Biomol. NMR 2010, 48, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger, L. The PyMOL Molecular Graphics System, Version 1.8; Schrodinger, LLC: New York, NY, USA, 2015. [Google Scholar]
- Robustelli, P.; Piana, S.; Shaw, D.E. Developing a Molecular Dynamics Force Field for Both Folded and Disordered Protein States. Proc. Natl. Acad. Sci. USA 2018, 115, E4758–E4766. [Google Scholar] [CrossRef]
- Shen, Y.; Bax, A. Protein Backbone and Sidechain Torsion Angles Predicted from NMR Chemical Shifts Using Artificial Neural Networks. J. Biomol. NMR 2013, 56, 227–241. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Description | Modification Type | Length | Initial Structure | Ions | Number of Waters | Simulation Time (ns) | Number of Tested Force Fields | Number of Trajectories |
|---|---|---|---|---|---|---|---|---|---|
| Dipeptide | |||||||||
| GpSG [52] | Phosphoserine dipeptide | pSer | 3 | Extended | 2 Na+ | 962 | 500 | 4 | 1 |
| GpTG [52] | Phosphothreonine dipeptide | pThr | 3 | Extended | 2 Na+ | 811 | 500 | 4 | 1 |
| GpYG [52] | Phosphotyrosine dipeptide | pTyr | 3 | Extended | 2 Na+ | 1089 | 500 | 4 | 1 |
| Disordered Protein | |||||||||
| TF [53] | Cytoplasmic Tail of Tissue Factor | 2*pSer | 19 | 2CEF | 2 Na+ | 2511 | 100 | 6 | 5 |
| Vg [54] | Vitellogenin | pSer | 35 | 2LID | 7 Na+ | 6316 | 100 | 4 | 5 |
| Folded Protein | |||||||||
| β3 [55] | β3 cytoplasmic tail | pSer | 24 | 2LKJ | 1 Na+ | 3303 | 100 | 4 | 5 |
| αM [56] | αM cytoplasmic tail | 2*pTyr | 47 | 2LJE | 2 Na+ | 3828 | 100 | 4 | 5 |
| Ets1 [57] | Ets1 | pSer, pThr | 111 | 2KMD | 9 Na+ | 7644 | 100 | 6 | 5 |
| 1000 | 4 | 1 | |||||||
| p-Ubiquitin [58] | Phosphorylated ubiquitin | pSer | 76 | 5XK4 | 2 Na+ | 3613 | 100 | 4 | 5 |
| Complex | |||||||||
| SH2 [59] | SH2 domain of Csk in complex with a phosphopeptide from Cbp | pTyr | 137 | 2RSY | 7 Na+ | 8267 | 100 | 4 | 5 |
| TrkB [60] | FRS2a PTB domain with neurotrophin receptor TrkB | pTyr | 138 | 2MFQ | 7 Na+ | 8756 | 100 | 4 | 5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhong, B.; Song, G.; Chen, H.-F. Balanced Force Field ff03CMAP Improving the Dynamics Conformation Sampling of Phosphorylation Site. Int. J. Mol. Sci. 2022, 23, 11285. https://doi.org/10.3390/ijms231911285
Zhong B, Song G, Chen H-F. Balanced Force Field ff03CMAP Improving the Dynamics Conformation Sampling of Phosphorylation Site. International Journal of Molecular Sciences. 2022; 23(19):11285. https://doi.org/10.3390/ijms231911285
Chicago/Turabian StyleZhong, Bozitao, Ge Song, and Hai-Feng Chen. 2022. "Balanced Force Field ff03CMAP Improving the Dynamics Conformation Sampling of Phosphorylation Site" International Journal of Molecular Sciences 23, no. 19: 11285. https://doi.org/10.3390/ijms231911285
APA StyleZhong, B., Song, G., & Chen, H.-F. (2022). Balanced Force Field ff03CMAP Improving the Dynamics Conformation Sampling of Phosphorylation Site. International Journal of Molecular Sciences, 23(19), 11285. https://doi.org/10.3390/ijms231911285