

Hypoxia-Inducible Factors Signaling in Osteogenesis and Skeletal Repair
Abstract
1. Introduction
2. Hypoxia-Inducible Factors
Effect of HIFs on Bone
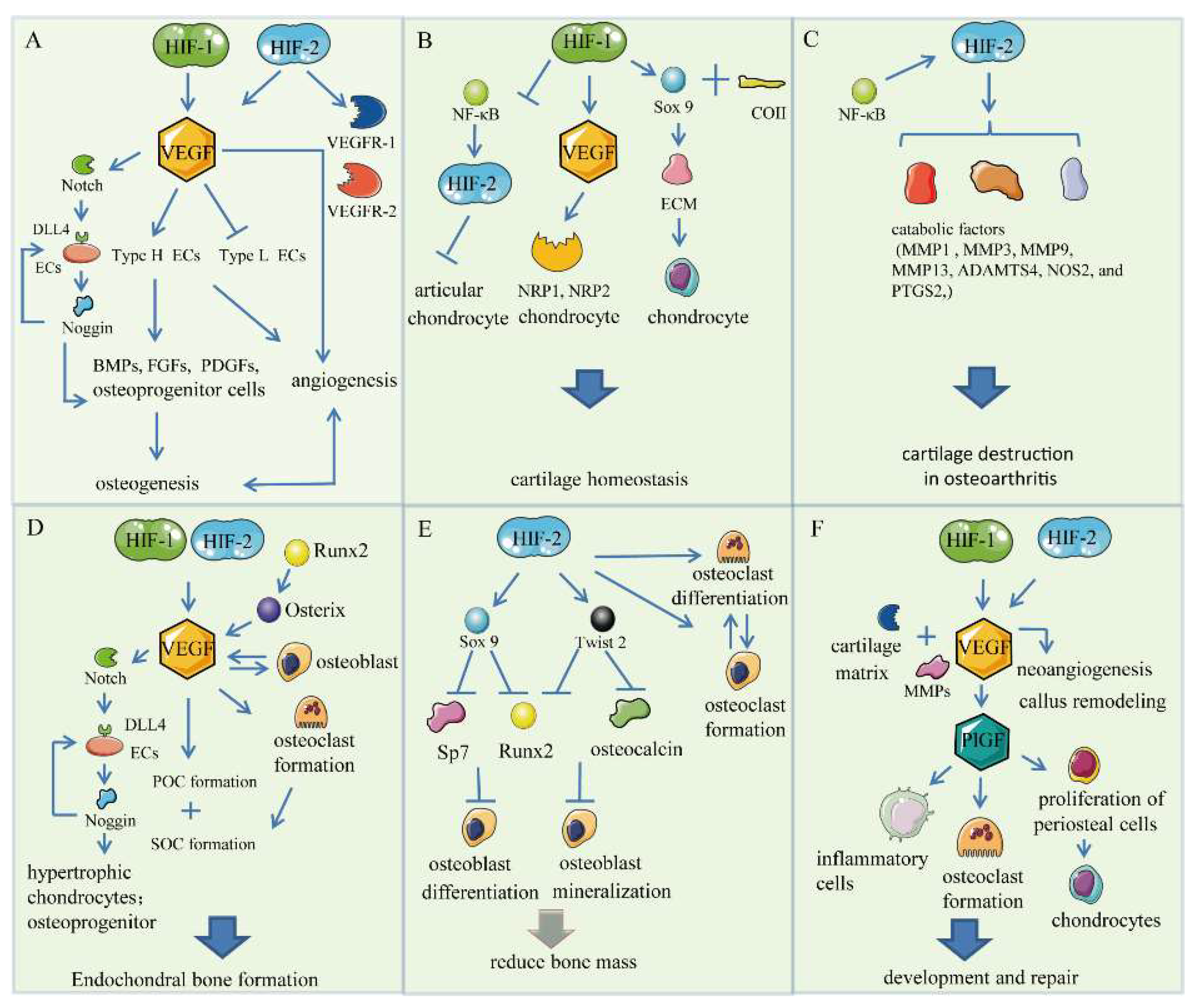
3. HIF/VEGF Pathways
3.1. Bone Angiogenesis
3.2. Chondrogenesis
3.3. Endochondral Bone Formation
3.4. Skeletal Repair
4. HIF/EPO Pathway
4.1. Bone Angiogenesis
4.2. Endochondral Bone Formation
4.3. Skeletal Repair
4.4. Controversial Role of EPO in Bone Formation
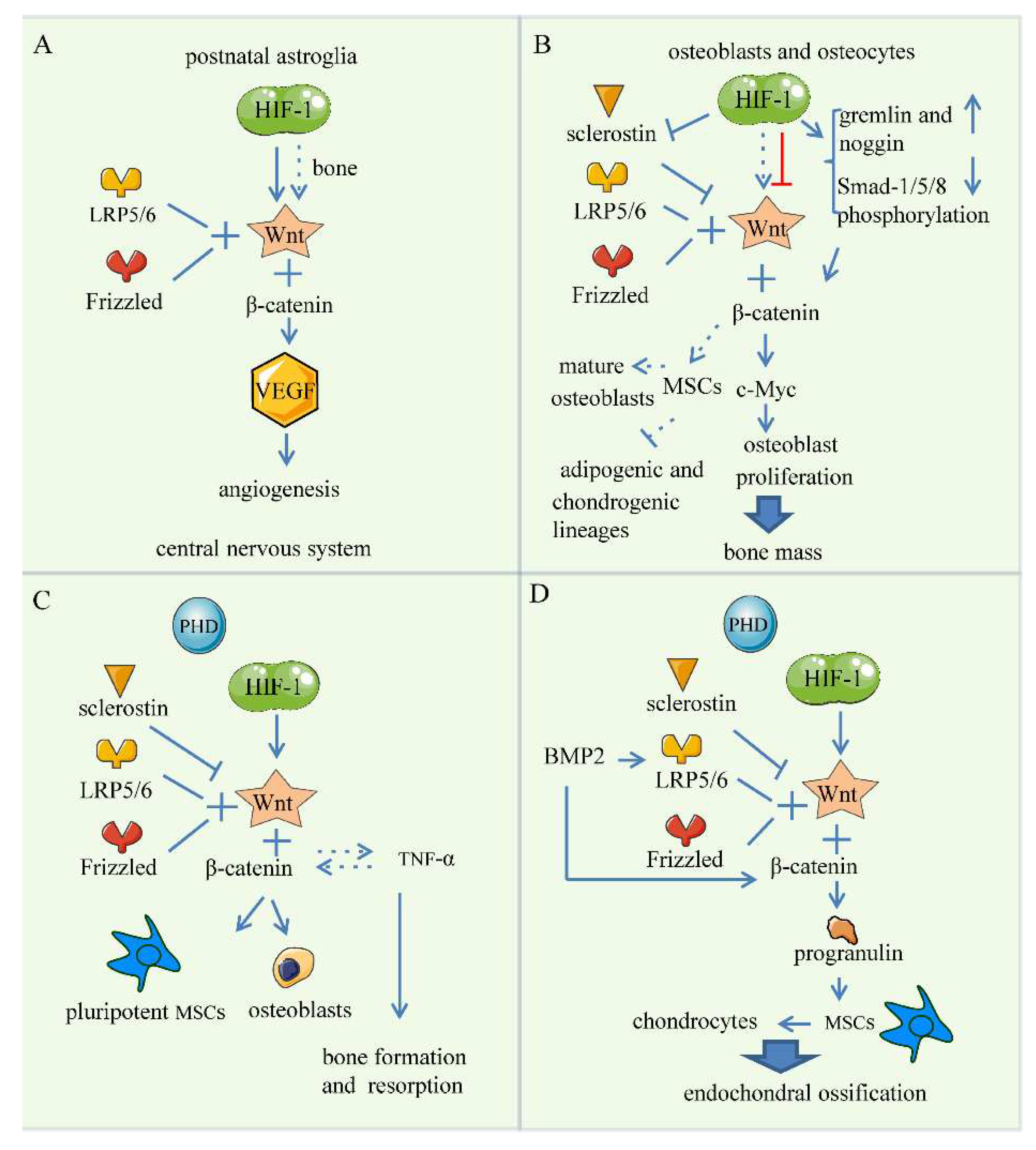
5. Wnt Signaling Pathway
5.1. Bone Angiogenesis
5.2. Endochondral Bone Formation
5.3. Skeletal Repair
6. SHIP-1
6.1. The Regulation in Osteoblasts and in MSCs
6.2. The Regulation in Osteoclasts
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Long, F.; Ornitz, D.M. Development of the Endochondral Skeleton. Cold Spring Harb. Perspect. Biol. 2013, 5, a008334. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-Q.; Tan, Y.-Y.; Wong, R.; Wenden, A.; Zhang, L.-K.; Rabie, A.B.M. The role of vascular endothelial growth factor in ossification. Int. J. Oral Sci. 2012, 4, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Kusumbe, A.P.; Ramasamy, S.K.; Adams, R.H. Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature 2014, 507, 323–328. [Google Scholar] [CrossRef]
- Yu, Y.; Ma, L.; Zhang, H.; Sun, W.; Zheng, L.; Liu, C.; Miao, L. EPO could be regulated by HIF-1 and promote osteogenesis and accelerate bone repair. Artif. Cells Nanomed. Biotechnol. 2020, 48, 206–217. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, J.A.; Partridge, N.C. Physiological Bone Remodeling: Systemic Regulation and Growth Factor Involvement. Physiology 2016, 31, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.; Olsen, B.R. The roles of vascular endothelial growth factor in bone repair and regeneration. Bone 2016, 91, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Sakadžić, S.; Roussakis, E.; Yaseen, M.A.; Mandeville, E.T.; Srinivasan, V.J.; Arai, K.; Ruvinskaya, S.; Devor, A.; Lo, E.H.; Vinogradov, S.A.; et al. Two-photon high-resolution measurement of partial pressure of oxygen in cerebral vasculature and tissue. Nat. Methods 2010, 7, 755–759. [Google Scholar] [CrossRef] [PubMed]
- Spencer, J.A.; Ferraro, F.; Roussakis, E.; Klein, A.; Wu, J.; Runnels, J.M.; Zaher, W.; Mortensen, L.J.; Alt, C.; Turcotte, R.; et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature 2014, 508, 269–273. [Google Scholar] [CrossRef]
- Choudhry, H.; Harris, A.L. Advances in Hypoxia-Inducible Factor Biology. Cell Metab. 2018, 27, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Frikha-Benayed, D.; Basta-Pljakic, J.; Majeska, R.J.; Schaffler, M.B. Regional differences in oxidative metabolism and mitochondrial activity among cortical bone osteocytes. Bone 2016, 90, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Grayson, W.L.; Zhao, F.; Bunnell, B.; Ma, T. Hypoxia enhances proliferation and tissue formation of human mesenchymal stem cells. Biochem. Biophys. Res. Commun. 2007, 358, 948–953. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Han, Z.; Jiang, W.; Huang, F.; Ren, C.; Wei, Q.; Zhou, N. Hypoxia improved vasculogenesis in distraction osteogenesis through Mesenchymal-Epithelial transition (MET), Wnt/β-catenin signaling pathway, and autophagy. Acta Histochem. 2020, 122, 151593. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jin, L.; Guo, J.; Bao, K.; Hu, J.; Zhang, Y.; Hou, Z.; Zhang, L. Chronic Intermittent Hypobaric Hypoxia Enhances Bone Fracture Healing. Front. Endocrinol. 2021, 11, 582670. [Google Scholar] [CrossRef]
- Stegen, S.; Carmeliet, G. The skeletal vascular system—Breathing life into bone tissue. Bone 2018, 115, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Jaakkola, P.; Mole, D.R.; Tian, Y.-M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-alpha to the von Hippel-Lindau Ubiquitylation Complex by O2-Regulated Prolyl Hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Yellowley, C.E.; Genetos, D.C. Hypoxia Signaling in the Skeleton: Implications for Bone Health. Curr. Osteoporos. Rep. 2019, 17, 26–35. [Google Scholar] [CrossRef]
- Park, K.M.; Gerecht, S. Hypoxia-inducible hydrogels. Nat. Commun. 2014, 5, 4075. [Google Scholar] [CrossRef]
- Pangou, E.; Befani, C.; Mylonis, I.; Samiotaki, M.; Panayotou, G.; Simos, G.; Liakos, P. HIF-2α phosphorylation by CK1δ pro motes erythropoietin secretion in liver cancer cells under hypoxia. J. Cell Sci. 2016, 129, 4213–4226. [Google Scholar] [CrossRef]
- Liang, K.; Ding, X.-Q.; Lin, C.; Kang, Y.J. Featured Article: Hypoxia-inducible factor-1α dependent nuclear entry of factor inhibiting HIF-1. Exp. Biol. Med. 2015, 240, 1446–1451. [Google Scholar] [CrossRef]
- Zelzer, E.; Levy, Y.; Kahana, C.; Shilo, B.; Rubinstein, M.; Cohen, B. Insulin induces transcription of target genes through the hypoxia-inducible factor HIF-1alpha/ARNT. EMBO J. 1998, 17, 5085–5094. [Google Scholar] [CrossRef]
- Rauner, M.; Franke, K.; Murray, M.; Singh, R.P.; Hiram-Bab, S.; Platzbecker, U.; Gassmann, M.; Socolovsky, M.; Neumann, D.; Gabet, Y.; et al. Increased EPO Levels Are Associated With Bone Loss in Mice Lacking PHD2 in EPO-Producing Cells. J. Bone Miner. Res. 2016, 31, 1877–1887. [Google Scholar] [CrossRef]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.-W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef]
- Gkotinakou, I.-M.; Befani, C.; Simos, G.; Liakos, P. ERK1/2 phosphorylates HIF-2α and regulates its activity by controlling its CRM1-dependent nuclear shuttling. J. Cell Sci. 2019, 132, jcs225698. [Google Scholar] [CrossRef]
- Suzuki, N.; Gradin, K.; Poellinger, L.; Yamamoto, M. Regulation of hypoxia-inducible gene expression after HIF activation. Exp. Cell Res. 2017, 356, 182–186. [Google Scholar] [CrossRef]
- Tolonen, J.-P.; Heikkilä, M.; Malinen, M.; Lee, H.-M.; Palvimo, J.J.; Wei, G.-H.; Myllyharju, J. A long hypoxia-inducible factor 3 isoform 2 is a transcription activator that regulates erythropoietin. Cell. Mol. Life Sci. 2020, 77, 3627–3642. [Google Scholar] [CrossRef]
- Jahangir, S.; Hosseini, S.; Mostafaei, F.; Sayahpour, F.A.; Eslaminejad, M.B. 3D-porous β-tricalcium phosphate–alginate–gelatin scaffold with DMOG delivery promotes angiogenesis and bone formation in rat calvarial defects. J. Mater. Sci. Mater. Med. 2018, 30, 1. [Google Scholar] [CrossRef]
- Zou, D.; He, J.; Zhang, K.; Dai, J.; Zhang, W.; Wang, S.; Zhou, J.; Huang, Y.; Zhang, Z.; Jiang, X. The Bone-Forming Effects of HIF-1α-Transduced BMSCs Promote Osseointegration with Dental Implant in Canine Mandible. PLoS ONE 2012, 7, e32355. [Google Scholar] [CrossRef]
- Nagai, K.; Ideguchi, H.; Kajikawa, T.; Li, X.; Chavakis, T.; Cheng, J.; Messersmith, P.B.; Heber-Katz, E.; Hajishengallis, G. An injectable hydrogel-formulated inhibitor of prolyl-4-hydroxylase promotes T regulatory cell recruitment and enhances alveolar bone regeneration during resolution of experimental periodontitis. FASEB J. 2020, 34, 13726–13740. [Google Scholar] [CrossRef]
- Cheng, S.; Xing, W.; Pourteymoor, S.; Schulte, J.; Mohan, S. Conditional Deletion of Prolyl Hydroxylase Domain-Containing Protein 2 (Phd2) Gene Reveals Its Essential Role in Chondrocyte Function and Endochondral Bone Formation. Endocrinology 2016, 157, 127–140. [Google Scholar] [CrossRef]
- Stegen, S.; Deprez, S.; Eelen, G.; Torrekens, S.; Van Looveren, R.; Goveia, J.; Ghesquière, B.; Carmeliet, P.; Carmeliet, G. Adequate hypoxia inducible factor 1α signaling is indispensable for bone regeneration. Bone 2016, 87, 176–186. [Google Scholar] [CrossRef]
- Tan, L.; Zhang, Y.; Huang, Y.; Luo, Y.; Liu, Y. Preservation of alveolar ridge after tooth extraction with hypoxia-inducible factor-1α protein in a dog model. Exp. Ther. Med. 2019, 17, 2913–2920. [Google Scholar] [CrossRef]
- Liu, W.; Li, L.; Rong, Y.; Qian, D.; Chen, J.; Zhou, Z.; Luo, Y.; Jiang, D.; Cheng, L.; Zhao, S.; et al. Hypoxic mesenchymal stem cell-derived exosomes promote bone fracture healing by the transfer of miR-126. Acta Biomater. 2020, 103, 196–212. [Google Scholar] [CrossRef]
- Zhang, J.; Pan, J.; Jing, W. Motivating role of type H vessels in bone regeneration. Cell Prolif. 2020, 53, e12874. [Google Scholar] [CrossRef]
- Ahn, G.-O.; Seita, J.; Hong, B.-J.; Kim, Y.-E.; Bok, S.; Lee, C.-J.; Kim, K.S.; Lee, J.C.; Leeper, N.J.; Cooke, J.P.; et al. Transcriptional activation of hypoxia-inducible factor-1 (HIF-1) in myeloid cells promotes angiogenesis through VEGF and S100A8. Proc. Natl. Acad. Sci. USA 2014, 111, 2698–2703. [Google Scholar] [CrossRef] [PubMed]
- Yoon, D.; Pastore, Y.D.; Divoky, V.; Liu, E.; Mlodnicka, A.E.; Rainey, K.; Ponka, P.; Semenza, G.L.; Schumacher, A.; Prchal, J.T. Hypoxia-inducible Factor-1 Deficiency Results in Dysregulated Erythropoiesis Signaling and Iron Homeostasis in Mouse Development. J. Biol. Chem. 2006, 281, 25703–25711. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-T.; Liu, J.-X.; Yu, B.; Liu, R.; Dong, C.; Li, S.-J. Notch signaling represses hypoxia-inducible factor-1α-induced activation of Wnt/β-catenin signaling in osteoblasts under cobalt-mimicked hypoxia. Mol. Med. Rep. 2016, 14, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Yeersheng, R.; Xia, Y.; Kang, P.; Wang, W. Hypoxia Enhanced Bone Regeneration Through the HIF-1α/β-Catenin Pathway in Femoral Head Osteonecrosis. Am. J. Med. Sci. 2021, 362, 78–91. [Google Scholar] [CrossRef] [PubMed]
- So, E.; Sun, C.; Wu, K.Q.; Driesman, A.; Leggett, S.; Isaac, M.; Spangler, T.; Dubielecka-Szczerba, P.M.; Reginato, A.M.; Liang, O.D. Lipid phosphatase SHIP-1 regulates chondrocyte hypertrophy and skeletal development. J. Cell. Physiol. 2020, 235, 1425–1437. [Google Scholar] [CrossRef]
- Hirai, K.; Furusho, H.; Hirota, K.; Sasaki, H. Activation of hypoxia-inducible factor 1 attenuates periapical inflammation and bone loss. Int. J. Oral Sci. 2018, 10, 12. [Google Scholar] [CrossRef]
- Meng, S.-S.; Xu, X.-P.; Chang, W.; Lu, Z.-H.; Huang, L.-L.; Xu, J.-Y.; Liu, L.; Qiu, H.-B.; Yang, Y.; Guo, F.-M. LincRNA-p21 promotes mesenchymal stem cell migration capacity and survival through hypoxic preconditioning. Stem Cell Res. Ther. 2018, 9, 280. [Google Scholar] [CrossRef]
- Tang, Y.; Hong, C.; Cai, Y.; Zhu, J.; Hu, X.; Tian, Y.; Song, X.; Song, Z.; Jiang, R.; Kang, F. HIF-1α Mediates Osteoclast-Induced Mandibular Condyle Growth via AMPK Signaling. J. Dent. Res. 2020, 99, 1377–1386. [Google Scholar] [CrossRef]
- Kapitsinou, P.P.; Liu, Q.; Unger, T.L.; Rha, J.; Davidoff, O.; Keith, B.; Epstein, J.A.; Moores, S.L.; Erickson-Miller, C.L.; Haase, V.H. Hepatic HIF-2 regulates erythropoietic responses to hypoxia in renal anemia. Blood 2010, 116, 3039–3048. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Fukai, A.; Mabuchi, A.; Ikeda, T.; Yano, F.; Ohba, S.; Nishida, N.; Akune, T.; Yoshimura, N.; Nakagawa, T.; et al. Transcriptional regulation of endochondral ossification by HIF-2α during skeletal growth and osteoarthritis development. Nat. Med. 2010, 16, 678–686. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Park, K.H.; Yu, H.-G.; Kook, E.; Song, W.-H.; Lee, G.; Koh, J.-T.; Shin, H.-I.; Choi, J.-Y.; Huh, Y.H.; et al. Controlling hypoxia-inducible factor-2α is critical for maintaining bone homeostasis in mice. Bone Res. 2019, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Merceron, C.; Ranganathan, K.; Wang, E.; Tata, Z.; Makkapati, S.; Khan, M.P.; Mangiavini, L.; Yao, Q.; Castellini, L.; Levi, B.; et al. Hypoxia-inducible factor 2α is a negative regulator of osteoblastogenesis and bone mass accrual. Bone Res. 2019, 7, 7. [Google Scholar] [CrossRef] [PubMed]
- Hannah, S.S.; McFadden, S.; McNeilly, A.; McClean, C. “Take My Bone Away?” Hypoxia and bone: A narrative review. J. Cell. Physiol. 2021, 236, 721–740. [Google Scholar] [CrossRef]
- Lee, S.Y.; Park, K.H.; Lee, G.; Kim, S.-J.; Song, W.-H.; Kwon, S.-H.; Koh, J.-T.; Huh, Y.H.; Ryu, J.-H. Hypoxia-inducible factor-2α mediates senescence-associated intrinsic mechanisms of age-related bone loss. Exp. Mol. Med. 2021, 53, 591–604. [Google Scholar] [CrossRef]
- Yang, S.; Kim, J.; Ryu, J.-H.; Oh, H.; Chun, C.-H.; Kim, B.J.; Min, B.H.; Chun, J.-S. Hypoxia-inducible factor-2α is a catabolic regulator of osteoarthritic cartilage destruction. Nat. Med. 2010, 16, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Kawaguchi, H. HIF-2α as a possible therapeutic target of osteoarthritis. Osteoarthr. Cartil. 2010, 18, 1552–1556. [Google Scholar] [CrossRef]
- Saito, T. NF-κB and HIF Signaling in Osteoarthritis. Encyclopedia of Bone Biology; Zaidi, M., Ed.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 605–608. ISBN 9780128140826. [Google Scholar]
- Bouaziz, W.; Sigaux, J.; Modrowski, D.; Devignes, C.-S.; Funck-Brentano, T.; Richette, P.; Ea, H.-K.; Provot, S.; Cohen-Solal, M.; Haÿ, E. Interaction of HIF1α and β-catenin inhibits matrix metalloproteinase 13 expression and prevents cartilage damage in mice. Proc. Natl. Acad. Sci. USA 2016, 113, 5453–5458. [Google Scholar] [CrossRef]
- Fröhlich, L.F. Micrornas at the Interface between Osteogenesis and Angiogenesis as Targets for Bone Regeneration. Cells 2019, 8, 121. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, G.; Subramanian, I.V.; Adhikari, N.; Zhang, X.; Joshi, H.P.; Basi, D.; Chandrashekhar, Y.; Hall, J.L.; Roy, S.; Zeng, Y.; et al. Hypoxia-induced microRNA-424 expression in human endothelial cells regulates HIF-α isoforms and promotes angiogenesis. J. Clin. Investig. 2010, 120, 4141–4154. [Google Scholar] [CrossRef] [PubMed]
- Maes, C.; Carmeliet, G.; Schipani, E. Hypoxia-driven pathways in bone development, regeneration and disease. Nat. Rev. Rheu matol. 2012, 8, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Haase, V.H. Regulation of erythropoiesis by hypoxia-inducible factors. Blood Rev. 2013, 27, 41–53. [Google Scholar] [CrossRef]
- Zhang, F.-J.; Luo, W.; Lei, G.-H. Role of HIF-1α and HIF-2α in osteoarthritis. Jt. Bone Spine 2015, 82, 144–147. [Google Scholar] [CrossRef]
- Bohensky, J.; Terkhorn, S.P.; Freeman, T.A.; Adams, C.S.; Garcia, J.A.; Shapiro, I.M.; Srinivas, V. Regulation of autophagy in human and murine cartilage: Hypoxia-inducible factor 2 suppresses chondrocyte autophagy. Arthritis Care Res. 2009, 60, 1406–1415. [Google Scholar] [CrossRef]
- Knowles, H.J. Distinct roles for the hypoxia-inducible transcription factors HIF-1α and HIF-2α in human osteoclast formation and function. Sci. Rep. 2020, 10, 21072. [Google Scholar] [CrossRef]
- Maes, C.; Araldi, E.; Haigh, K.; Khatri, R.; Van Looveren, R.; Giaccia, A.J.; Haigh, J.J.; Carmeliet, G.; Schipani, E. VEGF-independent cell-autonomous functions of HIF-1α regulating oxygen consumption in fetal cartilage are critical for chondrocyte survival. J. Bone Miner. Res. 2012, 27, 596–609. [Google Scholar] [CrossRef]
- Ferrara, N.; Gerber, H.-P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef]
- Watson, E.C.; Adams, R.H. Biology of Bone: The Vasculature of the Skeletal System. Cold Spring Harb. Perspect. Med. 2018, 8, a031559. [Google Scholar] [CrossRef]
- Duan, X.; Murata, Y.; Liu, Y.; Nicolae, C.; Olsen, B.R.; Berendsen, A.D. Vegfa regulates perichondrial vascularity and osteoblast differentiation in bone development. Development 2015, 142, 1984–1991. [Google Scholar] [CrossRef] [PubMed]
- Zavan, B.; Ferroni, L.; Gardin, C.; Sivolella, S.; Piattelli, A.; Mijiritsky, E. Release of VEGF from Dental Implant Improves Osteogenetic Process: Preliminary In Vitro Tests. Materials 2017, 10, 1052. [Google Scholar] [CrossRef] [PubMed]
- García, J.R.; Clark, A.Y.; García, A.J. Integrin-specific hydrogels functionalized with VEGF for vascularization and bone regeneration of critical-size bone defects. J. Biomed. Mater. Res. Part A 2016, 104, 889–900. [Google Scholar] [CrossRef]
- Ahluwalia, A.; Tarnawski, A.S. Critical Role of Hypoxia Sensor—HIF-1α in VEGF Gene Activation. Implications for Angiogen esis and Tissue Injury Healing. Curr. Med. Chem. 2012, 19, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Xie, J.; Zhong, J.; Cun, X.; Lin, S.; Lin, Y.; Cai, X. Hypoxia enhances angiogenesis in an adipose-derived stromal cell/endothelial cell co-culture 3D gel model. Cell Prolif. 2016, 49, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Yang, J.; Chen, Y.; Jin, X.; Li, Z.; Wen, X.; Xia, Q.; Wang, Y. Salidroside improves angiogenesis-osteogenesis coupling by regulating the HIF-1alpha/VEGF signaling pathway in the bone environment. Eur. J. Pharmacol. 2020, 884, 173394. [Google Scholar] [CrossRef]
- Chen, J.; Gu, Z.; Wu, M.; Yang, Y.; Zhang, J.; Ou, J.; Zuo, Z.; Wang, J.; Chen, Y. C-reactive protein can upregulate VEGF expression to promote ADSC-induced angiogenesis by activating HIF-1α via CD64/PI3k/Akt and MAPK/ERK signaling pathways. Stem Cell Res. Ther. 2016, 7, 114. [Google Scholar] [CrossRef]
- Jing, X.; Du, T.; Yang, X.; Zhang, W.; Wang, G.; Liu, X.; Li, T.; Jiang, Z. Desferoxamine protects against glucocorticoid-induced osteonecrosis of the femoral head via activating HIF-1α expression. J. Cell. Physiol. 2020, 235, 9864–9875. [Google Scholar] [CrossRef]
- Filipowska, J.; Tomaszewski, K.A.; Niedźwiedzki, L.; Walocha, J.A.; Niedźwiedzki, T. The role of vasculature in bone development, regeneration and proper systemic functioning. Angiogenesis 2017, 20, 291–302. [Google Scholar] [CrossRef]
- Peng, Y.; Wu, S.; Li, Y.; Crane, J.L. Type H blood vessels in bone modeling and remodeling. Theranostics 2020, 10, 426–436. [Google Scholar] [CrossRef]
- Sivaraj, K.K.; Adams, R.H. Blood vessel formation and function in bone. Development 2016, 143, 2706–2715. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xie, L. Unique bone marrow blood vessels couple angiogenesis and osteogenesis in bone homeostasis and diseases. Ann. N. Y. Acad. Sci. 2020, 1474, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Potente, M.; Gerhardt, H.; Carmeliet, P. Basic and Therapeutic Aspects of Angiogenesis. Cell 2011, 146, 873–887. [Google Scholar] [CrossRef]
- Ramasamy, S.K.; Kusumbe, A.P.; Wang, L.; Adams, R.H. Endothelial Notch activity promotes angiogenesis and osteogenesis in bone. Nature 2014, 507, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Li, C.-J.; Sun, X.; Guo, Q.; Xiao, Y.; Su, T.; Tu, M.-L.; Peng, H.; Lu, Q.; Liu, Q.; et al. MiR-497∼195 cluster regulates angiogenesis during coupling with osteogenesis by maintaining endothelial Notch and HIF-1α activity. Nat. Commun. 2017, 8, 16003. [Google Scholar] [CrossRef]
- Sasagawa, T.; Nagamatsu, T.; Morita, K.; Mimura, N.; Iriyama, T.; Fujii, T.; Shibuya, M. HIF-2α, but not HIF-1α, mediates hypoxia-induced up-regulation of Flt-1 gene expression in placental trophoblasts. Sci. Rep. 2018, 8, 17375. [Google Scholar] [CrossRef]
- Befani, C.; Liakos, P. The role of hypoxia-inducible factor-2 alpha in angiogenesis. J. Cell. Physiol. 2018, 233, 9087–9098. [Google Scholar] [CrossRef]
- Olsen, J.J.; Pohl, S.Ö.; Deshmukh, A.; Visweswaran, M.; Ward, N.C.; Arfuso, F.; Agostino, M.; Dharmarajan, A. The Role of Wnt Signalling in Angiogenesis. Clin. Biochem. Rev. 2017, 38, 131. [Google Scholar]
- Schipani, E.; Ryan, H.E.; Didrickson, S.; Kobayashi, T.; Knight, M.; Johnson, R.S. Hypoxia in cartilage: HIF-1α is essential for chondrocyte growth arrest and survival. Genome Res. 2001, 15, 2865–2876. [Google Scholar] [CrossRef]
- Ma, Z.; Jin, X.; Qian, Z.; Li, F.; Xu, M.; Zhang, Y.; Kang, X.; Li, H.; Gao, X.; Zhao, L.; et al. Deletion of clock gene Bmal1 impaired the chondrocyte function due to disruption of the HIF1α-VEGF signaling pathway. Cell Cycle 2019, 18, 1473–1489. [Google Scholar] [CrossRef]
- Taheem, D.K.; Jell, G.; Gentleman, E. Hypoxia Inducible Factor-1α in Osteochondral Tissue Engineering. Tissue Eng. Part B Rev. 2020, 26, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.-C.; Choi, W.H. Mithramycin A Alleviates Osteoarthritic Cartilage Destruction by Inhibiting HIF-2α Expression. Int. J. Mol. Sci. 2018, 19, 1411. [Google Scholar] [CrossRef] [PubMed]
- Okada, K.; Mori, D.; Makii, Y.; Nakamoto, H.; Murahashi, Y.; Yano, F.; Chang, S.H.; Taniguchi, Y.; Kobayashi, H.; Semba, H.; et al. Hypoxia-inducible factor-1 alpha maintains mouse articular cartilage through suppression of NF-κB signaling. Sci. Rep. 2020, 10, 5425. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Aghajanian, P.; Pourteymoor, S.; Alarcon, C.; Mohan, S. Prolyl Hydroxylase Domain-Containing Protein 2 (Phd2) Regulates Chondrocyte Differentiation and Secondary Ossification in Mice. Sci. Rep. 2016, 6, 35748. [Google Scholar] [CrossRef]
- Tang, W.; Yang, F.; Li, Y.; de Crombrugghe, B.; Jiao, H.; Xiao, G.; Zhang, C. Transcriptional Regulation of Vascular Endothelial Growth Factor (VEGF) by Osteoblast-specific Transcription Factor Osterix (Osx) in Osteoblasts. J. Biol. Chem. 2012, 287, 1671–1678. [Google Scholar] [CrossRef]
- Zelzer, E.; Glotzer, D.J.; Hartmann, C.; Thomas, D.; Fukai, N.; Soker, S.; Olsen, B.R. Tissue specific regulation of VEGF expression during bone development requires Cbfa1/Runx2. Mech. Dev. 2001, 106, 97–106. [Google Scholar] [CrossRef]
- Mevel, R.; Draper, J.E.; Lie-A-Ling, M.; Kouskoff, V.; Lacaud, G. RUNX transcription factors: Orchestrators of development. Development 2019, 146, dev148296. [Google Scholar] [CrossRef]
- Hu, K.; Olsen, B.R. Vascular endothelial growth factor control mechanisms in skeletal growth and repair. Dev. Dyn. 2017, 246, 227–234. [Google Scholar] [CrossRef]
- Niida, S.; Kaku, M.; Amano, H.; Yoshida, H.; Kataoka, H.; Nishikawa, S.; Tanne, K.; Maeda, N.; Nishikawa, S.-I.; Kodama, H. Vascular Endothelial Growth Factor Can Substitute for Macrophage Colony-Stimulating Factor in the Support of Osteoclastic Bone Resorption. J. Exp. Med. 1999, 190, 293–298. [Google Scholar] [CrossRef]
- Aldridge, S.E.; Lennard, T.W.; Williams, J.R.; Birch, M.A. Vascular endothelial growth factor receptors in osteoclast differentiation and function. Biochem. Biophys. Res. Commun. 2005, 335, 793–798. [Google Scholar] [CrossRef]
- Harper, J.; Gerstenfeld, L.C.; Klagsbrun, M. Neuropilin-1 Expression in Osteogenic Cells: Down-Regulation During Differentiation of Osteoblasts into Osteocytes. J. Cell. Biochem. 2001, 81, 82–92. [Google Scholar] [CrossRef]
- Zelzer, E.; Olsen, B.R. Multiple Roles of Vascular Endothelial Growth Factor (VEGF) in Skeletal Development, Growth, and Repair. Curr. Top. Dev. Biol. 2005, 65, 169–187. [Google Scholar] [CrossRef]
- Zelzer, E.; McLean, W.; Ng, Y.S.; Fukai, N.; Reginato, A.M.; Lovejoy, S.; D’Amore, P.A.; Olsen, B.R. Skeletal defects in VEGF120/120 mice reveal multiple roles for VEGF in skeletogenesis. Development 2002, 129, 1893–1904. [Google Scholar] [CrossRef]
- Raines, A.L.; Berger, M.B.; Patel, N.; Hyzy, S.L.; Boyan, B.D.; Schwartz, Z. VEGF-A regulates angiogenesis during osseointegration of Ti implants via paracrine/autocrine regulation of osteoblast response to hierarchical microstructure of the surface. J. Biomed. Mater. Res. Part A 2019, 107, 423–433. [Google Scholar] [CrossRef]
- Huang, B.; Wang, W.; Li, Q.; Wang, Z.; Yan, B.; Zhang, Z.; Wang, L.; Huang, M.; Jia, C.; Lu, J.; et al. Osteoblasts secrete Cxcl9 to regulate angiogenesis in bone. Nat. Commun. 2016, 7, 13885. [Google Scholar] [CrossRef]
- Riddle, R.C.; Khatri, R.; Schipani, E.; Clemens, T.L. Role of hypoxia-inducible factor-1α in angiogenic–osteogenic coupling. J. Mol. Med. 2009, 87, 583–590. [Google Scholar] [CrossRef]
- Komatsu, D.; Hadjiargyrou, M. Activation of the transcription factor HIF-1 and its target genes, VEGF, HO-1, iNOS, during fracture repair. Bone 2004, 34, 680–688. [Google Scholar] [CrossRef]
- Maes, C.; Coenegrachts, L.; Stockmans, I.; Daci, E.; Luttun, A.; Petryk, A.; Gopalakrishnan, R.; Moermans, K.; Smets, N.; Verfaillie, C.; et al. Placental growth factor mediates mesenchymal cell development, cartilage turnover, and bone remodeling during fracture repair. J. Clin. Investig. 2006, 116, 1230–1242. [Google Scholar] [CrossRef]
- Fan, L.; Li, J.; Yu, Z.; Dang, X.; Wang, K. The Hypoxia-Inducible Factor Pathway, Prolyl Hydroxylase Domain Protein Inhibitors, and Their Roles in Bone Repair and Regeneration. BioMed Res. Int. 2014, 2014, 239356. [Google Scholar] [CrossRef]
- Zhang, Y.; Hao, Z.; Wang, P.; Xia, Y.; Wu, J.; Xia, D.; Fang, S.; Xu, S. Exosomes from human umbilical cord mesenchymal stem cells enhance fracture healing through HIF-1α-mediated promotion of angiogenesis in a rat model of stabilized fracture. Cell Prolif. 2019, 52, e12570. [Google Scholar] [CrossRef]
- Hong, C.; Tang, Y.; Hu, X.; Song, X.; Cai, Y.; Song, Z.; Kang, F. Partial deficiency of HIF-1α in chondrocytes effected bone repair of mandibular condylar neck. Arch. Oral Biol. 2021, 122, 105023. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Chen, S.; Song, W.-Q.; Gao, Y.-S.; Guan, J.-J.; Wang, Y.; Sun, Y.; Zhang, C.-Q. Dimethyloxaloylglycine Improves Angiogenic Activity of Bone Marrow Stromal Cells in the Tissue-Engineered Bone. Int. J. Biol. Sci. 2014, 10, 746–756. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Feng, Z.; Wei, J.; Yu, Y.; Luo, J.; Zhou, J.; Li, Y.; Zheng, X.; Tang, W.; Liu, L.; et al. Repair of Critical-Sized Mandible Defects in Aged Rat Using Hypoxia Preconditioned BMSCs with Up-regulation of Hif-1α. Int. J. Biol. Sci. 2018, 14, 449–460. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, J.; Cai, F.; Liu, K.; Zhang, X.; Yusufu, A. Hypoxia During the Consolidation Phase of Distraction Osteogenesis Promotes Bone Regeneration. Front. Physiol. 2022, 13, 804469. [Google Scholar] [CrossRef]
- Schödel, J.; Ratcliffe, P.J. Mechanisms of hypoxia signaling: New implications for nephrology. Nat. Rev. Nephrol. 2019, 15, 641–659. [Google Scholar] [CrossRef]
- Jelkmann, W. Erythropoietin. Front. Horm. Res. 2016, 47, 115–127. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, L.; Dey, S.; Alnaeeli, M.; Suresh, S.; Rogers, H.; Teng, R.; Noguchi, C.T. Erythropoietin Action in Stress Response, Tissue Maintenance and Metabolism. Int. J. Mol. Sci. 2014, 15, 10296–10333. [Google Scholar] [CrossRef]
- Suresh, S.; Rajvanshi, P.K.; Noguchi, C.T. The Many Facets of Erythropoietin Physiologic and Metabolic Response. Front. Physiol. 2019, 10, 1534. [Google Scholar] [CrossRef]
- Liu, P.; Zhou, Y.; An, Q.; Song, Y.; Chen, X.; Yang, G.-Y.; Zhu, W. Erythropoietin Stimulates Endothelial Progenitor Cells to Induce Endothelialization in an Aneurysm Neck After Coil Embolization by Modulating Vascular Endothelial Growth Factor. Stem Cells Transl. Med. 2016, 5, 1182–1189. [Google Scholar] [CrossRef]
- Bahlmann, F.H.; Degroot, K.; Duckert, T.; Niemczyk, E.; Bahlmann, E.; Boehm, S.M.; Haller, H.; Fliser, D. Endothelial progenitor cell proliferation and differentiation is regulated by erythropoietin Rapid Communication. Kidney Int. 2003, 64, 1648–1652. [Google Scholar] [CrossRef]
- Kertesz, N.; Wu, J.; Chen, T.H.-P.; Sucov, H.M.; Wu, H. The role of erythropoietin in regulating angiogenesis. Dev. Biol. 2004, 276, 101–110. [Google Scholar] [CrossRef]
- Eggold, J.T.; Rankin, E.B. Erythropoiesis, EPO, macrophages, and bone. Bone 2019, 119, 36–41. [Google Scholar] [CrossRef]
- Suresh, S.; De Castro, L.F.; Dey, S.; Robey, P.G.; Noguchi, C.T. Erythropoietin modulates bone marrow stromal cell differentiation. Bone Res. 2019, 7, 21. [Google Scholar] [CrossRef]
- Su, J.; Li, Z.; Cui, S.; Ji, L.; Geng, H.; Chai, K.; Ma, X.; Bai, Z.; Yang, Y.; Wuren, T.; et al. The Local HIF-2α/EPO Pathway in the Bone Marrow is Associated with Excessive Erythrocytosis and the Increase in Bone Marrow Microvessel Density in Chronic Mountain Sickness. High Alt. Med. Biol. 2015, 16, 318–330. [Google Scholar] [CrossRef]
- Landau, D.; London, L.; Bandach, I.; Segev, Y. The hypoxia inducible factor/erythropoietin (EPO)/EPO receptor pathway is disturbed in a rat model of chronic kidney disease related anemia. PLoS ONE 2018, 13, e0196684. [Google Scholar] [CrossRef]
- Shiozawa, Y.; Jung, Y.; Ziegler, A.M.; Pedersen, E.A.; Wang, J.; Wang, Z.; Song, J.; Wang, J.; Lee, C.H.; Sud, S.; et al. Erythropoietin Couples Hematopoiesis with Bone Formation. PLoS ONE 2010, 5, e10853. [Google Scholar] [CrossRef]
- Wan, L.; Zhang, F.; He, Q.; Tsang, W.P.; Lu, L.; Li, Q.; Wu, Z.; Qiu, G.; Zhou, G.; Wan, C. EPO Promotes Bone Repair through Enhanced Cartilaginous Callus Formation and Angiogenesis. PLoS ONE 2014, 9, e102010. [Google Scholar] [CrossRef]
- Greenwald, A.; Licht, T.; Kumar, S.; Oladipupo, S.S.; Iyer, S.; Grunewald, M.; Keshet, E. VEGF expands erythropoiesis via hypoxia-independent induction of erythropoietin in noncanonical perivascular stromal cells. J. Exp. Med. 2019, 216, 215–230. [Google Scholar] [CrossRef]
- Rankin, E.B.; Wu, C.; Khatri, R.; Wilson, T.L.; Andersen, R.; Araldi, E.; Rankin, A.L.; Yuan, J.; Kuo, C.J.; Schipani, E.; et al. The HIF signaling pathway in osteoblasts directly modulates erythropoiesis through the production of EPO. Cell 2012, 149, 63–74. [Google Scholar] [CrossRef]
- Bogdanovski, D.A.; DiFazio, L.T.; Bogdanovski, A.K.; Csóka, B.; Jordan, G.B.; Paul, E.R.; Antonioli, L.; Pilip, S.A.; Nemeth, Z.H. Hypoxia-inducible-factor-1 in trauma and critical care. J. Crit. Care 2017, 42, 207–212. [Google Scholar] [CrossRef]
- Guo, L.; Luo, T.; Fang, Y.; Yang, L.; Wang, L.; Liu, J.; Shi, B. Effects of erythropoietin on osteoblast proliferation and function. Clin. Exp. Med. 2014, 14, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Suresh, S.; Lee, J.; Noguchi, C.T. Erythropoietin signaling in osteoblasts is required for normal bone formation and for bone loss during erythropoietin-stimulated erythropoiesis. FASEB J. 2020, 34, 11685–11697. [Google Scholar] [CrossRef] [PubMed]
- McGee, S.J.; Havens, A.M.; Shiozawa, Y.; Jung, Y.; Taichman, R.S. Effects of erythropoietin on the bone microenvironment. Growth Factors 2012, 30, 22–28. [Google Scholar] [CrossRef]
- He, Y.-B.; Liu, S.-Y.; Deng, S.-Y.; Kuang, L.-P.; Xu, S.-Y.; Li, Z.; Xu, L.; Liu, W.; Ni, G.-X. Mechanical Stretch Promotes the Osteogenic Differentiation of Bone Mesenchymal Stem Cells Induced by Erythropoietin. Stem Cells Int. 2019, 2019, 1839627. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Deng, L.; Xie, X.; Yang, Z.; Kang, P. Evaluation of the osteogenesis and angiogenesis effects of erythropoietin and the efficacy of deproteinized bovine bone/recombinant human erythropoietin scaffold on bone defect repair. J. Mater. Sci. Mater. Electron. 2016, 27, 101. [Google Scholar] [CrossRef] [PubMed]
- Stegen, S.; Carmeliet, G. Hypoxia, hypoxia-inducible transcription factors and oxygen-sensing prolyl hydroxylases in bone development and homeostasis. Curr. Opin. Nephrol. Hypertens. 2019, 28, 328–335. [Google Scholar] [CrossRef]
- Hanudel, M.R.; Laster, M.; Salusky, I.B. Non-renal-Related Mechanisms of FGF23 Pathophysiology. Curr. Osteoporos. Rep. 2018, 16, 724–729. [Google Scholar] [CrossRef]
- Roszko, K.L.; Brown, S.; Pang, Y.; Huynh, T.; Zhuang, Z.; Pacak, K.; Collins, M.T. C-Terminal, but Not Intact, FGF23 and EPO Are Strongly Correlatively Elevated in Patients With Gain-of-Function Mutations in HIF2A: Clinical Evidence for EPO Regulating FGF23. J. Bone Miner. Res. 2020, 36, 315–321. [Google Scholar] [CrossRef]
- Lang, F.; Leibrock, C.; Pandyra, A.A.; Stournaras, C.; Wagner, C.A.; Föller, M. Phosphate Homeostasis, Inflammation and the Regulation of FGF-23. Kidney Blood Press. Res. 2018, 43, 1742–1748. [Google Scholar] [CrossRef]
- Murali, S.K.; Roschger, P.; Zeitz, U.; Klaushofer, K.; Andrukhova, O.; Erben, R.G. FGF23 Regulates Bone Mineralization in a 1,25(OH)2D3and Klotho-Independent Manner. J. Bone Miner. Res. 2016, 31, 129–142. [Google Scholar] [CrossRef]
- Zhang, Q.; Doucet, M.; Tomlinson, R.E.; Han, X.; Quarles, L.D.; Collins, M.T.; Clemens, T.L. The hypoxia-inducible factor-1α activates ectopic production of fibroblast growth factor 23 in tumor-induced osteomalacia. Bone Res. 2016, 4, 16011. [Google Scholar] [CrossRef] [PubMed]
- Richmond, T.D.; Chohan, M.; Barber, D.L. Turning cells red: Signal transduction mediated by erythropoietin. Trends Cell Biol. 2005, 15, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Jung, Y.; Sun, H.; Joseph, J.; Mishra, A.; Shiozawa, Y.; Wang, J.; Krebsbach, P.H.; Taichman, R.S. Erythropoietin mediated bone formation is regulated by mTOR signaling. J. Cell. Biochem. 2012, 113, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Hiram-Bab, S.; Liron, T.; Deshet-Unger, N.; Mittelman, M.; Gassmann, M.; Rauner, M.; Franke, K.; Wielockx, B.; Neumann, D.; Gabet, Y. Erythropoietin directly stimulates osteoclast precursors and induces bone loss. FASEB J. 2015, 29, 1890–1900. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Shi, C.; Kim, J.; Chen, Y.; Ni, S.; Jiang, L.; Zheng, C.; Li, D.; Hou, J.; Taichman, R.S.; et al. Erythropoietin Promotes Bone Formation through EphrinB2/EphB4 Signaling. J. Dent. Res. 2015, 94, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wu, F.; Song, Y.; Duan, Y.; Jin, Z. Erythropoietin induces the osteogenesis of periodontal mesenchymal stem cells from healthy and periodontitis sources via activation of the p38 MAPK pathway. Int. J. Mol. Med. 2018, 41, 829–835. [Google Scholar] [CrossRef]
- Zheng, D.H.; Wang, X.X.; Ma, D.; Zhang, L.N.; Qiao, Q.F.; Zhang, J. Erythropoietin enhances osteogenic differentiation of human periodontal ligament stem cells via Wnt/beta-catenin signaling pathway. Drug Des Devel Ther. 2019, 13, 2543–2552. [Google Scholar] [CrossRef]
- Holstein, J.H.; Menger, M.D.; Scheuer, C.; Meier, C.; Culemann, U.; Wirbel, R.J.; Garcia, P.; Pohlemann, T. Erythropoietin (EPO) —EPO-receptor signaling improves early endochondral ossification and mechanical strength in fracture healing. Life Sci. 2007, 80, 893–900. [Google Scholar] [CrossRef]
- Omlor, G.W.; Kleinschmidt, K.; Gantz, S.; Speicher, A.; Guehring, T.; Richter, W. Increased bone formation in a rabbit long-bone defect model after single local and single systemic application of erythropoietin. Acta Orthop. 2016, 87, 425–431. [Google Scholar] [CrossRef]
- Garcia, P.; Speidel, V.; Scheuer, C.; Laschke, M.W.; Holstein, J.H.; Histing, T.; Pohlemann, T.; Menger, M. Low dose erythropoietin stimulates bone healing in mice. J. Orthop. Res. 2011, 29, 165–172. [Google Scholar] [CrossRef]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009, 1, a001651. [Google Scholar] [CrossRef]
- Oikonomidou, P.R.; Casu, C.; Yang, Z.; Crielaard, B.; Shim, J.H.; Rivella, S.; Vogiatzi, M.G. Polycythemia is associated with bone loss and reduced osteoblast activity in mice. Osteoporos. Int. 2016, 27, 1559–1568. [Google Scholar] [CrossRef]
- Orth, M.; Baudach, J.; Scheuer, C.; Osche, D.; Veith, N.; Braun, B.; Rollmann, M.; Herath, S.; Pohlemann, T.; Menger, M.; et al. Erythropoietin does not improve fracture healing in aged mice. Exp. Gerontol. 2019, 122, 1–9. [Google Scholar] [CrossRef]
- Deshet-Unger, N.; Kolomansky, A.; Ben-Califa, N.; Hiram-Bab, S.; Gilboa, D.; Liron, T.; Ibrahim, M.; Awida, Z.; Gorodov, A.; Oster, H.S.; et al. Erythropoietin receptor in B cells plays a role in bone remodeling in mice. Theranostics 2020, 10, 8744–8756. [Google Scholar] [CrossRef]
- Routledge, D.; Scholpp, S. Mechanisms of intercellular Wnt transport. Development 2019, 146, dev176073. [Google Scholar] [CrossRef]
- Anthony, C.C.; Robbins, D.J.; Ahmed, Y.; Lee, E. Nuclear Regulation of Wnt/β-Catenin Signaling: It’s a Complex Situation. Genes 2020, 11, 886. [Google Scholar] [CrossRef]
- Houschyar, K.S.; Tapking, C.; Borrelli, M.R.; Popp, D.; Duscher, D.; Maan, Z.N.; Chelliah, M.P.; Li, J.; Harati, K.; Wallner, C.; et al. Wnt Pathway in Bone Repair and Regeneration—What Do We Know So Far. Front. Cell Dev. Biol. 2018, 6, 170. [Google Scholar] [CrossRef]
- Baron, R.; Kneissel, M. WNT signaling in bone homeostasis and disease: From human mutations to treatments. Nat. Med. 2013, 19, 179–192. [Google Scholar] [CrossRef]
- Goodwin, A.; D’Amore, P. Wnt signaling in the vasculature. Angiogenesis 2002, 5, 1317–1323. [Google Scholar] [CrossRef]
- Yuen, T.J.; Silbereis, J.C.; Griveau, A.; Chang, S.M.; Daneman, R.; Fancy, S.P.J.; Zahed, H.; Maltepe, E.; Rowitch, D.H. Oligodendrocyte-encoded HIF function couples postnatal myelination and white matter angiogenesis. Cell 2014, 158, 383–396. [Google Scholar] [CrossRef]
- Zhang, S.; Kim, B.; Zhu, X.; Gui, X.; Wang, Y.; Lan, Z.; Prabhu, P.; Fond, K.; Wang, A.; Guo, F. Glial type specific regulation of CNS angiogenesis by HIFα-activated different signaling pathways. Nat. Commun. 2020, 11, 2027. [Google Scholar] [CrossRef] [PubMed]
- Dijke, P.T.; Krause, C.; de Gorter, D.J.J.; Löwik, C.W.; van Bezooijen, R.L. Osteocyte-Derived Sclerostin Inhibits Bone Formation: Its Role in Bone Morphogenetic Protein and Wnt Signaling. J. Bone Jt. Surg. 2008, 90, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Stegen, S.; Stockmans, I.; Moermans, K.; Thienpont, B.; Maxwell, P.H.; Carmeliet, P.; Carmeliet, G. Osteocytic oxygen sensing controls bone mass through epigenetic regulation of sclerostin. Nat. Commun. 2018, 9, 2557. [Google Scholar] [CrossRef] [PubMed]
- Kovács, B.; Vajda, E.; Nagy, E.E. Regulatory Effects and Interactions of the Wnt and OPG-RANKL-RANK Signaling at the Bone-Cartilage Interface in Osteoarthritis. Int. J. Mol. Sci. 2019, 20, 4653. [Google Scholar] [CrossRef] [PubMed]
- Genetos, D.C.; Toupadakis, C.A.; Raheja, L.F.; Wong, A.; Papanicolaou, S.E.; Fyhrie, D.P.; Loots, G.; Yellowley, C.E. Hypoxia decreases sclerostin expression and increases Wnt signaling in osteoblasts. J. Cell. Biochem. 2010, 110, 457–467. [Google Scholar] [CrossRef]
- Zhou, N.; Hu, N.; Liao, J.-Y.; Lin, L.-B.; Zhao, C.; Si, W.-K.; Yang, Z.; Yi, S.-X.; Fan, T.-X.; Bao, W.; et al. HIF-1α as a Regulator of BMP2-Induced Chondrogenic Differentiation, Osteogenic Differentiation, and Endochondral Ossification in Stem Cells. Cell. Physiol. Biochem. 2015, 36, 44–60. [Google Scholar] [CrossRef]
- Ullah, I.; Subbarao, R.B.; Rho, G.J. Human mesenchymal stem cells—Current trends and future prospective. Biosci. Rep. 2015, 35, e00191. [Google Scholar] [CrossRef]
- Bao, Q.; Chen, S.; Qin, H.; Feng, J.; Liu, H.; Liu, D.; Li, A.; Shen, Y.; Zhao, Y.; Li, J.; et al. An appropriate Wnt/β-catenin expression level during the remodeling phase is required for improved bone fracture healing in mice. Sci. Rep. 2017, 7, 2695. [Google Scholar] [CrossRef]
- Wu, S.; Zang, W.; Li, X.; Sun, H. Proepithelin Stimulates Growth Plate Chondrogenesis via Nuclear Factor-κB-p65-dependent Mechanisms. J. Biol. Chem. 2011, 286, 24057–24067. [Google Scholar] [CrossRef]
- Aslani, S.; Abhari, A.; Sakhinia, E.; Sanajou, D.; Rajabi, H.; Rahimzadeh, S. Interplay between microRNAs and Wnt, transforming growth factor-β, and bone morphogenic protein signaling pathways promote osteoblastic differentiation of mesenchymal stem cells. J. Cell. Physiol. 2019, 234, 8082–8093. [Google Scholar] [CrossRef]
- Zhang, M.; Yan, Y.; Lim, Y.-B.; Tang, D.; Xie, R.; Chen, A.; Tai, P.; Harris, S.E.; Xing, L.; Qin, Y.-X.; et al. BMP-2 modulates β-catenin signaling through stimulation of Lrp5 expression and inhibition of β-TrCP expression in osteoblasts. J. Cell. Biochem. 2009, 108, 896–905. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-B.; Leucht, P.; Lam, K.; Luppen, C.; Berge, D.T.; Nusse, R.; Helms, J.A. Bone Regeneration Is Regulated by Wnt Signaling. J. Bone Miner. Res. 2007, 22, 1913–1923. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Li, Y.; Zhou, Z.; Xing, Y.; Zhong, Y.; Zou, X.; Tian, W.; Zhang, C. Synergistic Inhibition of Wnt Pathway by HIF-1α and Osteoblast-Specific Transcription Factor Osterix (Osx) in Osteoblasts. PLoS ONE 2012, 7, e52948. [Google Scholar] [CrossRef]
- Chen, D.; Li, Y.; Zhou, Z.; Wu, C.; Xing, Y.; Zou, X.; Tian, W.; Zhang, C. HIF-1α Inhibits Wnt Signaling Pathway by Activating Sost Expression in Osteoblasts. PLoS ONE 2013, 8, e65940. [Google Scholar] [CrossRef]
- Mazumdar, J.; O’Brien, W.T.; Johnson, R.S.; LaManna, J.C.; Chavez, J.C.; Klein, P.S.; Simon, M.C. O2 regulates stem cells through Wnt/β-catenin signaling. Nat. Cell Biol. 2010, 12, 1007–1013. [Google Scholar] [CrossRef] [PubMed]
- Rohrschneider, L.R.; Fuller, J.F.; Wolf, I.; Liu, Y.; Lucas, D.M. Structure, function, and biology of SHIP proteins. Genes Dev. 2000, 14, 505–520. [Google Scholar] [CrossRef] [PubMed]
- Viernes, D.R.; Choi, L.B.; Kerr, W.; Chisholm, J.D. Discovery and Development of Small Molecule SHIP Phosphatase Modulators. Med. Res. Rev. 2014, 34, 795–824. [Google Scholar] [CrossRef]
- Iyer, S.; Margulies, B.S.; Kerr, W.G. Role of SHIP1 in bone biology. Ann. New York Acad. Sci. 2013, 1280, 11–14. [Google Scholar] [CrossRef]
- Hazen, A.L.; Smith, M.J.; Desponts, C.; Winter, O.; Moser, K.; Kerr, W.G. SHIP is required for a functional hematopoietic stem cell niche. Blood 2009, 113, 2924–2933. [Google Scholar] [CrossRef]
- Iyer, S.; Viernes, D.R.; Chisholm, J.D.; Margulies, B.S.; Kerr, W.G. SHIP1 Regulates MSC Numbers and Their Osteolineage Commitment by Limiting Induction of the PI3K/Akt/β-Catenin/Id2 Axis. Stem Cells Dev. 2014, 23, 2336–2351. [Google Scholar] [CrossRef]
- Takeshita, S.; Namba, N.; Zhao, J.J.; Jiang, Y.; Genant, H.K.; Silva, M.J.; Brodt, M.D.; Helgason, C.D.; Kalesnikoff, J.; Rauh, M.J.; et al. SHIP-deficient mice are severely osteoporotic due to increased numbers of hyper-resorptive osteoclasts. Nat. Med. 2002, 8, 943–949. [Google Scholar] [CrossRef] [PubMed]
- Kerr, W.G.; Pedicone, C.; Dormann, S.; Pacherille, A.; Chisholm, J.D. Small molecule targeting of SHIP1 and SHIP2. Biochem. Soc. Trans. 2020, 48, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Stiers, P.-J.; van Gastel, N.; Carmeliet, G. Targeting the hypoxic response in bone tissue engineering: A balance between supply and consumption to improve bone regeneration. Mol. Cell. Endocrinol. 2016, 432, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Greijer, A.; Van Der Groep, P.; Kemming, D.; Shvarts, A.; Semenza, G.; Meijer, G.; Van De Wiel, M.; Belien, J.; Van Diest, P.J.; Van Der Wall, E. Up-regulation of gene expression by hypoxia is mediated predominantly by hypoxia-inducible factor 1 (HIF-1). J. Pathol. 2005, 206, 291–304. [Google Scholar] [CrossRef]
- Knowles, H.J. Hypoxic regulation of osteoclast differentiation and bone resorption activity. Hypoxia 2015, 3, 73–82. [Google Scholar] [CrossRef]
- Zhang, P.; Ha, N.; Dai, Q.; Zhou, S.; Yu, C.; Jiang, L. Hypoxia suppresses osteogenesis of bone mesenchymal stem cells via the extracellular signal regulated 1/2 and p38mitogen activated protein kinase signaling pathways. Mol. Med. Rep. 2017, 16, 5515–5522. [Google Scholar] [CrossRef]
- Liu, Z.; Guo, L.; Li, R.; Xu, Q.; Yang, J.; Chen, J.; Deng, M. Transforming growth factor-beta1 and hypoxia inducible factor-1alpha synergistically inhibit the osteogenesis of periodontal ligament stem cells. Int. Immunopharmacol. 2019, 75, 105834. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HIFs |
Mouse Models /Cells | Signaling Pathway | Effects | Ref. |
|---|---|---|---|---|
| HIF-1 | EC-specific loss-of-function mice (Hif1aiΔEC) | HIF-1/VEGF | An increased number of type H vessels and enhanced endochondral angiogenesis and osteogenesis | [3,33] |
| Mature osteoblasts | HIF-1/VEGF | Contribute to the coordination of vascularization, ossification and matrix resorption in endochondral bone development | [16,24] | |
| The mouse model of hindlimb ischemia | HIF-1/VEGF | HIF-1 activation in myeloid cells promotes angiogenesis | [34] | |
| HIF-1α-deficient embryos | HIF-1/EPO | Affect embryonic development | [35] | |
| rat calvaria bone defect model | HIF-1/EPO | Promote osteogenesis and accelerate bone repair | [4] | |
| MC3T3-E1 | HIF-1/Wnt | Promote osteoblast proliferation | [36] | |
| BMSCs in osteonecrosis of the femoral head | HIF-1/β-Catenin | Reduce cellular apoptosis, lower empty lacunae rate, enhance bone formation, and stronger trabecular bone | [37] | |
| PDGFRα + Sca-1+(PαS) MSC | SHIP-1 | SHIP-1 maintains the stable expression of HIF-1α in Pαs MSC under hypoxia, and reduced the expression of HIF-1α inhibits the proliferation of SHIP- 1KOPαs MSC | [38] | |
| periapical lesions in mice | HIF-1/NF-κB | Attenuate periapical bone loss, inhibit osteoclasts | [39] | |
| MSCs | HIF-1/CXCR4 and CXCR7 | Promote MSCs migration and survival capacity | [40] | |
| 10-wk-old osteoclast-specific HIF-1α conditional knockout mice | HIF-1/AMPK | Maintain osteoclast-induced resorption of calcified cartilage matrix | [41] |
| HIFs |
Mouse Models /Cells | Signaling Pathway | Effects | Ref. |
|---|---|---|---|---|
| HIF-2 | Mature osteoblasts | HIF-2/VEGF | Contribute to the coordination of vascularization, ossification and matrix resorption in endochondral bone development | [54] |
| HIF-2α-ablated mice | HIF-2/EPO | Affect adult EPO synthesis | [55] | |
| N1511 mouse chondrocytes | HIF-2/Fas | Mediate chondrocyte apoptosis and regulates autophagy in maturing chondrocytes | [56,57] | |
| Human CD14+ monocytes | -- | Modulate osteoclast differentiation and formation | [58] | |
| Male mice | HIF-2/p16 and p21 | Act as a senescence-related intrinsic factor in age-related dysfunction of bone homeostasis | [47] | |
| Murine experimental OA models | NF-κB-HIF-2α pathway | Promote OA development | [48] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qin, Q.; Liu, Y.; Yang, Z.; Aimaijiang, M.; Ma, R.; Yang, Y.; Zhang, Y.; Zhou, Y. Hypoxia-Inducible Factors Signaling in Osteogenesis and Skeletal Repair. Int. J. Mol. Sci. 2022, 23, 11201. https://doi.org/10.3390/ijms231911201
Qin Q, Liu Y, Yang Z, Aimaijiang M, Ma R, Yang Y, Zhang Y, Zhou Y. Hypoxia-Inducible Factors Signaling in Osteogenesis and Skeletal Repair. International Journal of Molecular Sciences. 2022; 23(19):11201. https://doi.org/10.3390/ijms231911201
Chicago/Turabian StyleQin, Qiuyue, Yiping Liu, Zhen Yang, Maierhaba Aimaijiang, Rui Ma, Yixin Yang, Yidi Zhang, and Yanmin Zhou. 2022. "Hypoxia-Inducible Factors Signaling in Osteogenesis and Skeletal Repair" International Journal of Molecular Sciences 23, no. 19: 11201. https://doi.org/10.3390/ijms231911201
APA StyleQin, Q., Liu, Y., Yang, Z., Aimaijiang, M., Ma, R., Yang, Y., Zhang, Y., & Zhou, Y. (2022). Hypoxia-Inducible Factors Signaling in Osteogenesis and Skeletal Repair. International Journal of Molecular Sciences, 23(19), 11201. https://doi.org/10.3390/ijms231911201