
Integrating ATAC-seq and RNA-seq Reveals the Dynamics of Chromatin Accessibility and Gene Expression in Apple Response to Drought

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
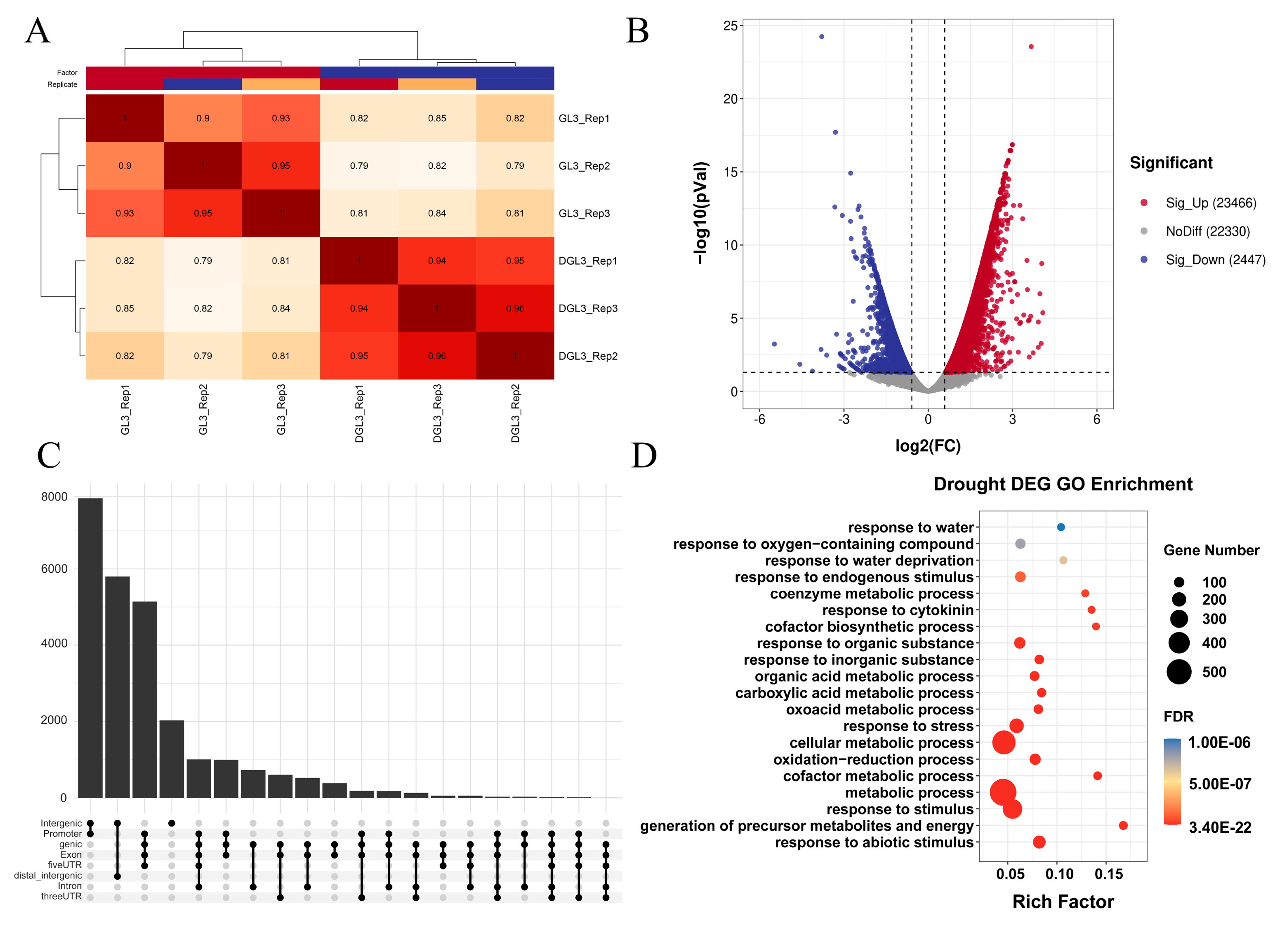
2.1. Landscape of Accessible Chromatin Regions and Phenotype in Apple
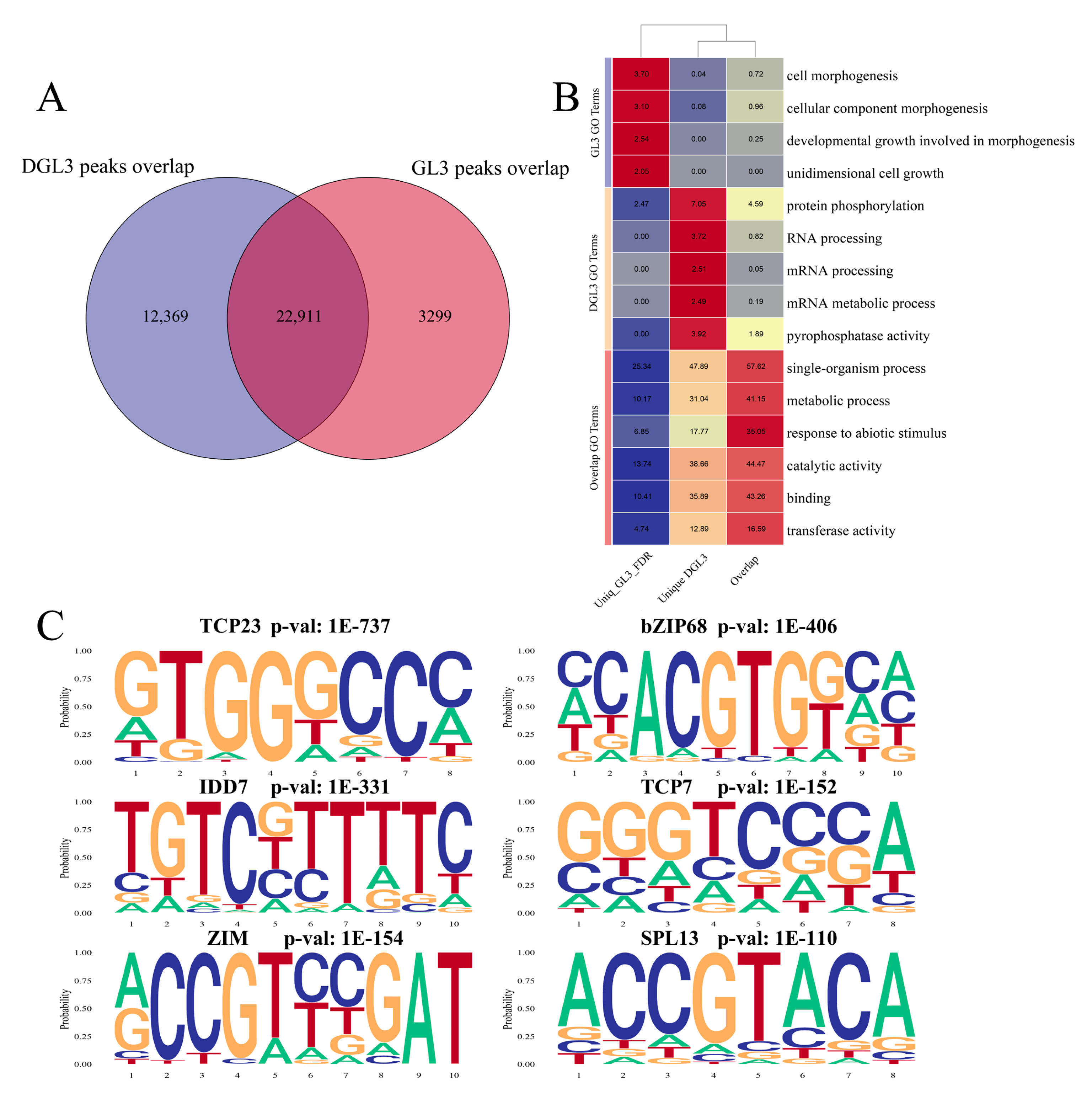
2.2. Enriched Motif Analysis and Functional Analysis of ATAC Peaks in Control and Drought Treatments
2.3. Differential Analysis Reveals Dynamic Changes in Chromatin Accessibility and Transcriptome after Drought Treatment
2.4. Combined Analysis Revealed Chromatin Opening Dependent Pathways and Associated Genes
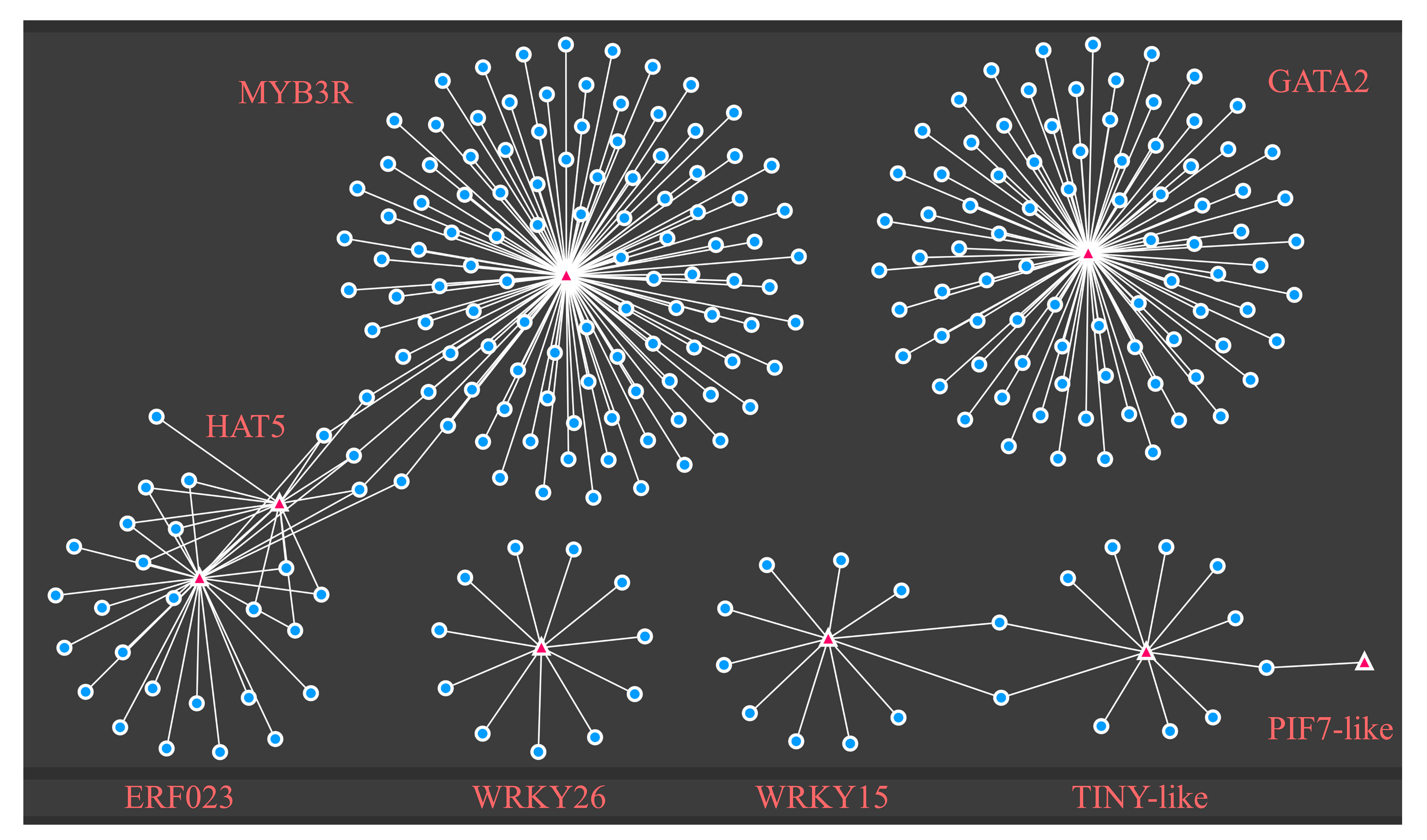
2.5. Identification of TFs and Enriched DEG Motif Analysis in Response to Drought Stress
2.6. Verification of Changes in Genes Expression in Response to Altered Chromatin Accessibility under Drought Treatment
3. Discussion
4. Material and Methods
4.1. Plant Materials and Drought Treatment
4.2. Library Construction and Analysis Process for RNA-seq Samples
4.3. Library Construction and Analysis Process for ATAC-seq
4.4. Differential Co-Expression Network Analysis and GO Enrichment
4.5. RNA Extraction and RT-qPCR Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, H.M.; Zhu, J.H.; Gong, Z.Z.; Zhu, J.K. Abiotic stress responses in plants. Nat. Rev. Genet. 2022, 23, 104–119. [Google Scholar] [CrossRef]
- Gupta, A.; Rico-Medina, A.; Cano-Delgado, A.I. The physiology of plant responses to drought. Science 2020, 368, 266–269. [Google Scholar] [CrossRef]
- Han, S.K.; Wagner, D. Role of chromatin in water stress responses in plants. J. Exp. Bot. 2014, 65, 2785–2799. [Google Scholar] [CrossRef]
- Asensi-Fabado, M.A.; Amtmann, A.; Perrella, G. Plant responses to abiotic stress: The chromatin context of transcriptional regulation. Biochim. Biophys. Acta-Gene Regul. Mech. 2017, 1860, 106–122. [Google Scholar] [CrossRef]
- Manna, M.; Thakur, T.; Chirom, O.; Mandlik, R.; Deshmukh, R.; Salvi, P. Transcription factors as key molecular target to strengthen the drought stress tolerance in plants. Physiol. Plant. 2021, 172, 847–868. [Google Scholar] [CrossRef]
- Gahlaut, V.; Jaiswal, V.; Kumar, A.; Gupta, P.K. Transcription factors involved in drought tolerance and their possible role in developing drought tolerant cultivars with emphasis on wheat (Triticum aestivum L.). Theor. Appl. Genet. 2016, 129, 2019–2042. [Google Scholar] [CrossRef]
- Rushton, D.L.; Tripathi, P.; Rabara, R.C.; Lin, J.; Ringler, P.; Boken, A.K.; Langum, T.J.; Smidt, L.; Boomsma, D.D.; Emme, N.J.; et al. WRKY transcription factors: Key components in abscisic acid signalling. Plant Biotechnol. J. 2012, 10, 2–11. [Google Scholar] [CrossRef]
- Hussain, Q.; Asim, M.; Zhang, R.; Khan, R.; Farooq, S.; Wu, J.S. Transcription Factors Interact with ABA through Gene Expression and Signaling Pathways to Mitigate Drought and Salinity Stress. Biomolecules 2021, 11, 1159. [Google Scholar] [CrossRef]
- Cai, H.S.; Tian, S.; Liu, C.L.; Dong, H.S. Identification of a MYB3R gene involved in drought, salt and cold stress in wheat (Triticum aestivum L.). Gene 2011, 485, 146–152. [Google Scholar] [CrossRef]
- Han, S.K.; Sang, Y.; Rodrigues, A.; Wu, M.F.; Rodriguez, P.L.; Wagner, D.; Biol, F. The SWI2/SNF2 Chromatin Remodeling ATPase BRAHMA Represses Abscisic Acid Responses in the Absence of the Stress Stimulus in Arabidopsis. Plant Cell 2012, 24, 4892–4906. [Google Scholar] [CrossRef]
- Peirats-Llobet, M.; Han, S.K.; Gonzalez-Guzman, M.; Jeong, C.W.; Rodriguez, L.; Belda-Palazon, B.; Wagner, D.; Rodriguez, P.L. A Direct Link between Abscisic Acid Sensing and the Chromatin-Remodeling ATPase BRAHMA via Core ABA Signaling Pathway Components. Mol. Plant 2016, 9, 136–147. [Google Scholar] [CrossRef]
- Saez, A.; Rodrigues, A.; Santiago, J.; Rubio, S.; Rodriguez, P.L. HAB1-SWI3B Interaction Reveals a Link between Abscisic Acid Signaling and Putative SWI/SNF Chromatin-Remodeling Complexes in Arabidopsis. Plant Cell 2008, 20, 2972–2988. [Google Scholar] [CrossRef]
- Mlynarova, L.; Nap, J.P.; Bisseling, T. The SWI/SNF chromatin-remodeling gene AtCHR12 mediates temporary growth arrest in Arabidopsis thaliana upon perceiving environmental stress. Plant J. 2007, 51, 874–885. [Google Scholar] [CrossRef]
- Ramirez-Prado, J.S.; Latrasse, D.; Rodriguez-Granados, N.Y.; Huang, Y.; Manza-Mianza, D.; Brik-Chaouche, R.; Jaouannet, M.; Citerne, S.; Bendahmane, A.; Hirt, H.; et al. The Polycomb protein LHP1 regulates Arabidopsis thaliana stress responses through the repression of the MYC2-dependent branch of immunity. Plant J. 2019, 100, 1118–1131. [Google Scholar] [CrossRef]
- Perruc, E.; Kinoshita, N.; Lopez-Molina, L. The role of chromatin-remodeling factor PKL in balancing osmotic stress responses during Arabidopsis seed germination. Plant J. 2007, 52, 927–936. [Google Scholar] [CrossRef]
- Lu, Z.F.; Marand, A.P.; Ricci, W.A.; Ethridge, C.L.; Zhang, X.Y.; Schmitz, R.J. The prevalence, evolution and chromatin signatures of plant regulatory elements. Nat. Plants 2019, 5, 1250–1259. [Google Scholar] [CrossRef]
- Maher, K.A.; Bajic, M.; Kajala, K.; Reynoso, M.; Pauluzzi, G.; West, D.A.; Zumstein, K.; Woodhouse, M.; Bubb, K.; Dorrity, M.W.; et al. Profiling of Accessible Chromatin Regions across Multiple Plant Species and Cell Types Reveals Common Gene Regulatory Principles and New Control Modules. Plant Cell 2018, 30, 15–36. [Google Scholar] [CrossRef]
- Schwope, R.; Magris, G.; Miculan, M.; Paparelli, E.; Celii, M.; Tocci, A.; Marroni, F.; Fornasiero, A.; De Paoli, E.; Morgante, M. Open chromatin in grapevine marks candidate CREs and with other chromatin features correlates with gene expression. Plant J. 2021, 107, 1631–1647. [Google Scholar] [CrossRef]
- Ren, C.; Li, H.Y.; Wang, Z.M.; Dai, Z.W.; Lecourieux, F.; Kuang, Y.F.; Xin, H.P.; Li, S.H.; Liang, Z.C. Characterization of Chromatin Accessibility and Gene Expression upon Cold Stress Reveals that the RAV1 Transcription Factor Functions in Cold Response in Vitis Amurensis. Plant Cell Physiol. 2021, 62, 1615–1629. [Google Scholar] [CrossRef]
- Liang, Z.; Zhang, Q.; Ji, C.M.; Hu, G.H.; Zhang, P.X.; Wang, Y.F.; Yang, L.W.; Gu, X.F. Reorganization of the 3D chromatin architecture of rice genomes during heat stress. BMC Biol. 2021, 19, 53. [Google Scholar] [CrossRef]
- Kim, S.; Piquerez, S.J.M.; Ramirez-Prado, J.S.; Mastorakis, E.; Veluchamy, A.; Latrasse, D.; Manza-Mianza, D.; Brik-Chaouche, R.; Huang, Y.; Rodriguez-Granados, N.Y.; et al. GCN5 modulates salicylic acid homeostasis by regulating H3K14ac levels at the 5 ’ and 3 ’ ends of its target genes. Nucleic Acids Res. 2020, 48, 5953–5966. [Google Scholar] [CrossRef]
- Yang, J.; Chang, Y.; Qin, Y.H.; Chen, D.J.; Zhu, T.; Peng, K.Q.; Wang, H.J.; Tang, N.; Li, X.K.; Wang, Y.S.; et al. A lamin-like protein OsNMCP1 regulates drought resistance and root growth through chromatin accessibility modulation by interacting with a chromatin remodeller OsSWI3C in rice. New Phytol. 2020, 227, 65–83. [Google Scholar] [CrossRef]
- Xu, J.; Zhou, S.; Gong, X.; Song, Y.; Nocker, S.V.; Ma, F.; Guan, Q. Single-base methylome analysis reveals dynamic epigenomic differences associated with water deficit in apple. Plant Biotechnol. J. 2018, 16, 672–687. [Google Scholar] [CrossRef]
- Li, X.; Xie, Y.; Lu, L.; Yan, M.; Fang, N.; Xu, J.; Wang, L.; Yan, Y.; Zhao, T.; van Nocker, S.; et al. Contribution of methylation regulation of MpDREB2A promoter to drought resistance of Mauls prunifolia. Plant Soil 2019, 441, 15–32. [Google Scholar] [CrossRef]
- Geng, D.L.; Chen, P.X.; Shen, X.X.; Zhang, Y.; Li, X.W.; Jiang, L.J.; Xie, Y.P.; Niu, C.D.; Zhang, J.; Huang, X.H.; et al. MdMYB88 and MdMYB124 Enhance Drought Tolerance by Modulating Root Vessels and Cell Walls in Apple. Plant Physiol. 2018, 178, 1296–1309. [Google Scholar] [CrossRef]
- Lu, P.T.; Yu, S.; Zhu, N.; Chen, Y.R.; Zhou, B.Y.; Pan, Y.; Tzeng, D.; Fabi, J.P.; Argyris, J.; Garcia-Mas, J.; et al. Genome encode analyses reveal the basis of convergent evolution of fleshy fruit ripening. Nat. Plants 2018, 4, 784–791. [Google Scholar] [CrossRef]
- Xiang, Y.; Huang, Y.M.; Xiong, L.Z. Characterization of stress-responsive CIPK genes in rice for stress tolerance improvement. Plant Physiol. 2007, 144, 1416–1428. [Google Scholar] [CrossRef]
- Ye, J.Q.; Yang, H.; Shi, H.T.; Wei, Y.X.; Tie, W.W.; Ding, Z.H.; Yan, Y.; Luo, Y.; Xia, Z.Q.; Wang, W.Q.; et al. The MAPKKK gene family in cassava: Genome-wide identification and expression analysis against drought stress. Sci. Rep. 2017, 7, 14939. [Google Scholar] [CrossRef]
- Aguilar-Martinez, J.A.; Sinha, N. Analysis of the role of Arabidopsis class I TCP genes At TCP7, At TCP8, At TCP22, and At TCP23 in leaf development. Front. Plant Sci. 2013, 4, 406. [Google Scholar] [CrossRef]
- Balsemao-Pires, E.; Andrade, L.R.; Sachetto-Martins, G. Functional study of TCP23 in Arabidopsis thaliana during plant development. Plant Physiol. Biochem. 2013, 67, 120–125. [Google Scholar] [CrossRef]
- Sun, B.Y.; Fan, Y.H.; Duan, H.C.; Liu, X.M.; Chen, Y.L.; Shang, G.X.; Liu, Y.N.; Yang, H.; Qu, C.M.; Li, J.N.; et al. Genome-wide characterization of Brassica napus INDETERMINATE DOMAIN genes reveals a negative role for BnA08.IDD7 in plant development. Ind. Crops Prod. 2022, 175, 114263. [Google Scholar] [CrossRef]
- Gao, R.M.; Gruber, M.Y.; Amyot, L.; Hannoufa, A. SPL13 regulates shoot branching and flowering time in Medicago sativa. Plant Mol. Biol. 2018, 96, 119–133. [Google Scholar] [CrossRef]
- Arshad, M.; Feyissa, B.A.; Amyot, L.; Aung, B.; Hannoufa, A. MicroRNA156 improves drought stress tolerance in alfalfa (Medicago sativa) by silencing SPL13. Plant Sci. 2017, 258, 122–136. [Google Scholar] [CrossRef]
- Li, Y.M.; Liu, W.Z.; Zhong, H.; Zhang, H.L.; Xia, Y.J. Redox-sensitive bZIP68 plays a role in balancing stress tolerance with growth in Arabidopsis. Plant J. 2019, 100, 768–783. [Google Scholar] [CrossRef]
- Song, S.S.; Qi, T.C.; Huang, H.; Ren, Q.C.; Wu, D.W.; Chang, C.Q.; Peng, W.; Liu, Y.L.; Peng, J.R.; Xie, D.X. The Jasmonate-ZIM Domain Proteins Interact with the R2R3-MYB Transcription Factors MYB21 and MYB24 to Affect Jasmonate-Regulated Stamen Development in Arabidopsis. Plant Cell 2011, 23, 1000–1013. [Google Scholar] [CrossRef]
- Li, S.J.; Fu, Q.T.; Chen, L.G.; Huang, W.D.; Yu, D.Q. Arabidopsis thaliana WRKY25, WRKY26, and WRKY33 coordinate induction of plant thermotolerance. Planta 2011, 233, 1237–1252. [Google Scholar] [CrossRef]
- Mukhopadhyay, P.; Tyagi, A.K. OsTCP19 influences developmental and abiotic stress signaling by modulating ABI4-mediated pathways. Sci. Rep. 2015, 5, 12381. [Google Scholar] [CrossRef]
- Pandey, G.K.; Grant, J.J.; Cheong, Y.H.; Kim, B.G.; Li, L.G.; Luan, S. ABR1, an APETALA2-domain transcription factor that functions as a repressor of ABA response in Arabidopsis. Plant Physiol. 2005, 139, 1185–1193. [Google Scholar] [CrossRef]
- Ashrafi-Dehkordi, E.; Alemzadeh, A.; Tanaka, N.; Razi, H. Meta-analysis of transcriptomic responses to biotic and abiotic stress in tomato. Peerj 2018, 6, e4631. [Google Scholar] [CrossRef]
- Guo, M.; Liu, J.H.; Ma, X.; Luo, D.X.; Gong, Z.H.; Lu, M.H. The Plant Heat Stress Transcription Factors (HSFs): Structure, Regulation, and Function in Response to Abiotic Stresses. Front. Plant Sci. 2016, 7, 114. [Google Scholar] [CrossRef]
- Ullah, A.; Manghwar, H.; Shaban, M.; Khan, A.H.; Akbar, A.; Ali, U.; Ali, E.; Fahad, S. Phytohormones enhanced drought tolerance in plants: A coping strategy. Environ. Sci. Pollut. Res. 2018, 25, 33103–33118. [Google Scholar] [CrossRef]
- Cabane, M.; Afif, D.; Hawkins, S. Lignins and Abiotic Stresses. In Lignins: Biosynthesis, Biodegradation and Bioengineering; Jouann, L., Lapierre, C., Eds.; Academic Press: Cambridge, MA, USA, 2012; Volume 61, pp. 219–262. [Google Scholar]
- Valdes, A.E.; Overnas, E.; Johansson, H.; Rada-Iglesias, A.; Engstrom, P. The homeodomain-leucine zipper (HD-Zip) class I transcription factors ATHB7 and ATHB12 modulate abscisic acid signalling by regulating protein phosphatase 2C and abscisic acid receptor gene activities. Plant Mol. Biol. 2012, 80, 405–418. [Google Scholar] [CrossRef]
- Zhou, Y.Y.; He, R.; Guo, Y.L.; Liu, K.K.; Huang, G.M.; Peng, C.X.; Liu, Y.G.; Zhang, M.C.; Li, Z.H.; Duan, L.S. A novel ABA functional analogue B2 enhances drought tolerance in wheat. Sci. Rep. 2019, 9, 2887. [Google Scholar] [CrossRef]
- Bielsa, B.; Hewitt, S.; Reyes-Chin-Wo, S.; Dhingra, A.; Rubio-Cabetas, M.J. Identification of water use efficiency related genes in ’Garnem’ almond-peach rootstock using time-course transcriptome analysis. PLoS ONE 2018, 13, e0205493. [Google Scholar] [CrossRef]
- Jiang, J.J.; Ma, S.H.; Ye, N.H.; Jiang, M.; Cao, J.S.; Zhang, J.H. WRKY transcription factors in plant responses to stresses. J. Integr. Plant Biol. 2017, 59, 86–101. [Google Scholar] [CrossRef]
- Wu, J.; Chen, J.B.; Wang, L.F.; Wang, S.M. Genome-Wide Investigation of WRKY Transcription Factors Involved in Terminal Drought Stress Response in Common Bean. Front. Plant Sci. 2016, 8, 380. [Google Scholar] [CrossRef]
- Kidokoro, S.; Maruyama, K.; Nakashima, K.; Imura, Y.; Narusaka, Y.; Shinwari, Z.K.; Osakabe, Y.; Fujita, Y.; Mizoi, J.; Shinozaki, K.; et al. The Phytochrome-Interacting Factor PIF7 Negatively Regulates DREB1 Expression under Circadian Control in Arabidopsis. Plant Physiol. 2009, 151, 2046–2057. [Google Scholar] [CrossRef]
- Xie, Z.L.; Nolan, R.O.R.; Jiang, H.; Tang, B.Y.; Zhang, M.C.; Li, Z.H.; Yin, Y.H. The AP2/ERF Transcription Factor TINY Modulates Brassinosteroid-Regulated Plant Growth and Drought Responses in Arabidopsis. Plant Cell 2019, 31, 1788–1806. [Google Scholar] [CrossRef] [PubMed]
- Verbsky, M.L.; Richards, E.J. Chromatin remodeling in plants. Curr. Opin. Plant Biol. 2001, 4, 494–500. [Google Scholar] [CrossRef]
- Chang, Y.N.; Zhu, C.; Jiang, J.; Zhang, H.M.; Zhu, J.K.; Duan, C.G. Epigenetic regulation in plant abiotic stress responses. J. Integr. Plant Biol. 2020, 62, 563–580. [Google Scholar] [CrossRef]
- Chinnusamy, V.; Gong, Z.Z.; Zhu, J.K. Abscisic acid-mediated epigenetic processes in plant development and stress responses. J. Integr. Plant Biol. 2008, 50, 1187–1195. [Google Scholar] [CrossRef] [PubMed]
- Sijacic, P.; Bajic, M.; McKinney, E.C.; Meagher, R.B.; Deal, R.B. Changes in chromatin accessibility between Arabidopsis stem cells and mesophyll cells illuminate cell type-specific transcription factor networks. Plant J. 2018, 94, 215–231. [Google Scholar] [CrossRef] [PubMed]
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 2013, 10, 1213–1218. [Google Scholar] [CrossRef] [PubMed]
- Geng, D.L.; Lu, L.Y.; Yan, M.J.; Shen, X.X.; Jiang, L.J.; Li, H.Y.; Wang, L.P.; Yan, Y.; Xu, J.D.; Li, C.Y.; et al. Physiological and transcriptomic analyses of roots from Malus sieversii under drought stress. J. Integr. Agric. 2019, 18, 1280–1294. [Google Scholar] [CrossRef]
- Jiang, K.N.; Yung, V.; Chiba, T.; Feldman, L.J. Longitudinal patterning in roots: A GATA2-auxin interaction underlies and maintains the root transition domain. Planta 2018, 247, 831–843. [Google Scholar] [CrossRef]
- Luo, X.M.; Lin, W.H.; Zhu, S.W.; Zhu, J.Y.; Sun, Y.; Fan, X.Y.; Cheng, M.L.; Hao, Y.Q.; Oh, E.; Tian, M.M.; et al. Integration of Light- and Brassinosteroid-Signaling Pathways by a GATA Transcription Factor in Arabidopsis. Dev. Cell 2010, 19, 872–883. [Google Scholar] [CrossRef]
- Capella, M.; Ribone, P.A.; Arce, A.L.; Chan, R.L. Arabidopsis thaliana HomeoBox 1 (AtHB1), a Homedomain-Leucine Zipper I (HD-Zip I) transcription factor, is regulated by PHYTOCHROME-INTERACTING FACTOR 1 to promote hypocotyl elongation. New Phytol. 2015, 207, 669–682. [Google Scholar] [CrossRef]
- Bogamuwa, S.P.; Jang, J.C. Tandem CCCH Zinc Finger Proteins in Plant Growth, Development and Stress Response. Plant Cell Physiol. 2014, 55, 1367–1375. [Google Scholar] [CrossRef]
- Daccord, N.; Celton, J.M.; Linsmith, G.; Becker, C.; Choisne, N.; Schijlen, E.; van de Geest, H.; Bianco, L.; Micheletti, D.; Velasco, R.; et al. High-quality de novo assembly of the apple genome and methylome dynamics of early fruit development. Nat. Genet. 2017, 49, 1099. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data, P. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Bajic, M.; Maher, K.A.; Deal, R.B. Identification of Open Chromatin Regions in Plant Genomes Using ATAC-Seq. Methods Mol. Biol. (Clifton N.J.) 2018, 1675, 183–201. [Google Scholar]
- Jiang, H.S.; Lei, R.; Ding, S.W.; Zhu, S.F. Skewer: A fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinform. 2014, 15, 182. [Google Scholar] [CrossRef]
- Tarasov, A.; Vilella, A.J.; Cuppen, E.; Nijman, I.J.; Prins, P. Sambamba: Fast processing of NGS alignment formats. Bioinformatics 2015, 31, 2032–2034. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nussbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based Analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef]
- Stark, R.; Brown, G. DiffBind: Differential binding analysis of ChIP-Seq peak data. R package version 2011, 100. [Google Scholar]
- Yu, G.C.; Wang, L.G.; He, Q.Y. ChIPseeker: An R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 2015, 31, 2382–2383. [Google Scholar] [CrossRef]
- Khan, A.; Mathelier, A. Intervene: A tool for intersection and visualization of multiple gene or genomic region sets. BMC Bioinform. 2017, 18, 287. [Google Scholar] [CrossRef]
- Chen, C.J.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.H.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Saito, R.; Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.L.; Lotia, S.; Pico, A.R.; Bader, G.D.; Ideker, T. A travel guide to Cytoscape plugins. Nat. Methods 2012, 9, 1069–1076. [Google Scholar] [CrossRef] [PubMed]
- Perini, P.; Pasquali, G.; Margis-Pinheiro, M.; de Oliviera, P.R.; Revers, L. Reference genes for transcriptional analysis of flowering and fruit ripening stages in apple (Malus × domestica Borkh.). Mol. Breed. 2014, 34, 829–842. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; He, J.; Deng, M.; Wang, C.; Wang, R.; Yan, J.; Luo, M.; Ma, F.; Guan, Q.; Xu, J. Integrating ATAC-seq and RNA-seq Reveals the Dynamics of Chromatin Accessibility and Gene Expression in Apple Response to Drought. Int. J. Mol. Sci. 2022, 23, 11191. https://doi.org/10.3390/ijms231911191
Wang S, He J, Deng M, Wang C, Wang R, Yan J, Luo M, Ma F, Guan Q, Xu J. Integrating ATAC-seq and RNA-seq Reveals the Dynamics of Chromatin Accessibility and Gene Expression in Apple Response to Drought. International Journal of Molecular Sciences. 2022; 23(19):11191. https://doi.org/10.3390/ijms231911191
Chicago/Turabian StyleWang, Shicong, Jieqiang He, Mengting Deng, Caixia Wang, Ruifeng Wang, Jinjiao Yan, Minrong Luo, Fengwang Ma, Qingmei Guan, and Jidi Xu. 2022. "Integrating ATAC-seq and RNA-seq Reveals the Dynamics of Chromatin Accessibility and Gene Expression in Apple Response to Drought" International Journal of Molecular Sciences 23, no. 19: 11191. https://doi.org/10.3390/ijms231911191
APA StyleWang, S., He, J., Deng, M., Wang, C., Wang, R., Yan, J., Luo, M., Ma, F., Guan, Q., & Xu, J. (2022). Integrating ATAC-seq and RNA-seq Reveals the Dynamics of Chromatin Accessibility and Gene Expression in Apple Response to Drought. International Journal of Molecular Sciences, 23(19), 11191. https://doi.org/10.3390/ijms231911191