
Estrogen Receptor Subtypes Elicit a Distinct Gene Expression Profile of Endothelial-Derived Factors Implicated in Atherosclerotic Plaque Vulnerability

 ,
,  , ,
, ,  ,
,  ,
,
Abstract
1. Introduction
2. Result
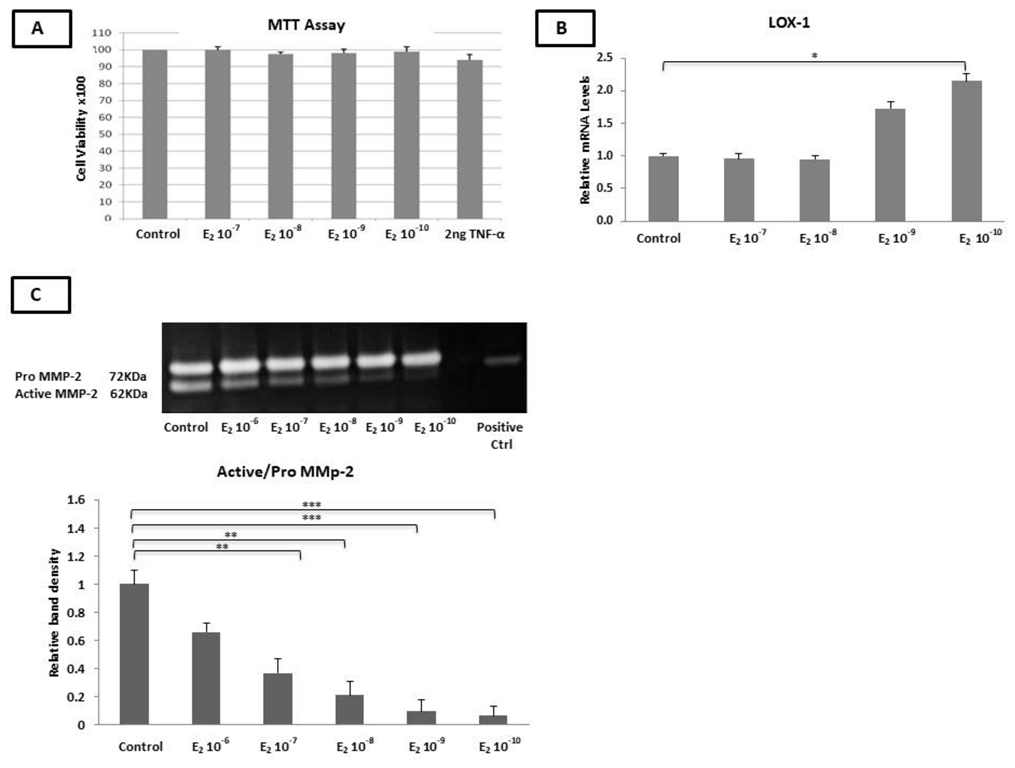
2.1. The Incubation of HAEC Cells with Estradiol and TNF-α Had No Effect on Cell Viability
2.2. Estradiol Did Not Alter the mRNA Levels of RANK, OPG, and MCP-1 and the TIMP-1, TIMP-2, and MCP-1 Protein Levels
2.3. Estradiol Reduced the MMP-2 Gelatinase Activity
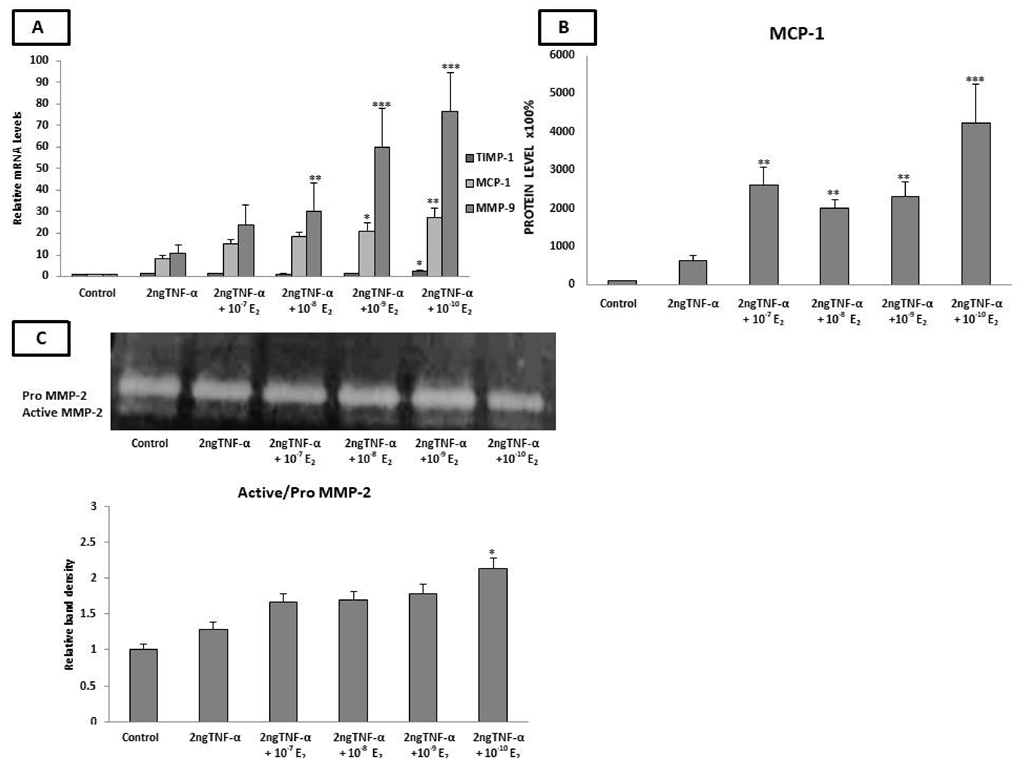
2.4. Estradiol Altered the mRNA Expression of LOX-1, TIMP-1, MMP-9, and MCP-1 and MCP-1 Protein Levels under Inflammatory Conditions
2.5. Estradiol Induced MMP-2 Activity under Low-Grade Inflammatory Conditions
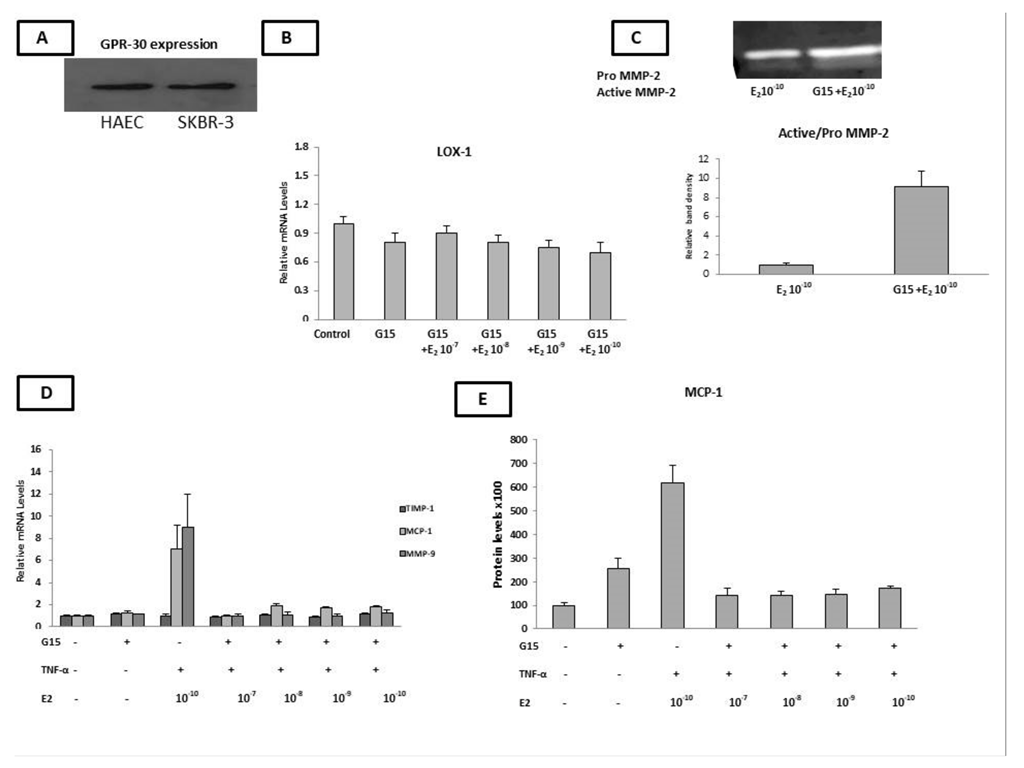
2.6. HAECs Express the GPR30 Estrogen Receptor
2.7. G15 (GPR-30 Antagonist) Countered the Estradiol-Induced Expression of LOX, MCP-1,TIMP-1, MMP-9, and MCP-1 as Well as the Decreased MMP-2 Gelatinase Activity
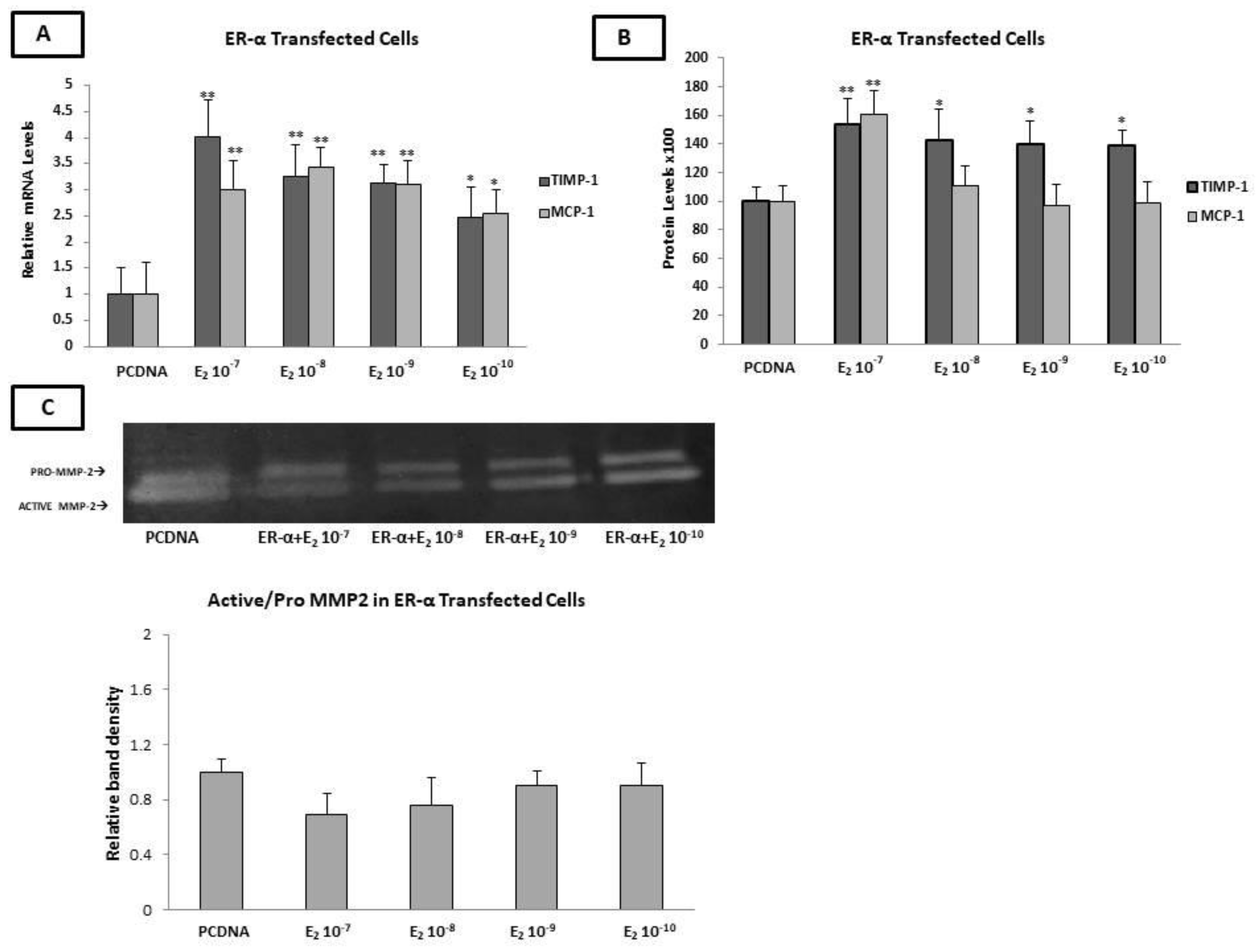
2.8. Estradiol Increased the Expression of MCP-1 and TIMP-1, as Well as MMP-2 Enzymatic Activity, through ERα
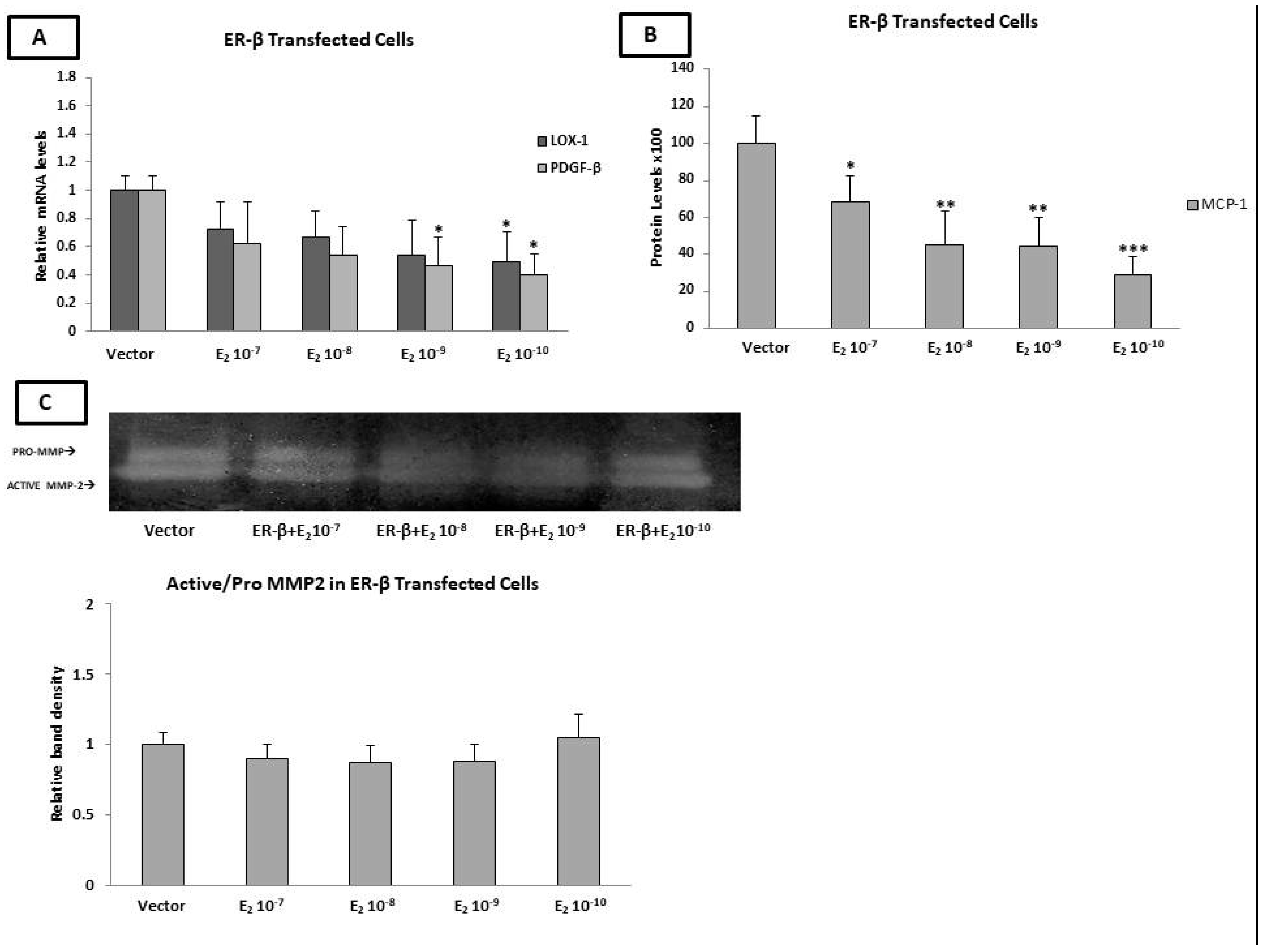
2.9. Estradiol Reduced the PDGF-β mRNA Levels and MCP-1 Protein Levels through ERβ
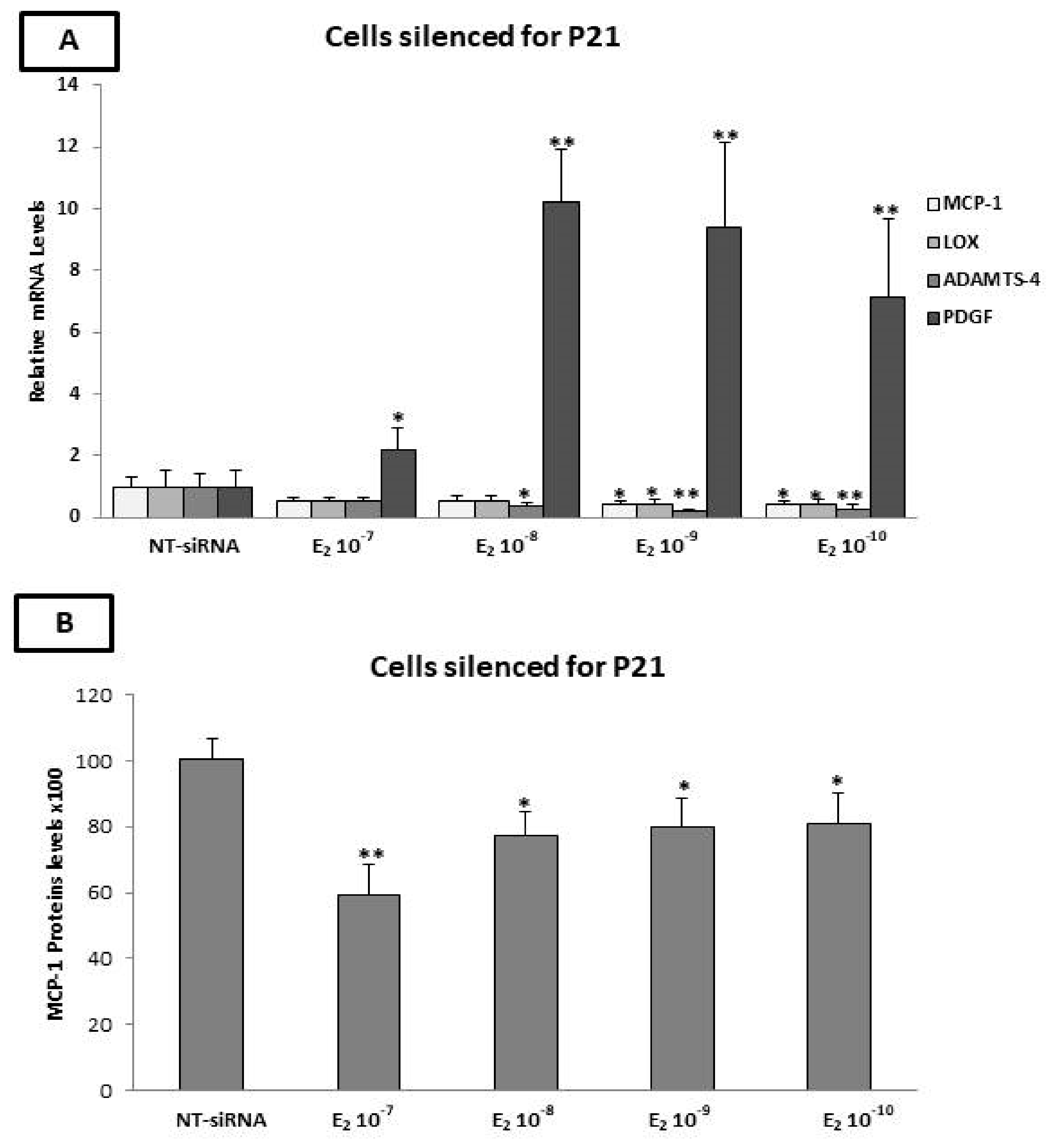
2.10. The p21 Silencing Reduced the Estradiol-Mediated Upregulation of MCP-1 and LOX-1, while It Increased the PDGF-β mRNA Levels
2.11. Evaluation of MCP-1, TIMP-1, TIMP-2, and OPG Protein Levels by ELISA after p21 Silencing
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatment
4.2. MTT Assay
4.3. Transfection with Small Interfering RNA
4.4. Transient Transfection Assays
4.5. RNA Isolation and qPCR
4.6. SDS-PAGE and Western-Blot Analysis
4.7. ELISA
4.8. Zymography
4.9. Statistical
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Garcia, M.; Mulvagh, S.L.; Merz, C.N.; Buring, J.E.; Manson, J.E. Cardiovascular Disease in Women: Clinical Perspectives. Circ. Res. 2016, 118, 1273–1293. [Google Scholar] [CrossRef]
- Kim, H.; Kim, S.; Han, S.; Rane, P.P.; Fox, K.M.; Qian, Y.; Suh, H.S. Prevalence and incidence of atherosclerotic cardiovascular disease and its risk factors in Korea: A nationwide population-based study. BMC Public Health 2019, 19, 1112. [Google Scholar] [CrossRef]
- Chen, G.; Farris, M.S.; Cowling, T.; Pinto, L.; Rogoza, R.M.; MacKinnon, E.; Champsi, S.; Anderson, T.J. Prevalence of atherosclerotic cardiovascular disease and subsequent major adverse cardiovascular events in Alberta, Canada: A real-world evidence study. Clin. Cardiol. 2021, 44, 1613–1620. [Google Scholar] [CrossRef]
- Sima, P.; Vannucci, L.; Vetvicka, V. Atherosclerosis as autoimmune disease. Ann. Transl. Med. 2018, 6, 116. [Google Scholar] [CrossRef]
- Spring, B.; Moller, A.C.; Colangelo, L.A.; Siddique, J.; Roehrig, M.; Daviglus, M.L.; Polak, J.F.; Reis, J.P.; Sidney, S.; Liu, K. Healthy lifestyle change and subclinical atherosclerosis in young adults: Coronary Artery Risk Development in Young Adults (CARDIA) study. Circulation 2014, 130, 10–17. [Google Scholar] [CrossRef]
- Barton, M. Cholesterol and atherosclerosis: Modulation by oestrogen. Curr. Opin. Lipidol. 2013, 24, 214–220. [Google Scholar] [CrossRef]
- Stampfer, M.J.; Colditz, G.A.; Willett, W.C.; Manson, J.E.; Rosner, B.; Speizer, F.E.; Hennekens, C.H. Postmenopausal estrogen therapy and cardiovascular disease. Ten-year follow-up from the nurses’ health study. N. Engl. J. Med. 1991, 325, 756–762. [Google Scholar] [CrossRef]
- Meng, Q.; Li, Y.; Ji, T.; Chao, Y.; Li, J.; Fu, Y.; Wang, S.; Chen, Q.; Chen, W.; Huang, F.; et al. Estrogen prevent atherosclerosis by attenuating endothelial cell pyroptosis via activation of estrogen receptor alpha-mediated autophagy. J. Adv. Res. 2021, 28, 149–164. [Google Scholar] [CrossRef]
- Fashe, M.; Yi, M.; Sueyoshi, T.; Negishi, M. Sex-specific expression mechanism of hepatic estrogen inactivating enzyme and transporters in diabetic women. Biochem. Pharmacol. 2021, 190, 114662. [Google Scholar] [CrossRef]
- Fonseca, M.I.H.; da Silva, I.T.; Ferreira, S.R.G. Impact of menopause and diabetes on atherogenic lipid profile: Is it worth to analyse lipoprotein subfractions to assess cardiovascular risk in women? Diabetol. Metab. Syndr. 2017, 9, 22. [Google Scholar] [CrossRef]
- Sato, A.; Watanabe, H.; Yamazaki, M.; Sakurai, E.; Ebina, K. Estrogen Sulfotransferase is Highly Expressed in Vascular Endothelial Cells Overlying Atherosclerotic Plaques. Protein. J. 2022, 41, 179–188. [Google Scholar] [CrossRef]
- Fait, T. Menopause hormone therapy: Latest developments and clinical practice. Drugs Context 2019, 8, 212551. [Google Scholar] [CrossRef]
- Hansson, G.K.; Libby, P. The immune response in atherosclerosis: A double-edged sword. Nat. Rev. Immunol. 2006, 6, 508–519. [Google Scholar] [CrossRef]
- Seo, D.W.; Saxinger, W.C.; Guedez, L.; Cantelmo, A.R.; Albini, A.; Stetler-Stevenson, W.G. An integrin-binding N-terminal peptide region of TIMP-2 retains potent angio-inhibitory and anti-tumorigenic activity in vivo. Peptides 2011, 32, 1840–1848. [Google Scholar] [CrossRef]
- Botts, S.R.; Fish, J.E.; Howe, K.L. Dysfunctional Vascular Endothelium as a Driver of Atherosclerosis: Emerging Insights Into Pathogenesis and Treatment. Front. Pharmacol. 2021, 12, 787541. [Google Scholar] [CrossRef]
- Salaminia, S.; Mohsenzadeh, Y.; Motedayen, M.; Sayehmiri, F.; Dousti, M. Hormone Replacement Therapy and Postmenopausal Cardiovascular Events: A Meta-Analysis. Iran. Red. Crescent. Med. 2019, 21, e82298. [Google Scholar] [CrossRef]
- Wolters, M.; Dejanovic, G.M.; Asllanaj, E.; Gunther, K.; Pohlabeln, H.; Bramer, W.M.; Ahrens, J.; Nagrani, R.; Pigeot, I.; Franco, O.H.; et al. Effects of phytoestrogen supplementation on intermediate cardiovascular disease risk factors among postmenopausal women: A meta-analysis of randomized controlled trials. Menopause 2020, 27, 1081–1092. [Google Scholar] [CrossRef]
- Adam, S.K.; Das, S.; Soelaiman, I.N.; Umar, N.A.; Jaarin, K. Consumption of repeatedly heated soy oil increases the serum parameters related to atherosclerosis in ovariectomized rats. Tohoku J. Exp. Med. 2008, 215, 219–226. [Google Scholar] [CrossRef]
- Hassan, H.A.; Abdel-Wahhab, M.A. Effect of soybean oil on atherogenic metabolic risks associated with estrogen deficiency in ovariectomized rats: Dietary soybean oil modulate atherogenic risks in overiectomized rats. J. Physiol. Biochem. 2012, 68, 247–253. [Google Scholar] [CrossRef]
- Hulley, S.; Grady, D.; Bush, T.; Furberg, C.; Herrington, D.; Riggs, B.; Vittinghoff, E. Randomized trial of estrogen plus progestin for secondary prevention of coronary heart disease in postmenopausal women. Heart and Estrogen/progestin Replacement Study (HERS) Research Group. JAMA 1998, 280, 605–613. [Google Scholar] [CrossRef]
- Kim, J.E.; Chang, J.H.; Jeong, M.J.; Choi, J.; Park, J.; Baek, C.; Shin, A.; Park, S.M.; Kang, D.; Choi, J.Y. A systematic review and meta-analysis of effects of menopausal hormone therapy on cardiovascular diseases. Sci. Rep. 2020, 10, 20631. [Google Scholar] [CrossRef] [PubMed]
- Rossouw, J.E.; Anderson, G.L.; Prentice, R.L.; LaCroix, A.Z.; Kooperberg, C.; Stefanick, M.L.; Jackson, R.D.; Beresford, S.A.; Howard, B.V.; Johnson, K.C.; et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: Principal results From the Women’s Health Initiative randomized controlled trial. JAMA 2002, 288, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Mehta, J.; Kling, J.M.; Manson, J.E. Risks, Benefits, and Treatment Modalities of Menopausal Hormone Therapy: Current Concepts. Front. Endocrinol. 2021, 12, 564781. [Google Scholar] [CrossRef] [PubMed]
- Denti, L. The hormone replacement therapy (HRT) of menopause: Focus on cardiovascular implications. Acta Biomed. 2010, 81 (Suppl. S1), 73–76. [Google Scholar] [PubMed]
- Hodis, H.N.; Mack, W.J. The timing hypothesis and hormone replacement therapy: A paradigm shift in the primary prevention of coronary heart disease in women. Part 1: Comparison of therapeutic efficacy. J. Am. Geriatr. Soc. 2013, 61, 1005–1010. [Google Scholar] [CrossRef]
- Arnal, J.F.; Fontaine, C.; Billon-Gales, A.; Favre, J.; Laurell, H.; Lenfant, F.; Gourdy, P. Estrogen receptors and endothelium. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1506–1512. [Google Scholar] [CrossRef]
- Nofer, J.R. Estrogens and atherosclerosis: Insights from animal models and cell systems. J. Mol. Endocrinol. 2012, 48, R13–R29. [Google Scholar] [CrossRef]
- Trenti, A.; Tedesco, S.; Boscaro, C.; Trevisi, L.; Bolego, C.; Cignarella, A. Estrogen, Angiogenesis, Immunity and Cell Metabolism: Solving the Puzzle. Int. J. Mol. Sci. 2018, 19, 859. [Google Scholar] [CrossRef]
- Lenfant, F.; Tremollieres, F.; Gourdy, P.; Arnal, J.F. Timing of the vascular actions of estrogens in experimental and human studies: Why protective early, and not when delayed? Maturitas 2011, 68, 165–173. [Google Scholar] [CrossRef]
- Davezac, M.; Buscato, M.; Zahreddine, R.; Lacolley, P.; Henrion, D.; Lenfant, F.; Arnal, J.F.; Fontaine, C. Estrogen Receptor and Vascular Aging. Front. Aging 2021, 2, 727380. [Google Scholar] [CrossRef]
- Arnal, J.F.; Lenfant, F.; Metivier, R.; Flouriot, G.; Henrion, D.; Adlanmerini, M.; Fontaine, C.; Gourdy, P.; Chambon, P.; Katzenellenbogen, B.; et al. Membrane and Nuclear Estrogen Receptor Alpha Actions: From Tissue Specificity to Medical Implications. Physiol. Rev. 2017, 97, 1045–1087. [Google Scholar] [CrossRef] [PubMed]
- Kassi, E.; Spilioti, E.; Nasiri-Ansari, N.; Adamopoulos, C.; Moutsatsou, P.; Papapanagiotou, A.; Siasos, G.; Tousoulis, D.; Papavassiliou, A.G. Vascular Inflammation and Atherosclerosis: The Role of Estrogen Receptors. Curr. Med. Chem. 2015, 22, 2651–2665. [Google Scholar] [CrossRef] [PubMed]
- Shioi, A.; Ikari, Y. Plaque Calcification During Atherosclerosis Progression and Regression. J. Atheroscler. Thromb. 2018, 25, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Georgakis, M.K.; Gill, D.; Rannikmae, K.; Traylor, M.; Anderson, C.D.; Lee, J.M.; Kamatani, Y.; Hopewell, J.C.; Worrall, B.B.; Bernhagen, J.; et al. Genetically Determined Levels of Circulating Cytokines and Risk of Stroke. Circulation 2019, 139, 256–268. [Google Scholar] [CrossRef] [PubMed]
- Georgakis, M.K.; Malik, R.; Bjorkbacka, H.; Pana, T.A.; Demissie, S.; Ayers, C.; Elhadad, M.A.; Fornage, M.; Beiser, A.S.; Benjamin, E.J.; et al. Circulating Monocyte Chemoattractant Protein-1 and Risk of Stroke: Meta-Analysis of Population-Based Studies Involving 17 180 Individuals. Circ. Res. 2019, 125, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Georgakis, M.K.; van der Laan, S.W.; Asare, Y.; Mekke, J.M.; Haitjema, S.; Schoneveld, A.H.; de Jager, S.C.A.; Nurmohamed, N.S.; Kroon, J.; Stroes, E.S.G.; et al. Monocyte-Chemoattractant Protein-1 Levels in Human Atherosclerotic Lesions Associate With Plaque Vulnerability. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 2038–2048. [Google Scholar] [CrossRef]
- Ho, F.M.; Liu, S.H.; Lin, W.W.; Liau, C.S. Opposite effects of high glucose on MMP-2 and TIMP-2 in human endothelial cells. J. Cell Biochem. 2007, 101, 442–450. [Google Scholar] [CrossRef]
- Olejarz, W.; Lacheta, D.; Kubiak-Tomaszewska, G. Matrix Metalloproteinases as Biomarkers of Atherosclerotic Plaque Instability. Int. J. Mol. Sci. 2020, 21, 3946. [Google Scholar] [CrossRef]
- Vacek, T.P.; Rehman, S.; Neamtu, D.; Yu, S.; Givimani, S.; Tyagi, S.C. Matrix metalloproteinases in atherosclerosis: Role of nitric oxide, hydrogen sulfide, homocysteine, and polymorphisms. Vasc. Health Risk Manag. 2015, 11, 173–183. [Google Scholar] [CrossRef]
- Wang, X.; Khalil, R.A. Matrix Metalloproteinases, Vascular Remodeling, and Vascular Disease. Adv. Pharmacol. 2018, 81, 241–330. [Google Scholar] [CrossRef]
- Nasiri-Ansari, N.; Dimitriadis, G.K.; Agrogiannis, G.; Perrea, D.; Kostakis, I.D.; Kaltsas, G.; Papavassiliou, A.G.; Randeva, H.S.; Kassi, E. Canagliflozin attenuates the progression of atherosclerosis and inflammation process in APOE knockout mice. Cardiovasc. Diabetol. 2018, 17, 106. [Google Scholar] [CrossRef] [PubMed]
- Perez-Martinez, L.; Jaworski, D.M. Tissue inhibitor of metalloproteinase-2 promotes neuronal differentiation by acting as an anti-mitogenic signal. J. Neurosci. 2005, 25, 4917–4929. [Google Scholar] [CrossRef] [PubMed]
- Seo, D.W.; Li, H.; Qu, C.K.; Oh, J.; Kim, Y.S.; Diaz, T.; Wei, B.; Han, J.W.; Stetler-Stevenson, W.G. Shp-1 mediates the antiproliferative activity of tissue inhibitor of metalloproteinase-2 in human microvascular endothelial cells. J. Biol. Chem. 2006, 281, 3711–3721. [Google Scholar] [CrossRef]
- Wessely, R. Atherosclerosis and cell cycle: Put the brakes on! Critical role for cyclin-dependent kinase inhibitors. J. Am. Coll. Cardiol. 2010, 55, 2269–2271. [Google Scholar] [CrossRef]
- Novak, R.; Hrkac, S.; Salai, G.; Bilandzic, J.; Mitar, L.; Grgurevic, L. The Role of ADAMTS-4 in Atherosclerosis and Vessel Wall Abnormalities. J. Vasc. Res. 2022, 59, 69–77. [Google Scholar] [CrossRef]
- Kumar, S.; Chen, M.; Li, Y.; Wong, F.H.; Thiam, C.W.; Hossain, M.Z.; Poh, K.K.; Hirohata, S.; Ogawa, H.; Angeli, V.; et al. Loss of ADAMTS4 reduces high fat diet-induced atherosclerosis and enhances plaque stability in ApoE(-/-) mice. Sci. Rep. 2016, 6, 31130. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.; Zhou, C.; Ji, L.; Pan, B.; Zheng, L. AG1296 enhances plaque stability via inhibiting inflammatory responses and decreasing MMP-2 and MMP-9 expression in ApoE−/− mice. Biochem. Biophys. Res. Commun. 2017, 489, 426–431. [Google Scholar] [CrossRef]
- Jover, E.; Silvente, A.; Marin, F.; Martinez-Gonzalez, J.; Orriols, M.; Martinez, C.M.; Puche, C.M.; Valdes, M.; Rodriguez, C.; Hernandez-Romero, D. Inhibition of enzymes involved in collagen cross-linking reduces vascular smooth muscle cell calcification. FASEB J. 2018, 32, 4459–4469. [Google Scholar] [CrossRef]
- Martinez-Gonzalez, J.; Varona, S.; Canes, L.; Galan, M.; Briones, A.M.; Cachofeiro, V.; Rodriguez, C. Emerging Roles of Lysyl Oxidases in the Cardiovascular System: New Concepts and Therapeutic Challenges. Biomolecules 2019, 9, 610. [Google Scholar] [CrossRef]
- Kim, M.S.; Day, C.J.; Morrison, N.A. MCP-1 is induced by receptor activator of nuclear factor-{kappa}B ligand, promotes human osteoclast fusion, and rescues granulocyte macrophage colony-stimulating factor suppression of osteoclast formation. J. Biol. Chem. 2005, 280, 16163–16169. [Google Scholar] [CrossRef]
- Montecucco, F.; Steffens, S.; Mach, F. The immune response is involved in atherosclerotic plaque calcification: Could the RANKL/RANK/OPG system be a marker of plaque instability? Clin. Dev. Immunol. 2007, 2007, 75805. [Google Scholar] [CrossRef]
- Rochette, L.; Meloux, A.; Rigal, E.; Zeller, M.; Cottin, Y.; Vergely, C. The Role of Osteoprotegerin and Its Ligands in Vascular Function. Int. J. Mol. Sci. 2019, 20, 705. [Google Scholar] [CrossRef] [PubMed]
- Schoppet, M.; Preissner, K.T.; Hofbauer, L.C. RANK ligand and osteoprotegerin: Paracrine regulators of bone metabolism and vascular function. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Malyankar, U.M.; Scatena, M.; Suchland, K.L.; Yun, T.J.; Clark, E.A.; Giachelli, C.M. Osteoprotegerin is an alpha vbeta 3-induced, NF-kappa B-dependent survival factor for endothelial cells. J. Biol. Chem. 2000, 275, 20959–20962. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, Y.; Huang, Z.; Chen, X.; Zhang, B. The roles of osteoprotegerin in cancer, far beyond a bone player. Cell Death Discov. 2022, 8, 252. [Google Scholar] [CrossRef]
- Boraldi, F.; Lofaro, F.D.; Quaglino, D. Apoptosis in the Extraosseous Calcification Process. Cells 2021, 10, 131. [Google Scholar] [CrossRef]
- Ono, T.; Hayashi, M.; Sasaki, F.; Nakashima, T. RANKL biology: Bone metabolism, the immune system, and beyond. Inflamm. Regen. 2020, 40, 2. [Google Scholar] [CrossRef]
- Omland, T.; Ueland, T.; Jansson, A.M.; Persson, A.; Karlsson, T.; Smith, C.; Herlitz, J.; Aukrust, P.; Hartford, M.; Caidahl, K. Circulating osteoprotegerin levels and long-term prognosis in patients with acute coronary syndromes. J. Am. Coll. Cardiol. 2008, 51, 627–633. [Google Scholar] [CrossRef]
- Gu, J.H.; Tong, X.S.; Chen, G.H.; Liu, X.Z.; Bian, J.C.; Yuan, Y.; Liu, Z.P. Regulation of matrix metalloproteinase-9 protein expression by 1alpha, 25-(OH)(2)D(3) during osteoclast differentiation. J. Vet. Sci. 2014, 15, 133–140. [Google Scholar] [CrossRef]
- Rochette, L.; Meloux, A.; Rigal, E.; Zeller, M.; Cottin, Y.; Vergely, C. The role of osteoprotegerin in the crosstalk between vessels and bone: Its potential utility as a marker of cardiometabolic diseases. Pharmacol. Ther. 2018, 182, 115–132. [Google Scholar] [CrossRef]
- Sandberg, W.J.; Yndestad, A.; Oie, E.; Smith, C.; Ueland, T.; Ovchinnikova, O.; Robertson, A.K.; Muller, F.; Semb, A.G.; Scholz, H.; et al. Enhanced T-cell expression of RANK ligand in acute coronary syndrome: Possible role in plaque destabilization. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 857–863. [Google Scholar] [CrossRef] [PubMed]
- Kiechl, S.; Schett, G.; Schwaiger, J.; Seppi, K.; Eder, P.; Egger, G.; Santer, P.; Mayr, A.; Xu, Q.; Willeit, J. Soluble receptor activator of nuclear factor-kappa B ligand and risk for cardiovascular disease. Circulation 2007, 116, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Mohammadpour, A.H.; Shamsara, J.; Nazemi, S.; Ghadirzadeh, S.; Shahsavand, S.; Ramezani, M. Evaluation of RANKL/OPG Serum Concentration Ratio as a New Biomarker for Coronary Artery Calcification: A Pilot Study. Thrombosis 2012, 2012, 306263. [Google Scholar] [CrossRef] [PubMed]
- Ovchinnikova, O.; Gylfe, A.; Bailey, L.; Nordstrom, A.; Rudling, M.; Jung, C.; Bergstrom, S.; Waldenstrom, A.; Hansson, G.K.; Nordstrom, P. Osteoprotegerin promotes fibrous cap formation in atherosclerotic lesions of ApoE-deficient mice-brief report. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1478–1480. [Google Scholar] [CrossRef]
- Ovchinnikova, O.A.; Folkersen, L.; Persson, J.; Lindeman, J.H.; Ueland, T.; Aukrust, P.; Gavrisheva, N.; Shlyakhto, E.; Paulsson-Berne, G.; Hedin, U.; et al. The collagen cross-linking enzyme lysyl oxidase is associated with the healing of human atherosclerotic lesions. J. Intern. Med. 2014, 276, 525–536. [Google Scholar] [CrossRef]
- Merched, A.J.; Chan, L. Absence of p21Waf1/Cip1/Sdi1 modulates macrophage differentiation and inflammatory response and protects against atherosclerosis. Circulation 2004, 110, 3830–3841. [Google Scholar] [CrossRef]
- Secchiero, P.; Corallini, F.; Rimondi, E.; Chiaruttini, C.; di Iasio, M.G.; Rustighi, A.; Del Sal, G.; Zauli, G. Activation of the p53 pathway down-regulates the osteoprotegerin expression and release by vascular endothelial cells. Blood 2008, 111, 1287–1294. [Google Scholar] [CrossRef][Green Version]
- Suzuki, M.; Minami, A.; Nakanishi, A.; Kobayashi, K.; Matsuda, S.; Ogura, Y.; Kitagishi, Y. Atherosclerosis and tumor suppressor molecules (review). Int. J. Mol. Med. 2014, 34, 934–940. [Google Scholar] [CrossRef]
- Berger, C.; Qian, Y.; Chen, X. The p53-estrogen receptor loop in cancer. Curr. Mol. Med. 2013, 13, 1229–1240. [Google Scholar] [CrossRef]
- Chimento, A.; De Luca, A.; Avena, P.; De Amicis, F.; Casaburi, I.; Sirianni, R.; Pezzi, V. Estrogen Receptors-Mediated Apoptosis in Hormone-Dependent Cancers. Int. J. Mol. Sci. 2022, 23, 1242. [Google Scholar] [CrossRef]
- Wright, J.W.; Stouffer, R.L.; Rodland, K.D. High-dose estrogen and clinical selective estrogen receptor modulators induce growth arrest, p21, and p53 in primate ovarian surface epithelial cells. J. Clin. Endocrinol. Metab. 2005, 90, 3688–3695. [Google Scholar] [CrossRef] [PubMed][Green Version]
- McRobb, L.S.; McGrath, K.C.Y.; Tsatralis, T.; Liong, E.C.; Tan, J.T.M.; Hughes, G.; Handelsman, D.J.; Heather, A.K. Estrogen Receptor Control of Atherosclerotic Calcification and Smooth Muscle Cell Osteogenic Differentiation. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1127–1137. [Google Scholar] [CrossRef] [PubMed]
- Villablanca, A.C.; Tenwolde, A.; Lee, M.; Huck, M.; Mumenthaler, S.; Rutledge, J.C. 17beta-estradiol prevents early-stage atherosclerosis in estrogen receptor-alpha deficient female mice. J. Cardiovasc. Transl. Res. 2009, 2, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, C.; Morfoisse, F.; Tatin, F.; Zamora, A.; Zahreddine, R.; Henrion, D.; Arnal, J.-F.; Lenfant, F.; Garmy-Susini, B. The Impact of Estrogen Receptor in Arterial and Lymphatic Vascular Diseases. Int. J. Mol. Sci. 2020, 21, 3244. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Cao, L.; Ping, N.N.; Wu, Y.; Liu, D.Z.; Cao, Y.X. Formononetin upregulates nitric oxide synthase in arterial endothelium through estrogen receptors and MAPK pathways. J. Pharm. Pharmacol. 2016, 68, 342–351. [Google Scholar] [CrossRef]
- Ahmad, N.; Chen, S.; Wang, W.; Kapila, S. 17beta-estradiol Induces MMP-9 and MMP-13 in TMJ Fibrochondrocytes via Estrogen Receptor alpha. J. Dent. Res. 2018, 97, 1023–1030. [Google Scholar] [CrossRef]
- Ghisletti, S.; Meda, C.; Maggi, A.; Vegeto, E. 17beta-estradiol inhibits inflammatory gene expression by controlling NF-kappaB intracellular localization. Mol. Cell. Biol. 2005, 25, 2957–2968. [Google Scholar] [CrossRef]
- Hirano, S.; Furutama, D.; Hanafusa, T. Physiologically high concentrations of 17beta-estradiol enhance NF-kappaB activity in human T cells. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R1465–R1471. [Google Scholar] [CrossRef]
- Abu-Taha, M.; Rius, C.; Hermenegildo, C.; Noguera, I.; Cerda-Nicolas, J.M.; Issekutz, A.C.; Jose, P.J.; Cortijo, J.; Morcillo, E.J.; Sanz, M.J. Menopause and ovariectomy cause a low grade of systemic inflammation that may be prevented by chronic treatment with low doses of estrogen or losartan. J. Immunol. 2009, 183, 1393–1402. [Google Scholar] [CrossRef] [PubMed]
- Chan, Q.K.; Lam, H.M.; Ng, C.F.; Lee, A.Y.; Chan, E.S.; Ng, H.K.; Ho, S.M.; Lau, K.M. Activation of GPR30 inhibits the growth of prostate cancer cells through sustained activation of Erk1/2, c-jun/c-fos-dependent upregulation of p21, and induction of G(2) cell-cycle arrest. Cell Death Differ. 2010, 17, 1511–1523. [Google Scholar] [CrossRef]
- Wei, W.; Chen, Z.J.; Zhang, K.S.; Yang, X.L.; Wu, Y.M.; Chen, X.H.; Huang, H.B.; Liu, H.L.; Cai, S.H.; Du, J.; et al. The activation of G protein-coupled receptor 30 (GPR30) inhibits proliferation of estrogen receptor-negative breast cancer cells in vitro and in vivo. Cell Death Dis. 2014, 5, e1428. [Google Scholar] [CrossRef] [PubMed]
- Obikane, H.; Abiko, Y.; Ueno, H.; Kusumi, Y.; Esumi, M.; Mitsumata, M. Effect of endothelial cell proliferation on atherogenesis: A role of p21(Sdi/Cip/Waf1) in monocyte adhesion to endothelial cells. Atherosclerosis 2010, 212, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Hodis, H.N.; Mack, W.J.; Henderson, V.W.; Shoupe, D.; Budoff, M.J.; Hwang-Levine, J.; Li, Y.; Feng, M.; Dustin, L.; Kono, N.; et al. Vascular Effects of Early versus Late Postmenopausal Treatment with Estradiol. N. Engl. J. Med. 2016, 374, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Naftolin, F.; Friedenthal, J.; Nachtigall, R.; Nachtigall, L. Cardiovascular health and the menopausal woman: The role of estrogen and when to begin and end hormone treatment. F1000Research 2019, 8, 1576. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Yang, D.; Yin, Y.Z.; Xiao, J. Estrogenic and anti-inflammatory effects of pseudoprotodioscin in atherosclerosis-prone mice: Insights into endothelial cells and perivascular adipose tissues. Eur. J. Pharmacol. 2020, 869, 172887. [Google Scholar] [CrossRef]
- Kuntz, S.; Asseburg, H.; Dold, S.; Rompp, A.; Frohling, B.; Kunz, C.; Rudloff, S. Inhibition of low-grade inflammation by anthocyanins from grape extract in an in vitro epithelial-endothelial co-culture model. Food Funct. 2015, 6, 1136–1149. [Google Scholar] [CrossRef]
- Lee, H.; Jee, Y.; Hong, K.; Hwang, G.S.; Chun, K.H. MicroRNA-494, upregulated by tumor necrosis factor-alpha, desensitizes insulin effect in C2C12 muscle cells. PLoS ONE 2013, 8, e83471. [Google Scholar] [CrossRef]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte chemoattractant protein-1 (MCP-1): An overview. J. Interferon. Cytokine Res. 2009, 29, 313–326. [Google Scholar] [CrossRef]
- Liu, X.L.; Zhang, P.F.; Ding, S.F.; Wang, Y.; Zhang, M.; Zhao, Y.X.; Ni, M.; Zhang, Y. Local gene silencing of monocyte chemoattractant protein-1 prevents vulnerable plaque disruption in apolipoprotein E-knockout mice. PLoS ONE 2012, 7, e33497. [Google Scholar] [CrossRef]
- Cho, K.Y.; Miyoshi, H.; Kuroda, S.; Yasuda, H.; Kamiyama, K.; Nakagawara, J.; Takigami, M.; Kondo, T.; Atsumi, T. The phenotype of infiltrating macrophages influences arteriosclerotic plaque vulnerability in the carotid artery. J. Stroke Cerebrovasc. Dis. 2013, 22, 910–918. [Google Scholar] [CrossRef]
- Ariazi, E.A.; Brailoiu, E.; Yerrum, S.; Shupp, H.A.; Slifker, M.J.; Cunliffe, H.E.; Black, M.A.; Donato, A.L.; Arterburn, J.B.; Oprea, T.I.; et al. The G protein-coupled receptor GPR30 inhibits proliferation of estrogen receptor-positive breast cancer cells. Cancer Res. 2010, 70, 1184–1194. [Google Scholar] [CrossRef] [PubMed]
- Xing, D.; Feng, W.; Miller, A.P.; Weathington, N.M.; Chen, Y.F.; Novak, L.; Blalock, J.E.; Oparil, S. Estrogen modulates TNF-alpha-induced inflammatory responses in rat aortic smooth muscle cells through estrogen receptor-beta activation. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H2607–H2612. [Google Scholar] [CrossRef] [PubMed]
- Xing, D.; Oparil, S.; Yu, H.; Gong, K.; Feng, W.; Black, J.; Chen, Y.F.; Nozell, S. Estrogen modulates NFkappaB signaling by enhancing IkappaBalpha levels and blocking p65 binding at the promoters of inflammatory genes via estrogen receptor-beta. PLoS ONE 2012, 7, e36890. [Google Scholar] [CrossRef] [PubMed]
- Iorga, A.; Umar, S.; Ruffenach, G.; Aryan, L.; Li, J.; Sharma, S.; Motayagheni, N.; Nadadur, R.D.; Bopassa, J.C.; Eghbali, M. Estrogen rescues heart failure through estrogen receptor Beta activation. Biol. Sex. Differ. 2018, 9, 48. [Google Scholar] [CrossRef]
- Zong, W.; Jiang, Y.; Zhao, J.; Zhang, J.; Gao, J.G. Estradiol plays a role in regulating the expression of lysyl oxidase family genes in mouse urogenital tissues and human Ishikawa cells. J. Zhejiang Univ. Sci. B. 2015, 16, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Newby, A.C. Metalloproteinases and vulnerable atherosclerotic plaques. Trends Cardiovasc. Med. 2007, 17, 253–258. [Google Scholar] [CrossRef]
- Stoneman, V.E.; Bennett, M.R. Role of apoptosis in atherosclerosis and its therapeutic implications. Clin. Sci. 2004, 107, 343–354. [Google Scholar] [CrossRef]
- Dhillon, O.S.; Khan, S.Q.; Narayan, H.K.; Ng, K.H.; Mohammed, N.; Quinn, P.A.; Squire, I.B.; Davies, J.E.; Ng, L.L. Matrix metalloproteinase-2 predicts mortality in patients with acute coronary syndrome. Clin. Sci. 2009, 118, 249–257. [Google Scholar] [CrossRef]
- Potier, M.; Karl, M.; Elliot, S.J.; Striker, G.E.; Striker, L.J. Response to sex hormones differs in atherosclerosis-susceptible and -resistant mice. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E1237–E1245. [Google Scholar] [CrossRef][Green Version]
- Wingrove, C.S.; Garr, E.; Godsland, I.F.; Stevenson, J.C. 17beta-oestradiol enhances release of matrix metalloproteinase-2 from human vascular smooth muscle cells. Biochim. Biophys. Acta 1998, 1406, 169–174. [Google Scholar] [CrossRef]
- Inoue, S.; Nakazawa, T.; Cho, A.; Dastvan, F.; Shilling, D.; Daum, G.; Reidy, M. Regulation of arterial lesions in mice depends on differential smooth muscle cell migration: A role for sphingosine-1-phosphate receptors. J. Vasc. Surg. 2007, 46, 756–763. [Google Scholar] [CrossRef][Green Version]
- Johnson, C.; Galis, Z.S. Matrix metalloproteinase-2 and -9 differentially regulate smooth muscle cell migration and cell-mediated collagen organization. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 54–60. [Google Scholar] [CrossRef]
- Zanger, D.; Yang, B.K.; Ardans, J.; Waclawiw, M.A.; Csako, G.; Wahl, L.M.; Cannon, R.O., 3rd. Divergent effects of hormone therapy on serum markers of inflammation in postmenopausal women with coronary artery disease on appropriate medical management. J. Am. Coll. Cardiol. 2000, 36, 1797–1802. [Google Scholar] [CrossRef]
- Koh, K.K.; Ahn, J.Y.; Kang, M.H.; Kim, D.S.; Jin, D.K.; Sohn, M.S.; Park, G.S.; Choi, I.S.; Shin, E.K. Effects of hormone replacement therapy on plaque stability, inflammation, and fibrinolysis in hypertensive or overweight postmenopausal women. Am. J. Cardiol. 2001, 88, 1423–1426. [Google Scholar] [CrossRef]
- Amin, M.; Pushpakumar, S.; Muradashvili, N.; Kundu, S.; Tyagi, S.C.; Sen, U. Regulation and involvement of matrix metalloproteinases in vascular diseases. Front. Biosci. 2016, 21, 89–118. [Google Scholar] [CrossRef]
- Romero, J.R.; Vasan, R.S.; Beiser, A.S.; Polak, J.F.; Benjamin, E.J.; Wolf, P.A.; Seshadri, S. Association of carotid artery atherosclerosis with circulating biomarkers of extracellular matrix remodeling: The Framingham Offspring Study. J. Stroke Cerebrovasc. Dis. 2008, 17, 412–417. [Google Scholar] [CrossRef][Green Version]
- Lewandowski, K.C.; Komorowski, J.; Mikhalidis, D.P.; Bienkiewicz, M.; Tan, B.K.; O’Callaghan, C.J.; Lewinski, A.; Prelevic, G.; Randeva, H.S. Effects of hormone replacement therapy type and route of administration on plasma matrix metalloproteinases and their tissue inhibitors in postmenopausal women. J. Clin. Endocrinol. Metab. 2006, 91, 3123–3130. [Google Scholar] [CrossRef]
- Wang, H.; Zhao, Z.; Lin, M.; Groban, L. Activation of GPR30 inhibits cardiac fibroblast proliferation. Mol. Cell Biochem. 2015, 405, 135–148. [Google Scholar] [CrossRef]
- Hwang, J.; Hodis, H.N.; Hsiai, T.K.; Asatryan, L.; Sevanian, A. Role of annexin II in estrogen-induced macrophage matrix metalloproteinase-9 activity: The modulating effect of statins. Atherosclerosis 2006, 189, 76–82. [Google Scholar] [CrossRef]
- Lappano, R.; De Marco, P.; De Francesco, E.M.; Chimento, A.; Pezzi, V.; Maggiolini, M. Cross-talk between GPER and growth factor signaling. J. Steroid Biochem Mol. Biol. 2013, 137, 50–56. [Google Scholar] [CrossRef]
- Palmieri, D.; Perego, P.; Palombo, D. Apigenin inhibits the TNFalpha-induced expression of eNOS and MMP-9 via modulating Akt signalling through oestrogen receptor engagement. Mol. Cell Biochem. 2012, 371, 129–136. [Google Scholar] [CrossRef]
- Voloshenyuk, T.G.; Larkin, K.; Fournett, A.; Gardner, J.D. Estrogen receptor dependence of lysyl oxidase expression and activity in cardiac fibroblasts. FASEB J. 2012, 26, 1059.16. [Google Scholar] [CrossRef]
- Li, S.Y.; Yan, J.Q.; Song, Z.; Liu, Y.F.; Song, M.J.; Qin, J.W.; Yang, Z.M.; Liang, X.H. Molecular characterization of lysyl oxidase-mediated extracellular matrix remodeling during mouse decidualization. FEBS Lett. 2017, 591, 1394–1407. [Google Scholar] [CrossRef]
- Dasgupta, S.; Eudaly, J. Estrogen receptor-alpha mediates Toll-like receptor-2 agonist-induced monocyte chemoattractant protein-1 production in mesangial cells. Results Immunol. 2012, 2, 196–203. [Google Scholar] [CrossRef]
- Zhou, Y.; Liu, X. The role of estrogen receptor beta in breast cancer. Biomark. Res. 2020, 8, 39. [Google Scholar] [CrossRef]
- Kanda, N.; Watanabe, S. 17Beta-estradiol inhibits MCP-1 production in human keratinocytes. J. Investig. Dermatol. 2003, 120, 1058–1066. [Google Scholar] [CrossRef]
- Condorelli, G.; Aycock, J.K.; Frati, G.; Napoli, C. Mutated p21/WAF/CIP transgene overexpression reduces smooth muscle cell proliferation, macrophage deposition, oxidation-sensitive mechanisms, and restenosis in hypercholesterolemic apolipoprotein E knockout mice. FASEB J. 2001, 15, 2162–2170. [Google Scholar] [CrossRef]
- Smith, R.C.; Branellec, D.; Gorski, D.H.; Guo, K.; Perlman, H.; Dedieu, J.F.; Pastore, C.; Mahfoudi, A.; Denefle, P.; Isner, J.M.; et al. p21CIP1-mediated inhibition of cell proliferation by overexpression of the gax homeodomain gene. Genes. Dev. 1997, 11, 1674–1689. [Google Scholar] [CrossRef]
- Matsuda, S.; Umemoto, S.; Yoshimura, K.; Itoh, S.; Murata, T.; Fukai, T.; Matsuzaki, M. Angiotensin Activates MCP-1 and Induces Cardiac Hypertrophy and Dysfunction via Toll-like Receptor 4. J. Atheroscler. Thromb. 2015, 22, 833–844. [Google Scholar] [CrossRef]
- Doi, T.; Yoshino, T.; Fuse, N.; Boku, N.; Yamazaki, K.; Koizumi, W.; Shimada, K.; Takinishi, Y.; Ohtsu, A. Phase I study of TAS-102 and irinotecan combination therapy in Japanese patients with advanced colorectal cancer. Investig. New Drugs 2015, 33, 1068–1077. [Google Scholar] [CrossRef]
- Kassi, E.; Nasiri-Ansari, N.; Spilioti, E.; Kalotychou, V.; Apostolou, P.E.; Moutsatsou, P.; Papavassiliou, A.G. Vitamin D interferes with glucocorticoid responsiveness in human peripheral blood mononuclear target cells. Cell Mol. Life Sci. 2016, 73, 4341–4354. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Forward | Reverse |
|---|---|---|
| MMP-2 | 5′-TGGCAAGTACGGCTTCTGTC-3′ | 5′-TTCTTGTCGCGGTCGTAGTC-3′ |
| MMP-9 | 5′-TGCGCTACCACCTCGAACTT-3′ | 5′-GATGCCATTGACGTCGTCCT-3′ |
| TIMP-1 | 5′-TGCGGATACTTCCACAGGTC-3′ | 5′-GCATTCCTCACAGCCAACAG-3′ |
| TIMP-2 | 5′-AAGAGCCTGAACCACAGGTA-3′ | 5′-GAGCCGTCACTTCTCTTGAT-3′ |
| MCP-1 | 5′-AATAGGAAGATCTCAGTGCA-3′ | 5′-TCAAGTCTTCGGAGTTTGGG-3′ |
| OPG | 5′-GGAACCCCAGAGCGAAATACA-3′ | 5′-CCTGAAGAATGCCTCCTCACA-3′ |
| RANK | 5′-CCCGTTGCAGCTCAACAAG-3′ | 5′-GCATTTGTCCGTGGAGGAA-3′ |
| RANKL | 5′ -ACGCAGTGAAAACACAGTT-3′ | 5′-TGCCTCTGGCTGGAAACC-3′ |
| LOX-1 | 5′ -CCAGAGGAGAGTGGCTGAAG-3′ | 5′-CCAGGTAGCTGGGGTTTACA-3′ |
| PDGF- β | 5′-CCATTCCCGAGGAGCTTTATG-3′ | 5′-CAGCAGGCGTTGGAGATCAT-3′ |
| P21 | 5′-ATGAAATTCACCCCCTTTCC-3′ | 5′-CCCTAGGCTGTGCTCACTTC-3′ |
| ER-α | 5′-TGGGCTTACTGACCAACCTG-3′ | 5′-CCTGATCATGGAGGGTCAAA-3′ |
| ΕR-β | 5′-AGAGTCCCTGGTGTGAAGCA-3′ | 5′-GACAGCGCAGAAGTGAGCATC-3′ |
| GPR-30 | 5′-TCACGGGCCACATTGTCAACCTC | 5′-GCTGAACCTCACATCTGACTGCTC |
| GAPDH | 5′-GGGTGTGAACCATGAGAAGT-3′ | 5′-CATGCCAGTGAGCTTCCCGTT-3′ |
| ADAMTS-4 | 5′-GACACTGGTGGTGGCAGATG-3′ | 5′-TCACTGTTAGCAGGTAGCGCTTTA-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nasiri-Ansari, N.; Spilioti, E.; Kyrou, I.; Kalotychou, V.; Chatzigeorgiou, A.; Sanoudou, D.; Dahlman-Wright, K.; Randeva, H.S.; Papavassiliou, A.G.; Moutsatsou, P.; et al. Estrogen Receptor Subtypes Elicit a Distinct Gene Expression Profile of Endothelial-Derived Factors Implicated in Atherosclerotic Plaque Vulnerability. Int. J. Mol. Sci. 2022, 23, 10960. https://doi.org/10.3390/ijms231810960
Nasiri-Ansari N, Spilioti E, Kyrou I, Kalotychou V, Chatzigeorgiou A, Sanoudou D, Dahlman-Wright K, Randeva HS, Papavassiliou AG, Moutsatsou P, et al. Estrogen Receptor Subtypes Elicit a Distinct Gene Expression Profile of Endothelial-Derived Factors Implicated in Atherosclerotic Plaque Vulnerability. International Journal of Molecular Sciences. 2022; 23(18):10960. https://doi.org/10.3390/ijms231810960
Chicago/Turabian StyleNasiri-Ansari, Narjes, Eliana Spilioti, Ioannis Kyrou, Vassiliki Kalotychou, Antonios Chatzigeorgiou, Despina Sanoudou, Karin Dahlman-Wright, Harpal S. Randeva, Athanasios G. Papavassiliou, Paraskevi Moutsatsou, and et al. 2022. "Estrogen Receptor Subtypes Elicit a Distinct Gene Expression Profile of Endothelial-Derived Factors Implicated in Atherosclerotic Plaque Vulnerability" International Journal of Molecular Sciences 23, no. 18: 10960. https://doi.org/10.3390/ijms231810960
APA StyleNasiri-Ansari, N., Spilioti, E., Kyrou, I., Kalotychou, V., Chatzigeorgiou, A., Sanoudou, D., Dahlman-Wright, K., Randeva, H. S., Papavassiliou, A. G., Moutsatsou, P., & Kassi, E. (2022). Estrogen Receptor Subtypes Elicit a Distinct Gene Expression Profile of Endothelial-Derived Factors Implicated in Atherosclerotic Plaque Vulnerability. International Journal of Molecular Sciences, 23(18), 10960. https://doi.org/10.3390/ijms231810960