

Antisense Morpholino-Based In Vitro Correction of a Pseudoexon-Generating Variant in the SGCB Gene


 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
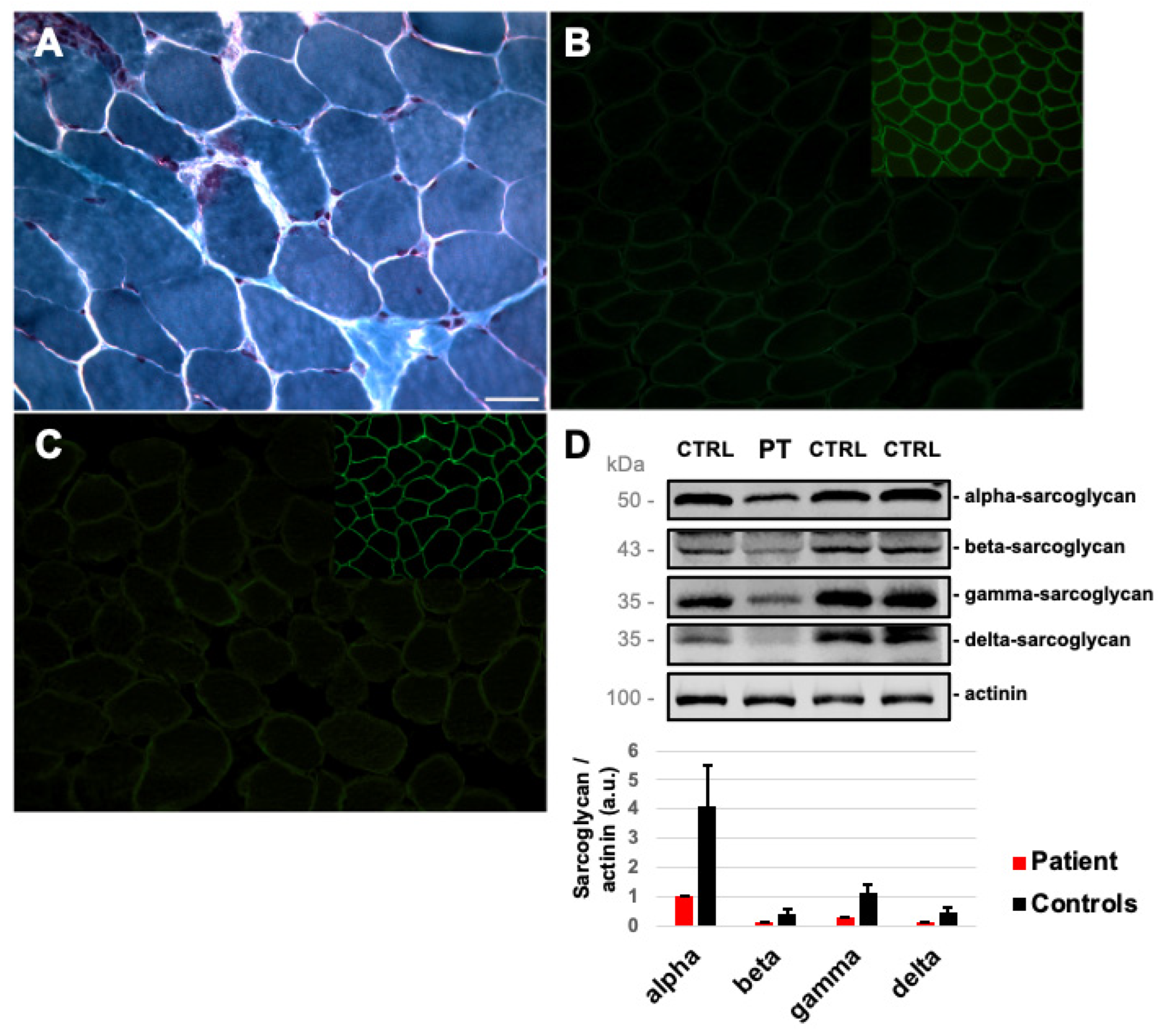
2.1. Case Report
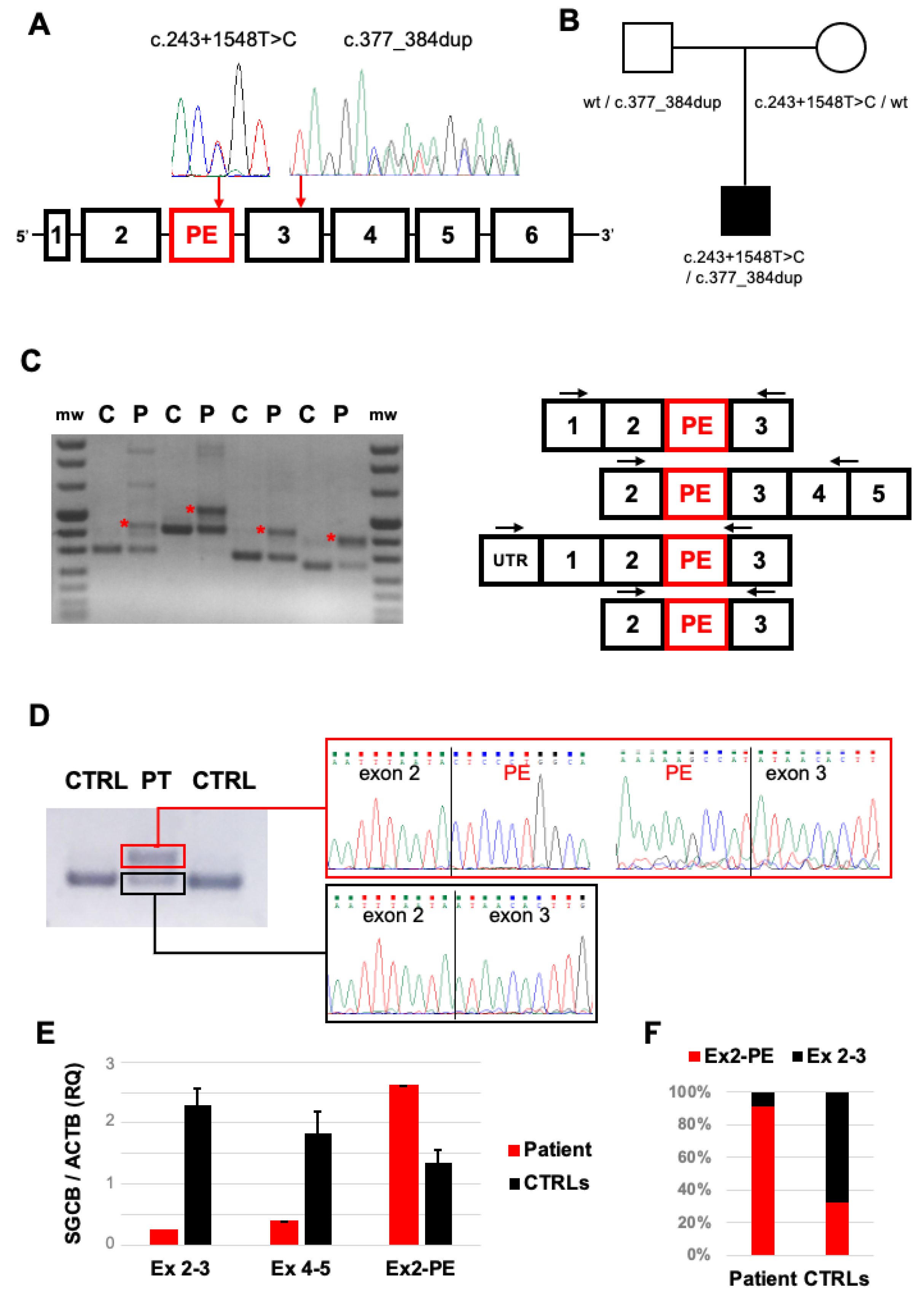
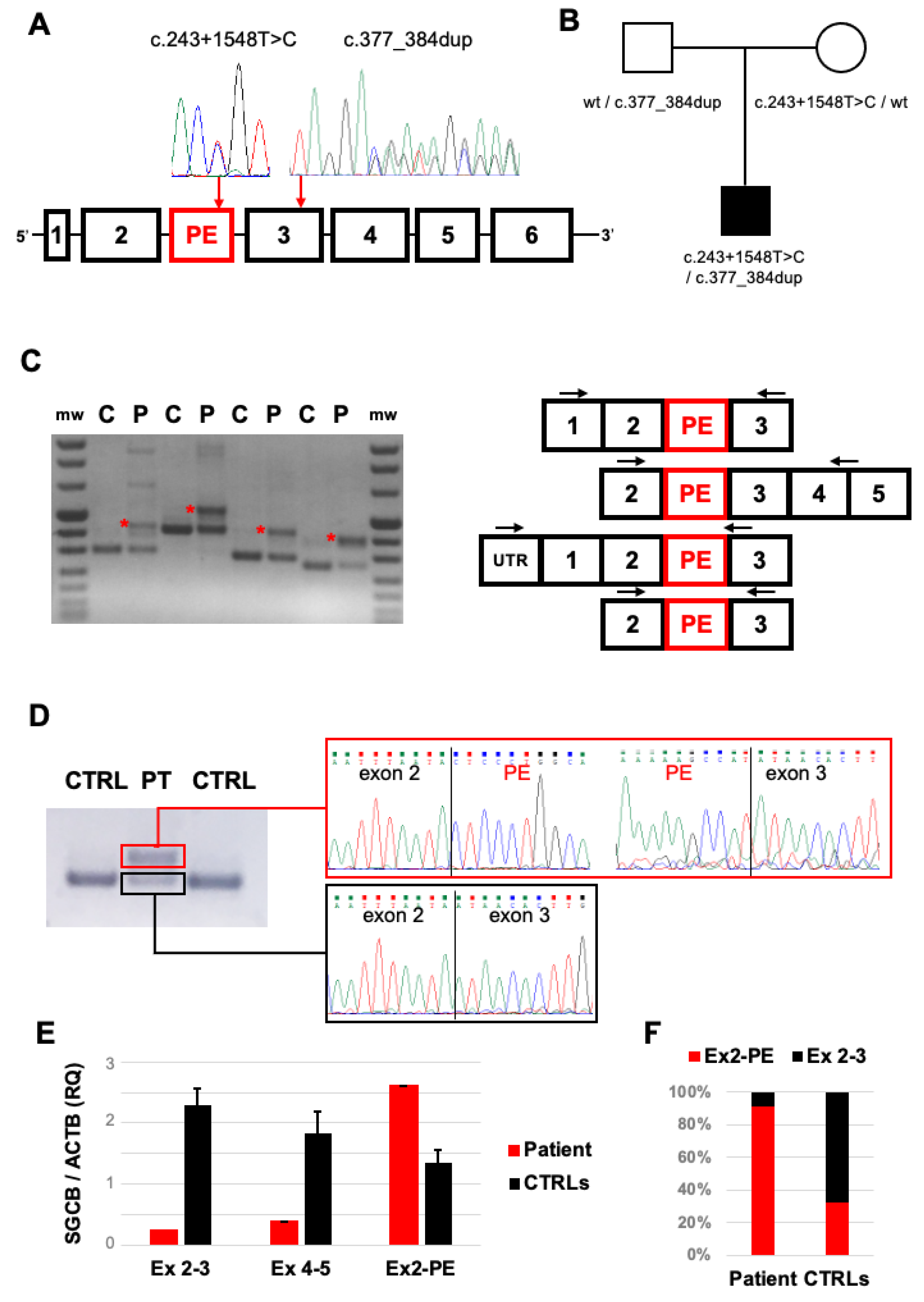
2.2. Genetic Studies
2.3. Transcript Analysis
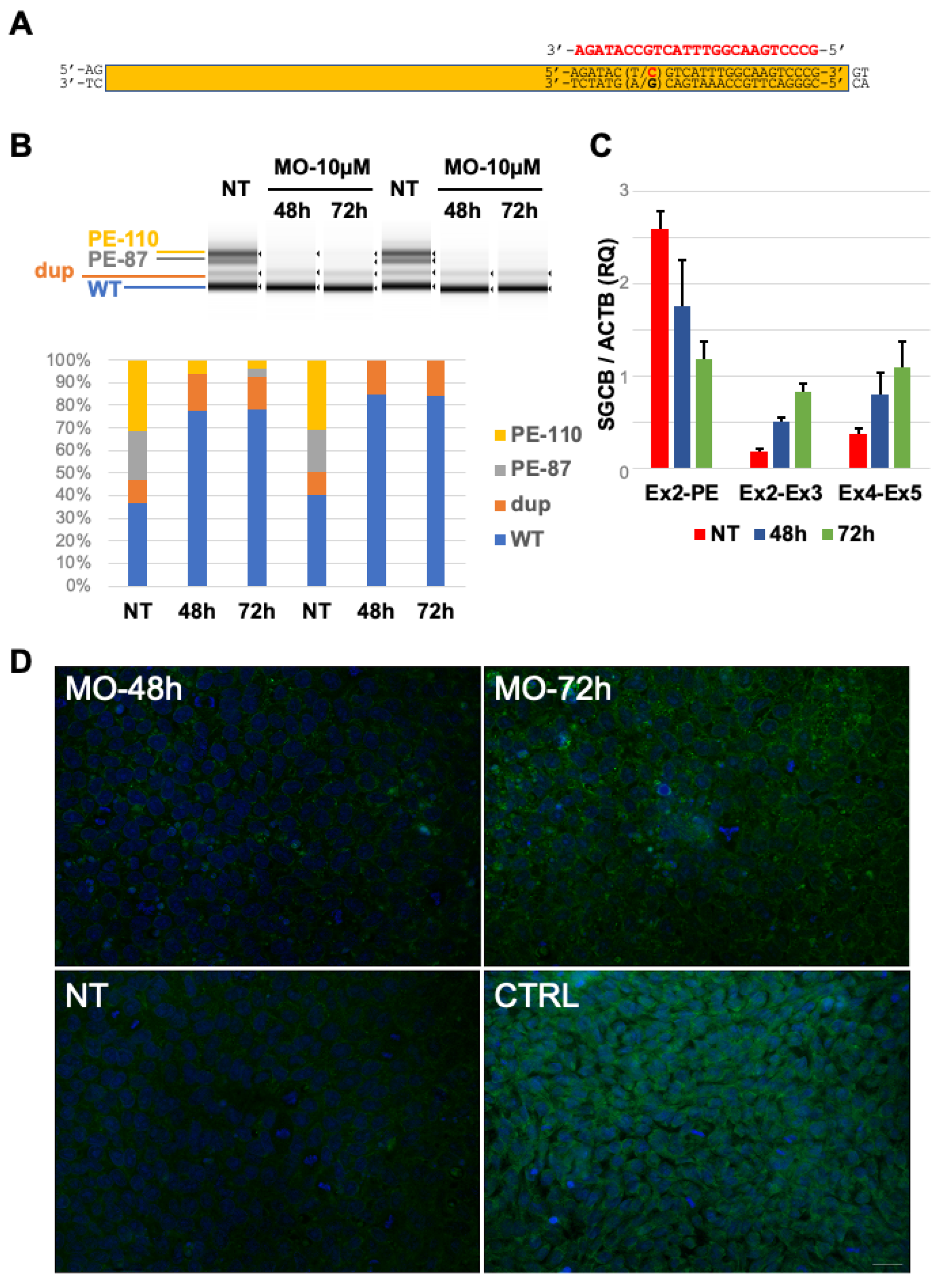
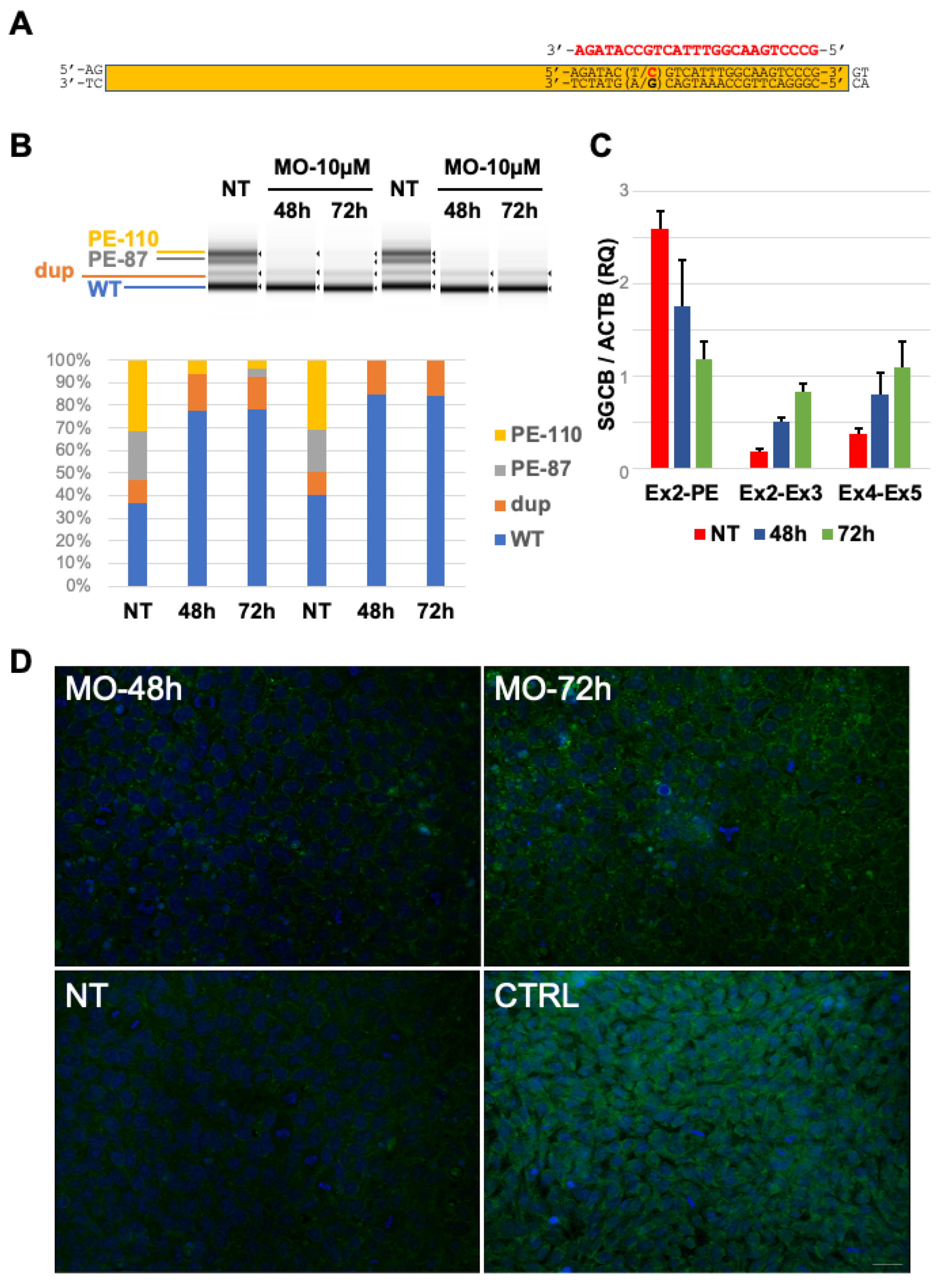
2.4. In Vitro Delivery of Antisense Morpholino Oligomer
3. Discussion
4. Materials and Methods
4.1. Muscle Tissue Analysis
4.2. Molecular Studies
4.3. Cell Cultures
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Angelini, C. LGMD. Identification, description and classification. Acta Myol. 2020, 39, 207–217. [Google Scholar]
- Vainzof, M.; Souza, L.S.; Gurgel-Giannetti, J.; Zatz, M. Sarcoglycanopathies: An update. Neuromuscul. Disord. 2021, 31, 1021–1027. [Google Scholar] [CrossRef]
- Straub, V.; Murphy, A.; Udd, B.; LGMD workshop study group. 229th ENMC international workshop: Limb girdle muscular dystrophies—Nomenclature and reformed classification Naarden, The Netherlands, 17–19 March 2017. Neuromuscul. Disord. 2018, 28, 702–710. [Google Scholar]
- Tarakci, H.; Berger, J. The sarcoglycan complex in skeletal muscle. Front. Biosci. (Landmark Ed) 2016, 21, 744–756. [Google Scholar]
- Lim, L.E.; Duclos, F.; Broux, O.; Bourg, N.; Sunada, Y.; Allamand, V.; Meyer, J.; Richard, I.; Moomaw, C.; Slaughter, C.; et al. Beta-sarcoglycan: Characterization and role in limb-girdle muscular dystrophy linked to 4q12. Nat. Genet. 1995, 11, 257–265. [Google Scholar] [CrossRef]
- Magri, F.; Nigro, V.; Angelini, C.; Mongini, T.; Mora, M.; Moroni, I.; Toscano, A.; D’angelo, M.G.; Tomelleri, G.; Siciliano, G.; et al. The italian limb girdle muscular dystrophy registry: Relative frequency, clinical features, and differential diagnosis. Muscle Nerve. 2017, 55, 55–68. [Google Scholar] [CrossRef]
- Alonso-Pérez, J.; González-Quereda, L.; Bello, L.; Guglieri, M.; Straub, V.; Gallano, P.; Semplicini, C.; Pegoraro, E.; Zangaro, V.; Nascimento, A.; et al. New genotype-phenotype correlations in a large European cohort of patients with sarcoglycanopathy. Brain 2020, 143, 2696–2708. [Google Scholar] [CrossRef] [PubMed]
- Schade van Westrum, S.M.; Dekker, L.R.; de Voogt, W.G.; Wilde, A.A.; Ginjaar, I.B.; de Visser, M.; van der Kooi, A.J. Cardiac involvement in Dutch patients with sarcoglycanopathy: A cross-sectional cohort and follow-up study. Muscle Nerve. 2014, 50, 909–913. [Google Scholar] [PubMed]
- Semplicini, C.; Vissing, J.; Dahlqvist, J.R.; Stojkovic, T.; Bello, L.; Witting, N.; Duno, M.; Leturcq, F.; Bertolin, C.; D’Ambrosio, P.; et al. Clinical and genetic spectrum in limb-girdle muscular dystrophy type 2E. Neurology 2015, 84, 1772–1781. [Google Scholar] [CrossRef] [PubMed]
- Barresi, R.; Di Blasi, C.; Negri, T.; Brugnoni, R.; Vitali, A.; Felisari, G.; Salandi, A.; Daniel, S.; Cornelio, F.; Morandi, L.; et al. Disruption of heart sarcoglycan complex and severe cardiomyopathy caused by beta sarcoglycan mutations. J. Med. Genet. 2000, 37, 102–107. [Google Scholar] [CrossRef]
- Gandolla, M.; Antonietti, A.; Longatelli, V.; Biffi, E.; Diella, E.; Delle Fave, M.; Rossini, M.; Molteni, F.; D’Angelo, G.; Bocciolone, M.; et al. Test-retest reliability of the Performance of Upper Limb (PUL) module for muscular dystrophy patients. PLoS ONE 2020, 15, e0239064. [Google Scholar] [CrossRef] [PubMed]
- Bérard, C.; Payan, C.; Hodgkinson, I.; Fermanian, J.; MFM Collaborative Study Group. A motor function measure for neuromuscular diseases. Construction and validation study. Neuromuscul. Disord. 2005, 15, 463–470. [Google Scholar]
- Jacobs, M.B.; James, M.K.; Lowes, L.P.; Alfano, L.N.; Eagle, M.; Muni Lofra, R.; Moore, U.; Feng, J.; Rufibach, L.E.; Rose, K.; et al. Assessing Dysferlinopathy Patients Over Three Years With a New Motor Scale. Ann. Neurol. 2021, 89, 967–978. [Google Scholar] [CrossRef]
- Fanin, M.; Hoffman, E.P.; Angelini, C.; Pegoraro, E. Private beta- and gamma-sarcoglycan gene mutations: Evidence of a founder effect in Northern Italy. Hum. Mutat. 2000, 16, 13–17. [Google Scholar] [CrossRef]
- Douglas, A.G.; Wood, M.J. Splicing therapy for neuromuscular disease. Mol. Cell Neurosci. 2013, 56, 169–185. [Google Scholar] [CrossRef]
- Petri, H.; Sveen, M.L.; Thune, J.J.; Vissing, C.; Dahlqvist, J.R.; Witting, N.; Bundgaard, H.; Køber, L.; Vissing, J. Progression of cardiac involvement in patients with limb-girdle type 2 and Becker muscular dystrophies: A 9-year follow-up study. Int. J. Cardiol. 2015, 182, 403–411. [Google Scholar] [CrossRef]
- Marchetti, G.B.; Valenti, L.; Torrente, Y. Clinical Determinants of Disease Progression in Patients with Beta-Sarcoglycan Gene Mutations. Front. Neurol. 2021, 12, 657949. [Google Scholar] [CrossRef]
- Aoki, Y.; Wood, M.J.A. Emerging Oligonucleotide Therapeutics for Rare Neuromuscular Diseases. J. Neuromuscul. Dis. 2021, 8, 869–884. [Google Scholar] [CrossRef]
- Matsuo, M. Antisense Oligonucleotide-Mediated Exon-skipping Therapies: Precision Medicine Spreading from Duchenne Muscular Dystrophy. JMA J. 2021, 4, 232–240. [Google Scholar]
- Dominov, J.A.; Uyan, Ö.; McKenna-Yasek, D.; Nallamilli, B.R.R.; Kergourlay, V.; Bartoli, M.; Levy, N.; Hudson, J.; Evangelista, T.; Lochmuller, H.; et al. Correction of pseudoexon splicing caused by a novel intronic dysferlin mutation. Ann. Clin. Transl. Neurol. 2019, 6, 642–654. [Google Scholar] [CrossRef]
- Pozsgai, E.R.; Griffin, D.A.; Heller, K.N.; Mendell, J.R.; Rodino-Klapac, L.R. Systemic AAV-Mediated β-Sarcoglycan Delivery Targeting Cardiac and Skeletal Muscle Ameliorates Histological and Functional Deficits in LGMD2E Mice. Mol. Ther. 2017, 25, 855–869. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, E.J.; Demonbreun, A.R.; Kim, E.Y.; Puckelwartz, M.J.; Vo, A.H.; Dellefave-Castillo, L.M.; Gao, Q.Q.; Vainzof, M.; Pavanello, R.C.M.; Zatz, M.; et al. Efficient exon skipping of SGCG mutations mediated by phosphorodiamidate morpholino oligomers. JCI Insight 2018, 3, e99357. [Google Scholar] [CrossRef]
- Prelle, A.; Comi, G.P.; Tancredi, L.; Rigoletto, C.; Ciscato, P.; Fortunato, F.; Nesti, S.; Sciacco, M.; Robotti, M.; Bazzi, P.; et al. Sarcoglycan deficiency in a large Italian population of myopathic patients. Acta Neuropathol. 1998, 96, 509–514. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magri, F.; Zanotti, S.; Salani, S.; Fortunato, F.; Ciscato, P.; Gerevini, S.; Maggi, L.; Sciacco, M.; Moggio, M.; Corti, S.; et al. Antisense Morpholino-Based In Vitro Correction of a Pseudoexon-Generating Variant in the SGCB Gene. Int. J. Mol. Sci. 2022, 23, 9817. https://doi.org/10.3390/ijms23179817
Magri F, Zanotti S, Salani S, Fortunato F, Ciscato P, Gerevini S, Maggi L, Sciacco M, Moggio M, Corti S, et al. Antisense Morpholino-Based In Vitro Correction of a Pseudoexon-Generating Variant in the SGCB Gene. International Journal of Molecular Sciences. 2022; 23(17):9817. https://doi.org/10.3390/ijms23179817
Chicago/Turabian StyleMagri, Francesca, Simona Zanotti, Sabrina Salani, Francesco Fortunato, Patrizia Ciscato, Simonetta Gerevini, Lorenzo Maggi, Monica Sciacco, Maurizio Moggio, Stefania Corti, and et al. 2022. "Antisense Morpholino-Based In Vitro Correction of a Pseudoexon-Generating Variant in the SGCB Gene" International Journal of Molecular Sciences 23, no. 17: 9817. https://doi.org/10.3390/ijms23179817
APA StyleMagri, F., Zanotti, S., Salani, S., Fortunato, F., Ciscato, P., Gerevini, S., Maggi, L., Sciacco, M., Moggio, M., Corti, S., Bresolin, N., Comi, G. P., & Ronchi, D. (2022). Antisense Morpholino-Based In Vitro Correction of a Pseudoexon-Generating Variant in the SGCB Gene. International Journal of Molecular Sciences, 23(17), 9817. https://doi.org/10.3390/ijms23179817