
Molecular Basis of the Schuurs–Hoeijmakers Syndrome: What We Know about the Gene and the PACS-1 Protein and Novel Therapeutic Approaches

 , , , , ,
, , , , ,  , , , and
, , , and
Abstract
1. Introduction
2. Clinical Characteristics of PACS1-NDD
3. Molecular Basis of the Disease
3.1. Genetic Update
3.2. PACS1 Gene Regulation
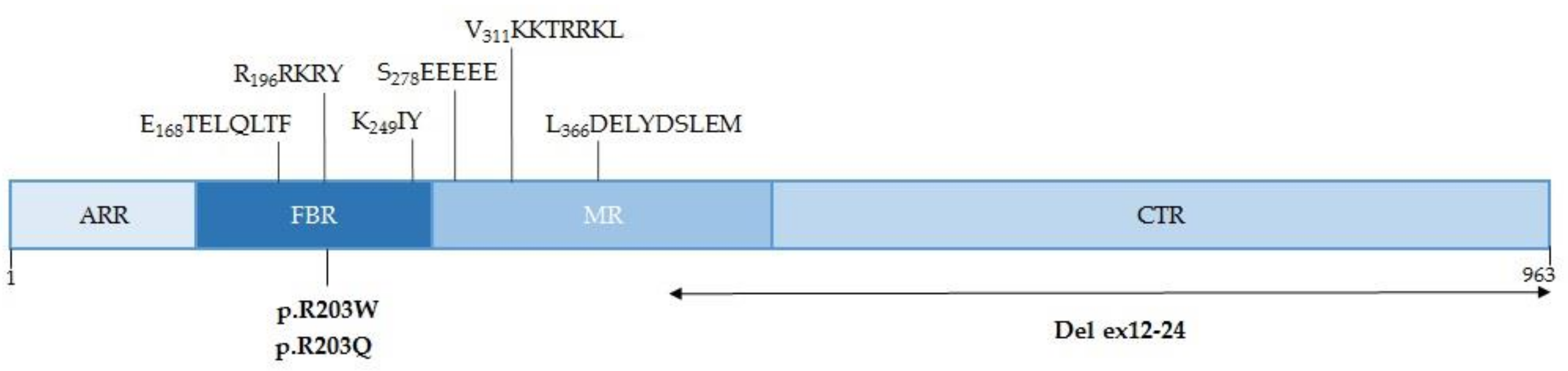
3.3. Characteristics of the PACS-1 Protein
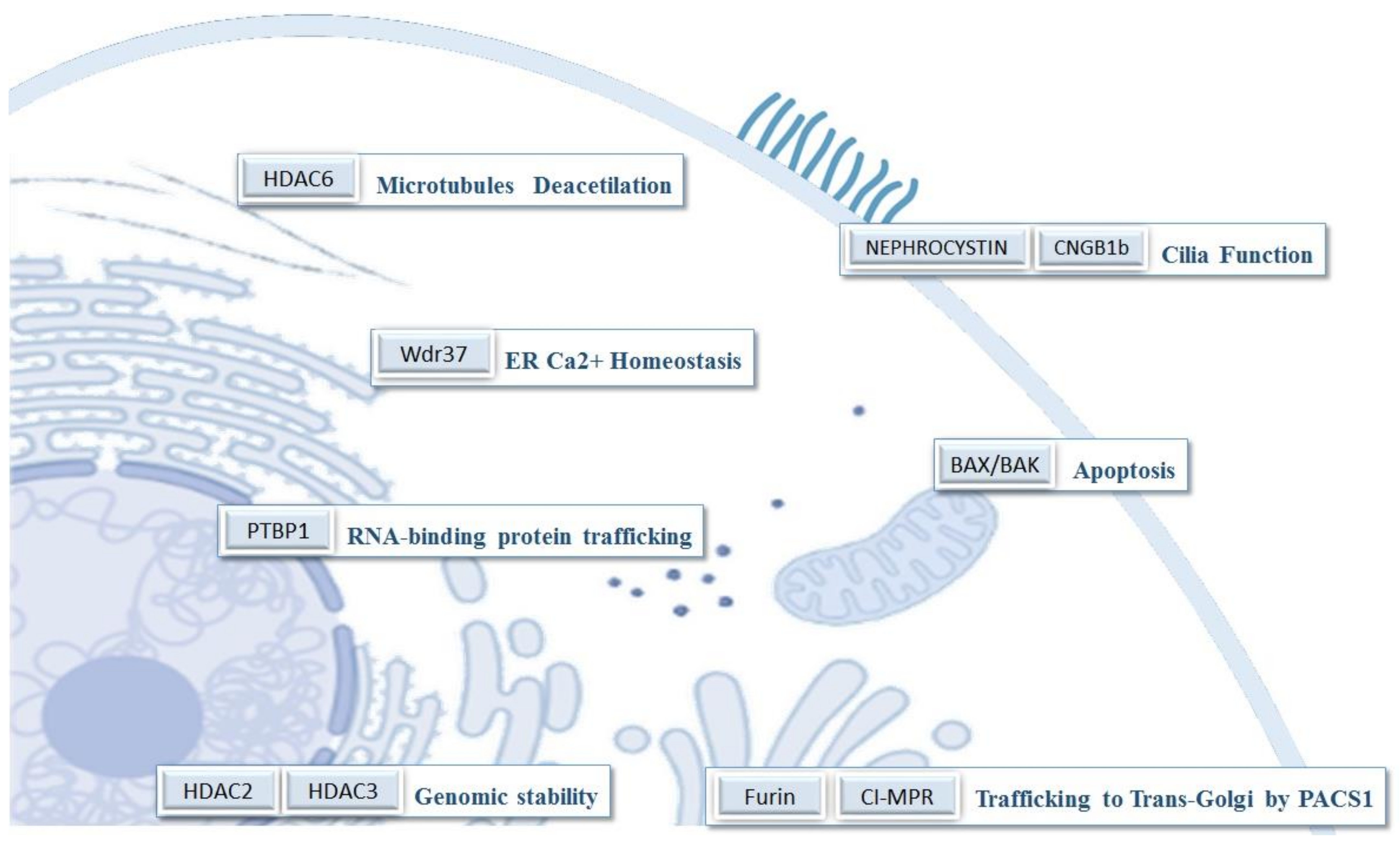
3.4. Functions of PACS-1
4. Relationship between PACS-1 Function and the PACS1-NDD Patient’s Phenotype
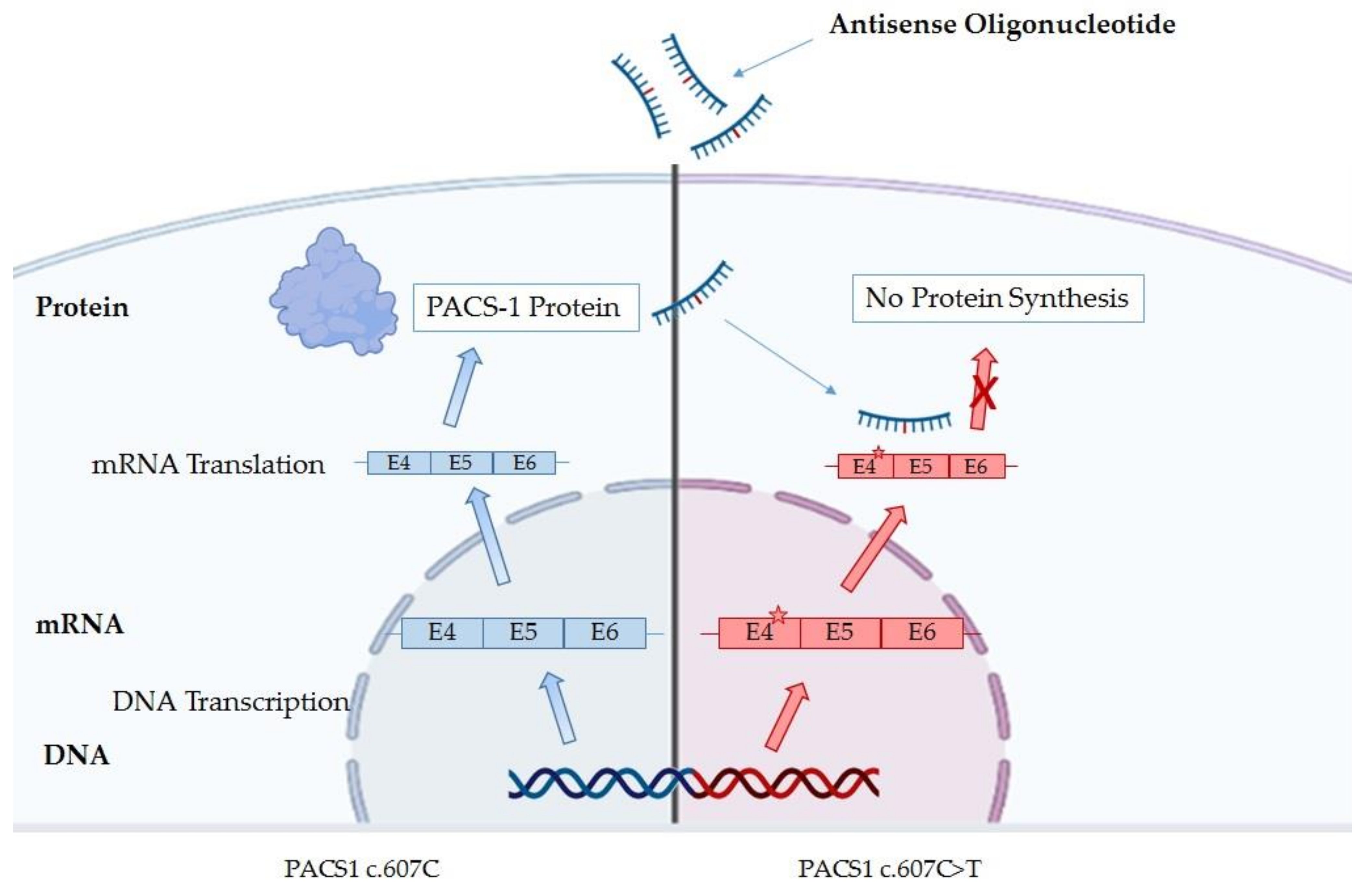
5. Therapy for PACS1 Deficiency Patients
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Schuurs-Hoeijmakers, J.H.; Oh, E.C.; Vissers, L.E.; Swinkels, M.E.; Gilissen, C.; Willemsen, M.A.; Holvoet, M.; Steehouwer, M.; Veltman, J.A.; de Vries, B.B.; et al. Recurrent De Novo Mutations in PACS1 Cause Defective Cranial-Neural-Crest Migration and Define a Recognizable Intellectual-Disability Syndrome. Am. J. Hum. Genet. 2012, 91, 1122–1127. [Google Scholar] [CrossRef] [PubMed]
- García-Cazorla, A.; Oyarzábal, A.; Saudubray, J.-M.; Martinelli, D.; Dionisi-Vici, C. Genetic disorders of cellular trafficking. Trends Genet. 2022, 38, 724–751. [Google Scholar] [CrossRef] [PubMed]
- Schuurs-Hoeijmakers, J.H.M.; Landsverk, M.L.; Foulds, N.; Kukolich, M.K.; Gavrilova, R.H.; Greville-Heygate, S.; Hanson-Kahn, A.; Bernstein, J.A.; Glass, J.; Chitayat, D.; et al. Clinical delineation of thePACS1-related syndrome-Report on 19 patients. Am. J. Med. Genet. Part A 2016, 170, 670–675. [Google Scholar] [CrossRef] [PubMed]
- Tenorio-Castaño, J.; Morte, B.; Nevado, J.; Martinez-Glez, V.; Santos-Simarro, F.; García-Miñaúr, S.; Palomares-Bralo, M.; Pacio-Míguez, M.; Gómez, B.; Arias, P.; et al. Schuurs–Hoeijmakers Syndrome (PACS1 Neurodevelopmental Disorder): Seven Novel Patients and a Review. Genes 2021, 12, 738. [Google Scholar] [CrossRef]
- Miyake, N.; Ozasa, S.; Mabe, H.; Kimura, S.; Shiina, M.; Imagawa, E.; Miyatake, S.; Nakashima, M.; Mizuguchi, T.; Takata, A.; et al. A novel missense mutation affecting the same amino acid as the recurrent PACS1 mutation in Schuurs-Hoeijmakers syndrome. Clin. Genet. 2017, 93, 929–930. [Google Scholar] [CrossRef]
- Liu, Y.; Ding, H.; Yan, T.; Liu, L.; Yu, L.; Huang, Y.; Li, F.; Zeng, Y.; Huang, W.; Zhang, Y.; et al. A Novel Multi-Exon Deletion of PACS1 in a Three-Generation Pedigree: Supplements to PACS1 Neurodevelopmental Disorder Spectrum. Front. Genet. 2021, 12, 690216. [Google Scholar] [CrossRef]
- Youker, R.T.; Shinde, U.; Day, R.; Thomas, G. At the crossroads of homoeostasis and disease: Roles of the PACS proteins in membrane traffic and apoptosis. Biochem. J. 2009, 421, 1–15. [Google Scholar] [CrossRef]
- Wan, L.; Molloy, S.S.; Thomas, L.; Liu, G.; Xiang, Y.; Rybak, S.L.; Thomas, G. PACS-1 Defines a Novel Gene Family of Cytosolic Sorting Proteins Required for trans-Golgi Network Localization. Cell 1998, 94, 205–216. [Google Scholar] [CrossRef]
- Sieburth, D.; Ch’Ng, Q.; Dybbs, M.; Tavazoie, M.; Kennedy, S.; Wang, D.; Dupuy, D.; Rual, J.-F.; Hill, D.E.; Vidal, M.; et al. Systematic analysis of genes required for synapse structure and function. Nature 2005, 436, 510–517. [Google Scholar] [CrossRef]
- Thomas, G.; Aslan, J.; Thomas, L.; Shinde, P.; Shinde, U.; Simmen, T. Caught in the act—Protein adaptation and the expanding roles of the PACS proteins in tissue homeostasis and disease. J. Cell Sci. 2017, 130, 1865–1876. [Google Scholar] [CrossRef]
- Nair-Gill, E.; Bonora, M.; Zhong, X.; Liu, A.; Miranda, A.; Stewart, N.; Ludwig, S.; Russell, J.; Gallagher, T.; Pinton, P.; et al. Calcium flux control by PACS1-Wdr37 promotes lymphocyte quiescence and lymphoproliferative diseases. EMBO J. 2021, 40, e104888. [Google Scholar] [CrossRef]
- Mani, C.; Tripathi, K.; Luan, S.; Clark, D.W.; Andrews, J.F.; Vindigni, A.; Thomas, G.; Palle, K. The multifunctional protein PACS-1 is required for HDAC2- and HDAC3-dependent chromatin maturation and genomic stability. Oncogene 2020, 39, 2583–2596. [Google Scholar] [CrossRef]
- Lusk, L.; Smith, S.; Martin, C.; Taylor, C.; Chung, W. PACS1 Neurodevelopmental Disorder. In GeneReviews®; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. Available online: http://www.ncbi.nlm.nih.gov/books/NBK559434/ (accessed on 23 June 2022).
- Stern, D.; Cho, M.; Chikarmane, R.; Willaert, R.; Retterer, K.; Kendall, F.; Deardorff, M.; Hopkins, S.; Bedoukian, E.; Slavotinek, A.; et al. Association of the missense variant p.Arg203Trp in PACS1 as a cause of intellectual disability and seizures. Clin. Genet. 2017, 92, 221–223. [Google Scholar] [CrossRef]
- Martinez-Monseny, A.; Bolasell, M.; Arjona, C.; Martorell, L.; Yubero, D.; Arsmtrong, J.; Maynou, J.; Fernandez, G.; Salgado, M.D.C.; Palau, F.; et al. Mutation of PACS1: The milder end of the spectrum. Clin. Dysmorphol. 2018, 27, 148–150. [Google Scholar] [CrossRef]
- Pefkianaki, M.; Schneider, A.; Capasso, J.E.; Wasserman, B.N.; Bardakjian, T.; Levin, A.V. Ocular manifestations of PACS1 mutation. J. Am. Assoc. Pediatr. Ophthalmol. Strabismus 2018, 22, 323–325. [Google Scholar] [CrossRef]
- Dutta, A.K. Schuurs-Hoeijmakers syndrome in a patient from India. Am. J. Med. Genet. Part A 2019, 179, 522–524. [Google Scholar] [CrossRef]
- Hoshino, Y.; Enokizono, T.; Imagawa, K.; Tanaka, R.; Suzuki, H.; Fukushima, H.; Arai, J.; Sumazaki, R.; Uehara, T.; Takenouchi, T.; et al. Schuurs-Hoeijmakers syndrome in two patients from Japan. Am. J. Med. Genet. Part A 2018, 179, 341–343. [Google Scholar] [CrossRef]
- Seto, M.T.; Bertoli-Avella, A.M.; Cheung, K.W.; Chan, K.Y.; Yeung, K.S.; Fung, J.L.; Beetz, C.; Bauer, P.; Luk, H.M.; Lo, I.F.; et al. Prenatal and postnatal diagnosis of Schuurs-Hoeijmakers syndrome: Case series and review of the literature. Am. J. Med. Genet. Part A 2020, 185, 384–389. [Google Scholar] [CrossRef]
- Hay, E.; Henderson, R.H.; Mansour, S.; Deshpande, C.; Jones, R.; Nutan, S.; Mankad, K.; Young, R.M.; Moosajee, M.; Genomics England Research Consortium; et al. Expanding the phenotypic spectrum consequent upon de novo WDR37 missense variants. Clin. Genet. 2020, 98, 191–197. [Google Scholar] [CrossRef]
- Kanca, O.; Andrews, J.C.; Lee, P.-T.; Patel, C.; Braddock, S.R.; Slavotinek, A.M.; Cohen, J.S.; Gubbels, C.S.; Aldinger, K.; Williams, J.; et al. De Novo Variants in WDR37 Are Associated with Epilepsy, Colobomas, Dysmorphism, Developmental Delay, Intellectual Disability, and Cerebellar Hypoplasia. Am. J. Hum. Genet. 2019, 105, 413–424. [Google Scholar] [CrossRef]
- Reis, L.M.; Sorokina, E.A.; Thompson, S.; Muheisen, S.; Velinov, M.; Zamora, C.; Aylsworth, A.S.; Semina, E.V. De Novo Missense Variants in WDR37 Cause a Severe Multisystemic Syndrome. Am. J. Hum. Genet. 2019, 105, 425–433. [Google Scholar] [CrossRef]
- Dentici, M.L.; Barresi, S.; Niceta, M.; Ciolfi, A.; Trivisano, M.; Bartuli, A.; Digilio, M.C.; Specchio, N.; Dallapiccola, B.; Tartaglia, M. Expanding the clinical spectrum associated with PACS2 mutations. Clin. Genet. 2019, 95, 525–531. [Google Scholar] [CrossRef]
- Olson, H.E.; Jean-Marçais, N.; Yang, E.; Heron, D.; Tatton-Brown, K.; van der Zwaag, P.A.; Bijlsma, E.K.; Krock, B.L.; Backer, E.; Kamsteeg, E.-J.; et al. A Recurrent De Novo PACS2 Heterozygous Missense Variant Causes Neonatal-Onset Developmental Epileptic Encephalopathy, Facial Dysmorphism, and Cerebellar Dysgenesis. Am. J. Hum. Genet. 2018, 102, 995–1007. [Google Scholar] [CrossRef]
- Terrone, G.; Marchese, F.; Vari, M.S.; Severino, M.; Madia, F.; Amadori, E.; Del Giudice, E.; Romano, A.; Gennaro, E.; Zara, F.; et al. A further contribution to the delineation of epileptic phenotype in PACS2-related syndrome. Seizure 2020, 79, 53–55. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.-L.; Kim, J. Craniofacial Diseases Caused by Defects in Intracellular Trafficking. Genes 2021, 12, 726. [Google Scholar] [CrossRef] [PubMed]
- ClinVar. Available online: https://www.ncbi.nlm.nih.gov/clinvar/?term=PACS1%5Bgene%5D&redir=gene (accessed on 22 August 2022).
- Lim, E.T.; Uddin, M.; De Rubeis, S.; Chan, Y.; Kamumbu, A.S.; Zhang, X.; D’Gama, A.M.; Kim, S.N.; Hill, R.S.; Goldberg, A.P.; et al. Rates, distribution and implications of postzygotic mosaic mutations in autism spectrum disorder. Nat. Neurosci. 2017, 20, 1217–1224. [Google Scholar] [CrossRef] [PubMed]
- DECIPHER. Available online: https://www.deciphergenomics.org/gene/PACS1/overview/clinical-info (accessed on 22 August 2022).
- GTEx. Available online: https://gtexportal.org/home/gene/PACS1 (accessed on 22 August 2022).
- Cardoso-Moreira, M.; Halbert, J.; Valloton, D.; Velten, B.; Chen, C.; Shao, Y.; Liechti, A.; Ascenção, K.; Rummel, C.; Ovchinnikova, S.; et al. Gene expression across mammalian organ development. Nature 2019, 571, 505–509. [Google Scholar] [CrossRef] [PubMed]
- EvoDevo. Available online: https://apps.kaessmannlab.org/evodevoapp/ (accessed on 22 August 2022).
- Ensembl. Available online: https://www.ensembl.org/Homo_sapiens/Gene/Summary?db=core;g=ENSG00000175115;r=11:66070272-66244744 (accessed on 22 August 2022).
- Brasacchio, D.; Alsop, A.E.; Noori, T.; Lufti, M.; Iyer, S.; Simpson, K.J.; Bird, P.I.; Kluck, R.M.; Johnstone, R.W.; Trapani, J.A. Epigenetic control of mitochondrial cell death through PACS1-mediated regulation of BAX/BAK oligomerization. Cell Death Differ. 2017, 24, 961–970. [Google Scholar] [CrossRef] [PubMed]
- Veena, M.S.; Raychaudhuri, S.; Basak, S.K.; Venkatesan, N.; Kumar, P.; Biswas, R.; Chakrabarti, R.; Lu, J.; Su, T.; Gallagher-Jones, M.; et al. Dysregulation of hsa-miR-34a and hsa-miR-449a leads to overexpression of PACS-1 and loss of DNA damage response (DDR) in cervical cancer. J. Biol. Chem. 2020, 295, 17169–17186. [Google Scholar] [CrossRef]
- He, C.; Su, C.; Zhang, W.; Wan, Q. miR-485-5p alleviates Alzheimer’s disease progression by targeting PACS1. Transl. Neurosci. 2021, 12, 335–345. [Google Scholar] [CrossRef]
- Trothen, S.M.; Zang, R.X.; Lurie, A.; Dikeakos, J.D. PACS-1 contains distinct motifs for nuclear-cytoplasmic transport and interacts with the RNA-binding protein PTBP1 in the nucleus and cytosol. FEBS Lett. 2021, 596, 232–248. [Google Scholar] [CrossRef]
- Zheng, W.; Wuyun, Q.; Zhou, X.; Li, Y.; Freddolino, P.L.; Zhang, Y. LOMETS3: Integrating deep learning and profile alignment for advanced protein template recognition and function annotation. Nucleic Acids Res. 2022, 50, W454–W464. [Google Scholar] [CrossRef]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef]
- Schmidt, V.; Sporbert, A.; Rohe, M.; Reimer, T.; Rehm, A.; Andersen, O.M.; Willnow, T.E. SorLA/LR11 Regulates Processing of Amyloid Precursor Protein via Interaction with Adaptors GGA and PACS-1. J. Biol. Chem. 2007, 282, 32956–32964. [Google Scholar] [CrossRef]
- Scott, G.K.; Fei, H.; Thomas, L.; Medigeshi, G.R.; Thomas, G. A PACS-1, GGA3 and CK2 complex regulates CI-MPR trafficking. EMBO J. 2006, 25, 4423–4435. [Google Scholar] [CrossRef]
- Schermer, B.; Höpker, K.; Omran, H.; Ghenoiu, C.; Fliegauf, M.; Fekete, A.; Horvath, J.; Köttgen, M.; Hackl, M.; Zschiedrich, S.; et al. Phosphorylation by casein kinase 2 induces PACS-1 binding of nephrocystin and targeting to cilia. EMBO J. 2005, 24, 4415–4424. [Google Scholar] [CrossRef]
- Jenkins, P.M.; Zhang, L.; Thomas, G.; Martens, J.R. PACS-1 Mediates Phosphorylation-Dependent Ciliary Trafficking of the Cyclic-Nucleotide-Gated Channel in Olfactory Sensory Neurons. J. Neurosci. 2009, 29, 10541–10551. [Google Scholar] [CrossRef]
- Seidel, C.; Schnekenburger, M.; Dicato, M.; Diederich, M. Histone deacetylase 6 in health and disease. Epigenomics 2015, 7, 103–118. [Google Scholar] [CrossRef]
- Thomas, G.; Thomas, L.; Villar-Pazos, S. Methods of Treating PACS1 and Pacs2 Syndromes. WO 2020/018647 A1. Available online: https://patents.google.com/patent/WO2020018647A1/en (accessed on 19 August 2022).
- Brasacchio, D.; Noori, T.; House, C.; Brennan, A.J.; Simpson, K.; Susanto, O.; Bird, P.; Johnstone, R.W.; Trapani, J.A. A functional genomics screen identifies PCAF and ADA3 as regulators of human granzyme B-mediated apoptosis and Bid cleavage. Cell Death Differ. 2014, 21, 748–760. [Google Scholar] [CrossRef]
- Brasacchio, D.; Busuttil, R.A.; Noori, T.; Johnstone, R.; Boussioutas, A.; Trapani, J.A. Down-regulation of a pro-apoptotic pathway regulated by PCAF/ADA3 in early stage gastric cancer. Cell Death Dis. 2018, 9, 442. [Google Scholar] [CrossRef]
- Rylaarsdam, L.; Guemez-Gamboa, A. A gain-of-function recurrent missense variant leads to a GABAergic/glutamatergic imbalance in a forebrain organoid model of PACS1 syndrome. bioRxiv 2022. [Google Scholar] [CrossRef]
- Van Nuland, A.; Reddy, T.; Quassem, F.; Vassalli, J.-D.; Berg, A.T. PACS1-Neurodevelopmental disorder: Clinical features and trial readiness. Orphanet J. Rare Dis. 2021, 16, 386. [Google Scholar] [CrossRef]
- Rylaarsdam, L.; Reddy, T.; Guemez-Gamboa, A. In search of a cure: PACS1 Research Foundation as a model of rare disease therapy development. Trends Genet. 2021, 38, 109–112. [Google Scholar] [CrossRef]
- PACS1 Foundation. Available online: https://www.PACS1foundation.org/research (accessed on 18 July 2022).
- Rinaldi, C.; Wood, M.J.A. Antisense oligonucleotides: The next frontier for treatment of neurological disorders. Nat. Rev. Neurol. 2017, 14, 9–21. [Google Scholar] [CrossRef]
- Mendonça, M.C.P.; Kont, A.; Aburto, M.R.; Cryan, J.F.; O’Driscoll, C.M. Advances in the Design of (Nano)Formulations for Delivery of Antisense Oligonucleotides and Small Interfering RNA: Focus on the Central Nervous System. Mol. Pharm. 2021, 18, 1491–1506. [Google Scholar] [CrossRef]
- Deshaies, R. Prime time for PROTACs. Nat. Chem. Biol. 2015, 11, 634–635. [Google Scholar] [CrossRef]
- Sun, M.; Zhang, H. Par3 and aPKC regulate BACE1 endosome-to-TGN trafficking through PACS1. Neurobiol. Aging 2017, 60, 129–140. [Google Scholar] [CrossRef]
- Hinners, I.; Wendler, F.; Fei, H.; Thomas, L.; Thomas, G.; Tooze, S.A. AP-1 recruitment to VAMP4 is modulated by phosphorylation-dependent binding of PACS-1. EMBO Rep. 2003, 4, 1182–1189. [Google Scholar] [CrossRef]
- Piguet, V.; Wan, L.; Borel, C.; Mangasarian, A.; Demaurex, N.; Thomas, G.; Trono, D. HIV-1 Nef protein binds to the cellular protein PACS-1 to downregulate class I major histocompatibility complexes. Nat. Cell Biol. 2000, 2, 163–167. [Google Scholar] [CrossRef]
- Liu, H.; Hu, P.-W.; Budhiraja, S.; Misra, A.; Couturier, J.; Lloyd, R.E.; Lewis, D.E.; Kimata, J.T.; Rice, A.P. PACS1 is an HIV-1 cofactor that functions in Rev-mediated nuclear export of viral RNA. Virology 2019, 540, 88–96. [Google Scholar] [CrossRef]
- Chiu, Y.-F.; Sugden, B.; Chang, P.-J.; Chen, L.-W.; Lin, Y.-J.; Lan, Y.-C.; Lai, C.-H.; Liou, J.-Y.; Liu, S.-T.; Hung, C.-H. Characterization and Intracellular Trafficking of Epstein-Barr Virus BBLF1, a Protein Involved in Virion Maturation. J. Virol. 2012, 86, 9647–9655. [Google Scholar] [CrossRef] [PubMed]
- Köttgen, M.; Benzing, T.; Simmen, T.; Tauber, R.; Buchholz, B.; Feliciangeli, S.; Huber, T.B.; Schermer, B.; Kramer-Zucker, A.; Höpker, K.; et al. Trafficking of TRPP2 by PACS proteins represents a novel mechanism of ion channel regulation. EMBO J. 2005, 24, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, J.; Meyers, K.R.; Enns, C.A. Transferrin-Directed Internalization and Cycling of Transferrin Receptor 2. Traffic 2009, 10, 1488–1501. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Molloy, S.S.; Thomas, L.; Thomas, G. The PC6B Cytoplasmic Domain Contains Two Acidic Clusters That Direct Sorting to distinct trans-Golgi Network/Endosomal Compartments. Mol. Biol. Cell 2000, 11, 1257–1273. [Google Scholar] [CrossRef][Green Version]




{kind=link}
{kind=link}
{kind=link}
| Clinical Feature | HPO ID * | PACS1-NDD | PACS2 Syndrome | Wdr37 Syndrome | Kabuki Syndrome | CdLS |
|---|---|---|---|---|---|---|
| Neurodevelopmental features | ||||||
| Intellectual disability | 0001249 | Obligate | Very frequent | Obligate | Obligate | Very frequent |
| Autism spectrum disorder | 000729 | Occasional | Occasional | Occasional | Occasional | Frequent |
| Development delay | 0012758 | Obligate | Very frequent | Obligate | Obligate | Occasional |
| Speech delay | 0000750 | Very frequent | Very frequent | Frequent | Occasional | Frequent |
| Hypotonia | 0001252 | Frequent | Frequent | Frequent | Frequent | Occasional |
| Seizures | 0001250 | Frequent | Very frequent | Very frequent | Occasional | Occasional |
| Congenital malformations | ||||||
| Dysmorphic facial features | ||||||
| Full and arched eyebrows | 0002553 | Frequent | Frequent | Frequent | Very frequent | Very frequent |
| Hypertelorism | 0000316 | Frequent | Frequent | Frequent | Occasional | Very rare |
| Downslanting palpebral fissures | 0000494 | Frequent | Frequent | Frequent | Very frequent | Very rare |
| Bulbous nasal tip | 0000414 | Frequent | Very frequent | Obligate | Frequent | Very frequent |
| Downturned mouth | 0002714 | Frequent | Frequent | Frequent | Very rare | Very frequent |
| Thin upper lip | 0000219 | Frequent | Very frequent | Very frequent | Occasional | Very frequent |
| Brain abnormalities | ||||||
| Hypoplasia or partial agenesis of the cerebellar dermis | 0006817 | Frequent | Frequent | Obligate | Occasional | Occasional |
| Ophthalmologic | ||||||
| Coloboma | 0000589 | Occasional | Occasional | Very frequent | Occasional | Very rare |
| Congenital heart anomalies | ||||||
| Atrial or ventricular septal defects | 0001671 | Frequent | Occasional | Frequent | Frequent | Frequent |
| Others | ||||||
| Feeding/GI issues | 0011968 | Occasional | Occasional | Very frequent | Frequent | Frequent |
| Skeletal anomalies | 0000924 | Occasional | Frequent | Frequent | Frequent/ very frequent | Frequent |
| Cryptorchidism | 0000028 | Frequent | Frequent | Very frequent | Occasional | frequent |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arnedo, M.; Ascaso, Á.; Latorre-Pellicer, A.; Lucia-Campos, C.; Gil-Salvador, M.; Ayerza-Casas, A.; Pablo, M.J.; Gómez-Puertas, P.; Ramos, F.J.; Bueno-Lozano, G.; et al. Molecular Basis of the Schuurs–Hoeijmakers Syndrome: What We Know about the Gene and the PACS-1 Protein and Novel Therapeutic Approaches. Int. J. Mol. Sci. 2022, 23, 9649. https://doi.org/10.3390/ijms23179649
Arnedo M, Ascaso Á, Latorre-Pellicer A, Lucia-Campos C, Gil-Salvador M, Ayerza-Casas A, Pablo MJ, Gómez-Puertas P, Ramos FJ, Bueno-Lozano G, et al. Molecular Basis of the Schuurs–Hoeijmakers Syndrome: What We Know about the Gene and the PACS-1 Protein and Novel Therapeutic Approaches. International Journal of Molecular Sciences. 2022; 23(17):9649. https://doi.org/10.3390/ijms23179649
Chicago/Turabian StyleArnedo, María, Ángela Ascaso, Ana Latorre-Pellicer, Cristina Lucia-Campos, Marta Gil-Salvador, Ariadna Ayerza-Casas, María Jesús Pablo, Paulino Gómez-Puertas, Feliciano J. Ramos, Gloria Bueno-Lozano, and et al. 2022. "Molecular Basis of the Schuurs–Hoeijmakers Syndrome: What We Know about the Gene and the PACS-1 Protein and Novel Therapeutic Approaches" International Journal of Molecular Sciences 23, no. 17: 9649. https://doi.org/10.3390/ijms23179649
APA StyleArnedo, M., Ascaso, Á., Latorre-Pellicer, A., Lucia-Campos, C., Gil-Salvador, M., Ayerza-Casas, A., Pablo, M. J., Gómez-Puertas, P., Ramos, F. J., Bueno-Lozano, G., Pié, J., & Puisac, B. (2022). Molecular Basis of the Schuurs–Hoeijmakers Syndrome: What We Know about the Gene and the PACS-1 Protein and Novel Therapeutic Approaches. International Journal of Molecular Sciences, 23(17), 9649. https://doi.org/10.3390/ijms23179649