
Bioenergetic and Metabolic Impairments in Induced Pluripotent Stem Cell-Derived Cardiomyocytes Generated from Duchenne Muscular Dystrophy Patients
 , , , ,
, , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
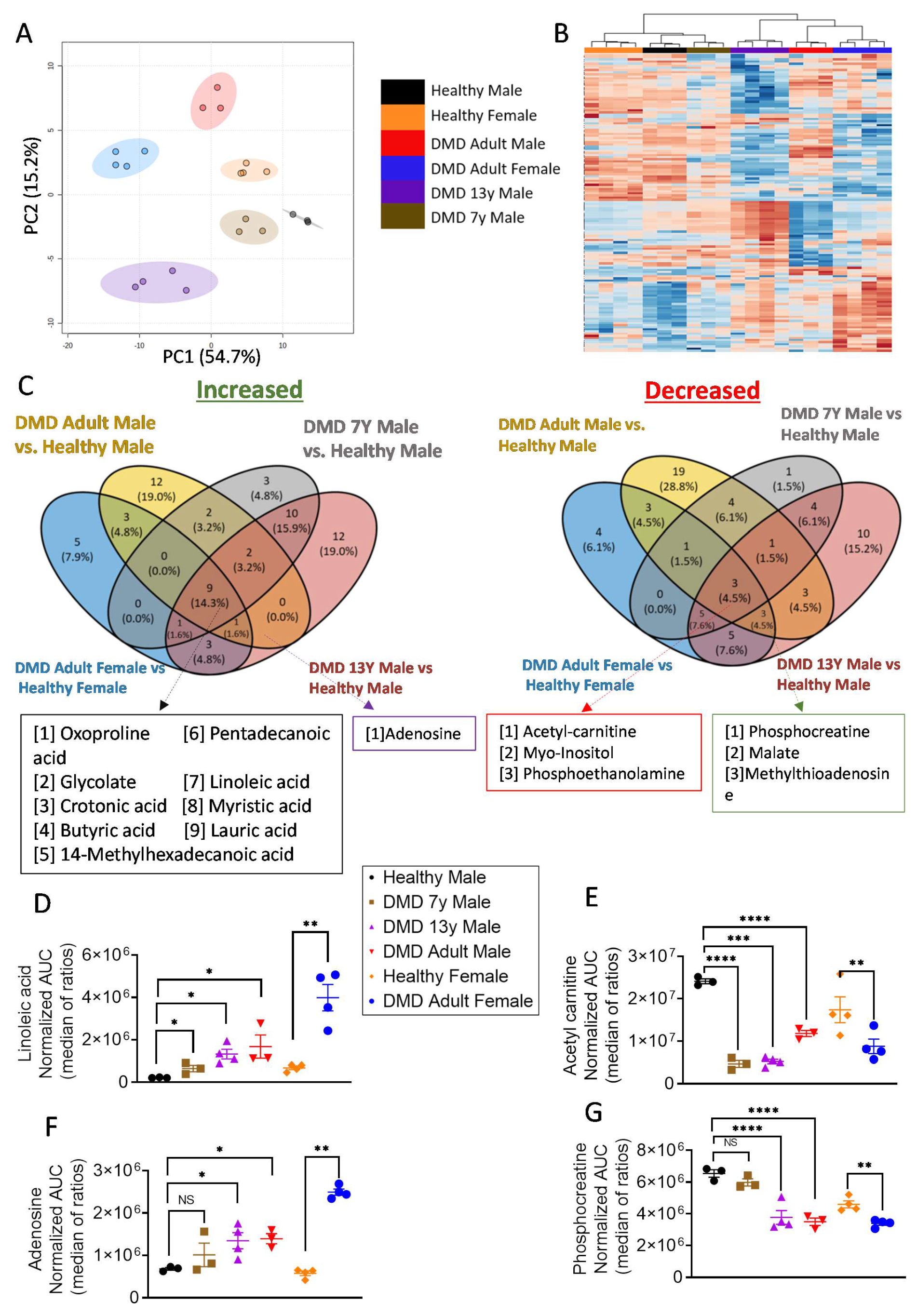
2.1. Metabolic Profiling Revealed a Bioenergetic Impairment of DMD iPSC-CMs
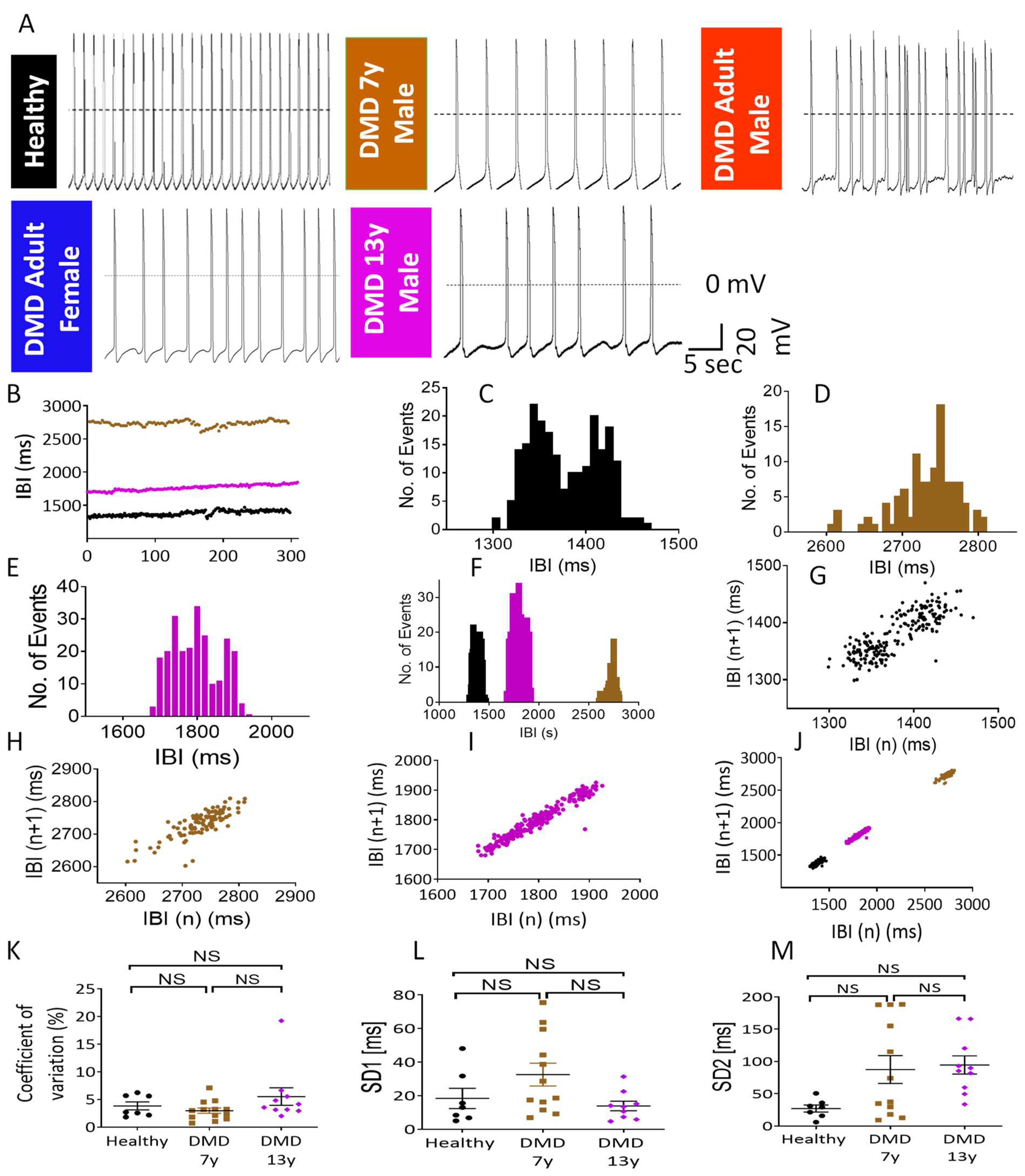
2.2. The Metabolic/Energy Status of the iPSC-CMs Correlates with Their Electrophysiological Characteristics
2.3. Stable-Isotope Glucose Tracing Reveals Decreased Oxidative Status in DMD Cardiomyocytes
2.4. DMD-Derived iPSC-CMs Demonstrate Oxidative Phosphorylation Deficiency
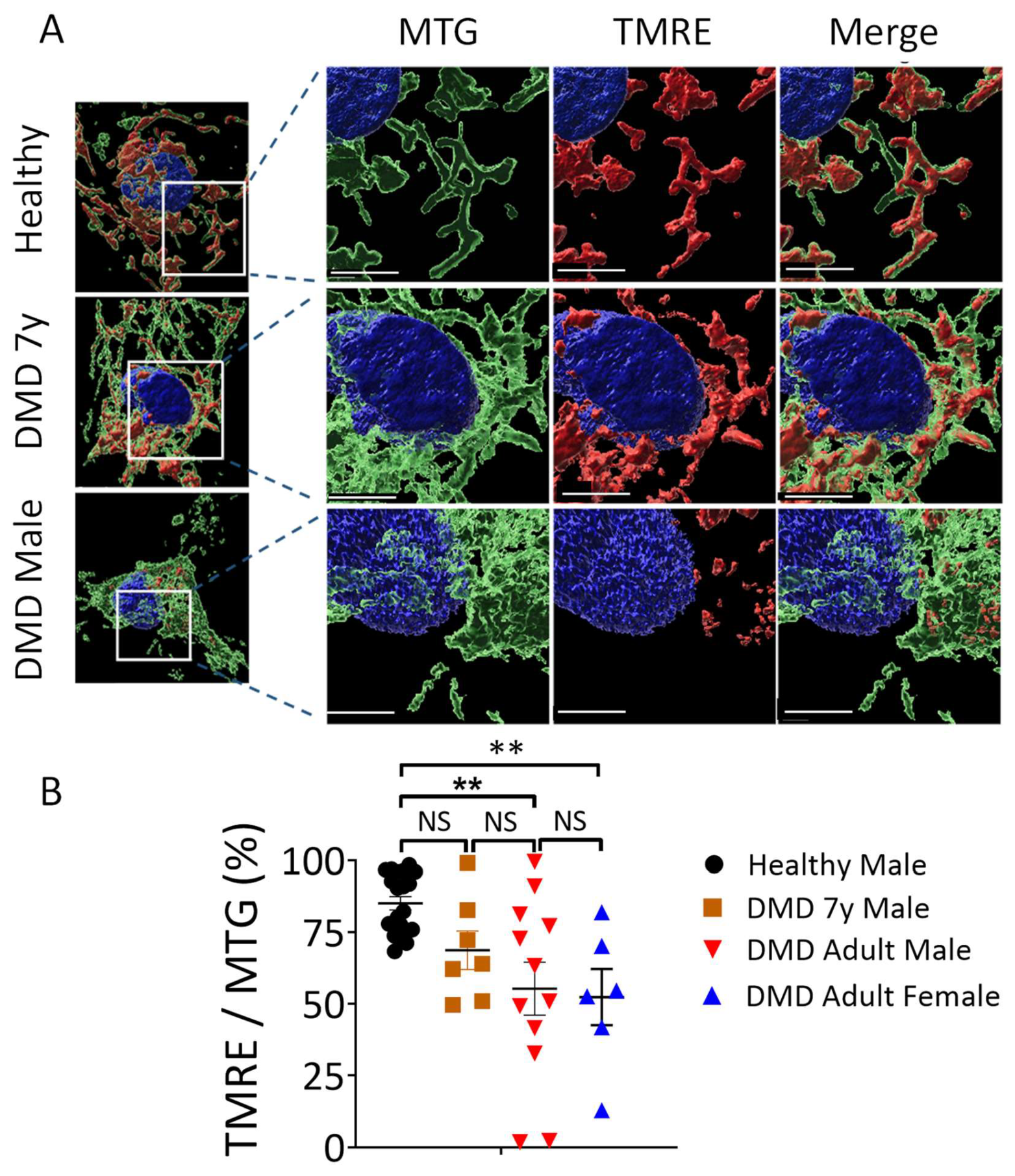
2.5. Mitochondria Structural and Functional Alterations in Energy-Depleted DMD iPSC-CMs
3. Discussion
3.1. Decreased PCr in DMD CMs
3.2. Mitochondrial Dysfunction in DMD CMs
3.3. Changes in Mitochondrial Content and Morphology in DMD CMs
3.4. The Link between Dystrophin Mutation and Mitochondrial Structural and Functional Aberrations
3.5. The Arrhythmias in DMD Cardiomyocytes
3.6. Do Fibroblasts “Remember” the Age of the DMD Patient?
4. Materials and Methods
4.1. The Experimental Groups
4.2. Experimental Protocols
4.3. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mavrogeni, S.; Markousis-Mavrogenis, G.; Papavasiliou, A.; Kolovou, G. Cardiac involvement in Duchenne and Becker muscular dystrophy. World J. Cardiol. 2015, 7, 410–414. [Google Scholar] [CrossRef]
- Bushby, K.; Finkel, R.; Birnkrant, D.J.; Case, L.E.; Clemens, P.R.; Cripe, L.; Kaul, A.; Kinnett, K.; McDonald, C.; Pandya, S.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: Implementation of multidisciplinary care. Lancet Neurol. 2010, 9, 177–189. [Google Scholar] [CrossRef]
- Eagle, M.; Baudouin, S.V.; Chandler, C.; Giddings, D.R.; Bollock, R.; Bushby, K. Survival in Duchenne muscular dystrophy: Improvements in life expectancy since 1967 and the impact of home nuctural ventilation. Neuromuscul. Disord. 2002, 12, 926–929. [Google Scholar] [CrossRef]
- McNally, E.M.; Kaltman, J.R.; Benson, D.W.; Canter, C.E.; Cripe, L.H.; Duan, D.; Finder, J.D.; Groh, W.J.; Hoffman, E.P.; Judge, D.P.; et al. Contemporary cardiac issues in Duchenne muscular dystrophy. Circulation 2015, 131, 1590–1598. [Google Scholar] [CrossRef]
- Allen, D.G.; Whitehead, N.P.; Froehner, S.C. Absence of Dystrophin Disrupts Skeletal Muscle Signaling: Roles of Ca 2+, Reactive Oxygen Species, and Nitric Oxide in the Development of Muscular Dystrophy. Physiol. Rev. 2016, 96, 253–305. [Google Scholar] [CrossRef]
- D’Amario, D.; Amodeo, A.; Adorisio, R.; Tiziano, F.D.; Leone, A.M.; Perri, G.; Bruno, P.; Massetti, M.; Ferlini, A.; Pane, M.; et al. A current approach to heart failure in Duchenne muscular dystrophy. Heart 2017, 103, 1770–1779. [Google Scholar] [CrossRef]
- Rajdev, A.; Groh, W.J. Arrhythmias in the muscular dystrophies. Card. Electrophysiol. Clin. 2015, 7, 303–308. [Google Scholar] [CrossRef]
- Finsterer, J.; Stöllberger, C. Cardiac involvement in primary myopathies. Cardiology 2000, 1, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Finsterer, J.; Stollberger, C. The heart in human dystrophinopathies. Cardiology 2003, 99, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Kamdar, F.; Garry, D.J. Dystrophin-Deficient Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 67, 2533–2546. [Google Scholar] [CrossRef] [PubMed]
- Ware, S.M. Genetics of paediatric cardiomyopathies. Curr. Opin. Pediatr. 2017, 29, 534–540. [Google Scholar] [CrossRef]
- Omairi, S.; Hau, K.L.; Collins-Hooper, H.; Scott, C.; Vaiyapuri, S.; Torelli, S.; Montanaro, F.; Matsakas, A.; Patel, K. Regulation of the dystrophin-associated glycoprotein complex composition by the metabolic properties of muscle fibres. Sci. Rep. 2019, 9, 2770. [Google Scholar] [CrossRef]
- Timpani, C.A.; Hayes, A.; Rybalka, E. Revisiting the dystrophin-ATP connection: How half a century of research still implicates mitochondrial dysfunction in Duchenne muscular dystrophy aetiology. Med. Hypotheses 2015, 85, 1021–1033. [Google Scholar] [CrossRef]
- Gumerson, J.D.; Michele, D.E. The dystrophin-glycoprotein complex in the prevention of muscle damage. J. Biomed. Biotechnol. 2011, 2011, 210797. [Google Scholar] [CrossRef]
- Hughes, M.C.; Ramos, S.V.; Turnbull, P.C.; Rebalka, I.A.; Cao, A.; Monaco, C.M.F.; Varah, N.E.; Edgett, B.A.; Huber, J.S.; Tadi, P.; et al. Early myopathy in Duchenne muscular dystrophy is associated with elevated mitochondrial H2O2 emission during impaired oxidative phosphorylation. J. Cachexia Sarcopenia Muscle 2019, 10, 643–661. [Google Scholar] [CrossRef]
- Esposito, G.; Carsana, A. Clinical Medicine Metabolic Alterations in Cardiomyocytes of Patients with Duchenne and Becker Muscular Dystrophies. J. Clin. Med. 2019, 8, 2151. [Google Scholar] [CrossRef]
- Groh, W.J. Arrhythmias in the muscular dystrophies. Heart Rhythm 2012, 9, 1890–1895. [Google Scholar] [CrossRef]
- Yilmaz, A.; Sechtem, U. Cardiac involvement in muscular dystrophy: Advances in diagnosis and therapy. Heart 2012, 98, 420–429. [Google Scholar] [CrossRef]
- Rybalka, E.; Timpani, C.A.; Cooke, M.B.; Williams, A.D.; Hayes, A. Defects in mitochondrial ATP synthesis in dystrophin-deficient Mdx skeletal muscles may be caused by complex I insufficiency. PLoS ONE 2014, 9, 1–16. [Google Scholar]
- Gambardella, J.; Trimarco, B.; Iaccarino, G.; Santulli, G. New Insights in Cardiac Calcium Handling and Excitation-Contraction Coupling. Adv. Exp. Med. Biol. 2018, 1067, 373. [Google Scholar]
- Gambardella, J.; Sorriento, D.; Ciccarelli, M.; Del Giudice, C.; Fiordelisi, A.; Napolitano, L.; Trimarco, B.; Iaccarino, G.; Santulli, G. Functional Role of Mitochondria in Arrhythmogenesis. Adv. Exp. Med. Biol. 2017, 982, 191. [Google Scholar] [PubMed]
- Lindsay, A.; Chamberlain, C.M.; Witthuhn, B.A.; Lowe, D.A.l.; Ervast, J.M. Dystrophinopathy-associated dysfunction of Krebs cycle metabolism. Hum. Mol. Genet. 2019, 28, 942–951. [Google Scholar] [CrossRef] [PubMed]
- Ramos, S.V.; Hughes, M.C.; Delfinis, L.J.; Bellissimo, C.A.; Perry, C.G.R. Mitochondrial bioenergetic dysfunction in the D2.mdx model of Duchenne muscular dystrophy is associated with microtubule disorganization in skeletal muscle. PLoS ONE 2020, 15, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.C.Y.; Ong, S.G.; LaGory, E.L.; Kraft, P.E.; Giaccia, A.J.; Wu, J.C.; Blau, H.M. Telomere shortening and metabolic compromise underlie dystrophic cardiomyopathy. Proc. Natl. Acad. Sci. USA 2016, 113, 13120–13125. [Google Scholar] [CrossRef]
- Stapleton, D.I.; Lau, X.; Flores, M.; Trieu, J.; Gehrig, S.M.; Chee, A.; Naim, T.; Lynch, G.S.; Koopman, R. Dysfunctional muscle and liver glycogen metabolism in mdx dystrophic mice. PLoS ONE 2014, 9, 1–9. [Google Scholar]
- Strakova, J.; Kamdar, F.; Kulhanek, D.; Razzoli, M.; Garry, D.J.; Ervasti, J.M.; Bartolomucci, A.; Townsend, D. Integrative effects of dystrophin loss on metabolic function of the mdx mouse. Sci. Rep. 2018, 8, 13624. [Google Scholar] [CrossRef]
- McConell, G.K.; Rattigan, S.; Lee-Young, R.S.; Wadley, G.D.; Merry, T.L. Skeletal muscle nitric oxide signaling and exercise: A focus on glucose metabolism. AJP Endocrinol. Metab. 2012, 303, 301–307. [Google Scholar] [CrossRef]
- Cole, M.A.; Rafael, J.A.; Taylor, D.J.; Lodi, R.; Davies, K.E.; Styles, P.A. Quantitative study of bioenergetics in skeletal muscle lacking utrophin and dystrophin. Neuromuscul. Disord. 2002, 12, 247–257. [Google Scholar] [CrossRef]
- Eisen, B.; Ben Jehuda, R.; Cuttitta, A.J.; Mekies, L.N.; Shemer, Y.; Baskin, P.; Reiter, I.; Willi, L.; Freimark, D.; Gherghiceanu, M.; et al. Electrophysiological abnormalities in induced pluripotent stem cell-derived cardiomyocytes generated from Duchenne muscular dystrophy patients. J. Cell. Mol. Med. 2019, 23, 2125–2135. [Google Scholar] [CrossRef]
- Sala, L.; Bellin, M.; Mummery, C.L. Integrating cardiomyocytes from human pluripotent stem cells in safety pharmacology: Has the time come? Br. J. Pharmacol. 2017, 174, 3749–3765. [Google Scholar] [CrossRef]
- Neal, R.C.; Ferdinand, K.C.; Yčas, J.; Miller, E. Relationship of ethnic origin, gender, and age to blood creatine kinase levels. Am. J. Med. 2009, 122, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Ben-Ari, M.; Naor, S.; Zeevi-Levin, N.; Schick, R.; Ben Jehuda, R.; Reiter, I.; Raveh, A.; Grijnevitch, I.; Barak, O.; Rosen, M.R.; et al. Developmental changes in electrophysiological characteristics of human-induced pluripotent stem cell-derived cardiomyocytes. Heart Rhythm 2016, 13, 2379–2387. [Google Scholar] [CrossRef] [PubMed]
- Ben Jehuda, R.; Eisen, B.; Shemer, Y.; Mekies, L.N.; Szantai, A.; Reiter, I.; Cui, H.; Guan, K.; Haron-Khun, S.; Freimark, D.; et al. CRISPR correction of the PRKAG2 gene mutation in the patient’s iPSC-derived cardiomyocytes eliminates the electrophysiological and structural abnormalities. Heart Rhythm 2017, 15, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Piquereau, J.; Caffin, F.; Novotova, M.; Lemaire, C.; Veksler, V.; Garnier, A.; Ventura-Clapier, R.; Joubert, F. Mitochondrial dynamics in the adult cardiomyocytes: Which roles for a highly specialized cell? Front. Physiol. 2013, 4, 102. [Google Scholar] [CrossRef]
- Burelle, Y.; Khairallah, M.; Ascah, A.; Allen, B.G.; Deschepper, C.F.; Petrof, B.J.; Des Rosiers, C. Alterations in mitochondrial function as a harbinger of cardiomyopathy: Lessons from the dystrophic heart. J. Mol. Cell. Cardiol. 2010, 48, 310–321. [Google Scholar] [CrossRef]
- Kushmerick, M.J. Skeletal muscle: A paradigm for testing principles of bioenergetics. J. Bioenerg. Biomembr. 1995, 27, 555–569. [Google Scholar] [CrossRef]
- Kushmerick, M.J. Patterns in mammalian muscle energetics. J. Exp. Biol. 1985, 115, 165–177. [Google Scholar] [CrossRef]
- Kushmerick, M.J. Energetics studies of muscles of different types. Basic Res. Cardiol. 1987, 82 (Suppl. 2), 17–30. [Google Scholar]
- Pulido, S.M.; Passaquin, A.C.; Leijendekker, W.J.; Challet, C.; Wallimann, T.; Rüegg, U.T. Creatine supplementation improves intracellular calcium handling and survival in mdx skeletal muscle cells. FEBS Lett. 1998, 439, 357–362. [Google Scholar] [CrossRef]
- Zhang, W.; ten Hove, M.; Schneider, J.E.; Stuckey, D.J.; Sebag-Montefiore, L.; Bia, B.L.; Radda, G.K.; Davies, K.E.; Neubauer, S.; Clarke, K. Abnormal cardiac morphology, function and energy metabolism in the dystrophic mdx mouse: An MRI and MRS study. J. Mol. Cell. Cardiol. 2008, 45, 754–760. [Google Scholar] [CrossRef]
- Moore, T.M.; Lin, A.J.; Strumwasser, A.R.; Strumwasser, A.R.; Cory, K.; Whitney, K.; Ho, T.; Ho, T.; Lee, J.L.; Rucker, D.H.; et al. Mitochondrial Dysfunction Is an Early Consequence of Partial or Complete Dystrophin Loss in mdx Mice. Front. Physiol. 2020, 11, 690. [Google Scholar] [CrossRef]
- Kang, C.; Badr, M.A.; Kyrychenko, V.; Eskelinen, E.L.; Shirokova, N. Deficit in PINK1/PARKIN-mediated mitochondrial autophagy at late stages of dystrophic cardiomyopathy. Cardiovasc. Res. 2018, 114, 90–102. [Google Scholar] [CrossRef]
- Onopiuk, M.; Brutkowski, W.; Wierzbicka, K.; Wojciechowska, S.; Szczepanowska, J.; Fronk, J.; Lochmüller, H.; Górecki, D.C.; Zabłocki, K. Mutation in dystrophin-encoding gene affects energy metabolism in mouse myoblasts. Biochem. Biophys. Res. Commun. 2009, 386, 463–466. [Google Scholar] [CrossRef]
- Giacomotto, J.; Brouilly, N.; Walter, L.; Mariol, M.C.; Berger, J.; Ségalat, L.; Becker, T.S.; Currie, P.D.; Gieseler, K. Chemical genetics unveils a key role of mitochondrial dynamics, cytochrome c release and IP3R activity in muscular dystrophy. Hum. Mol. Genet. 2013, 22, 4562–4578. [Google Scholar] [CrossRef] [Green Version]
- Dubinin, M.V.; Starinets, V.S.; Talanov, E.Y.; Mikheeva, I.B.; Belosludtseva, N.V.; Serov, D.A.; Tenkov, K.S.; Belosludtseva, E.V.; Belosludtsev, K.N. Effect of the non-immunosuppressive mpt pore inhibitor alisporivir on the functioning of heart mitochondria in dystrophin-deficient mdx mice. Biomedicines 2021, 9, 1232. [Google Scholar] [CrossRef]
- Mareedu, S.; Million, E.D.; Duan, D.; Babu, G.J. Abnormal calcium handling in Duchenne muscular dystrophy: Mechanisms and potential therapies. Front. Physiol. 2021, 12, 647010. [Google Scholar] [CrossRef]
- Zabłocka, B.; Górecki, D.C.; Zabłocki, K. Disrupted calcium homeostasis in Duchenne muscular dystrophy: A common mechanism behind diverse consequences. Int. J. Mol. Sci. 2021, 22, 11040. [Google Scholar] [CrossRef]
- Reid, A.L.; Alexander, M.S. The interplay of mitophagy and inflammation in Duchenne muscular dystrophy. Life 2021, 11, 648. [Google Scholar] [CrossRef]
- Bellissimo, C.A.; Garibotti, M.C.; Perry, C.G.R. Mitochondrial stress responses in Duchenne muscular dystrophy: Metabolic dysfunction or adaptive reprogramming? Am. J. Physiol. Cell Physiol. 2022, 3, 1–37. [Google Scholar] [CrossRef]
- Kamdar, F.; Das, S.; Gong, W.; Kamdar, A.; Meyers, T.A.; Shah, P.; Ervasti, J.M.; Townsend, D.; Kamp, T.J.; Wu, J.C.; et al. Stem Cell–derived cardiomyocytes and beta-adrenergic receptor blockade in Duchenne Muscular Dystrophy cardiomyopathy. J. Am. Coll. Cardiol. 2020, 75, 1159–1174. [Google Scholar] [CrossRef]
- Dubinin, M.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Mikheeva, I.B.; Sharapov, M.G.; Belosludtsev, K.N. Duchenne muscular dystrophy is associated with the inhibition of calcium uniport in mitochondria and an increased sensitivity of the organelles to the calcium-induced permeability transition. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165674. [Google Scholar] [CrossRef] [PubMed]
- Law, M.L.; Cohen, H.; Martin, A.A.; Angulski, A.B.B.; Metzger, J.M. Dysregulation of calcium handling in duchenne muscular dystrophy-associated dilated cardiomyopathy: Mechanisms and experimental therapeutic strategies. J. Clin. Med. 2020, 9, 520. [Google Scholar] [CrossRef] [PubMed]
- Farini, A.; Sitzia, C.; Cassinelli, L.; Colleoni, F.; Parolini, D.; Giovanella, U.; Maciotta, S.; Colombo, A.; Meregalli, M.; Torrente, Y. Inositol 1,4,5-trisphosphate (IP3)-dependent Ca2+ signaling mediates delayed myogenesis in Duchenne muscular dystrophy fetal muscle. Development 2016, 143, 658–669. [Google Scholar] [CrossRef] [PubMed]
- Mekies, L.N.; Regev, D.; Eisen, B.; Fernandez-Gracia, J.; Baskin, P.; Ben Jehuda, R.; Shulman, R.; Reiter, I.; Palty, R.; Arad, M.; et al. Depressed β-adrenergic inotropic responsiveness and intracellular calcium handling abnormalities in Duchenne Muscular Dystrophy patients’ induced pluripotent stem derived cardiomyocytes. J. Cell Mol. Med. 2021, 2021, 3922–3934. [Google Scholar] [CrossRef]
- Fauconnier, J.; Thireau, J.; Reiken, S.; Cassan, C.; Richard, S.; Matecki, S.; Marks, A.R.; Lacampagne, A. Leaky RyR2 trigger ventricular arrhythmias in Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 2010, 107, 1559–1564. [Google Scholar] [CrossRef]
- Dubinin, M.V.; Talanov, E.Y.; Tenkov, K.S.; Talanov, E.Y.; Starinets, V.S.; Tenkov, K.S.; Zakharova, N.M.; Belosludtseva, N.V. Transport of calcium and calcium-dependent permeability transition in heart mitochondria in the early stages of Duchenne muscular dystrophy. Biochim. Biophys. Acta Bioenerg. 2020, 1861, 1–11. [Google Scholar] [CrossRef]
- Ascah, A.; Khairallah, M.; Daussin, F.; Bourcier-Lucas, C.; Godin, R.; Allen, B.G.; Petrof, B.J.; Des Rosiers, C.; Burelle, Y. Stress-induced opening of the permeability transition pore in the dystrophin-deficient heart is attenuated by acute treatment with sildenafil. Am. J. Physiol. Hearth Circ. Physiol. 2011, 300, 144–153. [Google Scholar] [CrossRef]
- Eisen, B.; Ben Jehuda, R.; Cuttitta, A.J.; Mekies, L.N.; Reiter, I.; Ramchandren, S.; Arad, M.; Michele, D.E.; Binah, O. Generation of Duchenne muscular dystrophy patient-specific induced pluripotent stem cell line lacking exons 45–50 of the dystrophin gene (IITi001-A). Stem Cell Res. 2018, 29, 111–114. [Google Scholar] [CrossRef]
- Yehezkel, S.; Rebibo-Sabbah, A.; Segev, Y.; Tzukerman, M.; Shaked, R.; Huber, I.; Gepstein, L.; Skorecki, K.; Selig, S. Reprogramming of telomeric regions during the generation of human induced pluripotent stem cells and subsequent differentiation into fibroblast-like derivatives. Epigenetics 2011, 6, 63–75. [Google Scholar] [CrossRef]
- Novak, A.; Barad, L.; Lorber, A.; Gherghiceanu, M.; Reiter, I.; Eisen, B.; Eldor, L.; Itskovitz-Eldor, J.; Eldar, M.; Arad, M.; et al. Functional abnormalities in iPSC-derived cardiomyocytes generated from CPVT1 and CPVT2 patients carrying ryanodine or calsequestrin mutations. J. Cell. Mol. Med. 2015, 19, 2006–2018. [Google Scholar] [CrossRef]
- Jimenez-Vazquez, E.N.; Arad, M.; Macías, Á.; Vera-Pedrosa, L.; Cruz-Uréndez, F.M.; Cuttitta, A.J.; Monteiro Da Rocha, A.; Herron, T.J.; Ponce-Balbuena, D.; Guerrero-Serna, G.; et al. SNTA1 Gene rescues ion channel function in cardiomyocytes derived from induced Pluripotent Stem Cells reprogrammed from Muscular Dystrophy patients with arrhythmias. eLife 2022, 11, 1–30. [Google Scholar] [CrossRef]
- Streckfuss-Bömeke, K.; Wolf, F.; Azizian, A.; Stauske, M.; Tiburcy, M.; Wagner, S.; Hübscher, D.; Dressel, R.; Chen, S.; Jende, J.; et al. Comparative study of human-induced pluripotent stem cells derived from bone marrow cells, hair keratinocytes, and skin fibroblasts. Eur. Heart J. 2013, 34, 2618–2629. [Google Scholar] [CrossRef]
- Novak, A.; Barad, L.; Zeevi-Levin, N.; Shick, R.; Shtrichman, R.; Lorber, A.; Itskovitz-Eldor, J.; Binah, O. Cardiomyocytes generated from CPVT D307H patients are arrhythmogenic in response to betha-adrenergic stimulation. J. Cell Mol. Med. 2012, 16, 468–482. [Google Scholar] [CrossRef]
- MacKay, G.M.; Zheng, L.; Broek, N.J.F.; Van Den Gottlieb, E. Analysis of cell metabolism using LC-MS and isotope tracers. Methods Enzymol. 2015, 561, 171–196. [Google Scholar]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Pietzke, M.; Vazquez, A. Metabolite AutoPlotter—An application to process and visualise metabolite data in the web browser. Cancer Metab. 2020, 8, 1–11. [Google Scholar] [CrossRef]
- Ben-Ari, M.; Schick, R.; Barad, L.; Novak, A.; Ben-Ari, E.; Lorber, A.; Itskovitz-Eldor, J.; Rosen, M.R.; Weissman, A.; Binah, O. From beat rate variability in induced pluripotent stem cell-derived pacemaker cells to heart rate variability in human subjects. Heart Rhythm. 2014, 11, 1808–1818. [Google Scholar] [CrossRef]
- Mandel, Y.; Weissman, A.; Schick, R.; Barad, L.; Novak, A.; Meiry, G.; Goldberg, S.; Lorber, A.; Rosen, M.R.; Itskovitz-Eldor, J.; et al. Human embryonic and induced pluripotent stem cell-derived cardiomyocytes exhibit beat rate variability and power-law behavior. Circulation 2012, 125, 883–893. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Willi, L.; Abramovich, I.; Fernandez-Garcia, J.; Agranovich, B.; Shulman, M.; Milman, H.; Baskin, P.; Eisen, B.; Michele, D.E.; Arad, M.; et al. Bioenergetic and Metabolic Impairments in Induced Pluripotent Stem Cell-Derived Cardiomyocytes Generated from Duchenne Muscular Dystrophy Patients. Int. J. Mol. Sci. 2022, 23, 9808. https://doi.org/10.3390/ijms23179808
Willi L, Abramovich I, Fernandez-Garcia J, Agranovich B, Shulman M, Milman H, Baskin P, Eisen B, Michele DE, Arad M, et al. Bioenergetic and Metabolic Impairments in Induced Pluripotent Stem Cell-Derived Cardiomyocytes Generated from Duchenne Muscular Dystrophy Patients. International Journal of Molecular Sciences. 2022; 23(17):9808. https://doi.org/10.3390/ijms23179808
Chicago/Turabian StyleWilli, Lubna, Ifat Abramovich, Jonatan Fernandez-Garcia, Bella Agranovich, Margarita Shulman, Helena Milman, Polina Baskin, Binyamin Eisen, Daniel E. Michele, Michael Arad, and et al. 2022. "Bioenergetic and Metabolic Impairments in Induced Pluripotent Stem Cell-Derived Cardiomyocytes Generated from Duchenne Muscular Dystrophy Patients" International Journal of Molecular Sciences 23, no. 17: 9808. https://doi.org/10.3390/ijms23179808
APA StyleWilli, L., Abramovich, I., Fernandez-Garcia, J., Agranovich, B., Shulman, M., Milman, H., Baskin, P., Eisen, B., Michele, D. E., Arad, M., Binah, O., & Gottlieb, E. (2022). Bioenergetic and Metabolic Impairments in Induced Pluripotent Stem Cell-Derived Cardiomyocytes Generated from Duchenne Muscular Dystrophy Patients. International Journal of Molecular Sciences, 23(17), 9808. https://doi.org/10.3390/ijms23179808