
A New in Silico Antibody Similarity Measure Both Identifies Large Sets of Epitope Binders with Distinct CDRs and Accurately Predicts Off-Target Reactivity

 , ,
, ,  ,
,
Abstract
1. Introduction
2. Results
2.1. Antibody Similarity Measurement
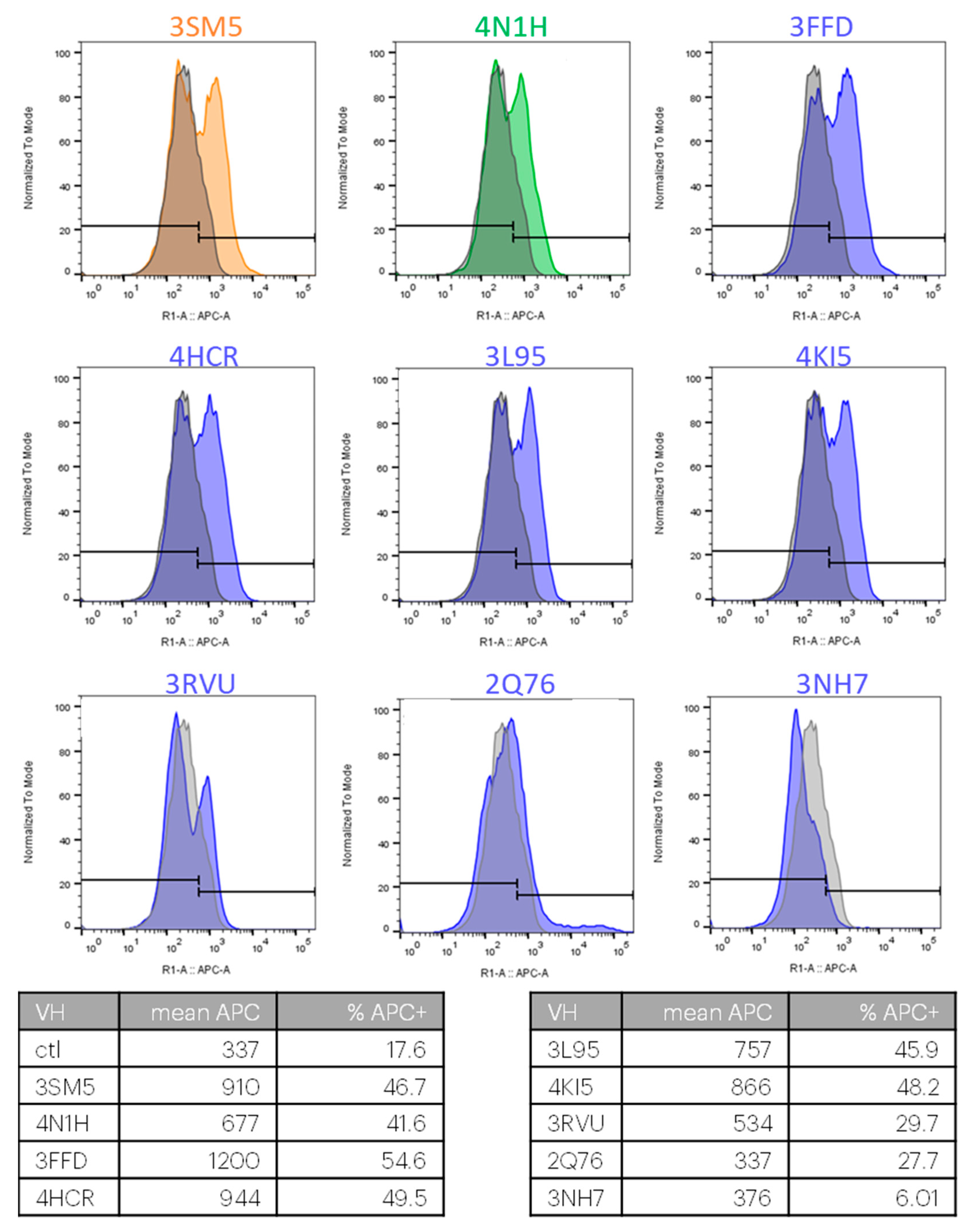
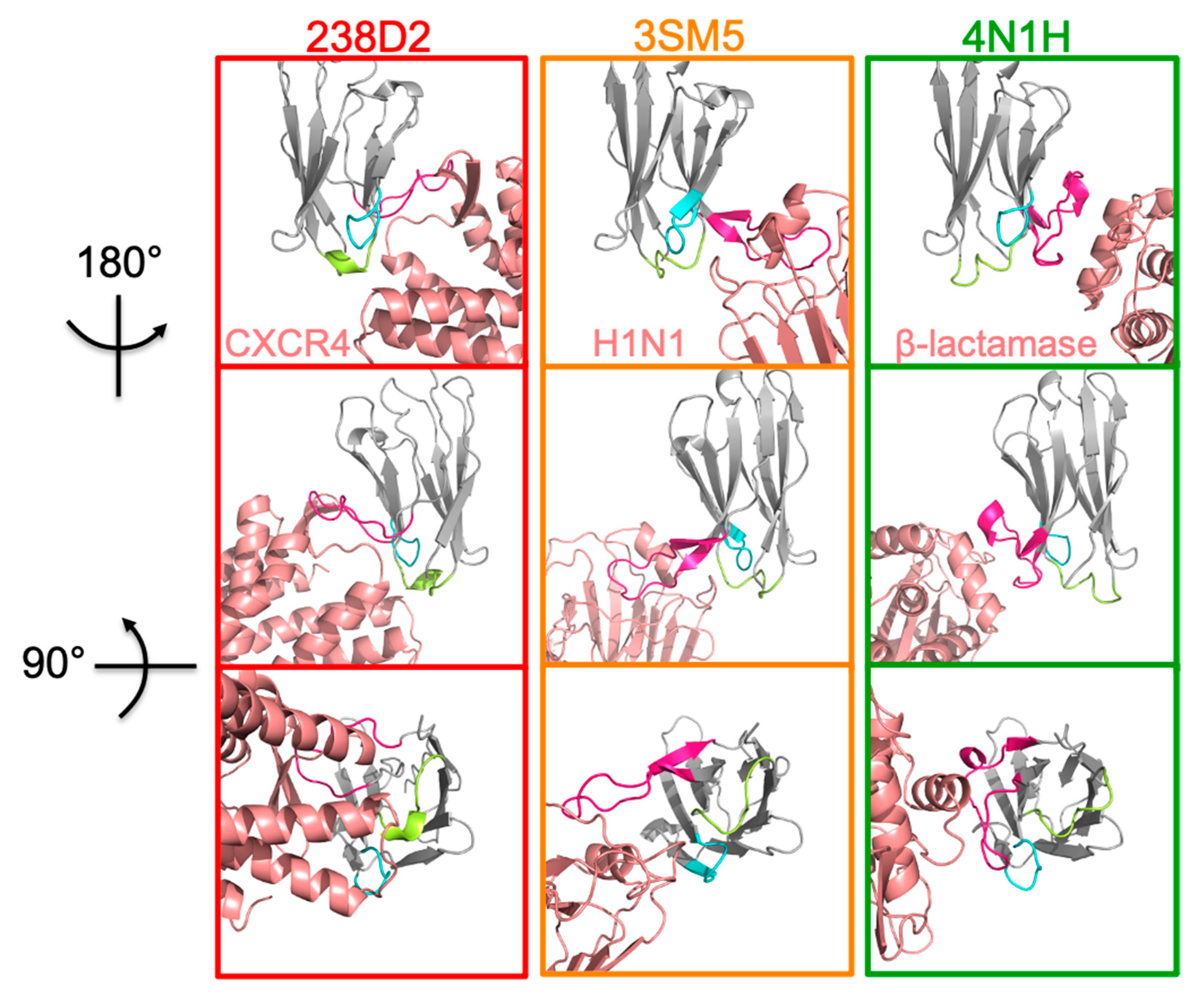
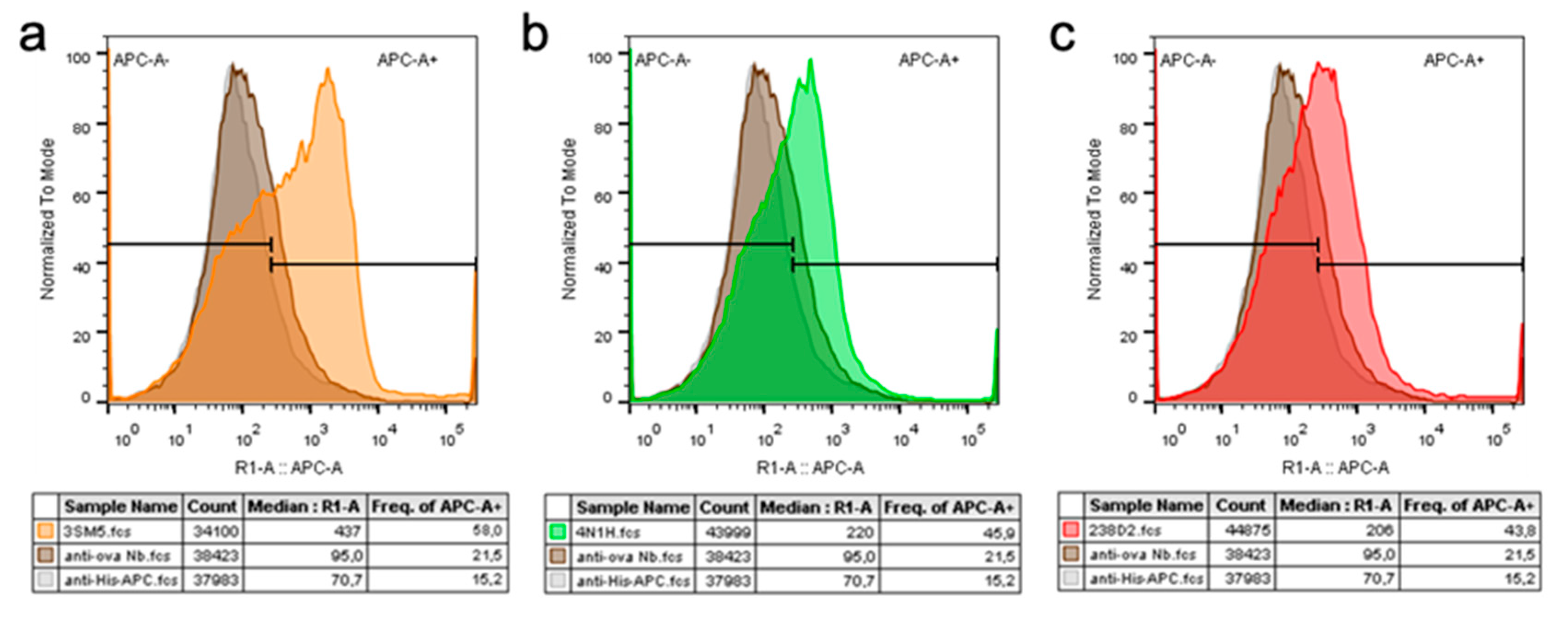
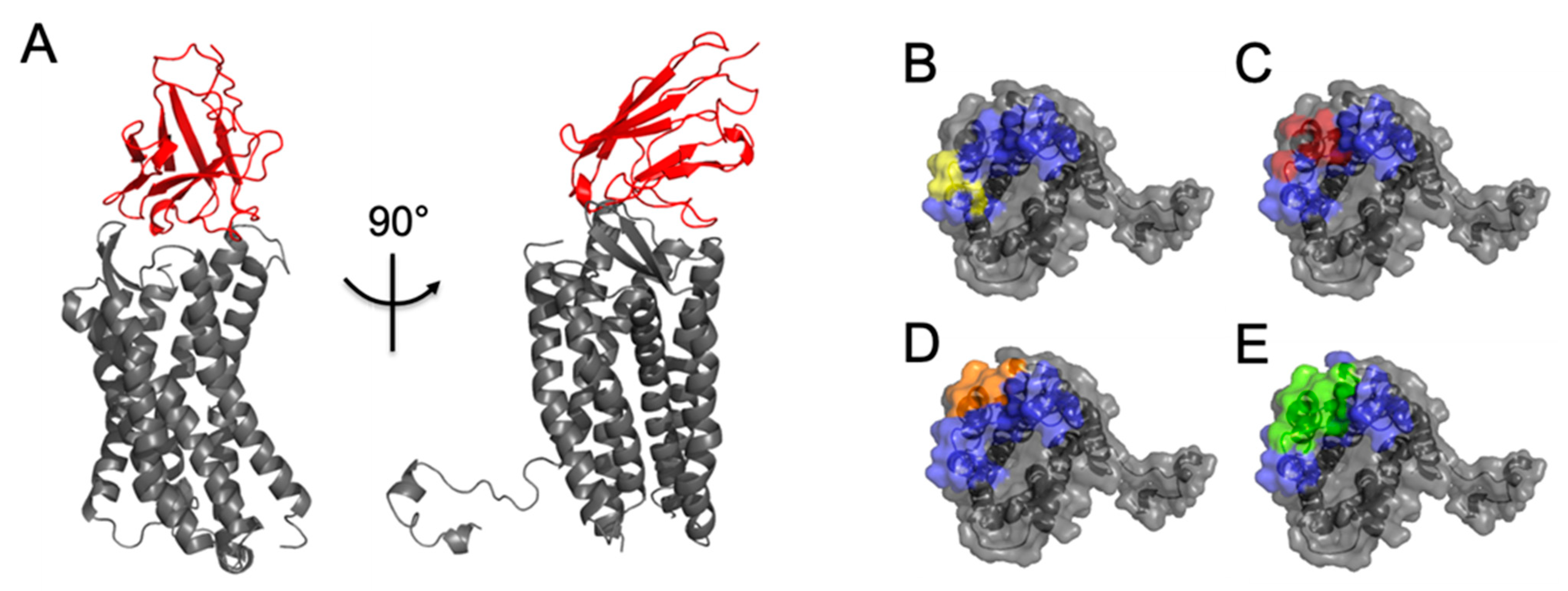
2.2. Selection of Antibody Fragments Similar to 238D2, An Anti-CXCR4 Nb
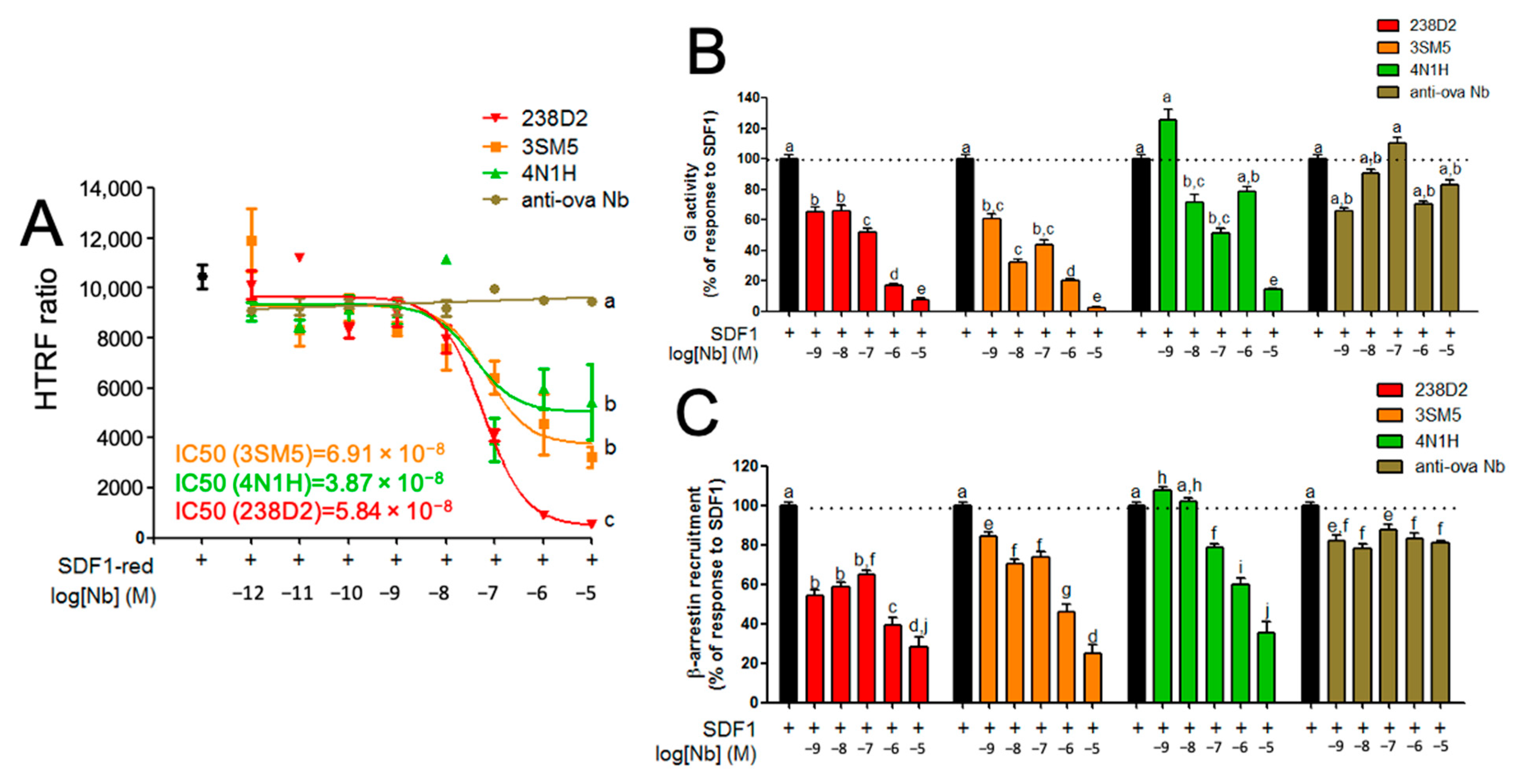
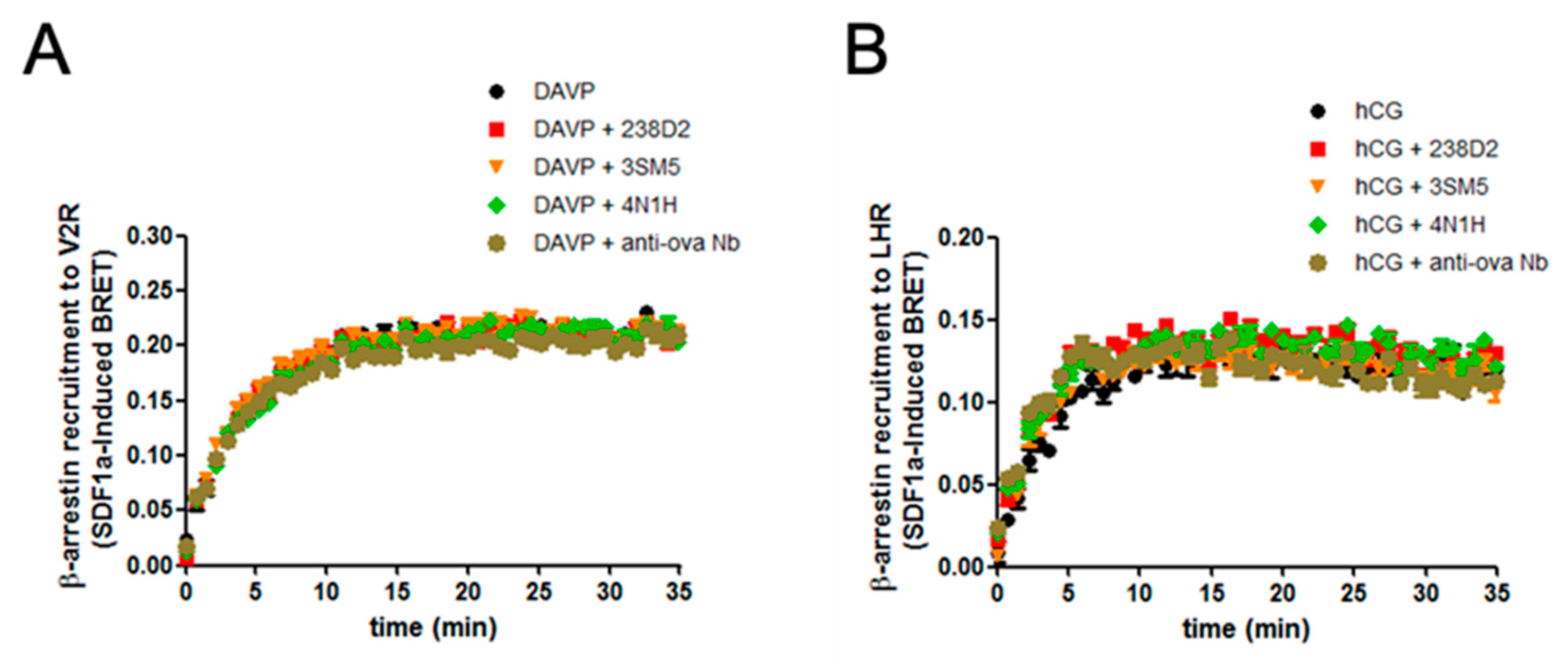
2.3. Biological Evidence for the Cross-Reactivity of Both 3SM5 and 4N1H on CXCR4 Function
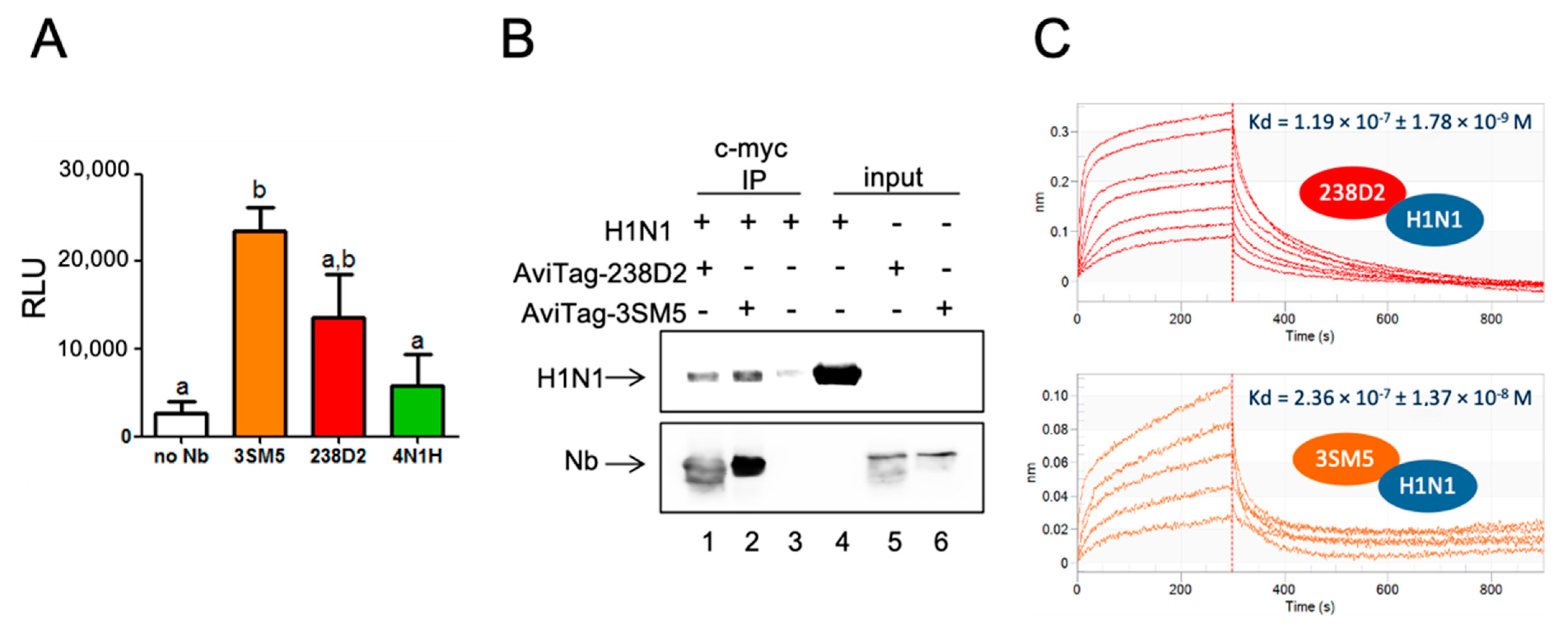
2.4. H1N1 Identified As a Target of 238D2
3. Discussion
4. Materials and Methods
4.1. MAbCross Itemsets Similarity
4.2. Nanobody Sequences
4.3. Bacterial Expression and Purification
4.4. Cell-Free Protein Expression and Purification
4.5. ELISA
4.6. Immuno-Precipitation and Western Blotting
4.7. Biolayer Interferometry (BLI)
4.8. Flow Cytometry
4.9. HTRF-Based Displacement Experiments
4.10. cAMP Production and β-Arrestin 2 Recruitment Assayed by BRET
4.11. Statistics
Author Contributions
Funding
Conflicts of Interest
Appendix A




References
- Leach, M.W.; Halpern, W.G.; Johnson, C.W.; Rojko, J.L.; MacLachlan, T.K.; Chan, C.M.; Galbreath, E.J.; Ndifor, A.M.; Blanset, D.L.; Polack, E.; et al. Use of Tissue Cross-Reactivity Studies in the Development of Antibody-Based Biopharmaceuticals: History, Experience, Methodology, and Future Directions. Toxicol. Pathol. 2010, 38, 1138–1166. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, O.; Scott, M.; Zhou, Z.S.; Finlay, W.J.J. Polyreactivity and Polyspecificity in Therapeutic Antibody Development: Risk Factors for Failure in Preclinical and Clinical Development Campaigns. mAbs 2021, 13, 1999195. [Google Scholar] [CrossRef] [PubMed]
- Lecerf, M.; Kanyavuz, A.; Lacroix-Desmazes, S.; Dimitrov, J.D. Sequence Features of Variable Region Determining Physicochemical Properties and Polyreactivity of Therapeutic Antibodies. Mol. Immunol. 2019, 112, 338–346. [Google Scholar] [CrossRef]
- Loberg, L.I.; Chhaya, M.; Ibraghimov, A.; Tarcsa, E.; Striebinger, A.; Popp, A.; Huang, L.; Oellien, F.; Barghorn, S. Off-Target Binding of an Anti-Amyloid Beta Monoclonal Antibody to Platelet Factor 4 Causes Acute and Chronic Toxicity in Cynomolgus Monkeys. mAbs 2021, 13, 1887628. [Google Scholar] [CrossRef] [PubMed]
- Seyhan, A.A. Lost in Translation: The Valley of Death across Preclinical and Clinical Divide—Identification of Problems and Overcoming Obstacles. Transl. Med. Commun. 2019, 4, 18. [Google Scholar] [CrossRef]
- Ahmed, L.; Gupta, P.; Martin, K.P.; Scheer, J.M.; Nixon, A.E.; Kumar, S. Intrinsic Physicochemical Profile of Marketed Antibody-Based Biotherapeutics. Proc. Natl. Acad. Sci. USA 2021, 118, e2020577118. [Google Scholar] [CrossRef]
- Bourquard, T.; Musnier, A.; Puard, V.; Tahir, S.; Ayoub, M.A.; Jullian, Y.; Boulo, T.; Gallay, N.; Watier, H.; Bruneau, G. MAbTope: A Method for Improved Epitope Mapping. J. Immunol. 2018, 201, 3096–3105. [Google Scholar]
- Tahir, S.; Bourquard, T.; Musnier, A.; Jullian, Y.; Corde, Y.; Omahdi, Z.; Mathias, L.; Reiter, E.; Crépieux, P.; Bruneau, G.; et al. Accurate Determination of Epitope for Antibodies with Unknown 3D Structures. mAbs 2021, 13, 1961349. [Google Scholar] [CrossRef]
- Chothia, C.; Lesk, A.M.; Tramontano, A.; Levitt, M.; Smith-Gill, S.J.; Air, G.; Sheriff, S.; Padlan, E.A.; Davies, D.; Tulip, W.R.; et al. Conformations of Immunoglobulin Hypervariable Regions. Nature 1989, 342, 877–883. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of Protein Secondary Structure: Pattern Recognition of Hydrogen-Bonded and Geometrical Features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Egho, E.; Raïssi, C.; Calders, T.; Jay, N.; Napoli, A. On Measuring Similarity for Sequences of Itemsets. Data Min. Knowl. Disc. 2015, 29, 732–764. [Google Scholar] [CrossRef][Green Version]
- Overington, J.P.; Al-Lazikani, B.; Hopkins, A.L. How Many Drug Targets Are There? Nat. Rev. Drug Discov. 2006, 5, 993–996. [Google Scholar] [CrossRef] [PubMed]
- Lataillade, J.-J.; Domenech, J.; Bousse-Kerdilès, M.-C.L. Stromal Cell-derived Factor-1 (SDF-1)\CXCR4 Couple Plays Multiple Roles on Haematopoietic Progenitors at the Border between the Old Cytokine and New Chemokine Worlds: Survival, Cell Cycling and Trafficking. Eur. Cytokine Netw. 2004, 15, 177–188. [Google Scholar] [PubMed]
- Juarez, J.; Bendall, L. SDF-1 and CXCR4 in Normal and Malignant Hematopoiesis. Histol. Histopathol. 2004, 19, 299–309. [Google Scholar]
- Jähnichen, S.; Blanchetot, C.; Maussang, D.; Gonzalez-Pajuelo, M.; Chow, K.Y.; Bosch, L.; De Vrieze, S.; Serruys, B.; Ulrichts, H.; Vandevelde, W.; et al. CXCR4 Nanobodies (VHH-Based Single Variable Domains) Potently Inhibit Chemotaxis and HIV-1 Replication and Mobilize Stem Cells. Proc. Natl. Acad. Sci. USA 2010, 107, 20565–20570. [Google Scholar] [CrossRef]
- Xu, J.L.; Davis, M.M. Diversity in the CDR3 Region of VH Is Sufficient for Most Antibody Specificities. Immunity 2000, 13, 37–45. [Google Scholar] [CrossRef]
- Whittle, J.R.R.; Zhang, R.; Khurana, S.; King, L.R.; Manischewitz, J.; Golding, H.; Dormitzer, P.R.; Haynes, B.F.; Walter, E.B.; Moody, M.A.; et al. Broadly Neutralizing Human Antibody That Recognizes the Receptor-Binding Pocket of Influenza Virus Hemagglutinin. Proc. Natl. Acad. Sci. USA 2011, 108, 14216–14221. [Google Scholar] [CrossRef]
- Busillo, J.M.; Benovic, J.L. Regulation of CXCR4 Signaling. Biochim. Et Biophys. Acta (BBA)-Biomembr. 2007, 1768, 952–963. [Google Scholar] [CrossRef]
- Talmage, D.W. Immunological Specificity. Science 1959, 129, 1643–1648. [Google Scholar] [CrossRef]
- Srinivasappa, J.; Saegusa, J.; Prabhakar, B.S.; Gentry, M.K.; Buchmeier, M.J.; Wiktor, T.J.; Koprowski, H.; Oldstone, M.B.; Notkins, A.L. Molecular Mimicry: Frequency of Reactivity of Monoclonal Antiviral Antibodies with Normal Tissues. J. Virol. 1986, 57, 397–401. [Google Scholar] [CrossRef]
- Stanfield, R.L.; Takimoto-Kamimura, M.; Rini, J.M.; Profy, A.T.; Wilson, I.A. Major Antigen-Induced Domain Rearrangements in an Antibody. Structure 1993, 1, 83–93. [Google Scholar] [CrossRef]
- Tormo, J.; Blaas, D.; Parry, N.R.; Rowlands, D.; Stuart, D.; Fita, I. Crystal Structure of a Human Rhinovirus Neutralizing Antibody Complexed with a Peptide Derived from Viral Capsid Protein VP2. EMBO J. 1994, 13, 2247–2256. [Google Scholar] [CrossRef] [PubMed]
- Arevalo, J.H.; Hassig, C.A.; Stura, E.A.; Sims, M.J.; Taussig, M.J.; Wilson, I.A. Structural Analysis of Antibody Specificity: Detailed Comparison of Five Fab′-Steroid Complexes. J. Mol. Biol. 1994, 241, 663–690. [Google Scholar] [CrossRef] [PubMed]
- Cusick, M.F.; Libbey, J.E.; Fujinami, R.S. Molecular Mimicry as a Mechanism of Autoimmune Disease. Clin. Rev. Allergy Immunol. 2012, 42, 102–111. [Google Scholar] [CrossRef] [PubMed]
- McKinstry, W.J.; Polekhina, G.; Diefenbach-Jagger, H.; Ho, P.W.M.; Sato, K.; Onuma, E.; Gillespie, M.T.; Martin, T.J.; Parker, M.W. Structural Basis for Antibody Discrimination between Two Hormones That Recognize the Parathyroid Hormone Receptor. J. Biol. Chem. 2009, 284, 15557–15563. [Google Scholar] [CrossRef]
- Nocton, J.J.; Dressler, F.; Rutledge, B.J.; Rys, P.N.; Persing, D.H.; Steere, A.C. Detection of Borrelia Burgdorferi DNA by Polymerase Chain Reaction in Synovial Fluid from Patients with Lyme Arthritis. N. Engl. J. Med. 1994, 330, 229–234. [Google Scholar] [CrossRef]
- Aspinall, G.O.; Fujimoto, S.; McDonald, A.G.; Pang, H.; Kurjanczyk, L.A.; Penner, J.L. Lipopolysaccharides from Campylobacter Jejuni Associated with Guillain-Barré Syndrome Patients Mimic Human Gangliosides in Structure. Infect. Immun. 1994, 62, 2122–2125. [Google Scholar] [CrossRef]
- Leon, J.S.; Engman, D.M. The Significance of Autoimmunity in the Pathogenesis of Chagas Heart Disease. Front. Biosci.-Landmark 2003, 8, 315–322. [Google Scholar] [CrossRef]
- Wu, Y.; Cain-Hom, C.; Choy, L.; Hagenbeek, T.J.; de Leon, G.P.; Chen, Y.; Finkle, D.; Venook, R.; Wu, X.; Ridgway, J.; et al. Therapeutic Antibody Targeting of Individual Notch Receptors. Nature 2010, 464, 1052–1057. [Google Scholar] [CrossRef]
- Walter, J.D.; Werther, R.A.; Polozova, M.S.; Pohlman, J.; Healey, J.F.; Meeks, S.L.; Lollar, P.; Spiegel, P.C. Characterization and Solution Structure of the Factor VIII C2 Domain in a Ternary Complex with Classical and Non-Classical Inhibitor Antibodies. J. Biol. Chem. 2013, 288, 9905–9914. [Google Scholar] [CrossRef]
- Suva, L.J.; Winslow, G.A.; Wettenhall, R.E.H.; Hammonds, R.G.; Moseley, J.M.; Diefenbach-Jagger, H.; Rodda, C.P.; Kemp, B.E.; Rodriguez, H.; Chen, E.Y.; et al. A Parathyroid Hormone-Related Protein Implicated in Malignant Hypercalcemia: Cloning and Expression. Science 1987, 237, 893–896. [Google Scholar] [CrossRef] [PubMed]
- Bigas, A.; Espinosa, L. The Multiple Usages of Notch Signaling in Development, Cell Differentiation and Cancer. Curr. Opin. Cell Biol. 2018, 55, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Yamakawa, Y.; Shizume, K.; Satoh, T.; Nohtomi, K.; Demura, H.; Akatsu, T.; Nagata, N.; Kasahara, T.; Ohkawa, H.; et al. Passive Immunization with Anti-Parathyroid Hormone-Related Protein Monoclonal Antibody Markedly Prolongs Survival Time of Hypercalcemic Nude Mice Bearing Transplanted Human PTHrP-Producing Tumors. J. Bone Miner. Res. 1993, 8, 849–860. [Google Scholar] [CrossRef] [PubMed]
- Sandborn, W.J.; Lee, S.D.; Tarabar, D.; Louis, E.; Klopocka, M.; Klaus, J.; Reinisch, W.; Hébuterne, X.; Park, D.-I.; Schreiber, S.; et al. Phase II Evaluation of Anti-MAdCAM Antibody PF-00547659 in the Treatment of Crohn’s Disease: Report of the OPERA Study. Gut 2018, 67, 1824–1835. [Google Scholar] [CrossRef] [PubMed]
- Fornoni, A.; Sageshima, J.; Wei, C.; Merscher-Gomez, S.; Robier, A.-P.; Jauregui, A.N.; Li, J.; Mattiazzi, A.; Ciancio, G.; Chen, L.; et al. Rituximab Targets Podocytes in Recurrent Focal Segmental Glomerulosclerosis. Sci. Transl. Med. 2011, 3, 85ra46. [Google Scholar] [CrossRef] [PubMed]
- Zhong, E.; Ghadiri, S.; Pai, A.; Marin, J.G.; Barbour, S.J. Rituximab for Adults With Multi-Drug Resistant Focal Segmental Glomerulosclerosis: A Case Series and Review of the Literature. Can. J. Kidney Health Dis. 2022, 9, 20543581221090010. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Thresholds | |||
| Precision = 1 | Precision = 0.95 | Precision = 0.75 | |
| This work | 73 | 55 | 34 |
| Sequence identity | 0.87 | 0.82 | 0.53 |
| CDR identity | 0.7 | 0.65 | 0.3 |
| Recall | |||
| This work | 0.05 | 0.11 | 0.7 |
| Sequence identity | 0.04 | 0.08 | 0.66 |
| CDR identity | 0.04 | 0.05 | 0.62 |
| Accession | Format | Score | Target Antigen |
|---|---|---|---|
| 3SM5 1 | Fab | 64.52 | Hemagglutinin |
| 3FFD | Fab | 59.88 | Parathyroid hormone-related protein |
| 4N1H | VHH | 59.87 | Beta-lactamase |
| 4KML | VHH | 57.58 | Human prion protein |
| 2Q76 | Fab | 57.58 | Hen egg white lysozyme |
| 4HCR | Fab | 56.03 | Mucosal addressin cell adhesion molecule 1 |
| 3L95 | Fab | 55.34 | Neurogenic locus Notch homolog protein 1 |
| 3RVU | Fab | 55.34 | Der f1 major allergen |
| 3NH7 | Fab | 55.28 | Bone morphogenetic protein receptor type-1A |
| 1SEQ | Fab | 55.28 | Neurotrophin receptor TrkA |
| 4KI5 | Fab | 55.27 | Coagulation factor VIII |
| Name | Format | Sequence |
|---|---|---|
| 238D2 1 | VHH | EVQLVESGGGLVQTGGSLRLSCAASGFTFSSYAMSWVRQAPGKGLEWVSGIKSSGDSTRYAGSVKGRFTISRDNAKNMLYLQMYSLKPEDTAVYYCAKSRVSRTGLYTYDNRGQGTQVTVSS |
| 3SM5 2 | VHH | MEVQLVQSGAEVKKPGASVKVSCKASGYTFTDYHINWVRQAPGQGLEWMGWIHPNSGDTNYAQKFQGWVTMTRDTAISTAYMEVNGLKSDDTAVYYCARGGLEPRSVDYYYYGMDVWGQGTTVTVSS |
| 3FFD 2 | Fab | EVQLVESGGDLVKPGGSLKLSCAASGFTFSSYGMSWIRQTPDKRLEWVATISSGGSYTYYPDSVKGRFTISRDNAKNTLYLQMSSLKSEDTAMFYCARQTTMTYFAYWGQGTLVTVSS |
| 4N1H 2 | Fab | MEVQLQESGGGLVQAGASLKLSCAASGRTFSSYAMGWFRQAPGKEREFVAAISRSGGDTKYADSVKGRFAISRDNDKNTVWLRMNSLKPEDTAVYYCAATTYASLSDTYIGEHIYDDWGQGTQVTVSS |
| 4KML 2 | VHH | AVQLQESGGGLVQPGGSLRLSCAASGRTFSSYNMGWFRQAPGKGREFVASITSSGDKSDYTDSVKGRFTISRDNAKNTMYLQMNNLKPEDTATYYCARGLGIYIIRARGGYDHWGQGTQVTVSS |
| 2Q76 2 | Fab | EVQLEQSGAELMKPGASVKISCKATGYTFTTYWIEWIKQRPGHSLEWIGEILPGSDSTYYNEKVKGKVTFTADASSNTAYMQLSSLTSEDSAVYYCARGDGFYVYWGQGTTLTVSS |
| 4HCR 2 | Fab | QVQLVQSGAEVKKPGASVKVSCKASGYTFTSYGINWVRQAPGQGLEWMGWISVYSGNTNYAQKVQGRVTMTADTSTSTAYMDLRSLRSDDTAVYYCAREGSSSSGDYYYGMDVWGQGTTVTVSS |
| 3L95 2 | Fab | EVQLVESGGGLVQPGGSLRLSCAASGFTFSSYWIHWVRQAPGKGLEWVARINPPNRSNQYADSVKGRFTISADTSKNTAYLQMNSLRAEDTAVYYCARGSGFRWVMDYWGQGTLVTVSS |
| 3RVU 2 | Fab | EVQLQESGPGLVKPSQSLSLTCTVTGYSITSDYAWNWIRQFPGNKLEWMGYISYSGTTSYNPSLKSRISITRDTSKNQFFLQLNSVTTEDTATYYCGRTGVYRYPERAPYWGQGTLVTVSA |
| 3NH7 2 | Fab | QVQLVESGGGLVQPGGSLRLSCAASGFTFSNYTLNWVRQAPGKGLEWVSYTSSSGSLTGYADSVKGRFTISRDNSKNTLYLQMNSLRAEDTAVYYCARERWHVRGYFDHWGQGTLVTVSS |
| 1SEQ 2 | Fab | EVKLVESGGGLVQPGGSLKLSCAASGFTFSTYTMSWARQTPEKKLEWVAYISKGGGSTYYPDTVKGRFTISRDNAKNTLYLQMSSLKSEDTALYYCARGAMFGNDFKYPMDRWGQGTSVTVSS |
| 4KI5 2 | Fab | QIQLVQSGPELKKPGKTVKISCKASDYTFTDYSLHWVKQAPGKGLKWMGWINTETGDPAYADDFKGRFAFSLETSVRTAYLQINNLKNEDTAIYFCAREDDGLASWGQGTTLTVSS |
| Anti-Ova 3 | VHH | EVQLQESGGSGQAGGSLRLSCAASGDTVRTMAWFRQAPGQEREGVAGFNLPISRPYYADGMKARFTISGDKSKNTVTLQMDNLAPEDTANYYCAATRYTLDLSSRIFQGDFDHWGHGTQVTVSS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Musnier, A.; Bourquard, T.; Vallet, A.; Mathias, L.; Bruneau, G.; Ayoub, M.A.; Travert, O.; Corde, Y.; Gallay, N.; Boulo, T.; et al. A New in Silico Antibody Similarity Measure Both Identifies Large Sets of Epitope Binders with Distinct CDRs and Accurately Predicts Off-Target Reactivity. Int. J. Mol. Sci. 2022, 23, 9765. https://doi.org/10.3390/ijms23179765
Musnier A, Bourquard T, Vallet A, Mathias L, Bruneau G, Ayoub MA, Travert O, Corde Y, Gallay N, Boulo T, et al. A New in Silico Antibody Similarity Measure Both Identifies Large Sets of Epitope Binders with Distinct CDRs and Accurately Predicts Off-Target Reactivity. International Journal of Molecular Sciences. 2022; 23(17):9765. https://doi.org/10.3390/ijms23179765
Chicago/Turabian StyleMusnier, Astrid, Thomas Bourquard, Amandine Vallet, Laetitia Mathias, Gilles Bruneau, Mohammed Akli Ayoub, Ophélie Travert, Yannick Corde, Nathalie Gallay, Thomas Boulo, and et al. 2022. "A New in Silico Antibody Similarity Measure Both Identifies Large Sets of Epitope Binders with Distinct CDRs and Accurately Predicts Off-Target Reactivity" International Journal of Molecular Sciences 23, no. 17: 9765. https://doi.org/10.3390/ijms23179765
APA StyleMusnier, A., Bourquard, T., Vallet, A., Mathias, L., Bruneau, G., Ayoub, M. A., Travert, O., Corde, Y., Gallay, N., Boulo, T., Cortes, S., Watier, H., Crépieux, P., Reiter, E., & Poupon, A. (2022). A New in Silico Antibody Similarity Measure Both Identifies Large Sets of Epitope Binders with Distinct CDRs and Accurately Predicts Off-Target Reactivity. International Journal of Molecular Sciences, 23(17), 9765. https://doi.org/10.3390/ijms23179765