
Monoclonal Antibody Engineering and Design to Modulate FcRn Activities: A Comprehensive Review




{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Antibody Modifications That Enhance IgG Half-Life and Biodistribution
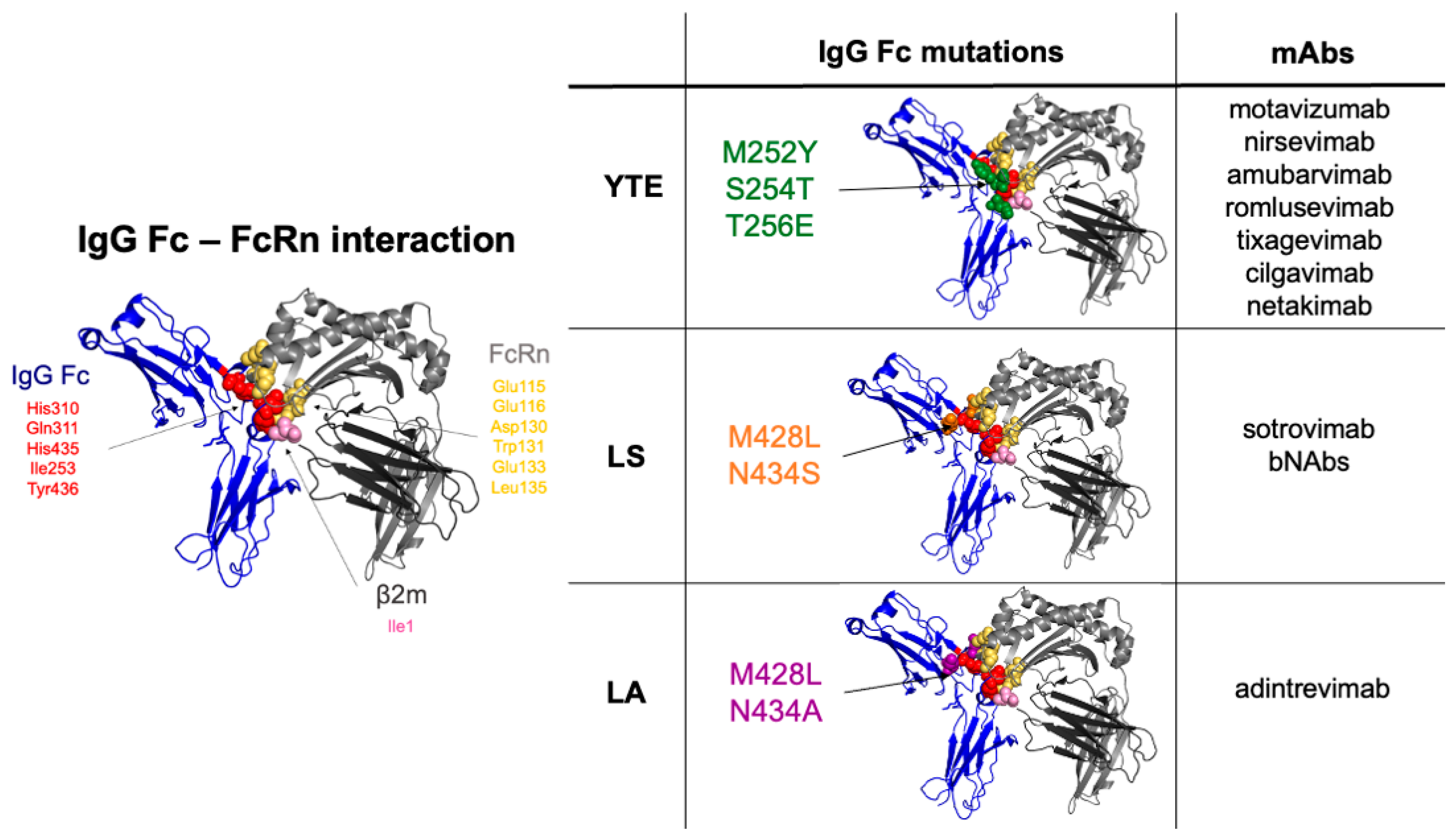
2.1. Mutations Close to the FcRn Binding Site, Increasing Affinity at Acidic pH
2.2. Modifications Outside FcRn Binding Site, Increasing Affinity for IgG Only at Acidic pH
2.3. Mutations in the Vicinity of FcRn Binding Site Associated with pH-Dependent Modulation of Antigen Binding
3. Antibody Modifications Targeting the FcRn Recycling Pathway to Modulate Endogenous IgG Levels including IgG Autoantibodies
Enhancing mAb Affinity to FcRn at Acidic and Neutral pH to Degrade Endogenous IgG
4. Anti-FcRn Antibodies to Block IgG Binding on FcRn Leading to IgG Degradation including IgG Autoantibodies
4.1. Full IgG Format
4.2. Other Formats
5. Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kandil, E.; Egashira, M.; Miyoshi, O.; Niikawa, N.; Ishibashi, T.; Kasahara, M.; Miyosi, O. The Human Gene Encoding the Heavy Chain of the Major Histocompatibility Complex Class I-like Fc Receptor (FCGRT) Maps to 19q13.3. Cytogenet. Cell Genet. 1996, 73, 97–98. [Google Scholar] [CrossRef] [PubMed]
- Brambell, F.W.; Hemmings, W.A.; Morris, I.G. A theoretical model of gamma-globulin catabolism. Nature 1964, 203, 1352–1354. [Google Scholar] [CrossRef] [PubMed]
- Kristoffersen, E.K. Human Placental Fc Gamma-Binding Proteins in the Maternofetal Transfer of IgG. APMIS Suppl. 1996, 64, 5–36. [Google Scholar] [CrossRef]
- Leach, J.L.; Sedmak, D.D.; Osborne, J.M.; Rahill, B.; Lairmore, M.D.; Anderson, C.L. Isolation from Human Placenta of the IgG Transporter, FcRn, and Localization to the Syncytiotrophoblast: Implications for Maternal-Fetal Antibody Transport. J. Immunol. 1996, 157, 3317–3322. [Google Scholar] [PubMed]
- Simister, N.E.; Story, C.M.; Chen, H.L.; Hunt, J.S. An IgG-Transporting Fc Receptor Expressed in the Syncytiotrophoblast of Human Placenta. Eur. J. Immunol. 1996, 26, 1527–1531. [Google Scholar] [CrossRef]
- Burmeister, W.P.; Gastinel, L.N.; Simister, N.E.; Blum, M.L.; Bjorkman, P.J. Crystal Structure at 2.2 A Resolution of the MHC-Related Neonatal Fc Receptor. Nature 1994, 372, 336–343. [Google Scholar] [CrossRef]
- Raghavan, M.; Chen, M.Y.; Gastinel, L.N.; Bjorkman, P.J. Investigation of the Interaction between the Class I MHC-Related Fc Receptor and Its Immunoglobulin G Ligand. Immunity 1994, 1, 303–315. [Google Scholar] [CrossRef]
- Roopenian, D.C.; Akilesh, S. FcRn: The Neonatal Fc Receptor Comes of Age. Nat. Rev. Immunol. 2007, 7, 715–725. [Google Scholar] [CrossRef]
- Pyzik, M.; Sand, K.M.K.; Hubbard, J.J.; Andersen, J.T.; Sandlie, I.; Blumberg, R.S. The Neonatal Fc Receptor (FcRn): A Misnomer? Front. Immunol. 2019, 10, 1540. [Google Scholar] [CrossRef]
- Raghavan, M.; Gastinel, L.N.; Bjorkman, P.J. The Class I Major Histocompatibility Complex Related Fc Receptor Shows PH-Dependent Stability Differences Correlating with Immunoglobulin Binding and Release. Biochemistry 1993, 32, 8654–8660. [Google Scholar] [CrossRef]
- Junghans, R.P.; Anderson, C.L. The Protection Receptor for IgG Catabolism Is the Beta2-Microglobulin-Containing Neonatal Intestinal Transport Receptor. Proc. Natl. Acad. Sci. USA 1996, 93, 5512–5516. [Google Scholar] [CrossRef] [PubMed]
- Chaudhury, C.; Brooks, C.L.; Carter, D.C.; Robinson, J.M.; Anderson, C.L. Albumin Binding to FcRn: Distinct from the FcRn-IgG Interaction. Biochemistry 2006, 45, 4983–4990. [Google Scholar] [CrossRef] [PubMed]
- Qi, T.; Cao, Y. In Translation: FcRn across the Therapeutic Spectrum. Int. J. Mol. Sci. 2021, 22, 3048. [Google Scholar] [CrossRef] [PubMed]
- Lamamy, J.; Boulard, P.; Brachet, G.; Tourlet, S.; Gouilleux-Gruart, V.; Ramdani, Y. Ways in Which the Neonatal Fc-Receptor Is Involved in Autoimmunity. J. Transl. Autoimmun. 2021, 4, 100122. [Google Scholar] [CrossRef]
- Kim, J.K.; Firan, M.; Radu, C.G.; Kim, C.H.; Ghetie, V.; Ward, E.S. Mapping the Site on Human IgG for Binding of the MHC Class I-Related Receptor, FcRn. Eur. J. Immunol. 1999, 29, 2819–2825. [Google Scholar] [CrossRef]
- Martin, W.L.; West, A.P.; Gan, L.; Bjorkman, P.J. Crystal Structure at 2.8 A of an FcRn/Heterodimeric Fc Complex: Mechanism of PH-Dependent Binding. Mol. Cell 2001, 7, 867–877. [Google Scholar] [CrossRef]
- Shields, R.L.; Namenuk, A.K.; Hong, K.; Meng, Y.G.; Rae, J.; Briggs, J.; Xie, D.; Lai, J.; Stadlen, A.; Li, B.; et al. High Resolution Mapping of the Binding Site on Human IgG1 for Fc Gamma RI, Fc Gamma RII, Fc Gamma RIII, and FcRn and Design of IgG1 Variants with Improved Binding to the Fc Gamma R. J. Biol. Chem. 2001, 276, 6591–6604. [Google Scholar] [CrossRef]
- Oganesyan, V.; Damschroder, M.M.; Cook, K.E.; Li, Q.; Gao, C.; Wu, H.; Dall’Acqua, W.F. Structural Insights into Neonatal Fc Receptor-Based Recycling Mechanisms. J. Biol. Chem. 2014, 289, 7812–7824. [Google Scholar] [CrossRef]
- Monnet, C.; Jorieux, S.; Urbain, R.; Fournier, N.; Bouayadi, K.; De Romeuf, C.; Behrens, C.K.; Fontayne, A.; Mondon, P. Selection of IgG Variants with Increased FcRn Binding Using Random and Directed Mutagenesis: Impact on Effector Functions. Front. Immunol. 2015, 6, 39. [Google Scholar] [CrossRef]
- Ghetie, V.; Popov, S.; Borvak, J.; Radu, C.; Matesoi, D.; Medesan, C.; Ober, R.J.; Ward, E.S. Increasing the Serum Persistence of an IgG Fragment by Random Mutagenesis. Nat. Biotechnol. 1997, 15, 637–640. [Google Scholar] [CrossRef]
- Burmeister, W.P.; Huber, A.H.; Bjorkman, P.J. Crystal Structure of the Complex of Rat Neonatal Fc Receptor with Fc. Nature 1994, 372, 379–383. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Bjorkman, P.J. Crystal Structure and Immunoglobulin G Binding Properties of the Human Major Histocompatibility Complex-Related Fc Receptor. Biochemistry 2000, 39, 9698–9708. [Google Scholar] [CrossRef] [PubMed]
- Huber, A.H.; Kelley, R.F.; Gastinel, L.N.; Bjorkman, P.J. Crystallization and Stoichiometry of Binding of a Complex between a Rat Intestinal Fc Receptor and Fc. J. Mol. Biol. 1993, 230, 1077–1083. [Google Scholar] [CrossRef]
- Ward, E.S.; Ober, R.J. Targeting FcRn to Generate Antibody-Based Therapeutics. Trends Pharmacol. Sci. 2018, 39, 892–904. [Google Scholar] [CrossRef] [PubMed]
- Qiao, S.-W.; Kobayashi, K.; Johansen, F.-E.; Sollid, L.M.; Andersen, J.T.; Milford, E.; Roopenian, D.C.; Lencer, W.I.; Blumberg, R.S. Dependence of Antibody-Mediated Presentation of Antigen on FcRn. Proc. Natl. Acad. Sci. USA 2008, 105, 9337–9342. [Google Scholar] [CrossRef]
- Wang, W.; Lu, P.; Fang, Y.; Hamuro, L.; Pittman, T.; Carr, B.; Hochman, J.; Prueksaritanont, T. Monoclonal Antibodies with Identical Fc Sequences Can Bind to FcRn Differentially with Pharmacokinetic Consequences. Drug Metab. Dispos. 2011, 39, 1469–1477. [Google Scholar] [CrossRef] [PubMed]
- Gurbaxani, B.; Dela Cruz, L.L.; Chintalacharuvu, K.; Morrison, S.L. Analysis of a Family of Antibodies with Different Half-Lives in Mice Fails to Find a Correlation between Affinity for FcRn and Serum Half-Life. Mol. Immunol. 2006, 43, 1462–1473. [Google Scholar] [CrossRef]
- Dall’Acqua, W.F.; Woods, R.M.; Ward, E.S.; Palaszynski, S.R.; Patel, N.K.; Brewah, Y.A.; Wu, H.; Kiener, P.A.; Langermann, S. Increasing the Affinity of a Human IgG1 for the Neonatal Fc Receptor: Biological Consequences. J. Immunol. 2002, 169, 5171–5180. [Google Scholar] [CrossRef]
- Yeung, Y.A.; Leabman, M.K.; Marvin, J.S.; Qiu, J.; Adams, C.W.; Lien, S.; Starovasnik, M.A.; Lowman, H.B. Engineering Human IgG1 Affinity to Human Neonatal Fc Receptor: Impact of Affinity Improvement on Pharmacokinetics in Primates. J. Immunol. 2009, 182, 7663–7671. [Google Scholar] [CrossRef]
- Datta-Mannan, A.; Witcher, D.R.; Tang, Y.; Watkins, J.; Wroblewski, V.J. Monoclonal Antibody Clearance. Impact of Modulating the Interaction of IgG with the Neonatal Fc Receptor. J. Biol. Chem. 2007, 282, 1709–1717. [Google Scholar] [CrossRef] [Green Version]
- Hinton, P.R.; Xiong, J.M.; Johlfs, M.G.; Tang, M.T.; Keller, S.; Tsurushita, N. An Engineered Human IgG1 Antibody with Longer Serum Half-Life. J. Immunol. 2006, 176, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Kuo, T.T.; Aveson, V.G. Neonatal Fc Receptor and IgG-Based Therapeutics. MAbs 2011, 3, 422–430. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tian, Z.; Thirumalai, D.; Zhang, X. Neonatal Fc Receptor (FcRn): A Novel Target for Therapeutic Antibodies and Antibody Engineering. J. Drug Target. 2014, 22, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Deng, R.; Loyet, K.M.; Lien, S.; Iyer, S.; DeForge, L.E.; Theil, F.-P.; Lowman, H.B.; Fielder, P.J.; Prabhu, S. Pharmacokinetics of Humanized Monoclonal Anti-Tumor Necrosis Factor-{alpha} Antibody and Its Neonatal Fc Receptor Variants in Mice and Cynomolgus Monkeys. Drug Metab. Dispos. 2010, 38, 600–605. [Google Scholar] [CrossRef]
- Zalevsky, J.; Chamberlain, A.K.; Horton, H.M.; Karki, S.; Leung, I.W.L.; Sproule, T.J.; Lazar, G.A.; Roopenian, D.C.; Desjarlais, J.R. Enhanced Antibody Half-Life Improves in Vivo Activity. Nat. Biotechnol. 2010, 28, 157–159. [Google Scholar] [CrossRef]
- Sievers, S.A.; Scharf, L.; West, A.P.; Bjorkman, P.J. Antibody Engineering for Increased Potency, Breadth and Half-Life. Curr Opin HIV AIDS 2015, 10, 151–159. [Google Scholar] [CrossRef]
- Gupta, A.; Gonzalez-Rojas, Y.; Juarez, E.; Crespo Casal, M.; Moya, J.; Falci, D.R.; Sarkis, E.; Solis, J.; Zheng, H.; Scott, N.; et al. Early Treatment for COVID-19 with SARS-CoV-2 Neutralizing Antibody Sotrovimab. N. Engl. J. Med. 2021, 385, 1941–1950. [Google Scholar] [CrossRef]
- Robbie, G.J.; Criste, R.; Dall’Acqua, W.F.; Jensen, K.; Patel, N.K.; Losonsky, G.A.; Griffin, M.P. A Novel Investigational Fc-Modified Humanized Monoclonal Antibody, Motavizumab-YTE, Has an Extended Half-Life in Healthy Adults. Antimicrob. Agents Chemother. 2013, 57, 6147–6153. [Google Scholar] [CrossRef]
- Kaplon, H.; Reichert, J.M. Antibodies to Watch in 2021. MAbs 2021, 13, 1860476. [Google Scholar] [CrossRef]
- IMGT/3Dstructure-DB Card. Available online: https://www.imgt.org/3Dstructure-DB/cgi/details.cgi?pdbcode=10387 (accessed on 1 August 2022).
- Puig, L.; Bakulev, A.L.; Kokhan, M.M.; Samtsov, A.V.; Khairutdinov, V.R.; Morozova, M.A.; Zolkin, N.A.; Kuryshev, I.V.; Petrov, A.N.; Artemeva, A.V.; et al. Efficacy and Safety of Netakimab, A Novel Anti-IL-17 Monoclonal Antibody, in Patients with Moderate to Severe Plaque Psoriasis. Results of A 54-Week Randomized Double-Blind Placebo-Controlled PLANETA Clinical Trial. Dermatol. Ther. 2021, 11, 1319–1332. [Google Scholar] [CrossRef]
- Adagio Therapeutics Provides Update for ADG20 COVID-19 Antibody Program and Reports Third Quarter 2021 Financial Results|Adagio Therapeutics, Inc. Available online: https://investors.adagiotx.com/news-releases/news-release-details/adagio-therapeutics-provides-update-adg20-covid-19-antibody/ (accessed on 5 February 2022).
- Griffin, M.P.; Yuan, Y.; Takas, T.; Domachowske, J.B.; Madhi, S.A.; Manzoni, P.; Simões, E.A.F.; Esser, M.T.; Khan, A.A.; Dubovsky, F.; et al. Single-Dose Nirsevimab for Prevention of RSV in Preterm Infants. N. Engl. J. Med. 2020, 383, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Grevys, A.; Bern, M.; Foss, S.; Bratlie, D.B.; Moen, A.; Gunnarsen, K.S.; Aase, A.; Michaelsen, T.E.; Sandlie, I.; Andersen, J.T. Fc Engineering of Human IgG1 for Altered Binding to the Neonatal Fc Receptor Affects Fc Effector Functions. J. Immunol. 2015, 194, 5497–5508. [Google Scholar] [CrossRef] [PubMed]
- Lobo, E.D.; Hansen, R.J.; Balthasar, J.P. Antibody Pharmacokinetics and Pharmacodynamics. J. Pharm. Sci. 2004, 93, 2645–2668. [Google Scholar] [CrossRef]
- Ternant, D.; Arnoult, C.; Pugnière, M.; Dhommée, C.; Drocourt, D.; Perouzel, E.; Passot, C.; Baroukh, N.; Mulleman, D.; Tiraby, G.; et al. IgG1 Allotypes Influence the Pharmacokinetics of Therapeutic Monoclonal Antibodies through FcRn Binding. J. Immunol. 2016, 196, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Datta-Mannan, A.; Thangaraju, A.; Leung, D.; Tang, Y.; Witcher, D.R.; Lu, J.; Wroblewski, V.J. Balancing Charge in the Complementarity-Determining Regions of Humanized MAbs without Affecting PI Reduces Non-Specific Binding and Improves the Pharmacokinetics. MAbs 2015, 7, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Piche-Nicholas, N.M.; Avery, L.B.; King, A.C.; Kavosi, M.; Wang, M.; O’Hara, D.M.; Tchistiakova, L.; Katragadda, M. Changes in Complementarity-Determining Regions Significantly Alter IgG Binding to the Neonatal Fc Receptor (FcRn) and Pharmacokinetics. MAbs 2018, 10, 81–94. [Google Scholar] [CrossRef]
- Li, B.; Tesar, D.; Boswell, C.A.; Cahaya, H.S.; Wong, A.; Zhang, J.; Meng, Y.G.; Eigenbrot, C.; Pantua, H.; Diao, J.; et al. Framework Selection Can Influence Pharmacokinetics of a Humanized Therapeutic Antibody through Differences in Molecule Charge. MAbs 2014, 6, 1255–1264. [Google Scholar] [CrossRef]
- Duncan, A.R.; Woof, J.M.; Partridge, L.J.; Burton, D.R.; Winter, G. Localization of the Binding Site for the Human High-Affinity Fc Receptor on IgG. Nature 1988, 332, 563–564. [Google Scholar] [CrossRef]
- Duncan, A.R.; Winter, G. The Binding Site for C1q on IgG. Nature 1988, 332, 738–740. [Google Scholar] [CrossRef]
- Monnet, C.; Jorieux, S.; Souyris, N.; Zaki, O.; Jacquet, A.; Fournier, N.; Crozet, F.; de Romeuf, C.; Bouayadi, K.; Urbain, R.; et al. Combined Glyco- and Protein-Fc Engineering Simultaneously Enhance Cytotoxicity and Half-Life of a Therapeutic Antibody. MAbs 2014, 6, 422–436. [Google Scholar] [CrossRef] [Green Version]
- Idusogie, E.E.; Wong, P.Y.; Presta, L.G.; Gazzano-Santoro, H.; Totpal, K.; Ultsch, M.; Mulkerrin, M.G. Engineered Antibodies with Increased Activity to Recruit Complement. J. Immunol. 2001, 166, 2571–2575. [Google Scholar] [CrossRef] [PubMed]
- Shields, R.L.; Lai, J.; Keck, R.; O’Connell, L.Y.; Hong, K.; Meng, Y.G.; Weikert, S.H.A.; Presta, L.G. Lack of Fucose on Human IgG1 N-Linked Oligosaccharide Improves Binding to Human Fcgamma RIII and Antibody-Dependent Cellular Toxicity. J. Biol. Chem. 2002, 277, 26733–26740. [Google Scholar] [CrossRef] [PubMed]
- Finkelman, F.D.; Madden, K.B.; Morris, S.C.; Holmes, J.M.; Boiani, N.; Katona, I.M.; Maliszewski, C.R. Anti-Cytokine Antibodies as Carrier Proteins. Prolongation of in Vivo Effects of Exogenous Cytokines by Injection of Cytokine-Anti-Cytokine Antibody Complexes. J. Immunol. 1993, 151, 1235–1244. [Google Scholar]
- Chaparro-Riggers, J.; Liang, H.; DeVay, R.M.; Bai, L.; Sutton, J.E.; Chen, W.; Geng, T.; Lindquist, K.; Casas, M.G.; Boustany, L.M.; et al. Increasing Serum Half-Life and Extending Cholesterol Lowering in Vivo by Engineering Antibody with PH-Sensitive Binding to PCSK9. J. Biol. Chem. 2012, 287, 11090–11097. [Google Scholar] [CrossRef]
- Devanaboyina, S.C.; Lynch, S.M.; Ober, R.J.; Ram, S.; Kim, D.; Puig-Canto, A.; Breen, S.; Kasturirangan, S.; Fowler, S.; Peng, L.; et al. The Effect of PH Dependence of Antibody-Antigen Interactions on Subcellular Trafficking Dynamics. MAbs 2013, 5, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Igawa, T.; Ishii, S.; Tachibana, T.; Maeda, A.; Higuchi, Y.; Shimaoka, S.; Moriyama, C.; Watanabe, T.; Takubo, R.; Doi, Y.; et al. Antibody Recycling by Engineered PH-Dependent Antigen Binding Improves the Duration of Antigen Neutralization. Nat. Biotechnol. 2010, 28, 1203–1207. [Google Scholar] [CrossRef]
- Sheridan, D.; Yu, Z.-X.; Zhang, Y.; Patel, R.; Sun, F.; Lasaro, M.A.; Bouchard, K.; Andrien, B.; Marozsan, A.; Wang, Y.; et al. Design and Preclinical Characterization of ALXN1210: A Novel Anti-C5 Antibody with Extended Duration of Action. PLoS ONE 2018, 13, e0195909. [Google Scholar] [CrossRef]
- Fukuzawa, T.; Sampei, Z.; Haraya, K.; Ruike, Y.; Shida-Kawazoe, M.; Shimizu, Y.; Gan, S.W.; Irie, M.; Tsuboi, Y.; Tai, H.; et al. Long Lasting Neutralization of C5 by SKY59, a Novel Recycling Antibody, Is a Potential Therapy for Complement-Mediated Diseases. Sci. Rep. 2017, 7, 1080. [Google Scholar] [CrossRef]
- Traboulsee, A.; Greenberg, B.M.; Bennett, J.L.; Szczechowski, L.; Fox, E.; Shkrobot, S.; Yamamura, T.; Terada, Y.; Kawata, Y.; Wright, P.; et al. Safety and Efficacy of Satralizumab Monotherapy in Neuromyelitis Optica Spectrum Disorder: A Randomised, Double-Blind, Multicentre, Placebo-Controlled Phase 3 Trial. Lancet Neurol. 2020, 19, 402–412. [Google Scholar] [CrossRef]
- Katagiri, R.; Ishihara-Hattori, K.; Frings, W.; Amano, J.; Fuchs, A.; Chiba, S. Effects of SA237, a Humanized Anti-Interleukin-6 Receptor Monoclonal Antibody, on Pre- and Postnatal Development in Cynomolgus Monkey. Birth Defects Res. 2017, 109, 843–856. [Google Scholar] [CrossRef]
- FDA. FDA Approves Ravulizumab-Cwvz for Paroxysmal Nocturnal Hemoglobinuria; FDA: Silver Spring, MD, USA, 2019. [Google Scholar]
- EMA Ultomiris. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/ultomiris (accessed on 20 April 2022).
- Commissioner, O. of the FDA Approves Treatment for Rare Disease Affecting Optic Nerves, Spinal Cord. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-treatment-rare-disease-affecting-optic-nerves-spinal-cord (accessed on 20 April 2022).
- EMA EU/3/16/1680. Available online: https://www.ema.europa.eu/en/medicines/human/orphan-designations/eu3161680 (accessed on 20 April 2022).
- Röth, A.; Egyed, M.; Ichikawa, S.; Kim, J.S.; Nagy, Z.; Gaàl Weisinger, J.; Panse, J.P.; Peffault De Latour, R.; Sicre De Fontbrune, F.; Sica, S.; et al. The SMART Anti-HC5 Antibody (SKY59/RO7112689) Shows Good Safety and Efficacy in Patients with Paroxysmal Nocturnal Hemoglobinuria (PNH). Blood 2018, 132, 535. [Google Scholar] [CrossRef]
- Röth, A.; Nishimura, J.-I.; Nagy, Z.; Gaàl-Weisinger, J.; Panse, J.; Yoon, S.-S.; Egyed, M.; Ichikawa, S.; Ito, Y.; Kim, J.S.; et al. The Complement C5 Inhibitor Crovalimab in Paroxysmal Nocturnal Hemoglobinuria. Blood 2020, 135, 912–920. [Google Scholar] [CrossRef] [PubMed]
- Igawa, T.; Maeda, A.; Haraya, K.; Tachibana, T.; Iwayanagi, Y.; Mimoto, F.; Higuchi, Y.; Ishii, S.; Tamba, S.; Hironiwa, N.; et al. Engineered Monoclonal Antibody with Novel Antigen-Sweeping Activity in Vivo. PLoS ONE 2013, 8, e63236. [Google Scholar] [CrossRef]
- Igawa, T.; Mimoto, F.; Hattori, K. PH-Dependent Antigen-Binding Antibodies as a Novel Therapeutic Modality. Biochim. Biophys. Acta 2014, 1844, 1943–1950. [Google Scholar] [CrossRef]
- Yang, D.; Giragossian, C.; Castellano, S.; Lasaro, M.; Xiao, H.; Saraf, H.; Hess Kenny, C.; Rybina, I.; Huang, Z.-F.; Ahlberg, J.; et al. Maximizing in Vivo Target Clearance by Design of PH-Dependent Target Binding Antibodies with Altered Affinity to FcRn. MAbs 2017, 9, 1105–1117. [Google Scholar] [CrossRef] [PubMed]
- Gan, Z.; Ram, S.; Vaccaro, C.; Ober, R.J.; Ward, E.S. Analyses of the Recycling Receptor, FcRn, in Live Cells Reveal Novel Pathways for Lysosomal Delivery. Traffic 2009, 10, 600–614. [Google Scholar] [CrossRef] [PubMed]
- Kroetsch, A.; Qiao, C.; Heavey, M.; Guo, L.; Shah, D.K.; Park, S. Engineered PH-Dependent Recycling Antibodies Enhance Elimination of Staphylococcal Enterotoxin B Superantigen in Mice. MAbs 2019, 11, 411–421. [Google Scholar] [CrossRef]
- Bogen, J.P.; Hinz, S.C.; Grzeschik, J.; Ebenig, A.; Krah, S.; Zielonka, S.; Kolmar, H. Dual Function PH Responsive Bispecific Antibodies for Tumor Targeting and Antigen Depletion in Plasma. Front. Immunol. 2019, 10, 1892. [Google Scholar] [CrossRef]
- Vaccaro, C.; Zhou, J.; Ober, R.J.; Ward, E.S. Engineering the Fc Region of Immunoglobulin G to Modulate in Vivo Antibody Levels. Nat. Biotechnol. 2005, 23, 1283–1288. [Google Scholar] [CrossRef]
- Peter, H.-H.; Ochs, H.D.; Cunningham-Rundles, C.; Vinh, D.C.; Kiessling, P.; Greve, B.; Jolles, S. Targeting FcRn for Immunomodulation: Benefits, Risks, and Practical Considerations. J. Allergy Clin. Immunol. 2020, 146, 479–491.e5. [Google Scholar] [CrossRef]
- Shock, A.; Humphreys, D.; Nimmerjahn, F. Dissecting the Mechanism of Action of Intravenous Immunoglobulin in Human Autoimmune Disease: Lessons from Therapeutic Modalities Targeting Fcγ Receptors. J. Allergy Clin. Immunol. 2020, 146, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Ulrichts, P.; Guglietta, A.; Dreier, T.; van Bragt, T.; Hanssens, V.; Hofman, E.; Vankerckhoven, B.; Verheesen, P.; Ongenae, N.; Lykhopiy, V.; et al. Neonatal Fc Receptor Antagonist Efgartigimod Safely and Sustainably Reduces IgGs in Humans. J. Clin. Investig. 2018, 128, 4372–4386. [Google Scholar] [CrossRef] [PubMed]
- Heo, Y.-A. Efgartigimod: First Approval. Drugs 2022, 82, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Efgartigimod. Available online: https://www.argenx.com/pipeline/efgartigimod (accessed on 3 April 2022).
- Goebeler, M.; Bata-Csörgő, Z.; De Simone, C.; Didona, B.; Remenyik, E.; Reznichenko, N.; Stoevesandt, J.; Ward, E.S.; Parys, W.; de Haard, H.; et al. Treatment of Pemphigus Vulgaris and Foliaceus with Efgartigimod, a Neonatal Fc Receptor Inhibitor: A Phase II Multicentre, Open-Label Feasibility Trial. Br. J. Derm. 2022, 186, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Briani, C.; Visentin, A. Therapeutic Monoclonal Antibody Therapies in Chronic Autoimmune Demyelinating Neuropathies. Neurotherapeutics 2022, 19, 874–884. [Google Scholar] [CrossRef] [PubMed]
- Devanaboyina, S.C.; Khare, P.; Challa, D.K.; Ober, R.J.; Ward, E.S. Engineered Clearing Agents for the Selective Depletion of Antigen-Specific Antibodies. Nat. Commun. 2017, 8, 15314. [Google Scholar] [CrossRef]
- Sun, W.; Khare, P.; Wang, X.; Challa, D.K.; Greenberg, B.M.; Ober, R.J.; Ward, E.S. Selective Depletion of Antigen-Specific Antibodies for the Treatment of Demyelinating Disease. Mol. Ther. 2021, 29, 1312–1323. [Google Scholar] [CrossRef]
- Schwab, I.; Nimmerjahn, F. Intravenous Immunoglobulin Therapy: How Does IgG Modulate the Immune System? Nat. Rev. Immunol. 2013, 13, 176–189. [Google Scholar] [CrossRef]
- Weissmann, G. Rheumatoid Arthritis and Systemic Lupus Erythematosus as Immune Complex Diseases. Bull. NYU Hosp. Jt Dis. 2009, 67, 251–253. [Google Scholar]
- Liebman, H.A.; Weitz, I.C. Autoimmune Hemolytic Anemia. Med. Clin. N. Am. 2017, 101, 351–359. [Google Scholar] [CrossRef]
- Zufferey, A.; Kapur, R.; Semple, J.W. Pathogenesis and Therapeutic Mechanisms in Immune Thrombocytopenia (ITP). J. Clin. Med. 2017, 6, 16. [Google Scholar] [CrossRef] [PubMed]
- Monnet, C.; Jacque, E.; de Romeuf, C.; Fontayne, A.; Abache, T.; Fournier, N.; Dupont, G.; Derache, D.; Engrand, A.; Bauduin, A.; et al. The Dual Targeting of FcRn and FcγRs via Monomeric Fc Fragments Results in Strong Inhibition of IgG-Dependent Autoimmune Pathologies. Front. Immunol. 2021, 12, 728322. [Google Scholar] [CrossRef] [PubMed]
- Nixon, A.E.; Chen, J.; Sexton, D.J.; Muruganandam, A.; Bitonti, A.J.; Dumont, J.; Viswanathan, M.; Martik, D.; Wassaf, D.; Mezo, A.; et al. Fully Human Monoclonal Antibody Inhibitors of the Neonatal Fc Receptor Reduce Circulating IgG in Non-Human Primates. Front. Immunol. 2015, 6, 176. [Google Scholar] [CrossRef]
- Kenniston, J.A.; Taylor, B.M.; Conley, G.P.; Cosic, J.; Kopacz, K.J.; Lindberg, A.P.; Comeau, S.R.; Atkins, K.; Bullen, J.; TenHoor, C.; et al. Structural Basis for PH-Insensitive Inhibition of Immunoglobulin G Recycling by an Anti-Neonatal Fc Receptor Antibody. J. Biol. Chem. 2017, 292, 17449–17460. [Google Scholar] [CrossRef] [PubMed]
- Zuercher, A.W.; Spirig, R.; Baz Morelli, A.; Rowe, T.; Käsermann, F. Next-Generation Fc Receptor–Targeting Biologics for Autoimmune Diseases. Autoimmun. Rev. 2019, 18, 102366. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.; Kiessling, A.; Lledo-Garcia, R.; Dixon, K.L.; Christodoulou, L.; Catley, M.C.; Atherfold, P.; D’Hooghe, L.E.; Finney, H.; Greenslade, K.; et al. Generation and Characterization of a High Affinity Anti-Human FcRn Antibody, Rozanolixizumab, and the Effects of Different Molecular Formats on the Reduction of Plasma IgG Concentration. MAbs 2018, 10, 1111–1130. [Google Scholar] [CrossRef]
- Orilanolimab-Alexion AstraZeneca Rare Disease-AdisInsight. Available online: https://adisinsight.springer.com/drugs/800045005 (accessed on 16 April 2022).
- Ling, L.E.; Hillson, J.L.; Tiessen, R.G.; Bosje, T.; van Iersel, M.P.; Nix, D.J.; Markowitz, L.; Cilfone, N.A.; Duffner, J.; Streisand, J.B.; et al. M281, an Anti-FcRn Antibody: Pharmacodynamics, Pharmacokinetics, and Safety Across the Full Range of IgG Reduction in a First-in-Human Study. Clin. Pharm. 2019, 105, 1031–1039. [Google Scholar] [CrossRef]
- Batoclimab-HanAll Biopharma/Harbour BioMed/Immunovant-AdisInsight. Available online: https://adisinsight.springer.com/drugs/800043060 (accessed on 16 April 2022).
- Blumberg, L.J.; Humphries, J.E.; Jones, S.D.; Pearce, L.B.; Holgate, R.; Hearn, A.; Cheung, J.; Mahmood, A.; Del Tito, B.; Graydon, J.S.; et al. Blocking FcRn in Humans Reduces Circulating IgG Levels and Inhibits IgG Immune Complex-Mediated Immune Responses. Sci. Adv. 2019, 5, eaax9586. [Google Scholar] [CrossRef]
- Rozanolixizumab (UCB7665)|UCB. Available online: https://www.ucb.com/clinical-studies/Clinical-studies-index/Rozanolixizumab-UCB7665 (accessed on 16 April 2022).
- Yan, C.; Duan, R.-S.; Yang, H.; Li, H.-F.; Zou, Z.; Zhang, H.; Zhou, H.; Li, X.-L.; Zhou, H.; Jiao, L.; et al. Therapeutic Effects of Batoclimab in Chinese Patients with Generalized Myasthenia Gravis: A Double-Blinded, Randomized, Placebo-Controlled Phase II Study. Neurol. Ther. 2022, 11, 815–834. [Google Scholar] [CrossRef]
- Momenta Pharmaceuticals, Inc. A Phase 2, Multicenter, Randomized, Double-Blind, Placebo-Controlled Study to Evaluate the Safety, Tolerability, Efficacy, Pharmacokinetics and Pharmacodynamics of M281 Administered to Adults with Generalized Myasthenia Gravis. 2021. Available online: clinicaltrials.gov (accessed on 16 April 2022).
- Bril, V.; Benatar, M.; Andersen, H.; Vissing, J.; Brock, M.; Greve, B.; Kiessling, P.; Woltering, F.; Griffin, L.; Van den Bergh, P. Efficacy and Safety of Rozanolixizumab in Moderate-to-Severe Generalised Myasthenia Gravis: A Phase 2 RCT. Neurology 2020, 96, e853–e865. [Google Scholar] [CrossRef]
- UCB Announces Positive Phase 3 Results for Rozanolixizumab in Generalized Myasthenia Gravis|UCB. Available online: https://www.ucb.com/stories-media/Press-Releases/article/UCB-announces-positive-Phase-3-results-for-rozanolixizumab-in-generalized-myasthenia-gravis (accessed on 16 April 2022).
- Roy, S.; Nanovskaya, T.; Patrikeeva, S.; Cochran, E.; Parge, V.; Guess, J.; Schaeck, J.; Choudhury, A.; Ahmed, M.; Ling, L.E. M281, an Anti-FcRn Antibody, Inhibits IgG Transfer in a Human Ex Vivo Placental Perfusion Model. Am. J. Obs. Gynecol. 2019, 220, 498.e1–498.e9. [Google Scholar] [CrossRef]
- Janssen Research & Development, LLC A Multicenter, Open-Label Study to Evaluate the Safety, Efficacy, Pharmacokinetics and Pharmacodynamics of M281 Administered to Pregnant Women at High Risk for Early Onset Severe Hemolytic Disease of the Fetus and Newborn (HDFN). 2022. Available online: clinicaltrials.gov (accessed on 16 April 2022).
- Wang, Z.; Fraley, C.; Mezo, A.R. Discovery and Structure-Activity Relationships of Small Molecules That Block the Human Immunoglobulin G-Human Neonatal Fc Receptor (HIgG-HFcRn) Protein-Protein Interaction. Bioorganic Med. Chem. Lett. 2013, 23, 1253–1256. [Google Scholar] [CrossRef] [PubMed]
- Mezo, A.R.; McDonnell, K.A.; Hehir, C.A.T.; Low, S.C.; Palombella, V.J.; Stattel, J.M.; Kamphaus, G.D.; Fraley, C.; Zhang, Y.; Dumont, J.A.; et al. Reduction of IgG in Nonhuman Primates by a Peptide Antagonist of the Neonatal Fc Receptor FcRn. Proc. Natl. Acad. Sci. USA 2008, 105, 2337–2342. [Google Scholar] [CrossRef]
- Seijsing, J.; Yu, S.; Frejd, F.Y.; Höiden-Guthenberg, I.; Gräslund, T. In Vivo Depletion of Serum IgG by an Affibody Molecule Binding the Neonatal Fc Receptor. Sci. Rep. 2018, 8, 5141. [Google Scholar] [CrossRef] [PubMed]
- Seijsing, J.; Lindborg, M.; Höidén-Guthenberg, I.; Bönisch, H.; Guneriusson, E.; Frejd, F.Y.; Abrahmsén, L.; Ekblad, C.; Löfblom, J.; Uhlén, M.; et al. An Engineered Affibody Molecule with PH-Dependent Binding to FcRn Mediates Extended Circulatory Half-Life of a Fusion Protein. Proc. Natl. Acad. Sci. USA 2014, 111, 17110–17115. [Google Scholar] [CrossRef] [PubMed]
- Affibody Announces Termination of ABY-039 (FcRn) Program. Available online: https://www.affibody.se/affibody-announces-termination-of-aby-039-fcrn-program/ (accessed on 16 April 2022).
- Swiercz, R.; Mo, M.; Khare, P.; Schneider, Z.; Ober, R.J.; Ward, E.S. Loss of Expression of the Recycling Receptor, FcRn, Promotes Tumor Cell Growth by Increasing Albumin Consumption. Oncotarget 2017, 8, 3528–3541. [Google Scholar] [CrossRef] [PubMed]
- Dalloneau, E.; Baroukh, N.; Mavridis, K.; Maillet, A.; Gueugnon, F.; Courty, Y.; Petit, A.; Kryza, T.; Del Rio, M.; Guyetant, S.; et al. Downregulation of the Neonatal Fc Receptor Expression in Non-Small Cell Lung Cancer Tissue Is Associated with a Poor Prognosis. Oncotarget 2016, 7, 54415–54429. [Google Scholar] [CrossRef] [PubMed]
- Baker, K.; Rath, T.; Flak, M.B.; Arthur, J.C.; Chen, Z.; Glickman, J.N.; Zlobec, I.; Karamitopoulou, E.; Stachler, M.D.; Odze, R.D.; et al. Neonatal Fc Receptor Expression in Dendritic Cells Mediates Protective Immunity against Colorectal Cancer. Immunity 2013, 39, 1095–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramdani, Y.; Lamamy, J.; Watier, H.; Gouilleux-Gruart, V. Monoclonal Antibody Engineering and Design to Modulate FcRn Activities: A Comprehensive Review. Int. J. Mol. Sci. 2022, 23, 9604. https://doi.org/10.3390/ijms23179604
Ramdani Y, Lamamy J, Watier H, Gouilleux-Gruart V. Monoclonal Antibody Engineering and Design to Modulate FcRn Activities: A Comprehensive Review. International Journal of Molecular Sciences. 2022; 23(17):9604. https://doi.org/10.3390/ijms23179604
Chicago/Turabian StyleRamdani, Yanis, Juliette Lamamy, Hervé Watier, and Valérie Gouilleux-Gruart. 2022. "Monoclonal Antibody Engineering and Design to Modulate FcRn Activities: A Comprehensive Review" International Journal of Molecular Sciences 23, no. 17: 9604. https://doi.org/10.3390/ijms23179604
APA StyleRamdani, Y., Lamamy, J., Watier, H., & Gouilleux-Gruart, V. (2022). Monoclonal Antibody Engineering and Design to Modulate FcRn Activities: A Comprehensive Review. International Journal of Molecular Sciences, 23(17), 9604. https://doi.org/10.3390/ijms23179604