
Comparative Metabolome and Transcriptome Analysis of Anthocyanin Biosynthesis in White and Pink Petals of Cotton (Gossypium hirsutum L.)

 ,
,  , , and
, , and 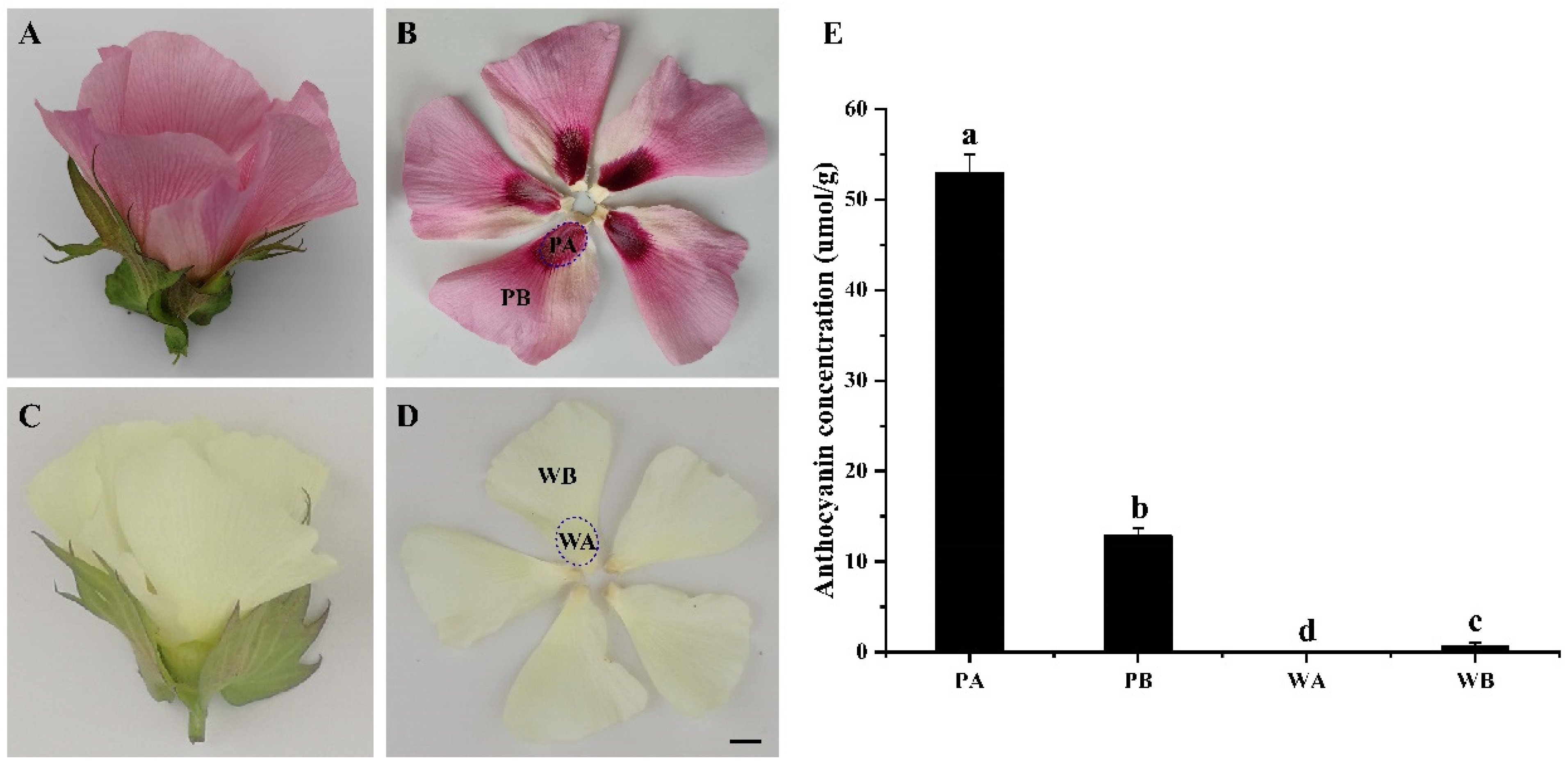{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
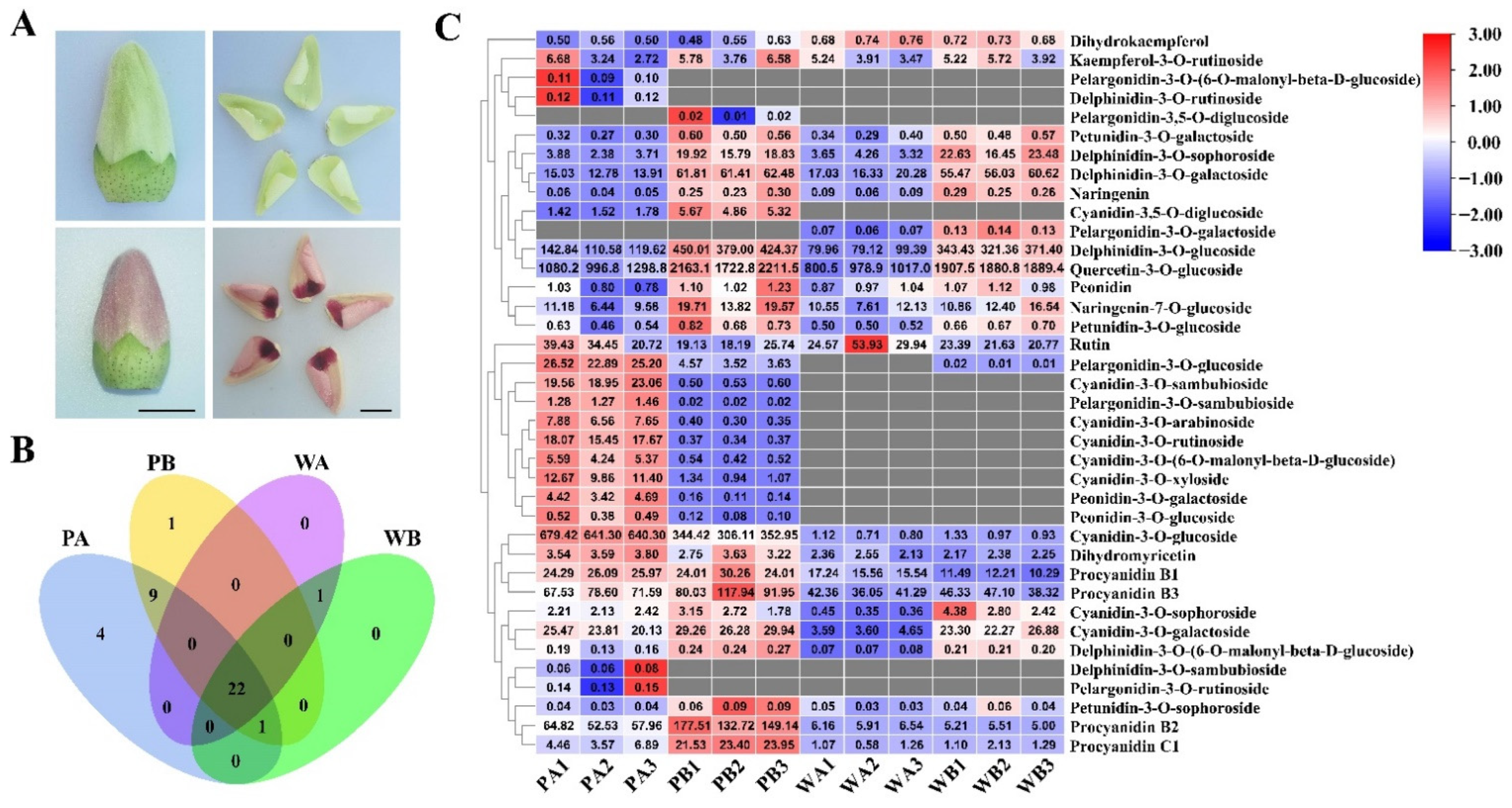
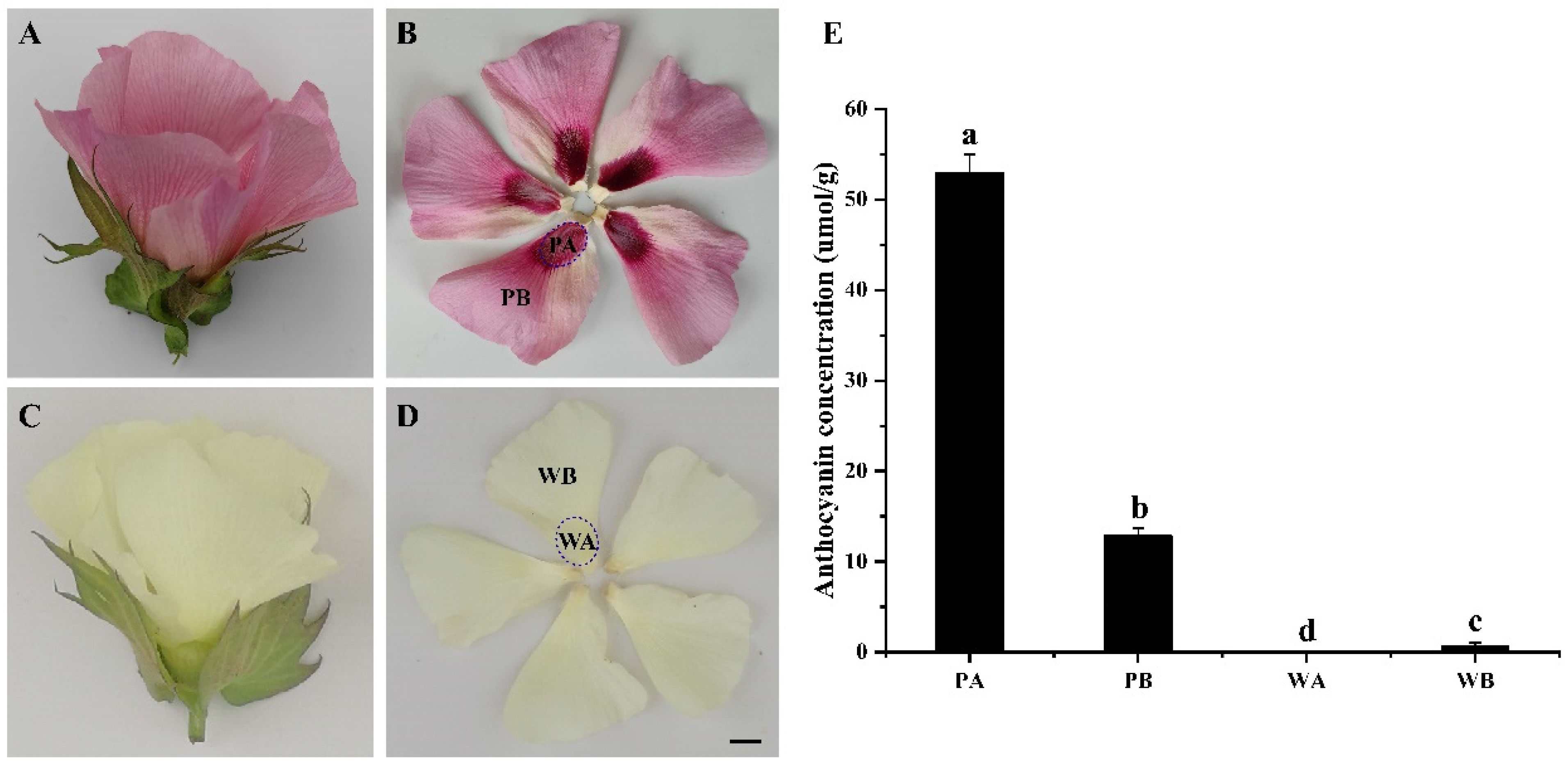
2.1. Analysis of Anthocyanin Composition in Petals of X74 and PF
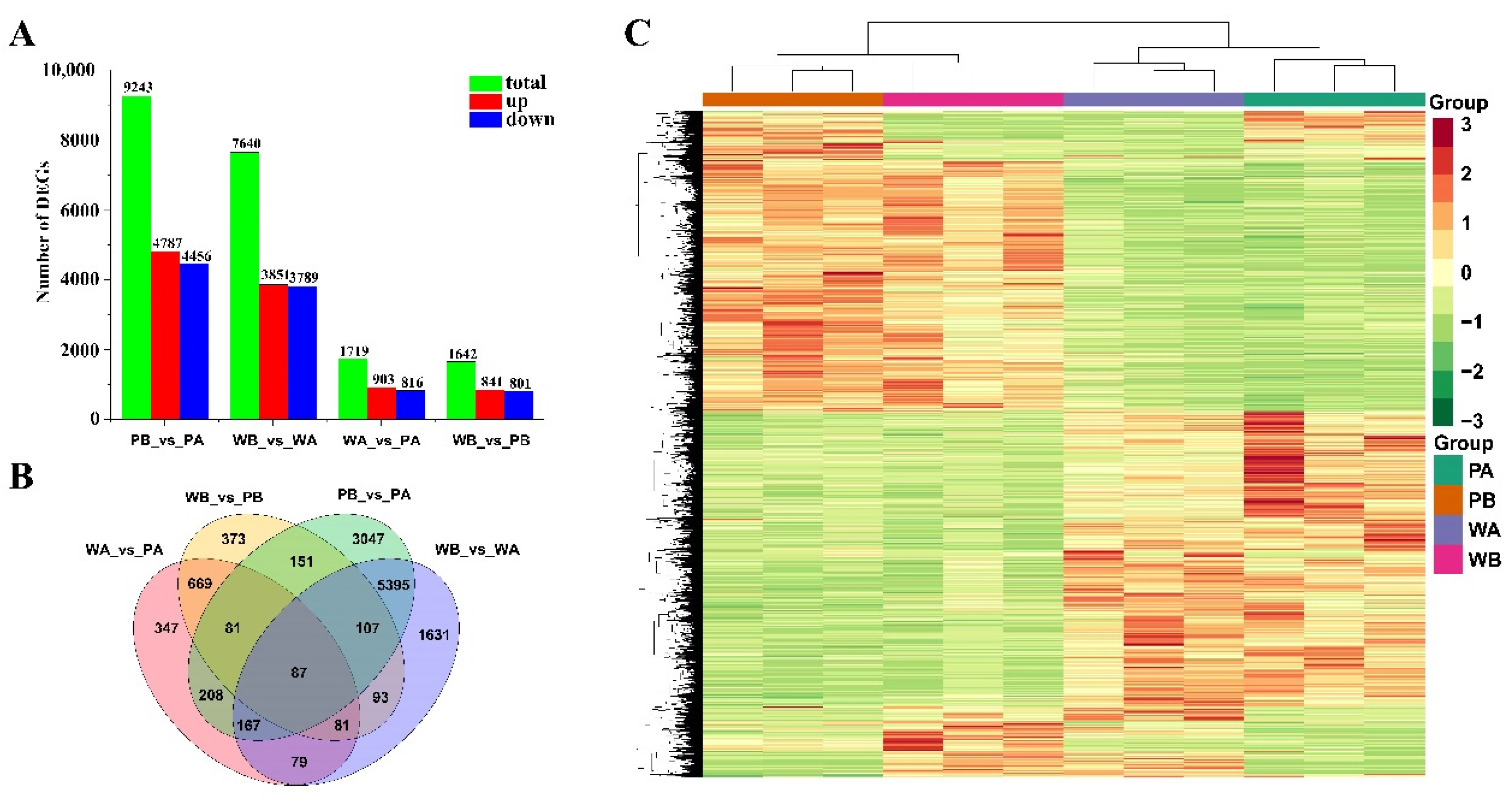
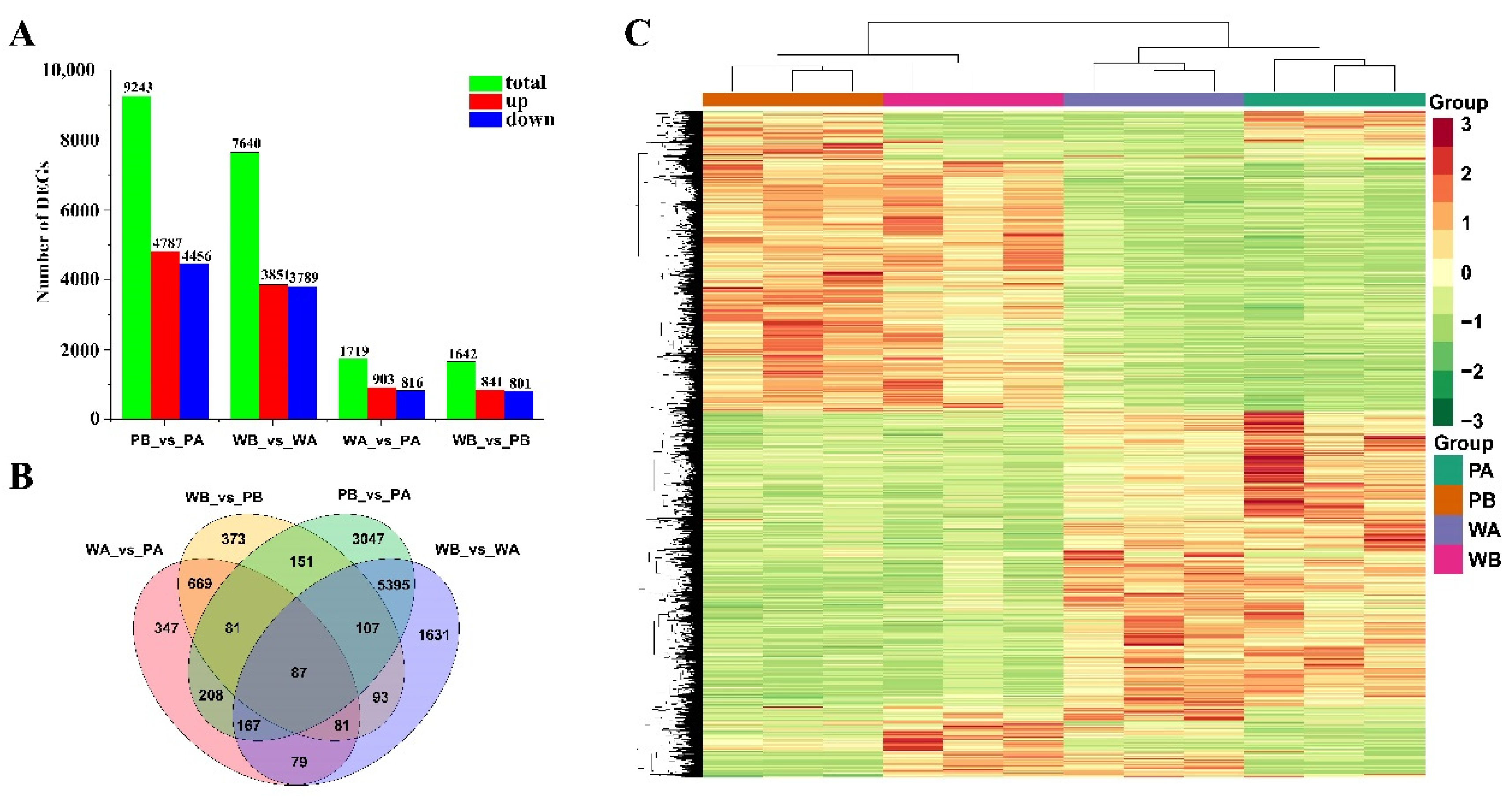
2.2. Overview of the RNA Sequencing Data
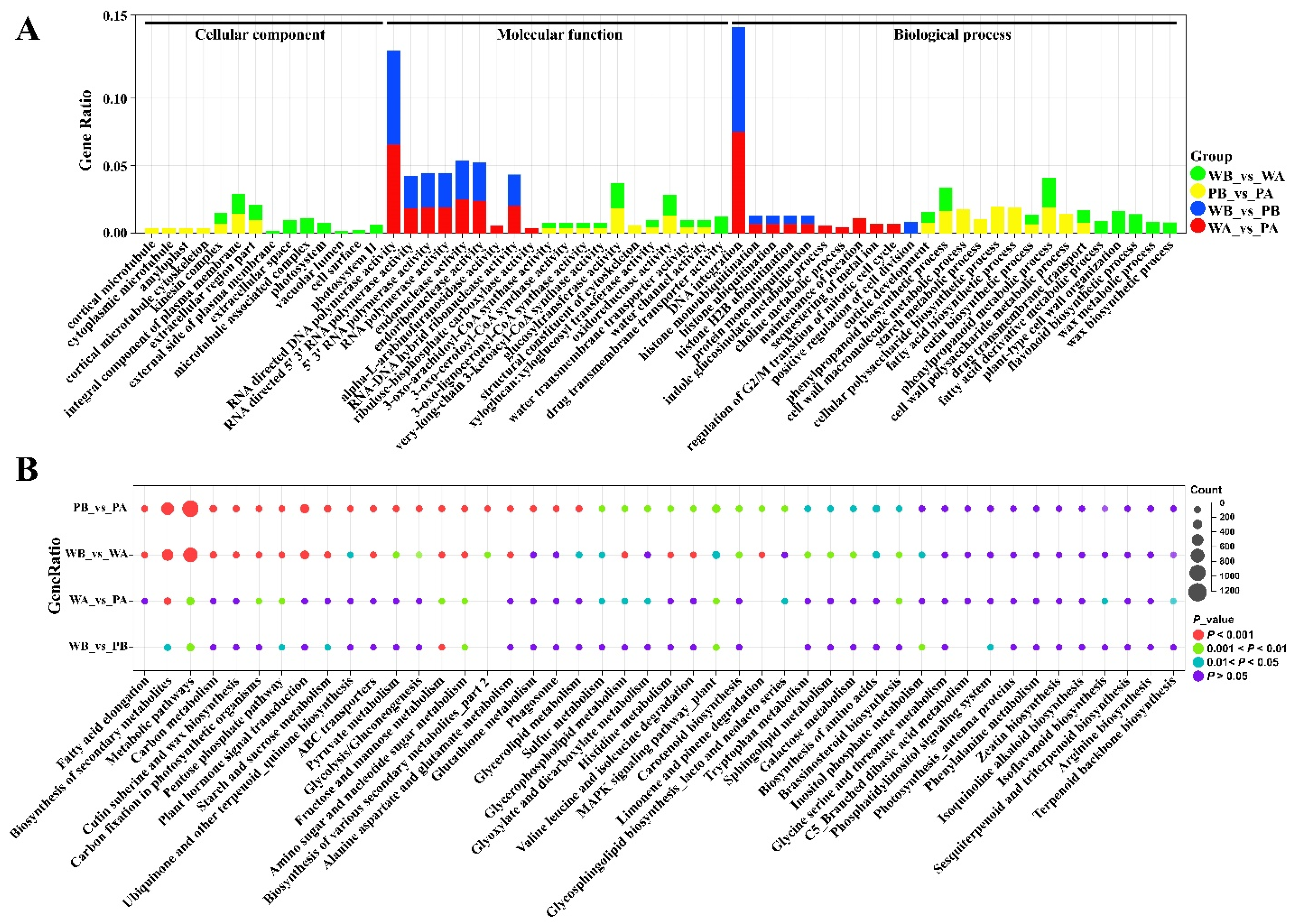
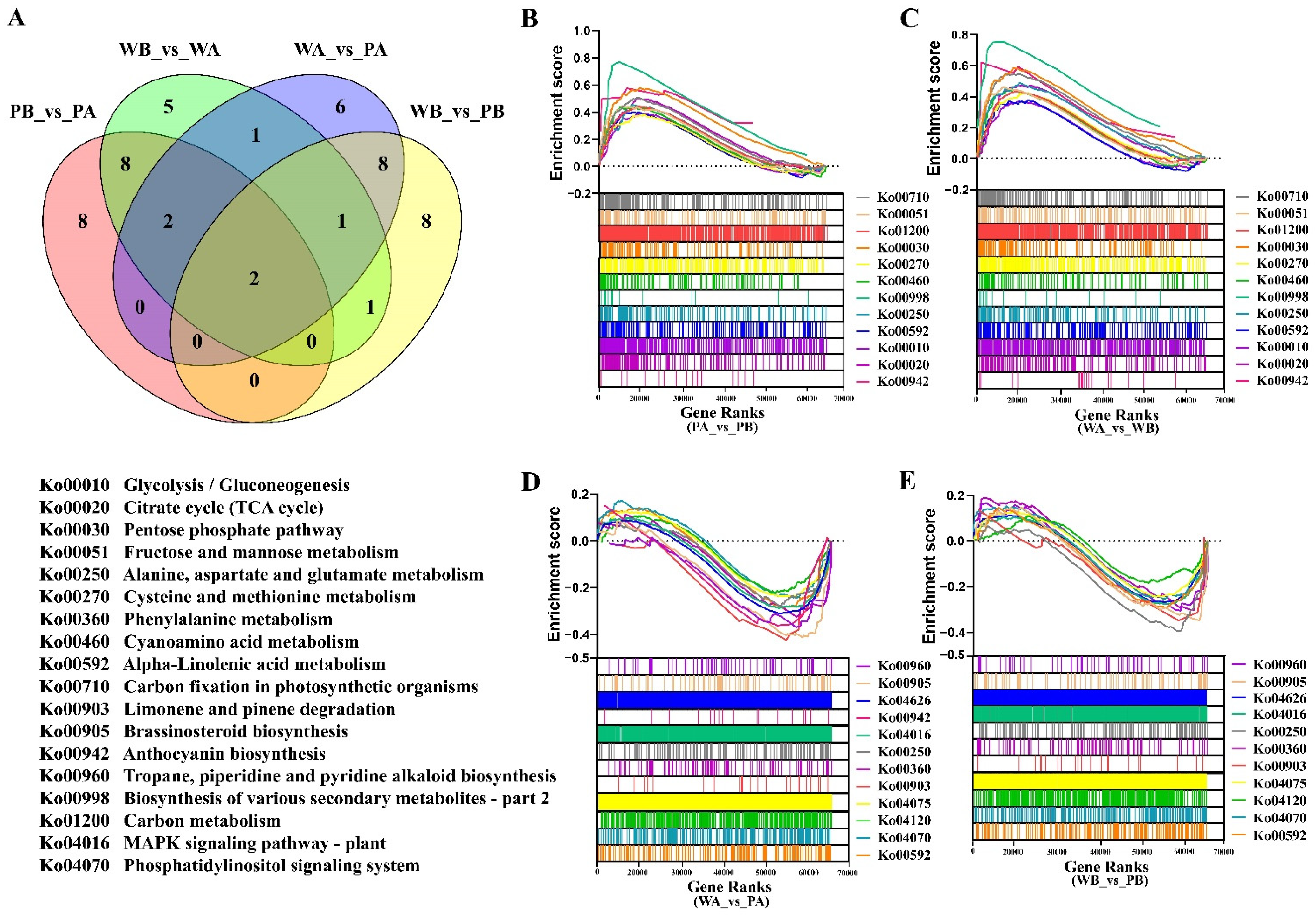
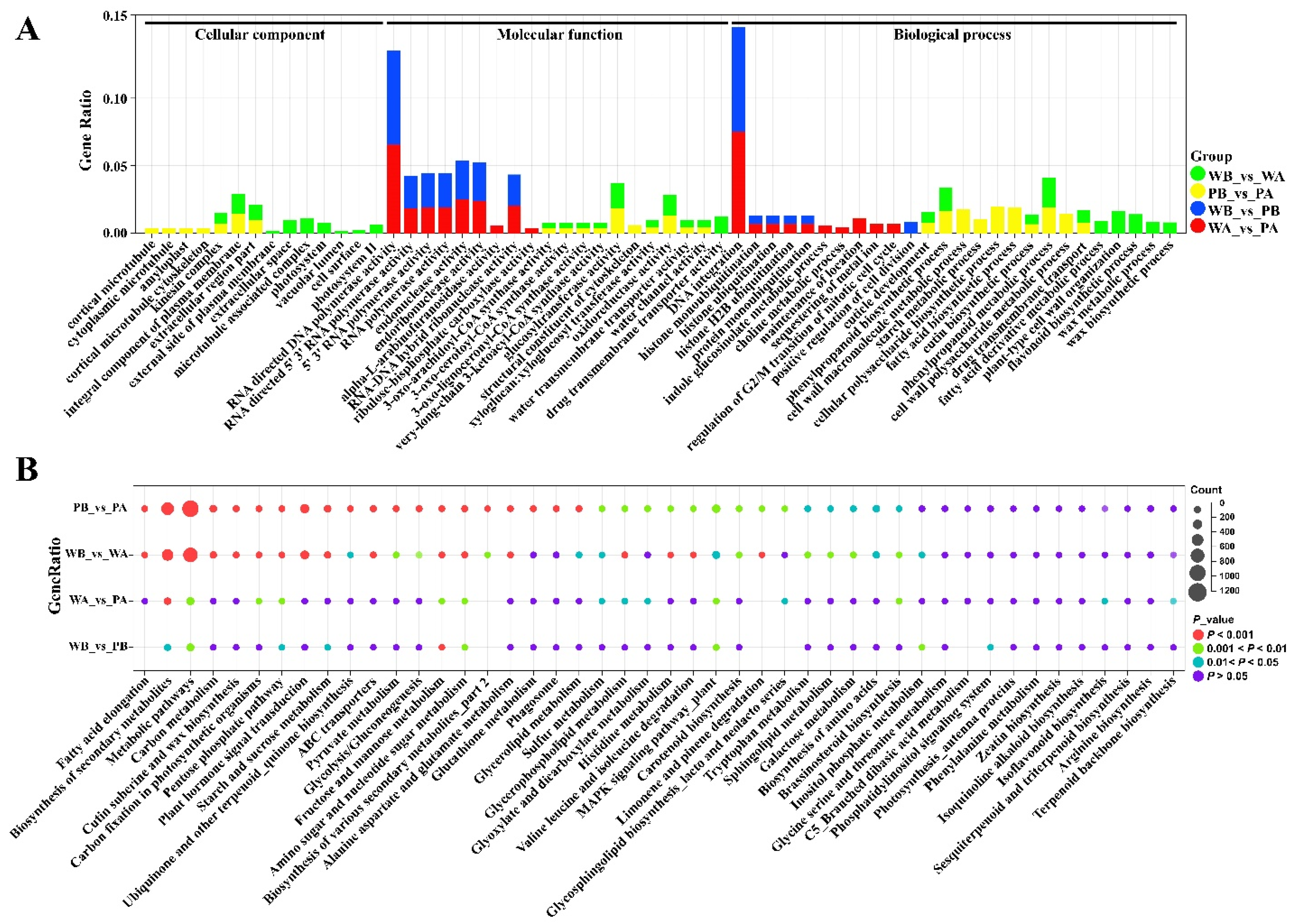
2.3. Analysis of Functional Enrichment of Differentially Expressed Genes
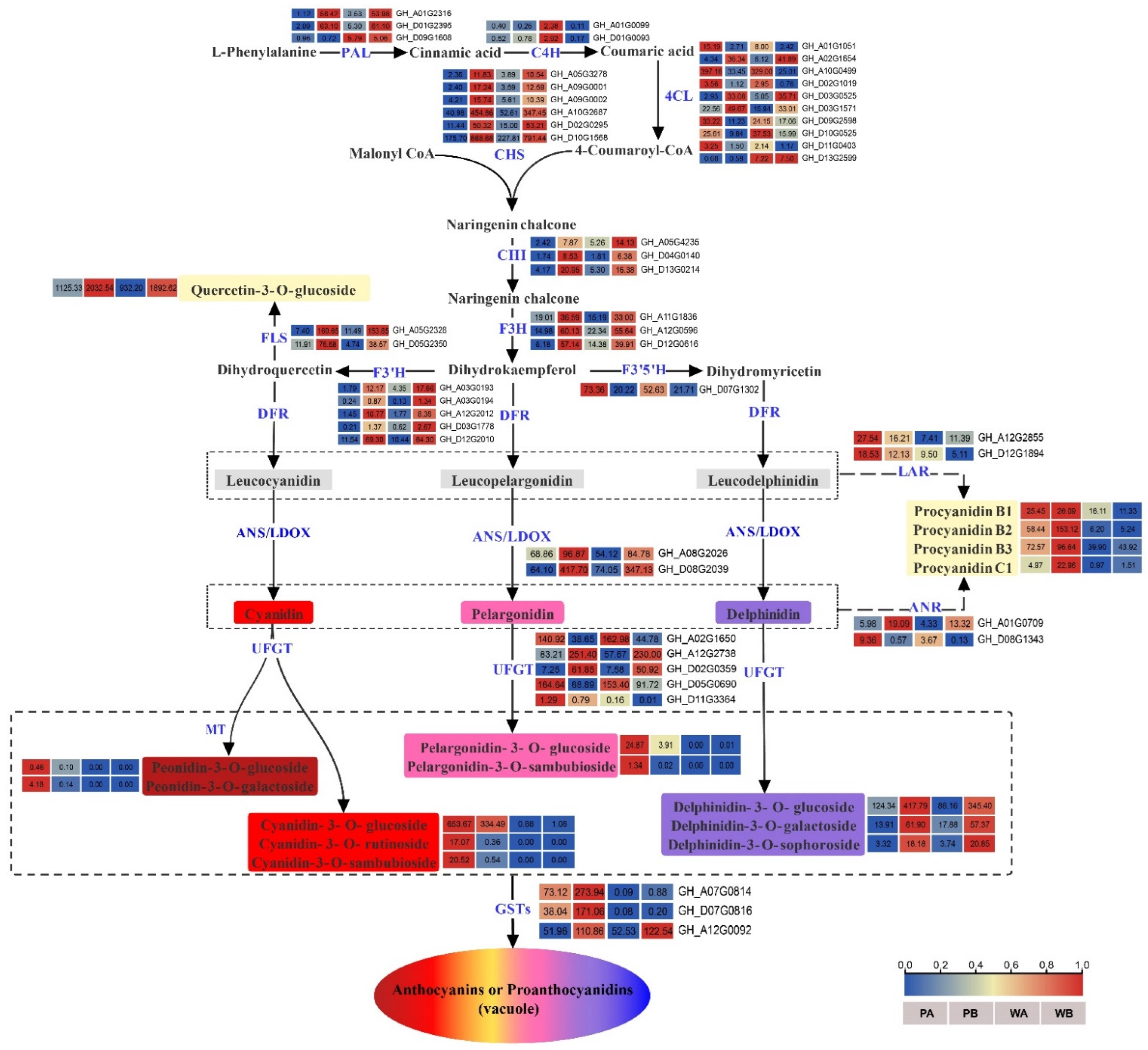
2.4. Analysis of DEGs Related to Biosynthesis and Transport of Anthocyanidins
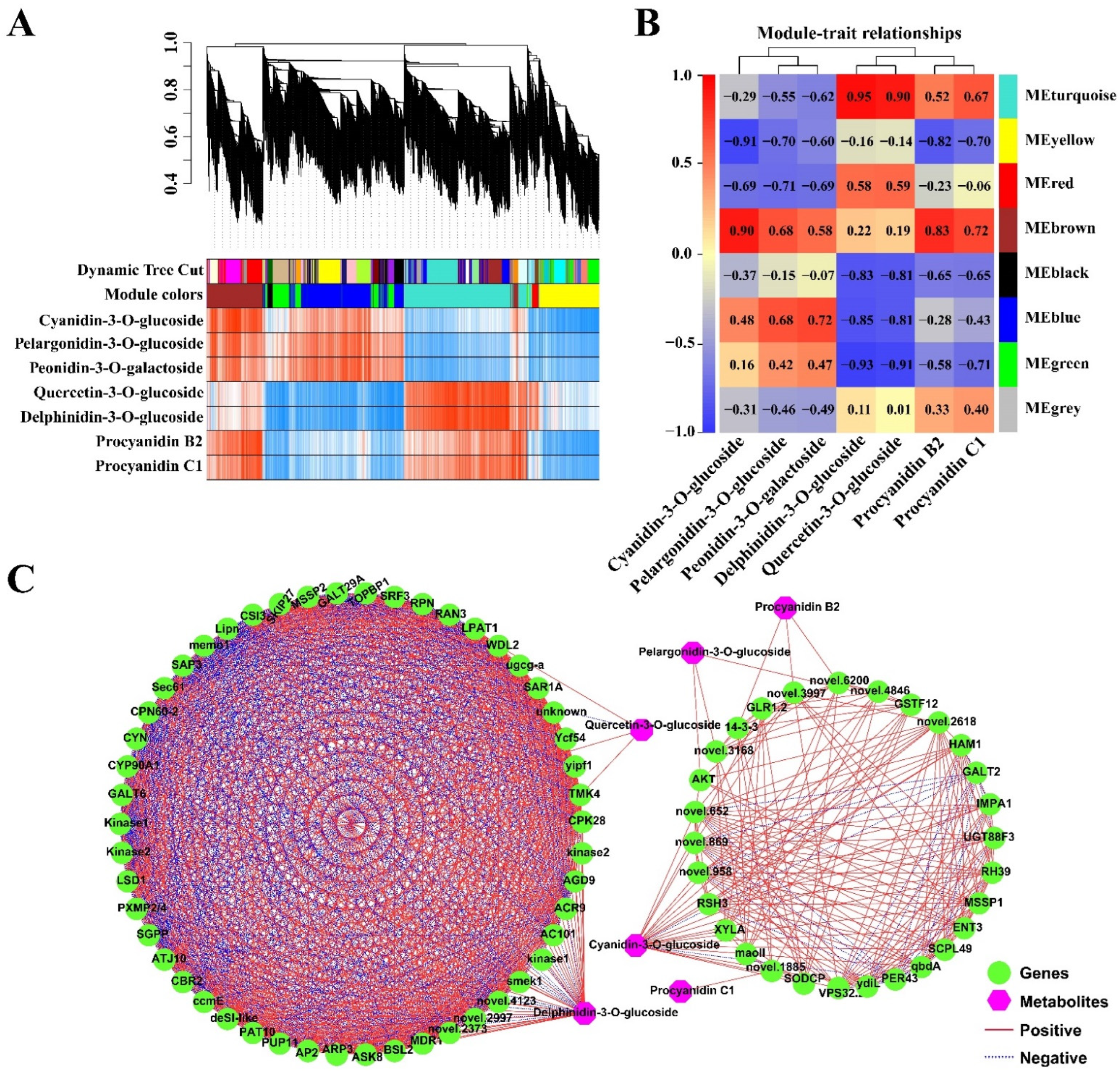
2.5. Co-Expression Analysis of the Genes Related to the Anthocyanidin Biosynthesis Pathway
3. Discussion
3.1. Cyanidin-3-O-Glucoside Is the Main Contributor of the Pink Color Observed in the PF Mutant
3.2. Key Genes Responsible for Biosynthesis and Accumulation of Anthocyanins in PF Petals
3.3. Outlook of Anthocyanin Biosynthesis in Upland Cotton
4. Materials and Methods
4.1. Plant Materials and Sampling
4.2. Extraction and Measurement of Total Anthocyanins
4.3. Qualitative and Quantitative Analysis of Anthocyanins
4.4. RNA Extraction and Sequencing
4.5. Transcriptome Data Processing
4.6. Differential Gene Expression and Pathway Analysis
4.7. Gene Set Enrichment Analysis (GSEA)
4.8. Gene Network Construction and Visualization
4.9. Quantitative Real-Time PCR Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tanaka, Y.; Tsuda, S.; Kusumi, T. Metabolic Engineering to Modify Flower Color. Plant Cell Physiol. 1998, 39, 1119–1126. [Google Scholar] [CrossRef]
- Li, Y.; Fang, J.; Qi, X.; Lin, M.; Zhong, Y.; Sun, L.; Cui, W. Combined Analysis of the Fruit Metabolome and Transcriptome Reveals Candidate Genes Involved in Flavonoid Biosynthesis in Actinidia arguta. Int. J. Mol. Sci. 2018, 19, 1471. [Google Scholar] [CrossRef]
- Albert, N.W.; Davies, K.M.; E Schwinn, K. Gene regulation networks generate diverse pigmentation patterns in plants. Plant Signal. Behav. 2014, 9, e29526. [Google Scholar] [CrossRef]
- Noman, A.; Aqeel, M.; Deng, J.; Khalid, N.; Sanaullah, T.; Shuilin, H. Biotechnological Advancements for Improving Floral Attributes in Ornamental Plants. Front. Plant Sci. 2017, 8, 530. [Google Scholar] [CrossRef]
- Lang, X.; Li, N.; Li, L.; Zhang, S. Integrated Metabolome and Transcriptome Analysis Uncovers the Role of Anthocyanin Metabolism in Michelia maudiae. J. Genom. 2019, 2019, 4393905. [Google Scholar] [CrossRef]
- Peng, J.; Dong, X.; Xue, C.; Liu, Z.; Cao, F. Exploring the Molecular Mechanism of Blue Flower Color Formation in Hydrangea macrophylla cv. “Forever Summer”. Front. Plant Sci. 2021, 12, 585665. [Google Scholar] [CrossRef]
- Wang, R.; Ren, C.; Dong, S.; Chen, C.; Xian, B.; Wu, Q.; Wang, J.; Pei, J.; Chen, J. Integrated Metabolomics and Transcriptome Analysis of Flavonoid Biosynthesis in Safflower (Carthamus tinctorius L.) with Different Colors. Front. Plant Sci. 2021, 12, 712038. [Google Scholar] [CrossRef]
- Petroni, K.; Tonelli, C. Recent advances on the regulation of anthocyanin synthesis in reproductive organs. Plant Sci. 2011, 181, 219–229. [Google Scholar] [CrossRef]
- Tanaka, Y.; Sasaki, N.; Ohmiya, A. Biosynthesis of plant pigments: Anthocyanins, betalains and carotenoids. Plant J. 2008, 54, 733–749. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, Q.; Wang, X.; Zuo, Z.; Oka, Y.; Lin, C. New insights into the mechanisms of phytochrome-cryptochrome coaction. New Phytol. 2018, 217, 547–551. [Google Scholar] [CrossRef] [Green Version]
- Zheng, T.; Tan, W.; Yang, H.; Zhang, L.; Li, T.; Liu, B.; Zhang, D.; Lin, H. Regulation of anthocyanin accumulation via MYB75/HAT1/TPL-mediated transcriptional repression. PLoS Genet. 2019, 15, e1007993. [Google Scholar] [CrossRef]
- Samkumar, A.; Jones, D.; Karppinen, K.; Dare, A.P.; Sipari, N.; Espley, R.V.; Martinussen, I.; Jaakola, L. Red and blue light treatments of ripening bilberry fruits reveal differences in signalling through abscisic acid-regulated anthocyanin biosynthesis. Plant Cell Environ. 2021, 44, 3227–3245. [Google Scholar] [CrossRef]
- Zhang, B.; Chopra, D.; Schrader, A.; Hülskamp, M. Evolutionary comparison of competitive protein-complex formation of MYB, bHLH, and WDR proteins in plants. J. Exp. Bot. 2019, 70, 3197–3209. [Google Scholar] [CrossRef]
- Zhao, J. Flavonoid transport mechanisms: How to go, and with whom. Trends Plant Sci. 2015, 20, 576–585. [Google Scholar] [CrossRef]
- Zhao, J.; Dixon, R.A. The ‘ins’ and ‘outs’ of flavonoid transport. Trends Plant Sci. 2010, 15, 72–80. [Google Scholar] [CrossRef]
- Kim, S.-J.; Brandizzi, F. News and Views into the SNARE Complexity in Arabidopsis. Front. Plant Sci. 2012, 3, 28. [Google Scholar] [CrossRef]
- Pecenkova, T.; Marković, V.; Sabol, P.; Kulich, I.; Žárský, V. Exocyst and autophagy-related membrane trafficking in plants. J. Exp. Bot. 2017, 69, 47–57. [Google Scholar] [CrossRef]
- Lloyd, A.; Brockman, A.; Aguirre, L.; Campbell, A.; Bean, A.; Cantero, A.; Gonzalez, A. Advances in the MYB–bHLH–WD Repeat (MBW) Pigment Regulatory Model: Addition of a WRKY Factor and Co-option of an Anthocyanin MYB for Betalain Regulation. Plant Cell Physiol. 2017, 58, 1431–1441. [Google Scholar] [CrossRef]
- Naing, A.H.; Kil Kim, C. Roles of R2R3-MYB transcription factors in transcriptional regulation of anthocyanin biosynthesis in horticultural plants. Plant Mol. Biol. 2018, 98, 1–18. [Google Scholar] [CrossRef]
- An, J.; Zhang, X.; You, C.; Bi, S.; Wang, X.; Hao, Y. Md WRKY 40 promotes wounding-induced anthocyanin biosynthesis in association with Md MYB 1 and undergoes Md BT 2-mediated degradation. New Phytol. 2019, 224, 380–395. [Google Scholar] [CrossRef]
- Bhatia, C.; Gaddam, S.R.; Pandey, A.; Trivedi, P.K. COP1 mediates light-dependent regulation of flavonol biosynthesis through HY5 in Arabidopsis. Plant Sci. 2021, 303, 110760. [Google Scholar] [CrossRef]
- Li, C.; Pei, J.; Yan, X.; Cui, X.; Tsuruta, M.; Liu, Y.; Lian, C. A poplar B-box protein PtrBBX23 modulates the accumulation of anthocyanins and proanthocyanidins in response to high light. Plant Cell Environ. 2021, 44, 3015–3033. [Google Scholar] [CrossRef]
- Liu, C.-C.; Chi, C.; Jin, L.-J.; Zhu, J.; Yu, J.-Q.; Zhou, Y.-H. The bZip transcription factor HY5 mediates CRY1a -induced anthocyanin biosynthesis in tomato. Plant Cell Environ. 2018, 41, 1762–1775. [Google Scholar] [CrossRef]
- Ma, H.; Yang, T.; Li, Y.; Zhang, J.; Wu, T.; Song, T.; Yao, Y.; Tian, J. The long noncoding RNA MdLNC499 bridges MdWRKY1 and MdERF109 function to regulate early-stage light-induced anthocyanin accumulation in apple fruit. Plant Cell 2021, 33, 3309–3330. [Google Scholar] [CrossRef]
- Maier, A.; Schrader, A.; Kokkelink, L.; Falke, C.; Welter, B.; Iniesto, E.; Rubio, V.; Uhrig, J.F.; Hülskamp, M.; Hoecker, U. Light and the E3 ubiquitin ligase COP1/SPA control the protein stability of the MYB transcription factors PAP1 and PAP2 involved in anthocyanin accumulation in Arabidopsis. Plant J. 2013, 74, 638–651. [Google Scholar] [CrossRef]
- Ou, C.; Zhang, X.; Wang, F.; Zhang, L.; Zhang, Y.; Fang, M.; Wang, J.; Wang, J.; Jiang, S.; Zhang, Z. A 14 nucleotide deletion mutation in the coding region of the PpBBX24 gene is associated with the red skin of “Zaosu Red” pear (Pyrus pyrifolia White Pear Group): A deletion in the PpBBX24 gene is associated with the red skin of pear. Hortic. Res. 2020, 7, 39. [Google Scholar] [CrossRef]
- Qian, M.; Ni, J.; Niu, Q.; Bai, S.; Bao, L.; Li, J.; Sun, Y.; Zhang, D.; Teng, Y. Response of miR156-SPL Module during the Red Peel Coloration of Bagging-Treated Chinese Sand Pear (Pyrus pyrifolia Nakai). Front. Physiol. 2017, 8, 550. [Google Scholar] [CrossRef]
- Shin, J.; Park, E.; Choi, G. PIF3 regulates anthocyanin biosynthesis in an HY5-dependent manner with both factors directly binding anthocyanin biosynthetic gene promoters in Arabidopsis. Plant J. 2007, 49, 981–994. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, W.; Wang, X.; Yang, R.; Wu, Z.; Wang, H.; Wang, L.; Hu, Z.; Guo, S.; Zhang, H.; et al. MiR156 regulates anthocyanin biosynthesis through SPL targets and other microRNAs in poplar. Hortic. Res. 2020, 7, 118. [Google Scholar] [CrossRef]
- Xie, Y.; Tan, H.; Ma, Z.; Huang, J. DELLA Proteins Promote Anthocyanin Biosynthesis via Sequestering MYBL2 and JAZ Suppressors of the MYB/bHLH/WD40 Complex in Arabidopsis thaliana. Mol. Plant 2016, 9, 711–721. [Google Scholar] [CrossRef] [Green Version]
- An, J.-P.; Zhang, X.-W.; Bi, S.-Q.; You, C.-X.; Wang, X.-F.; Hao, Y.-J. The ERF transcription factor MdERF38 promotes drought stress-induced anthocyanin biosynthesis in apple. Plant J. 2020, 101, 573–589. [Google Scholar] [CrossRef]
- Kim, M.; Kim, P.; Chen, Y.; Chen, B.; Yang, J.; Liu, X.; Kawabata, S.; Wang, Y.; Li, Y. Blue and UV-B light synergistically induce anthocyanin accumulation by co-activating nitrate reductase gene expression in Anthocyanin fruit (Aft) tomato. Plant Biol. 2020, 23, 210–220. [Google Scholar] [CrossRef]
- Tao, R.; Bai, S.; Ni, J.; Yang, Q.; Zhao, Y.; Teng, Y. The blue light signal transduction pathway is involved in anthocyanin accumulation in ‘Red Zaosu’ pear. Planta 2018, 248, 37–48. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, A.; Wu, X.; Zhu, Z.; Yang, Z.; Zhu, Y.; Zha, D. Transcriptome analysis revealed expression of genes related to anthocyanin biosynthesis in eggplant (Solanum melongena L.) under high-temperature stress. BMC Plant Biol. 2019, 19, 387. [Google Scholar] [CrossRef]
- Zhao, Y.; Min, T.; Chen, M.; Wang, H.; Zhu, C.; Jin, R.; Allan, A.C.; Lin-Wang, K.; Xu, C. The Photomorphogenic Transcription Factor PpHY5 Regulates Anthocyanin Accumulation in Response to UVA and UVB Irradiation. Front. Plant Sci. 2020, 11, 603178. [Google Scholar] [CrossRef]
- Zoratti, L.; Karppinen, K.; Escobar, A.L.; Hãggman, H.; Jaakola, L. Light-controlled flavonoid biosynthesis in fruits. Front. Plant Sci. 2014, 5, 534. [Google Scholar] [CrossRef]
- Tan, J.; Wang, M.; Tu, L.; Nie, Y.; Lin, Y.; Zhang, X. The Flavonoid Pathway Regulates the Petal Colors of Cotton Flower. PLoS ONE 2013, 8, e72364. [Google Scholar] [CrossRef]
- Ashraf, J.; Zuo, D.; Wang, Q.; Malik, W.; Zhang, Y.; Abid, M.A.; Cheng, H.; Yang, Q.; Song, G. Recent insights into cotton functional genomics: Progress and future perspectives. Plant Biotechnol. J. 2018, 16, 699–713. [Google Scholar] [CrossRef]
- Gao, Z.; Liu, C.; Zhang, Y.; Li, Y.; Yi, K.; Zhao, X.; Cui, M.-L. The Promoter Structure Differentiation of a MYB Transcription Factor RLC1 Causes Red Leaf Coloration in Empire Red Leaf Cotton under Light. PLoS ONE 2013, 8, e77891. [Google Scholar] [CrossRef]
- Liang, A.; Zhao, J.; Li, X.; Yan, F.; Chen, Z.; Chen, X.; Wang, Y.; Li, Y.; Wang, C.; Xiao, Y. Up-regulation of GhPAP1A results in moderate anthocyanin accumulation and pigmentation in sub-red cotton. Mol. Genet. Genom. 2020, 295, 1393–1400. [Google Scholar] [CrossRef]
- Shao, D.; Li, Y.; Zhu, Q.; Zhang, X.; Liu, F.; Xue, F.; Sun, J. GhGSTF12, a glutathione S-transferase gene, is essential for anthocyanin accumulation in cotton (Gossypium hirsutum L.). Plant Sci. 2021, 305, 110827. [Google Scholar] [CrossRef]
- Li, X.; Ouyang, X.; Zhang, Z.; He, L.; Wang, Y.; Li, Y.; Zhao, J.; Chen, Z.; Wang, C.; Ding, L.; et al. Over-expression of the red plant gene R1 enhances anthocyanin production and resistance to bollworm and spider mite in cotton. Mol. Genet. Genom. 2019, 294, 469–478. [Google Scholar] [CrossRef]
- Long, L.; Liu, J.; Gao, Y.; Xu, F.-C.; Zhao, J.-R.; Li, B.; Gao, W. Flavonoid accumulation in spontaneous cotton mutant results in red coloration and enhanced disease resistance. Plant Physiol. Biochem. 2019, 143, 40–49. [Google Scholar] [CrossRef]
- Abid, M.A.; Wei, Y.; Meng, Z.; Wang, Y.; Ye, Y.; Wang, Y.; He, H.; Zhou, Q.; Li, Y.; Wang, P.; et al. Increasing floral visitation and hybrid seed production mediated by beauty mark in Gossypium hirsutum. Plant Biotechnol. J. 2022, 20, 1274–1284. [Google Scholar] [CrossRef]
- He, Y.; Chen, H.; Zhou, L.; Liu, Y.; Chen, H. Comparative transcription analysis of photosensitive and non-photosensitive eggplants to identify genes involved in dark regulated anthocyanin synthesis. BMC Genom. 2019, 20, 678. [Google Scholar] [CrossRef]
- Zheng, X.; Zhu, K.; Sun, Q.; Zhang, W.; Wang, X.; Cao, H.; Tan, M.; Xie, Z.; Zeng, Y.; Ye, J.; et al. Natural Variation in CCD4 Promoter Underpins Species-Specific Evolution of Red Coloration in Citrus Peel. Mol. Plant 2019, 12, 1294–1307. [Google Scholar] [CrossRef]
- Espley, R.V.; Brendolise, C.; Chagné, D.; Kutty-Amma, S.; Green, S.; Volz, R.; Putterill, J.; Schouten, H.J.; Gardiner, S.E.; Hellens, R.P.; et al. Multiple Repeats of a Promoter Segment Causes Transcription Factor Autoregulation in Red Apples. Plant Cell 2009, 21, 168–183. [Google Scholar] [CrossRef]
- Sun, C.; Deng, L.; Du, M.; Zhao, J.; Chen, Q.; Huang, T.; Jiang, H.; Li, C.-B.; Li, C. A Transcriptional Network Promotes Anthocyanin Biosynthesis in Tomato Flesh. Mol. Plant 2020, 13, 42–58. [Google Scholar] [CrossRef]
- Zhang, J.; Lei, Y.; Wang, B.; Li, S.; Yu, S.; Wang, Y.; Li, H.; Liu, Y.; Ma, Y.; Dai, H.; et al. The high-quality genome of diploid strawberry (Fragaria nilgerrensis) provides new insights into anthocyanin accumulation. Plant Biotechnol. J. 2020, 18, 1908–1924. [Google Scholar] [CrossRef]
- Xiang, L.; Liu, X.; Li, H.; Yin, X.; Grierson, D.; Li, F.; Chen, K. CmMYB#7, an R3 MYB transcription factor, acts as a negative regulator of anthocyanin biosynthesis in chrysanthemum. J. Exp. Bot. 2019, 70, 3111–3123. [Google Scholar] [CrossRef]
- Zhang, B.; Xu, X.; Huang, R.; Yang, S.; Li, M.; Guo, Y. CRISPR/Cas9-mediated targeted mutation reveals a role for AN4 rather than DPL in regulating venation formation in the corolla tube of Petunia hybrida. Hortic. Res. 2021, 8, 116. [Google Scholar] [CrossRef]
- Zhang, Y.; Cheng, Y.; Ya, H.; Xu, S.; Han, J. Transcriptome sequencing of purple petal spot region in tree peony reveals differentially expressed anthocyanin structural genes. Front. Plant Sci. 2015, 6, 964. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Yang, X.; Zheng, J.; Wang, L.; Yang, X.; Tu, Y.; Ye, J.; Zhang, W.; Liao, Y.; Cheng, S.; et al. Unraveling the Regulatory Mechanism of Color Diversity in Camellia japonica Petals by Integrative Transcriptome and Metabolome Analysis. Front. Plant Sci. 2021, 12, 685136. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Mei, X.; Rothenberg, D.O.; Yang, Z.; Zhang, W.; Wan, S.; Yang, H.; Zhang, L. Metabolome and Transcriptome Analysis Reveals Putative Genes Involved in Anthocyanin Accumulation and Coloration in White and Pink Tea (Camellia sinensis) Flower. Molecules 2020, 25, 190. [Google Scholar] [CrossRef] [PubMed]
- Chaves-Silva, S.; dos Santos, A.L.; Chalfun-Júnior, A.; Zhao, J.; Peres, L.E.; Benedito, V.A. Understanding the genetic regulation of anthocyanin biosynthesis in plants—Tools for breeding purple varieties of fruits and vegetables. Phytochemistry 2018, 153, 11–27. [Google Scholar] [CrossRef] [PubMed]
- Yonekura-Sakakibara, K.; Fukushima, A.; Nakabayashi, R.; Hanada, K.; Matsuda, F.; Sugawara, S.; Inoue, E.; Kuromori, T.; Ito, T.; Shinozaki, K.; et al. Two glycosyltransferases involved in anthocyanin modification delineated by transcriptome independent component analysis in Arabidopsis thaliana. Plant J. 2011, 69, 154–167. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Li, Y.; Wang, S.; Zhang, L.; Liu, Y.; Xue, F.; Sun, Y.; Wang, Y.; Sun, J. Molecular analysis of proanthocyanidins related to pigmentation in brown cotton fibre (Gossypium hirsutum L.). J. Exp. Bot. 2014, 65, 5759–5769. [Google Scholar] [CrossRef]
- Sun, S.; Xiong, X.-P.; Zhu, Q.; Li, Y.-J.; Sun, J. Transcriptome Sequencing and Metabolome Analysis Reveal Genes Involved in Pigmentation of Green-Colored Cotton Fibers. Int. J. Mol. Sci. 2019, 20, 4838. [Google Scholar] [CrossRef]
- Yan, Q.; Wang, Y.; Li, Q.; Zhang, Z.; Ding, H.; Zhang, Y.; Liu, H.; Luo, M.; Liu, D.; Song, W.; et al. Up-regulation of GhTT2-3A in cotton fibres during secondary wall thickening results in brown fibres with improved quality. Plant Biotechnol. J. 2018, 16, 1735–1747. [Google Scholar] [CrossRef]
- Sun, J.; Sun, Y.; Zhu, Q.-H. Breeding Next-Generation Naturally Colored Cotton. Trends Plant Sci. 2021, 26, 539–542. [Google Scholar] [CrossRef]
- Wang, N.; Zhang, B.; Yao, T.; Shen, C.; Wen, T.; Zhang, R.; Li, Y.; Le, Y.; Li, Z.; Zhang, X.; et al. Re enhances anthocyanin and proanthocyanidin accumulation to produce red foliated cotton and brown fiber. Plant Physiol. 2022, 189, 1466–1481. [Google Scholar] [CrossRef] [PubMed]
- Perez-Prat, E.; van Lookeren Campagne, M.M. Hybrid seed production and the challenge of propagating male-sterile plants. Trends Plant Sci. 2002, 7, 199–203. [Google Scholar] [CrossRef]
- Kim, Y.-J.; Zhang, D. Molecular Control of Male Fertility for Crop Hybrid Breeding. Trends Plant Sci. 2018, 23, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Chang, Z.; Chen, Z.; Wang, N.; Xie, G.; Lu, J.; Yan, W.; Zhou, J.; Tang, X.; Deng, X.W. Construction of a male sterility system for hybrid rice breeding and seed production using a nuclear male sterility gene. Proc. Natl. Acad. Sci. USA 2016, 113, 14145–14150. [Google Scholar] [CrossRef] [PubMed]
- Du, M.; Zhou, K.; Liu, Y.; Deng, L.; Zhang, X.; Lin, L.; Zhou, M.; Zhao, W.; Wen, C.; Xing, J.; et al. A biotechnology-based male-sterility system for hybrid seed production in tomato. Plant J. 2020, 102, 1090–1100. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Fox, T.W.; Trimnell, M.R.; Wang, L.; Xu, R.; Cigan, A.M.; Huffman, G.A.; Garnaat, C.W.; Hershey, H.; Albertsen, M.C. Development of a novel recessive genetic male sterility system for hybrid seed production in maize and other cross-pollinating crops. Plant Biotechnol. J. 2015, 14, 1046–1054. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, P.; McDurmon, A.; Mattoon, E.; Keefe, M.; Carley, L.; Lee, C.; Bingham, R.; Anderson, J.T. Ecological causes and consequences of flower color polymorphism in a self-pollinating plant (Boechera stricta). New Phytol. 2018, 218, 380–392. [Google Scholar] [CrossRef]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Zheng, Y.; Jiao, C.; Sun, H.; Rosli, H.G.; Pombo, M.A.; Zhang, P.; Banf, M.; Dai, X.; Martin, G.B.; Giovannoni, J.J.; et al. iTAK: A Program for Genome-wide Prediction and Classification of Plant Transcription Factors, Transcriptional Regulators, and Protein Kinases. Mol. Plant 2016, 9, 1667–1670. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shao, D.; Liang, Q.; Wang, X.; Zhu, Q.-H.; Liu, F.; Li, Y.; Zhang, X.; Yang, Y.; Sun, J.; Xue, F. Comparative Metabolome and Transcriptome Analysis of Anthocyanin Biosynthesis in White and Pink Petals of Cotton (Gossypium hirsutum L.). Int. J. Mol. Sci. 2022, 23, 10137. https://doi.org/10.3390/ijms231710137
Shao D, Liang Q, Wang X, Zhu Q-H, Liu F, Li Y, Zhang X, Yang Y, Sun J, Xue F. Comparative Metabolome and Transcriptome Analysis of Anthocyanin Biosynthesis in White and Pink Petals of Cotton (Gossypium hirsutum L.). International Journal of Molecular Sciences. 2022; 23(17):10137. https://doi.org/10.3390/ijms231710137
Chicago/Turabian StyleShao, Dongnan, Qian Liang, Xuefeng Wang, Qian-Hao Zhu, Feng Liu, Yanjun Li, Xinyu Zhang, Yonglin Yang, Jie Sun, and Fei Xue. 2022. "Comparative Metabolome and Transcriptome Analysis of Anthocyanin Biosynthesis in White and Pink Petals of Cotton (Gossypium hirsutum L.)" International Journal of Molecular Sciences 23, no. 17: 10137. https://doi.org/10.3390/ijms231710137
APA StyleShao, D., Liang, Q., Wang, X., Zhu, Q.-H., Liu, F., Li, Y., Zhang, X., Yang, Y., Sun, J., & Xue, F. (2022). Comparative Metabolome and Transcriptome Analysis of Anthocyanin Biosynthesis in White and Pink Petals of Cotton (Gossypium hirsutum L.). International Journal of Molecular Sciences, 23(17), 10137. https://doi.org/10.3390/ijms231710137