

Factors and Pathways Modulating Endothelial Cell Senescence in Vascular Aging

 , and
, and {kind=link}
{kind=link}
Abstract
:1. Introduction
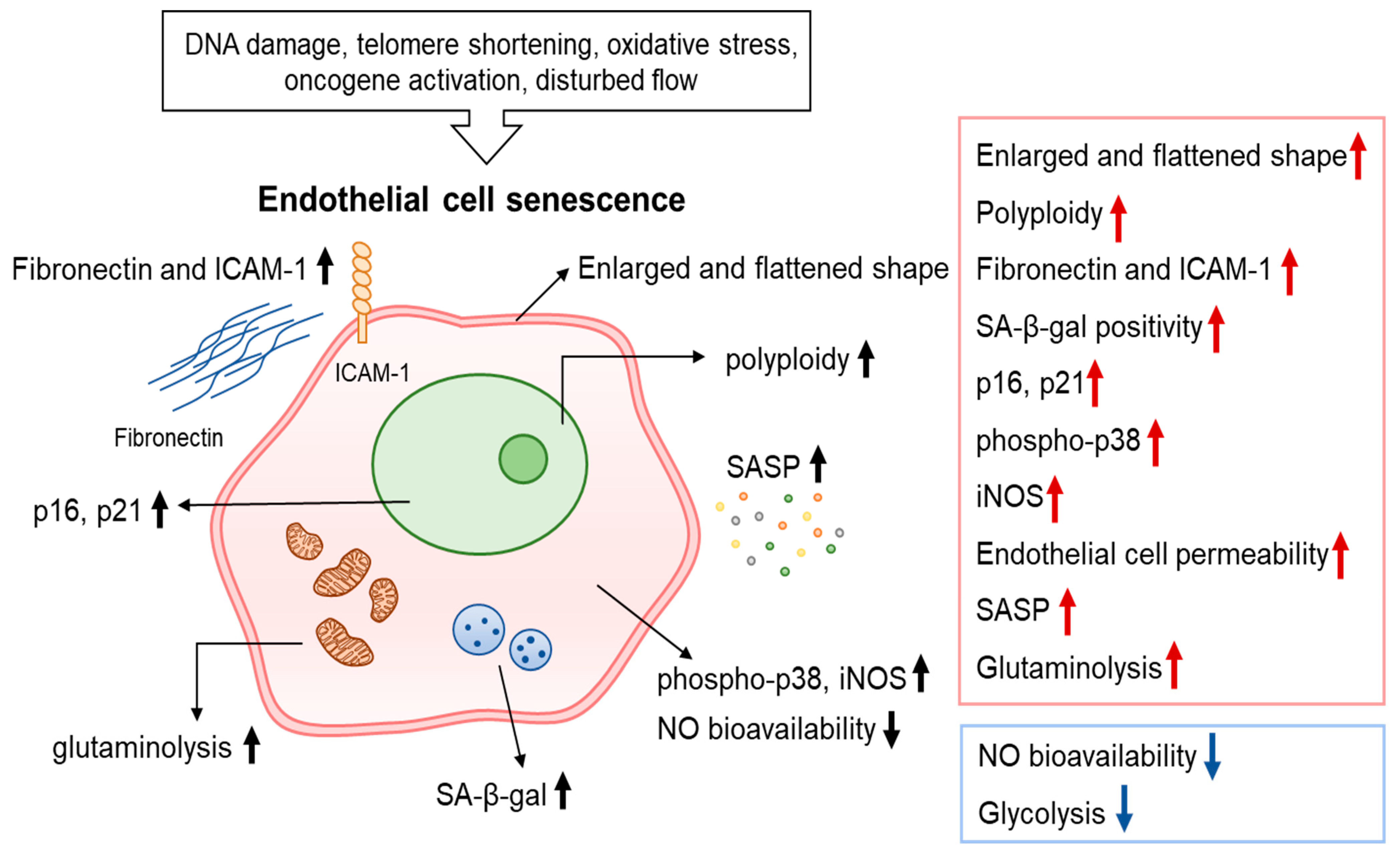
2. Characteristics of Endothelial Cell Senescence
3. Molecular Players and Pathways Associated with Endothelial Cell Senescence
3.1. Sirtuins
3.2. Klotho
3.3. Renin-Angiotensin-Aldosterone System
3.4. Insulin-like Growth Factor-Binding Proteins
3.5. Nuclear Factor-E2-Related Factor
3.6. Mammalian Target of Rapamycin
3.7. Others Molecules
4. Conclusions and Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Chicas, A.; Wang, X.; Zhang, C.; McCurrach, M.; Zhao, Z.; Mert, O.; Dickins, R.A.; Narita, M.; Zhang, M.; Lowe, S.W. Dissecting the unique role of the retinoblastoma tumor suppressor during cellular senescence. Cancer Cell 2010, 17, 376–387. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Espín, D.; Cañamero, M.; Maraver, A.; Gómez-López, G.; Contreras, J.; Murillo-Cuesta, S.; Rodríguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed cell senescence during mammalian embryonic development. Cell 2013, 155, 1104–1118. [Google Scholar] [CrossRef]
- Demaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.M.; Vijg, J.; Van Steeg, H.; Dollé, M.E.; et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733. [Google Scholar] [CrossRef]
- Feng, T.; Meng, J.; Kou, S.; Jiang, Z.; Huang, X.; Lu, Z.; Zhao, H.; Lau, L.F.; Zhou, B.; Zhang, H. CCN1-Induced Cellular Senescence Promotes Heart Regeneration. Circulation 2019, 139, 2495–2498. [Google Scholar] [CrossRef]
- Campisi, J. Senescent cells, tumor suppression, and organismal aging: Good citizens, bad neighbors. Cell 2005, 120, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Aroor, A.R.; DeMarco, V.G.; Martinez-Lemus, L.A.; Meininger, G.A.; Sowers, J.R. Vascular stiffness in insulin resistance and obesity. Front. Physiol. 2015, 6, 231. [Google Scholar] [CrossRef]
- Tian, X.L.; Li, Y. Endothelial cell senescence and age-related vascular diseases. J Genet Genomics 2014, 41, 485–495. [Google Scholar] [CrossRef]
- Xinghui, S.; Mark, W.F. Vascular Endothelial Senescence: Pathobiological Insights, Emerging Long Noncoding RNA Targets, Challenges and Therapeutic Opportunities. Front Physiol. 2021, 12, 693067. [Google Scholar]
- Jia, G.; Aroor, A.R.; Jia, C.; Sowers, J.R. Endothelial cell senescence in aging-related vascular dysfunction. Biochim. Biophys. Acta. Mol. Basis. Dis. 2019, 1865, 1802–1809. [Google Scholar] [CrossRef]
- Higashi, Y.; Kihara, Y.; Noma, K. Endothelial dysfunction and hypertension in aging. Hypertens Res. 2012, 35, 1039–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Wang, X.; Liu, T.; Zhu, X.; Pan, X. The multifaceted role of the SASP in atherosclerosis: From mechanisms to therapeutic opportunities. Cell Biosci. 2022, 12, 74. [Google Scholar] [CrossRef] [PubMed]
- Brown, W.R.; Thore, C.R. Review: Cerebral microvascular pathology in ageing and neurodegeneration. Neuropathol. Appl. Neurobiol. 2011, 37, 56–74. [Google Scholar] [CrossRef]
- Bonithon-Kopp, C.; Touboul, P.J.; Berr, C.; Leroux, C.; Mainard, F.; Courbon, D.; Ducimetière, P. Relation of intima-media thickness to atherosclerotic plaques in carotid arteries. The Vascular Aging (EVA) Study. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Behnke, B.J.; Prisby, R.D.; Lesniewski, L.A.; Donato, A.J.; Olin, H.M.; Delp, M.D. Influence of ageing and physical activity on vascular morphology in rat skeletal muscle. J. Physiol. 2006, 575, 617–626. [Google Scholar] [CrossRef]
- Kim, S.A.; Lee, K.H.; Won, H.Y.; Park, S.; Chung, J.H.; Jang, Y.; Ha, J.W. Quantitative assessment of aortic elasticity with aging using velocity-vector imaging and its histologic correlation. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1306–1312. [Google Scholar] [CrossRef]
- Wang, J.C.; Bennett, M. Aging and atherosclerosis: Mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ. Res. 2012, 111, 245–259. [Google Scholar] [CrossRef] [PubMed]
- Erusalimsky, J.D.; Kurz, D.J. Endothelial cell senescence. Handb. Exp. Pharmacol. 2006, 176, 213–248. [Google Scholar]
- Chen, J.; Goligorsky, M.S. Premature senescence of endothelial cells: Methusaleh’s dilemma. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, 1729–1739. [Google Scholar] [CrossRef]
- Kurz, D.J.; Hong, Y.; Trivier, E.; Huang, H.L.; Decary, S.; Zang, G.H.; Lüscher, T.F.; Erusalimsky, J.D. Fibroblast growth factor-2, but not vascular endothelial growth factor, upregulates telomerase activity in human endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 748–754. [Google Scholar] [CrossRef]
- Cho, K.A.; Ryu, S.J.; Oh, Y.S.; Park, J.H.; Lee, J.W.; Kim, H.P.; Kim, K.T.; Jang, I.S.; Park, S.C. Morphological adjustment of senescent cells by modulating caveolin-1 status. J. Biol. Chem. 2004, 279, 42270–42278. [Google Scholar] [CrossRef] [PubMed]
- Coleman, P.R.; Hahn, C.N.; Grimshaw, M.; Lu, Y.; Li, X.; Brautigan, P.J.; Beck, K.; Stocker, R.; Vadas, M.A.; Gamble, J.R. Stress-induced premature senescence mediated by a novel gene, SENEX, results in an anti-inflammatory phenotype in endothelial cells. Blood 2010, 116, 4016–4024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, H.; Pickering, J.G. Cellular Senescence and Vascular Disease: Novel Routes to Better Understanding and Therapy. Can. J. Cardiol. 2016, 32, 612–623. [Google Scholar] [CrossRef] [PubMed]
- Shelton, D.N.; Chang, E.; Whittier, P.S.; Choi, D.; Funk, W.D. Microarray analysis of replicative senescence. Curr. Biol. 1999, 9, 939–945. [Google Scholar] [CrossRef]
- Kumazaki, T.; Mitsui, Y. Alterations in transcription factor-binding activities to fibronectin promoter during aging of vascular endothelial cells. Mech. Ageing Dev. 1996, 88, 111–124. [Google Scholar] [CrossRef]
- Minamino, T.; Miyauchi, H.; Yoshida, T.; Ishida, Y.; Yoshida, H.; Komuro, I. Endothelial cell senescence in human atherosclerosis: Role of telomere in endothelial dysfunction. Circulation 2002, 105, 1541–1544. [Google Scholar] [CrossRef]
- Brandes, R.P.; Fleming, I.; Busse, R. Endothelial aging. Cardiovasc. Res. 2005, 66, 286–294. [Google Scholar] [CrossRef]
- Coppé, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef]
- Hampel, B.; Fortschegger, K.; Ressler, S.; Chang, M.W.; Unterluggauer, H.; Breitwieser, A.; Sommergruber, W.; Fitzky, B.; Lepperdinger, G.; Jansen-Dürr, P.; et al. Increased expression of extracellular proteins as a hallmark of human endothelial cell in vitro senescence. Exp. Gerontol. 2006, 41, 474–481. [Google Scholar] [CrossRef]
- Lin, J.R.; Shen, W.L.; Yan, C.; Gao, P.J. Downregulation of dynamin-related protein 1 contributes to impaired autophagic flux and angiogenic function in senescent endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1413–1422. [Google Scholar] [CrossRef]
- Barinda, A.J.; Ikeda, K.; Nugroho, D.B.; Wardhana, D.A.; Sasaki, N.; Honda, S.; Urata, R.; Matoba, S.; Hirata, K.I.; Emoto, N. Endothelial progeria induces adipose tissue senescence and impairs insulin sensitivity through senescence associated secretory phenotype. Nat. Commun. 2020, 11, 481. [Google Scholar] [CrossRef] [PubMed]
- Dominic, A.; Banerjee, P.; Hamilton, D.J.; Le, N.T.; Abe, J.I. Time-dependent replicative senescence vs. disturbed flow-induced pre-mature aging in atherosclerosis. Redox Biol. 2020, 37, 101614. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.Y.; Awad, E.M.; Oszwald, A.; Mayr, M.; Yin, X.; Waltenberger, B.; Stuppner, H.; Lipovac, M.; Uhrin, P.; Breuss, J.M. Premature senescence of endothelial cells upon chronic exposure to TNFα can be prevented by N-acetyl cysteine and plumericin. Sci. Rep. 2017, 7, 39501. [Google Scholar] [CrossRef]
- Hwang, H.J.; Lee, Y.R.; Kang, D.; Lee, H.C.; Seo, H.R.; Ryu, J.K.; Kim, Y.N.; Ko, Y.G.; Park, H.J.; Lee, J.S. Endothelial cells under therapy-induced senescence secrete CXCL11, which increases aggressiveness of breast cancer cells. Cancer Lett. 2020, 490, 100–110. [Google Scholar] [CrossRef]
- Mai, S.; Klinkenberg, M.; Auburger, G.; Bereiter-Hahn, J.; Jendrach, M. Decreased expression of Drp1 and Fis1 mediates mitochondrial elongation in senescent cells and enhances resistance to oxidative stress through PINK1. J. Cell Sci. 2010, 123, 917–926. [Google Scholar] [CrossRef]
- Kim, Y.M.; Youn, S.W.; Sudhahar, V.; Das, A.; Chandhri, R.; Cuervo Grajal, H.; Kweon, J.; Leanhart, S.; He, L.; Toth, P.T.; et al. Redox Regulation of Mitochondrial Fission Protein Drp1 by Protein Disulfide Isomerase Limits Endothelial Senescence. Cell Rep. 2018, 23, 3565–3578. [Google Scholar] [CrossRef] [PubMed]
- Saeedi Saravi, S.S.; Eroglu, E.; Waldeck-Weiermair, M.; Sorrentino, A.; Steinhorn, B.; Belousov, V.; Michel, T. Differential endothelial signaling responses elicited by chemogenetic H2O2 synthesis. Redox Biol. 2020, 36, 101605. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Habibi, J.; Bostick, B.P.; Ma, L.; DeMarco, V.G.; Aroor, A.R.; Hayden, M.R.; Whaley-Connell, A.T.; Sowers, J.R. Uric acid promotes left ventricular diastolic dysfunction in mice fed a Western diet. Hypertension 2015, 65, 531–539. [Google Scholar] [CrossRef]
- Aldosari, S.; Awad, M.; Harrington, E.O.; Sellke, F.W.; Abid, M.R. Subcellular reactive oxygen species (ROS) in cardiovascular pathophysiology. Antioxidants 2018, 7, 14. [Google Scholar] [CrossRef]
- Craige, S.M.; Chen, K.; Pei, Y.; Li, C.; Huang, X.; Chen, C. NADPH Oxidase 4 promotes endothelial angiogenesis through endothelial nitric oxide synthase activation. Circulation 2011, 124, 731–740. [Google Scholar] [CrossRef]
- Schröder, K.; Zhang, M.; Benkhoff, S.; Mieth, A.; Pliquett, R.; Kosowski, J. Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ. Res. 2012, 110, 1217–1225. [Google Scholar] [CrossRef] [PubMed]
- Shafique, E.; Choy, W.C.; Liu, Y.; Feng, J.; Arthur Lyra, B.C.; Arafah, M. Oxidative stress improves coronary endothelial function through activation of the pro-survival kinase AMPK. Aging 2013, 5, 515–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.M.; Kim, S.J.; Tatsunami, R.; Yamamura, H.; Fukai, T.; Fukai, M.U. ROS-induced ROS release orchestrated by Nox4, Nox2, and mitochondria in VEGF signaling and angiogenesis. Am. J. Physiol. Cell Physiol. 2017, 312, C749–C764. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Habibi, J.; Aroor, A.R.; Hill, M.A.; DeMarco, V.G.; Lee, L.E.; Ma, L.; Barron, B.J.; Whaley-Connell, A.; Sowers, J.R. Enhanced endothelium epithelial sodium channel signaling prompts left ventricular diastolic dysfunction in obese female mice. Metabolism 2018, 78, 69–79. [Google Scholar] [CrossRef]
- Chatterjee, S.; Ganini, D.; Tokar, E.J.; Kumar, A.; Das, S.; Corbett, J.; Kadiiska, M.B.; Waalkes, M.P.; Diehl, A.M.; Mason, R.P. Leptin is key to peroxynitrite-mediated oxidative stress and Kupffer cell activation in experimental non-alcoholic steatohepatitis. J. Hepatol. 2013, 58, 778–784. [Google Scholar] [CrossRef] [PubMed]
- Haendeler, J.; Hoffmann, J.; Diehl, J.F.; Vasa, M.; Spyridopoulos, I.; Zeiher, A.M.; Dimmeler, S. Antioxidants inhibit nuclear export of telomerase reverse transcriptase and delay replicative senescence of endothelial cells. Circ. Res. 2004, 94, 768–775. [Google Scholar] [CrossRef]
- Kurz, D.J.; Decary, S.; Hong, Y.; Trivier, E.; Akhmedov, A.; Erusalimsky, J.D. Chronic oxidative stress compromises telomere integrity and accelerates the onset of senescence in human endothelial cells. J. Cell. Sci. 2004, 117, 2417–2426. [Google Scholar] [CrossRef]
- Unterluggauer, H.; Mazurek, S.; Lener, B.; Hütter, E.; Eigenbrodt, E.; Zwerschke, W.; Jansen-Dürr, P. Premature senescence of human endothelial cells induced by inhibition of glutaminase. Biogerontology 2008, 9, 247–259. [Google Scholar] [CrossRef]
- Kuosmanen, S.M.; Sihvola, V.; Kansanen, E.; Kaikkonen, M.U.; Levonen, A.L. MicroRNAs mediate the senescence-associated decline of NRF2 in endothelial cells. Redox Biol. 2018, 18, 77–83. [Google Scholar] [CrossRef]
- Cheng, X.; Shihabudeen Haider Ali, M.S.; Moran, M.; Viana, M.P.; Schlichte, S.L.; Zimmerman, M.C.; Khalimonchuk, O.; Feinberg, M.W.; Sun, X. Long non-coding RNA Meg3 deficiency impairs glucose homeostasis and insulin signaling by inducing cellular senescence of hepatic endothelium in obesity. Redox Biol. 2021, 40, 101863. [Google Scholar] [CrossRef]
- Johmura, Y.; Yamanaka, T.; Omori, S.; Wang, T.W.; Sugiura, Y.; Matsumoto, M.; Suzuki, N.; Kumamoto, S.; Yamaguchi, K.; Hatakeyama, S.; et al. Senolysis by glutaminolysis inhibition ameliorates various age-associated disorders. Science 2021, 371, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Haspula, D.; Vallejos, A.K.; Moore, T.M.; Tomar, N.; Dash, R.K.; Hoffmann, B.R. Influence of a Hyperglycemic Microenvironment on a Diabetic Versus Healthy Rat Vascular Endothelium Reveals Distinguishable Mechanistic and Phenotypic Responses. Front. Physiol. 2019, 10, 558. [Google Scholar] [CrossRef] [PubMed]
- Liemburg-Apers, D.C.; Willems, P.H.; Koopman, W.J.; Grefte, S. Interactions between mitochondrial reactive oxygen species and cellular glucose metabolism. Arch. Toxicol. 2015, 89, 1209–1226. [Google Scholar] [CrossRef]
- Fenton, M.; Barker, S.; Kurz, D.J.; Erusalimsky, J.D. Cellular senescence after single and repeated balloon catheter denudations of rabbit carotid arteries. Arterioscler. Thromb. Vasc. Biol. 2001, 22, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Brodsky, S.V.; Goligorsky, D.M.; Hampel, D.J.; Li, H.; Gross, S.S.; Goligorsky, M.S. Glycated collagen I induces premature senescence-like phenotypic changes in endothelial cells. Circ. Res. 2002, 90, 1290–1298. [Google Scholar] [CrossRef] [PubMed]
- Brodsky, S.V.; Gealekman, O.; Chen, J.; Zhang, F.; Togashi, N.; Crabtree, M.; Gross, S.S.; Nasjletti, A.; Goligorsky, M.S. Prevention and reversal of premature endothelial cell senescence and vasculopathy in obesity-induced diabetes by ebselen. Circ. Res. 2004, 94, 377–384. [Google Scholar] [CrossRef]
- Kiss, T.; Nyúl-Tóth, Á.; Balasubramanian, P.; Tarantini, S.; Ahire, C.; DelFavero, J.; Yabluchanskiy, A.; Csipo, T.; Farkas, E.; Wiley, G.; et al. Single-cell RNA sequencing identifies senescent cerebromicrovascular endothelial cells in the aged mouse brain. Geroscience 2020, 42, 429–444. [Google Scholar] [CrossRef] [PubMed]
- Vasile, E.; Tomita, Y.; Brown, L.F.; Kocher, O.; Dvorak, H.F. Differential expression of thymosin beta-10 by early passage and senescent vascular endothelium is modulated by VPF/VEGF: Evidence for senescent endothelial cells in vivo at sites of atherosclerosis. FASEB J. 2001, 15, 458–466. [Google Scholar] [CrossRef]
- Villaret, A.; Galitzky, J.; Decaunes, P.; Estève, D.; Marques, M.A.; Sengenès, C.; Chiotasso, P.; Tchkonia, T.; Lafontan, M.; Kirkland, J.L.; et al. Adipose tissue endothelial cells from obese human subjects: Differences among depots in angiogenic, metabolic, and inflammatory gene expression and cellular senescence. Diabetes 2010, 59, 2755–2763. [Google Scholar] [CrossRef]
- Arunachalam, G.; Yao, H.; Sundar, I.K.; Caito, S.; Rahman, I. SIRT1 regulates oxidant- and cigarette smoke-induced eNOS acetylation in endothelial cells: Role of resveratrol. Biochem. Biophys. Res. Commun. 2010, 393, 66–72. [Google Scholar] [CrossRef]
- Carlomosti, F.; D’Agostino, M.; Beji, S.; Torcinaro, A.; Rizzi, R.; Zaccagnini, G.; Maimone, B.; Di Stefano, V.; De Santa, F.; Cordisco, S.; et al. Oxidative Stress-Induced miR-200c Disrupts the Regulatory Loop Among SIRT1, FOXO1, and eNOS. Antioxid. Redox Signal. 2017, 27, 328–344. [Google Scholar] [CrossRef] [PubMed]
- Menghini, R.; Casagrande, V.; Cardellini, M.; Martelli, E.; Terrinoni, A.; Amati, F.; Vasa-Nicotera, M.; Ippoliti, A.; Novelli, G.; Melino, G.; et al. MicroRNA 217 modulates endothelial cell senescence via silent information regulator 1. Circulation 2009, 120, 1524–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ota, H.; Eto, M.; Kano, M.R.; Ogawa, S.; Iijima, K.; Akishita, M.; Ouchi, Y. Cilostazol inhibits oxidative stress-induced premature senescence via upregulation of Sirt1 in human endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1634–1639. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Yu, H.; Huang, S.; Zhu, P. Resveratrol Protects against TNF-α-Induced Injury in Human Umbilical Endothelial Cells through Promoting Sirtuin-1-Induced Repression of NF-KB and p38 MAPK. PLoS ONE 2016, 11, e0147034. [Google Scholar] [CrossRef] [PubMed]
- Yeung, F.; Hoberg, J.E.; Ramsey, C.S.; Keller, M.D.; Jones, D.R.; Frye, R.A.; Mayo, M.W. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004, 23, 2369–2380. [Google Scholar] [CrossRef] [PubMed]
- Yoshizaki, T.; Schenk, S.; Imamura, T.; Babendure, J.L.; Sonoda, N.; Bae, E.J.; Oh, D.Y.; Lu, M.; Milne, J.C.; Westphal, C.; et al. SIRT1 inhibits inflammatory pathways in macrophages and modulates insulin sensitivity. Am. J. Physiol. Endocrinol. Metab. 2010, 298, 419–428. [Google Scholar] [CrossRef] [PubMed]
- D’Onofrio, N.; Vitiello, M.; Casale, R.; Servillo, L.; Giovane, A.; Balestrieri, M.L. Sirtuins in vascular diseases: Emerging roles and therapeutic potential. Biochim. Biophys. Acta 2015, 1852, 1311–1322. [Google Scholar] [CrossRef]
- Paschalaki, K.E.; Starke, R.D.; Hu, Y.; Mercado, N.; Margariti, A.; Gorgoulis, V.G.; Randi, A.M.; Barnes, P.J. Dysfunction of endothelial progenitor cells from smokers and chronic obstructive pulmonary disease patients due to increased DNA damage and senescence. Stem Cells 2013, 31, 2813–2826. [Google Scholar] [CrossRef]
- Wan, Y.Z.; Gao, P.; Zhou, S.; Zhang, Z.Q.; Hao, D.L.; Lian, L.S.; Li, Y.J.; Chen, H.Z.; Liu, D.P. SIRT1-mediated epigenetic downregulation of plasminogen activator inhibitor-1 prevents vascular endothelial replicative senescence. Aging Cell 2014, 13, 890–899. [Google Scholar] [CrossRef]
- Zhou, S.; Chen, H.Z.; Wan, Y.Z.; Zhang, Q.J.; Wei, Y.S.; Huang, S.; Liu, J.J.; Lu, Y.B.; Zhang, Z.Q.; Yang, R.F.; et al. Repression of P66Shc expression by SIRT1 contributes to the prevention of hyperglycemia-induced endothelial dysfunction. Circ. Res. 2011, 109, 639–648. [Google Scholar] [CrossRef]
- Kong, D.; Zhan, Y.; Liu, Z.; Ding, T.; Li, M.; Yu, H.; Zhang, L.; Li, H.; Luo, A.; Zhang, D.; et al. SIRT1-mediated ERβ suppression in the endothelium contributes to vascular aging. Aging Cell 2016, 15, 1092–1102. [Google Scholar] [CrossRef] [PubMed]
- Vasko, R.; Xavier, S.; Chen, J.; Lin, C.H.; Ratliff, B.; Rabadi, M.; Maizel, J.; Tanokuchi, R.; Zhang, F.; Cao, J.; et al. Endothelial sirtuin 1 deficiency perpetrates nephrosclerosis through downregulation of matrix metalloproteinase-14: Relevance to fibrosis of vascular senescence. J. Am. Soc. Nephrol. 2014, 25, 276–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, B.; Man, A.W.; Yang, K.; Guo, Y.; Xu, C.; Tse, H.F.; Han, W.; Bloksgaard, M.; De Mey, J.G.; Vanhoutte, P.M.; et al. Endothelial SIRT1 prevents adverse arterial remodeling by facilitating HERC2-mediated degradation of acetylated LKB1. Oncotarget 2016, 7, 39065–39081. [Google Scholar] [CrossRef] [PubMed]
- Maksin-Matveev, A.; Kanfi, Y.; Hochhauser, E.; Isak, A.; Cohen, H.Y.; Shainberg, A. Sirtuin 6 protects the heart from hypoxic damage. Exp. Cell Res. 2015, 330, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Liu, H.; Ha, Y.; Tilton, R.G.; Zhang, W. Oxidative stress induces endothelial cell senescence via downregulation of Sirt6. Biomed. Res. Int. 2014, 2014, 902842. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Li, Z.; Mu, W.; Li, L.; Liang, Y.; Lu, M.; Wang, Z.; Qiu, Y.; Wang, Z. Calorie restriction-induced SIRT6 activation delays aging by suppressing NF-κB signaling. Cell Cycle 2016, 15, 1009–1018. [Google Scholar] [CrossRef]
- Cardus, A.; Uryga, A.K.; Walters, G.; Erusalimsky, J.D. SIRT6 protects human endothelial cells from DNA damage, telomere dysfunction, and senescence. Cardiovasc. Res. 2013, 97, 571–579. [Google Scholar] [CrossRef]
- Balestrieri, M.L.; Rizzo, M.R.; Barbieri, M.; Paolisso, P.; D’Onofrio, N.; Giovane, A.; Siniscalchi, M.; Minicucci, F.; Sardu, C.; D’Andrea, D.; et al. Sirtuin 6 expression and inflammatory activity in diabetic atherosclerotic plaques: Effects of incretin treatment. Diabetes 2015, 64, 1395–1406. [Google Scholar] [CrossRef] [PubMed]
- Sundaresan, N.R.; Vasudevan, P.; Zhong, L.; Kim, G.; Samant, S.; Parekh, V.; Pillai, V.B.; Ravindra, P.V.; Gupta, M.; Jeevanandam, V.; et al. The sirtuin SIRT6 blocks IGF-Akt signaling and development of cardiac hypertrophy by targeting c-Jun. Nat. Med. 2012, 18, 1643–1650. [Google Scholar] [CrossRef]
- Tao, R.; Xiong, X.; DePinho, R.A.; Deng, C.X.; Dong, X.C. FoxO3 transcription factor and Sirt6 deacetylase regulate low density lipoprotein (LDL)-cholesterol homeostasis via control of the proprotein convertase subtilisin/kexin type 9 (Pcsk9) gene expression. J. Biol. Chem. 2013, 288, 29252–29259. [Google Scholar] [CrossRef]
- Kuro-o, M.; Matsumura, Y.; Aizawa, H.; Kawaguchi, H.; Suga, T.; Utsugi, T.; Ohyama, Y.; Kurabayashi, M.; Kaname, T.; Kume, E.; et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 1997, 390, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Yamagishi, T.; Nakamura, T.; Ohyama, Y.; Aizawa, H.; Suga, T.; Matsumura, Y.; Masuda, H.; Kurabayashi, M.; Kuro-o, M.; et al. Klotho protein protects against endothelial dysfunction. Biochem. Biophys. Res. Commun. 1998, 248, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Nakamura, T.; Ohyama, Y.; Suzuki, T.; Iida, A.; Shiraki-Iida, T.; Kuro-o, M.; Nabeshima, Y.; Kurabayashi, M.; Nagai, R. In vivo klotho gene delivery protects against endothelial dysfunction in multiple risk factor syndrome. Biochem. Biophys. Res. Commun. 2000, 276, 767–772. [Google Scholar] [CrossRef]
- Ikushima, M.; Rakugi, H.; Ishikawa, K.; Maekawa, Y.; Yamamoto, K.; Ohta, J.; Chihara, Y.; Kida, I.; Ogihara, T. Anti-apoptotic and anti-senescence effects of Klotho on vascular endothelial cells. Biochem. Biophys. Res. Commun. 2006, 339, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Clark, J.D.; Pastor, J.V.; Gurnani, P.; Nandi, A.; Kurosu, H.; Miyoshi, M.; Ogawa, Y.; Castrillon, D.H.; Rosenblatt, K.P.; et al. Regulation of oxidative stress by the anti-aging hormone klotho. J. Biol. Chem. 2005, 280, 38029–38034. [Google Scholar] [CrossRef]
- Kurosu, H.; Yamamoto, M.; Clark, J.D.; Pastor, J.V.; Nandi, A.; Gurnani, P.; McGuinness, O.P.; Chikuda, H.; Yamaguchi, M.; Kawaguchi, H.; et al. Suppression of aging in mice by the hormone Klotho. Science 2005, 309, 1829–1833. [Google Scholar] [CrossRef] [PubMed]
- Semba, R.D.; Cappola, A.R.; Sun, K.; Bandinelli, S.; Dalal, M.; Crasto, C.; Guralnik, J.M.; Ferrucci, L. Plasma klotho and cardiovascular disease in adults. J. Am. Geriatr. Soc. 2011, 59, 1596–1601. [Google Scholar] [CrossRef] [PubMed]
- Navarro-González, J.F.; Donate-Correa, J.; Muros de Fuentes, M.; Pérez-Hernández, H.; Martínez-Sanz, R.; Mora-Fernández, C. Reduced Klotho is associated with the presence and severity of coronary artery disease. Heart 2014, 100, 34–40. [Google Scholar] [CrossRef]
- Kitagawa, M.; Sugiyama, H.; Morinaga, H.; Inoue, T.; Takiue, K.; Ogawa, A.; Yamanari, T.; Kikumoto, Y.; Uchida, H.A.; Kitamura, S.; et al. A decreased level of serum soluble Klotho is an independent biomarker associated with arterial stiffness in patients with chronic kidney disease. PLoS ONE 2013, 8, e56695. [Google Scholar] [CrossRef]
- Lindberg, K.; Olauson, H.; Amin, R.; Ponnusamy, A.; Goetz, R.; Taylor, R.F.; Mohammadi, M.; Canfield, A.; Kublickiene, K.; Larsson, T.E. Arterial klotho expression and FGF23 effects on vascular calcification and function. PLoS ONE 2013, 8, e60658. [Google Scholar]
- Saravi, B.; Li, Z.; Lang, C.N.; Schmid, B.; Lang, F.K.; Grad, S.; Alini, M.; Richards, R.G.; Schmal, H.; Südkamp, N.; et al. The Tissue Renin-Angiotensin System and Its Role in the Pathogenesis of Major Human Diseases: Quo Vadis? Cells 2021, 10, 650. [Google Scholar] [CrossRef] [PubMed]
- Carey, R.M.; Siragy, H.M. Newly recognized components of the renin-angiotensin system: Potential roles in cardiovascular and renal regulation. Endocr. Rev. 2003, 24, 261–271. [Google Scholar] [CrossRef] [PubMed]
- de Gasparo, M.; Catt, K.J.; Inagami, T.; Wright, J.W.; Unger, T. International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol. Rev. 2000, 52, 415–472. [Google Scholar]
- Herbert, K.E.; Mistry, Y.; Hastings, R.; Poolman, T.; Niklason, L.; Williams, B. Angiotensin II-mediated oxidative DNA damage accelerates cellular senescence in cultured human vascular smooth muscle cells via telomere-dependent and independent pathways. Circ. Res. 2008, 102, 201–208. [Google Scholar] [CrossRef]
- Kunieda, T.; Minamino, T.; Nishi, J.; Tateno, K.; Oyama, T.; Katsuno, T.; Miyauchi, H.; Orimo, M.; Okada, S.; Takamura, M.; et al. Angiotensin II induces premature senescence of vascular smooth muscle cells and accelerates the development of atherosclerosis via a p21-dependent pathway. Circulation 2006, 114, 953–960. [Google Scholar] [CrossRef]
- Min, L.J.; Mogi, M.; Iwanami, J.; Li, J.M.; Sakata, A.; Fujita, T.; Tsukuda, K.; Iwai, M.; Horiuchi, M. Cross-talk between aldosterone and angiotensin II in vascular smooth muscle cell senescence. Cardiovasc. Res. 2007, 76, 506–516. [Google Scholar] [CrossRef]
- Shan, H.; Bai, X.; Chen, X. Angiotensin II induces endothelial cell senescence via the activation of mitogen-activated protein kinases. Cell Biochem. Funct. 2008, 26, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Mi, X.; Yang, S.; Yang, Y.; Zhang, S.; Hui, R.; Chen, Y.; Zhang, W. Long-term stimulation of angiotensin II induced endothelial senescence and dysfunction. Exp. Gerontol. 2019, 119, 212–220. [Google Scholar] [CrossRef]
- Basso, N.; Paglia, N.; Stella, I.; de Cavanagh, E.M.; Ferder, L.; del Rosario Lores Arnaiz, M.; Inserra, F. Protective effect of the inhibition of the renin-angiotensin system on aging. Regul. Pept. 2005, 128, 247–252. [Google Scholar] [CrossRef]
- de Cavanagh, E.M.; Piotrkowski, B.; Fraga, C.G. Concerted action of the renin-angiotensin system, mitochondria, and antioxidant defenses in aging. Mol. Aspects. Med. 2004, 25, 27–36. [Google Scholar] [CrossRef]
- Yoon, H.E.; Kim, E.N.; Kim, M.Y.; Lim, J.H.; Jang, I.A.; Ban, T.H.; Shin, S.J.; Park, C.W.; Chang, Y.S.; Choi, B.S. Age-Associated Changes in the Vascular Renin-Angiotensin System in Mice. Oxid. Med. Cell Longev. 2016, 6731093. [Google Scholar] [CrossRef] [PubMed]
- Valencia, 1st; Shamoon, L.; Romero, A.; De la Cuesta, F.; Sánchez-Ferrer, C.F.; Peiró, C. Angiotensin-(1-7), a protective peptide against vascular aging. Peptides 2022, 152, 170775. [Google Scholar] [CrossRef]
- Romero, A.; San Hipólito-Luengo, Á.; Villalobos, L.A.; Vallejo, S.; Valencia, I.; Michalska, P.; Pajuelo-Lozano, N.; Sánchez-Pérez, I.; León, R.; Bartha, J.L.; et al. The angiotensin-(1-7)/Mas receptor axis protects from endothelial cell senescence via klotho and Nrf2 activation. Aging Cell 2019, 18, e12913. [Google Scholar] [CrossRef]
- Romero, A.; Dongil, P.; Valencia, I.; Vallejo, S.; Hipólito-Luengo, Á.S.; Díaz-Araya, G.; Bartha, J.L.; González-Arlanzón, M.M.; Rivilla, F.; de la Cuesta, F.; et al. Pharmacological Blockade of NLRP3 Inflammasome/IL-1β-Positive Loop Mitigates Endothelial Cell Senescence and Dysfunction. Aging. Dis. 2022, 13, 284–297. [Google Scholar] [CrossRef]
- van der Kaay, D.; Deal, C.; de Kort, S.; Willemsen, R.; Leunissen, R.; Ester, W.; Paquette, J.; van Doorn, J.; Hokken-Koelega, A. Insulin-like growth factor-binding protein-1: Serum levels, promoter polymorphism, and associations with components of the metabolic syndrome in short subjects born small for gestational age. J. Clin. Endocrinol. Metab. 2009, 94, 1386–1392. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Pawlikowska, L.; Kanaya, A.; Hsueh, W.C.; Colbert, L.; Newman, A.B.; Satterfield, S.; Rosen, C.; Cummings, S.R.; Harris, T.B.; et al. Health, Aging, and Body Composition Study. Serum insulin-like growth factor-1 binding proteins 1 and 2 and mortality in older adults: The Health, Aging, and Body Composition Study. J. Am. Geriatr. Soc. 2009, 57, 1213–1218. [Google Scholar] [CrossRef]
- Panganiban, R.A.; Day, R.M. Inhibition of IGF-1R prevents ionizing radiation-induced primary endothelial cell senescence. PLoS ONE 2013, 8, e78589. [Google Scholar] [CrossRef] [PubMed]
- Higashi, Y.; Pandey, A.; Goodwin, B.; Delafontaine, P. Insulin-like growth factor-1 regulates glutathione peroxidase expression and activity in vascular endothelial cells: Implications for atheroprotective actions of insulin-like growth factor-1. Biochim. Biophys. Acta 2013, 1832, 391–399. [Google Scholar] [CrossRef]
- Grillari, J.; Hohenwarter, O.; Grabherr, R.M.; Katinger, H. Subtractive hybridization of mRNA from early passage and senescent endothelial cells. Exp. Gerontol. 2000, 35, 187–197. [Google Scholar] [CrossRef]
- Kim, K.S.; Kim, M.S.; Seu, Y.B.; Chung, H.Y.; Kim, J.H.; Kim, J.R. Regulation of replicative senescence by insulin-like growth factor-binding protein 3 in human umbilical vein endothelial cells. Aging Cell 2007, 6, 535–545. [Google Scholar] [CrossRef]
- Kim, K.S.; Seu, Y.B.; Baek, S.H.; Kim, M.J.; Kim, K.J.; Kim, J.H.; Kim, J.R. Induction of cellular senescence by insulin-like growth factor binding protein-5 through a p53-dependent mechanism. Mol. Biol. Cell 2007, 18, 4543–4552. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.S.; Kim, J.E.; Choi, K.J.; Bae, S.; Kim, D.H. Characterization of DNA damage-induced cellular senescence by ionizing radiation in endothelial cells. Int. J. Radiat. Biol. 2014, 90, 71–80. [Google Scholar] [PubMed]
- Rombouts, C.; Aerts, A.; Quintens, R.; Baselet, B.; El-Saghire, H.; Harms-Ringdahl, M.; Haghdoost, S.; Janssen, A.; Michaux, A.; Yentrapalli, R.; et al. Transcriptomic profiling suggests a role for IGFBP5 in premature senescence of endothelial cells after chronic low dose rate irradiation. Int. J. Radiat. Biol. 2014, 90, 560–574. [Google Scholar] [CrossRef] [PubMed]
- Niture, S.K.; Khatri, R.; Jaiswal, A.K. Regulation of Nrf2-an update. Free Radic. Biol. Med. 2014, 66, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Fulop, G.A.; Kiss, T.; Tarantini, S.; Balasubramanian, P.; Yabluchanskiy, A.; Farkas, E.; Bari, F.; Ungvari, Z.; Csiszar, A. Nrf2 deficiency in aged mice exacerbates cellular senescence promoting cerebrovascular inflammation. Geroscience 2018, 40, 513–521. [Google Scholar] [CrossRef]
- Tarantini, S.; Valcarcel-Ares, M.N.; Yabluchanskiy, A.; Tucsek, Z.; Hertelendy, P.; Kiss, T.; Gautam, T.; Zhang, X.A.; Sonntag, W.E.; de Cabo, R.; et al. Nrf2 Deficiency Exacerbates Obesity-Induced Oxidative Stress, Neurovascular Dysfunction, Blood-Brain Barrier Disruption, Neuroinflammation, Amyloidogenic Gene Expression, and Cognitive Decline in Mice, Mimicking the Aging Phenotype. J. Gerontol. A Biol. Sci. Med. Sci. 2018, 73, 853–863. [Google Scholar] [CrossRef]
- Ungvari, Z.; Bagi, Z.; Feher, A.; Recchia, F.A.; Sonntag, W.E.; Pearson, K.; de Cabo, R.; Csiszar, A. Resveratrol confers endothelial protection via activation of the antioxidant transcription factor Nrf2. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, 18–24. [Google Scholar] [CrossRef]
- Ungvari, Z.; Bailey-Downs, L.; Gautam, T.; Jimenez, R.; Losonczy, G.; Zhang, C.; Ballabh, P.; Recchia, F.A.; Wilkerson, D.C.; Sonntag, W.E.; et al. Adaptive induction of NF-E2-related factor-2-driven antioxidant genes in endothelial cells in response to hyperglycemia. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, 1133–1140. [Google Scholar] [CrossRef]
- El Assar, M.; Angulo, J.; Rodríguez-Mañas, L. Oxidative stress and vascular inflammation in aging. Free Radic. Biol. Med. 2013, 65, 380–401. [Google Scholar] [CrossRef]
- Ungvari, Z.; Bailey-Downs, L.; Gautam, T.; Sosnowska, D.; Wang, M.; Monticone, R.E.; Telljohann, R.; Pinto, J.T.; de Cabo, R.; Sonntag, W.E.; et al. Age-associated vascular oxidative stress, Nrf2 dysfunction, and NF-{kappa}B activation in the nonhuman primate Macaca mulatta. J. Gerontol. A Biol. Sci. Med. Sci. 2011, 66, 866–875. [Google Scholar] [CrossRef]
- Ungvari, Z.; Bailey-Downs, L.; Sosnowska, D.; Gautam, T.; Koncz, P.; Losonczy, G.; Ballabh, P.; de Cabo, R.; Sonntag, W.E.; Csiszar, A. Vascular oxidative stress in aging: A homeostatic failure due to dysregulation of NRF2-mediated antioxidant response. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Heitman, J.; Movva, N.R.; Hall, M.N. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 1991, 253, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Lipton, J.O.; Sahin, M. The neurology of mTOR. Neuron 2014, 84, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Khor, E.S.; Wong, P.F. Endothelial replicative senescence delayed by the inhibition of MTORC1 signaling involves MicroRNA-107. Int. J. Biochem. Cell Biol. 2018, 101, 64–73. [Google Scholar] [CrossRef]
- Kolesnichenko, M.; Hong, L.; Liao, R.; Vogt, P.K.; Sun, P. Attenuation of TORC1 signaling delays replicative and oncogenic RAS-induced senescence. Cell Cycle 2012, 11, 2391–2401. [Google Scholar] [CrossRef] [PubMed]
- Donato, A.J.; Walker, A.E.; Magerko, K.A.; Bramwell, R.C.; Black, A.D.; Henson, G.D.; Lawson, B.R.; Lesniewski, L.A.; Seals, D.R. Life-long caloric restriction reduces oxidative stress and preserves nitric oxide bioavailability and function in arteries of old mice. Aging Cell 2013, 12, 772–783. [Google Scholar] [CrossRef]
- Donato, A.J.; Morgan, R.G.; Walker, A.E.; Lesniewski, L.A. Cellular and molecular biology of aging endothelial cells. J. Mol. Cell Cardiol. 2015, 89, 122–135. [Google Scholar] [CrossRef]
- Rajapakse, A.G.; Yepuri, G.; Carvas, J.M.; Stein, S.; Matter, C.M.; Scerri, I.; Ruffieux, J.; Montani, J.P.; Ming, X.F.; Yang, Z. Hyperactive S6K1 mediates oxidative stress and endothelial dysfunction in aging: Inhibition by resveratrol. PLoS ONE 2011, 6, e19237. [Google Scholar] [CrossRef] [PubMed]
- Tsuji-Tamura, K.; Ogawa, M. Inhibition of the PI3K-Akt and mTORC1 signaling pathways promotes the elongation of vascular endothelial cells. J. Cell Sci. 2016, 129, 1165–1178. [Google Scholar] [CrossRef]
- Herranz, N.; Gallage, S.; Mellone, M.; Wuestefeld, T.; Klotz, S.; Hanley, C.J.; Raguz, S.; Acosta, J.C.; Innes, A.J.; Banito, A.; et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat. Cell Biol. 2015, 17, 1205–1217. [Google Scholar] [CrossRef]
- Laberge, R.M.; Sun, Y.; Orjalo, A.V.; Patil, C.K.; Freund, A.; Zhou, L.; Curran, S.C.; Davalos, A.R.; Wilson-Edell, K.A.; Liu, S.; et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat. Cell Biol. 2015, 17, 1049–1061. [Google Scholar] [CrossRef] [PubMed]
- Narita, M.; Young, A.R.; Arakawa, S.; Samarajiwa, S.A.; Nakashima, T.; Yoshida, S.; Hong, S.; Berry, L.S.; Reichelt, S.; Ferreira, M.; et al. Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science 2011, 332, 966–970. [Google Scholar] [CrossRef] [PubMed]
- Bent, E.H.; Gilbert, L.A.; Hemann, M.T. A senescence secretory switch mediated by PI3K/AKT/mTOR activation controls chemoprotective endothelial secretory responses. Genes Dev. 2016, 30, 1811–3021. [Google Scholar] [CrossRef]
- Yang, H.W.; Hong, H.L.; Luo, W.W.; Dai, C.M.; Chen, X.Y.; Wang, L.P.; Li, Q.; Li, Z.Q.; Liu, P.Q.; Li, Z.M. mTORC2 facilitates endothelial cell senescence by suppressing Nrf2 expression via the Akt/GSK-3β/C/EBPα signaling pathway. Acta Pharmacol. Sin. 2018, 39, 1837–1846. [Google Scholar] [CrossRef]
- Wang, X.M.; Song, S.S.; Xiao, H.; Gao, P.; Li, X.J.; Si, L.Y. Fibroblast growth factor 21 protects against high glucose induced cellular damage and dysfunction of endothelial nitric-oxide synthase in endothelial cells. Cell Physiol. Biochem. 2014, 34, 658–671. [Google Scholar] [CrossRef]
- Wang, X.M.; Xiao, H.; Liu, L.L.; Cheng, D.; Li, X.J.; Si, L.Y. FGF21 represses cerebrovascular aging via improving mitochondrial biogenesis and inhibiting p53 signaling pathway in an AMPK-dependent manner. Exp. Cell Res. 2016, 346, 147–156. [Google Scholar] [CrossRef]
- Giorgio, M.; Migliaccio, E.; Orsini, F.; Paolucci, D.; Moroni, M.; Contursi, C.; Pelliccia, G.; Luzi, L.; Minucci, S.; Marcaccio, M.; et al. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell 2005, 122, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Francia, P.; delli Gatti, C.; Bachschmid, M.; Martin-Padura, I.; Savoia, C.; Migliaccio, E.; Pelicci, P.G.; Schiavoni, M.; Lüscher, T.F.; Volpe, M.; et al. Deletion of p66shc gene protects against age-related endothelial dysfunction. Circulation 2004, 110, 2889–2895. [Google Scholar] [CrossRef]
- Yang, Z.; Li, H.; Luo, P.; Yan, D.; Yang, N.; Zhang, Y.; Huang, Y.; Liu, Y.; Zhang, L.; Yan, J.; et al. UNC5B Promotes Vascular Endothelial Cell Senescence via the ROS-Mediated P53 Pathway. Oxid. Med. Cell. Longev. 2021, 2021, 5546711. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, M.; Liu, W.; Hu, C.; Li, H.; Deng, J.; Cao, Q.; Wang, Y.; Hu, W.; Li, Q. Th17/IL-17 induces endothelial cell senescence via activation of NF-κB/p53/Rb signaling pathway. Lab. Invest. 2021, 101, 1418–1426. [Google Scholar] [CrossRef]
- Dou, F.; Wu, B.; Chen, J.; Liu, T.; Yu, Z.; Chen, C. PPARα Targeting GDF11 Inhibits Vascular Endothelial Cell Senescence in an Atherosclerosis Model. Oxid. Med. Cell. Longev. 2021, 2021, 2045259. [Google Scholar] [CrossRef] [PubMed]
- Le, A.N.; Park, S.S.; Le, M.X.; Lee, U.H.; Ko, B.K.; Lim, H.R.; Yu, R.; Choi, S.H.; Lee, B.J.; Ham, S.Y.; et al. DRG2 Depletion Promotes Endothelial Cell Senescence and Vascular Endothelial Dysfunction. Int. J. Mol. Sci. 2022, 23, 2877. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Han, B.; Zhang, R.; Liu, Q.; Wang, X.; Huang, X.; Liu, D.; Qiao, W.; Yang, M.; Luo, X.; et al. C1q/TNF-Related Protein 9 Attenuates Atherosclerosis by Inhibiting Hyperglycemia-Induced Endothelial Cell Senescence Through the AMPKα/KLF4 Signaling Pathway. Front. Pharmacol. 2021, 12, 758792. [Google Scholar] [CrossRef]
- Valencia, I.; Vallejo, S.; Dongil, P.; Romero, A.; San Hipólito-Luengo, Á.; Shamoon, L.; Posada, M.; García-Olmo, D.; Carraro, R.; Erusalimsky, J.D.; et al. DPP4 Promotes Human Endothelial Cell Senescence and Dysfunction via the PAR2-COX-2-TP Axis and NLRP3 Inflammasome Activation. Hypertension 2022, 79, 1361–1373. [Google Scholar] [CrossRef] [PubMed]
- Tsai, P.H.; Chen, L.Z.; Tseng, K.F.; Chen, F.Y.; Shen, M.Y. Apolipoprotein C3-Rich Low-Density Lipoprotein Induces Endothelial Cell Senescence via FBXO31 and Its Inhibition by Sesamol In Vitro and In Vivo. Biomedicines 2022, 10, 854. [Google Scholar] [CrossRef] [PubMed]
- Shamoon, L.; Espitia-Corredor, J.A.; Dongil, P.; Menéndez-Ribes, M.; Romero, A.; Valencia, I.; Díaz-Araya, G.; Sánchez-Ferrer, C.F.; Peiró, C. Resolvin E1 attenuates doxorubicin-induced endothelial senescence by modulating NLRP3 inflammasome activation. Biochem. Pharmacol. 2022, 201, 115078. [Google Scholar] [CrossRef]
- Foreman, K.E.; Tang, J. Molecular mechanisms of replicative senescence in endothelial cells. Exp. Gerontol. 2003, 38, 1251–1257. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hwang, H.J.; Kim, N.; Herman, A.B.; Gorospe, M.; Lee, J.-S. Factors and Pathways Modulating Endothelial Cell Senescence in Vascular Aging. Int. J. Mol. Sci. 2022, 23, 10135. https://doi.org/10.3390/ijms231710135
Hwang HJ, Kim N, Herman AB, Gorospe M, Lee J-S. Factors and Pathways Modulating Endothelial Cell Senescence in Vascular Aging. International Journal of Molecular Sciences. 2022; 23(17):10135. https://doi.org/10.3390/ijms231710135
Chicago/Turabian StyleHwang, Hyun Jung, Nayeon Kim, Allison B. Herman, Myriam Gorospe, and Jae-Seon Lee. 2022. "Factors and Pathways Modulating Endothelial Cell Senescence in Vascular Aging" International Journal of Molecular Sciences 23, no. 17: 10135. https://doi.org/10.3390/ijms231710135
APA StyleHwang, H. J., Kim, N., Herman, A. B., Gorospe, M., & Lee, J.-S. (2022). Factors and Pathways Modulating Endothelial Cell Senescence in Vascular Aging. International Journal of Molecular Sciences, 23(17), 10135. https://doi.org/10.3390/ijms231710135