
Autophagy Dysregulation in Metabolic Associated Fatty Liver Disease: A New Therapeutic Target

Abstract
:1. Introduction
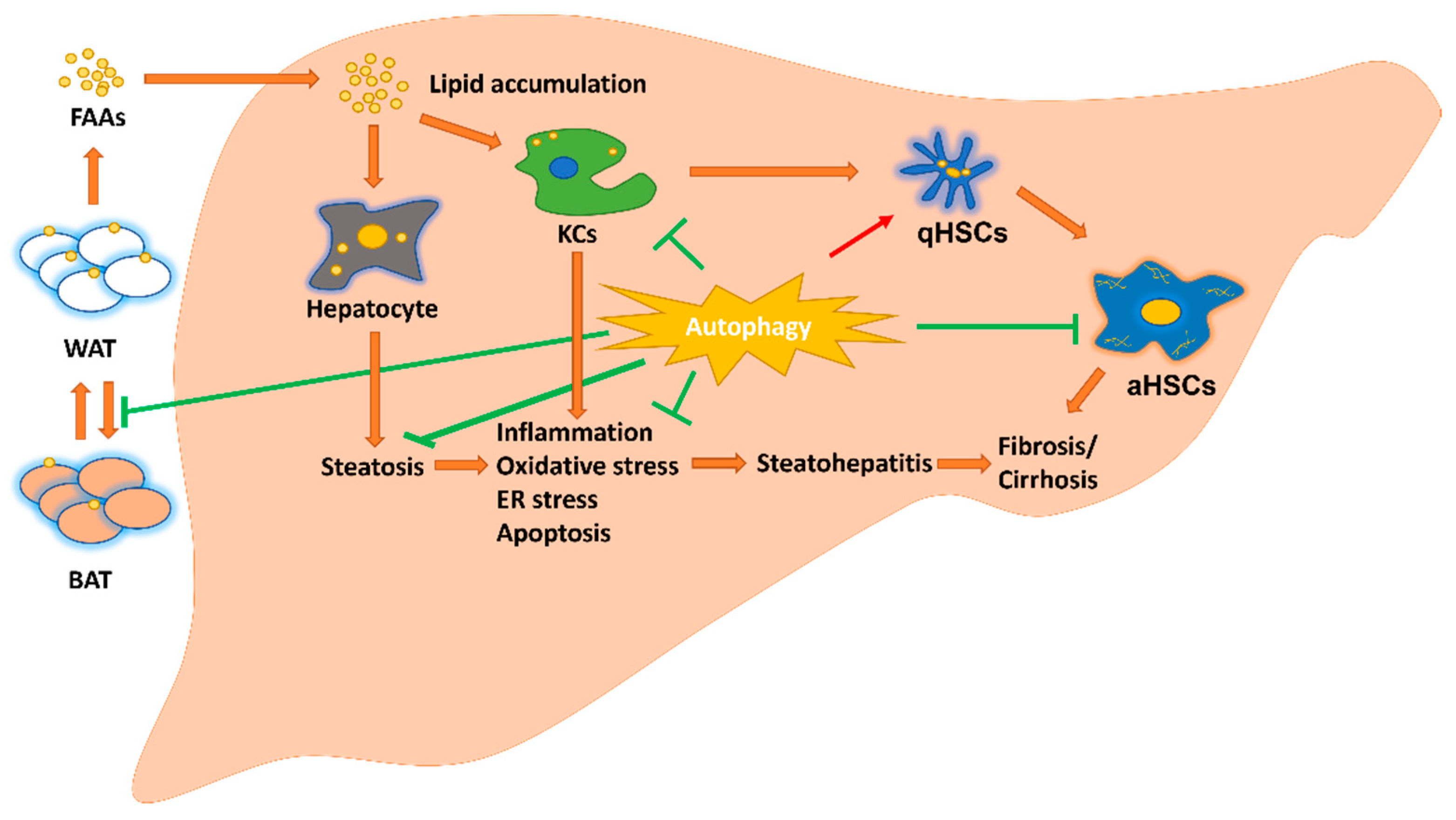
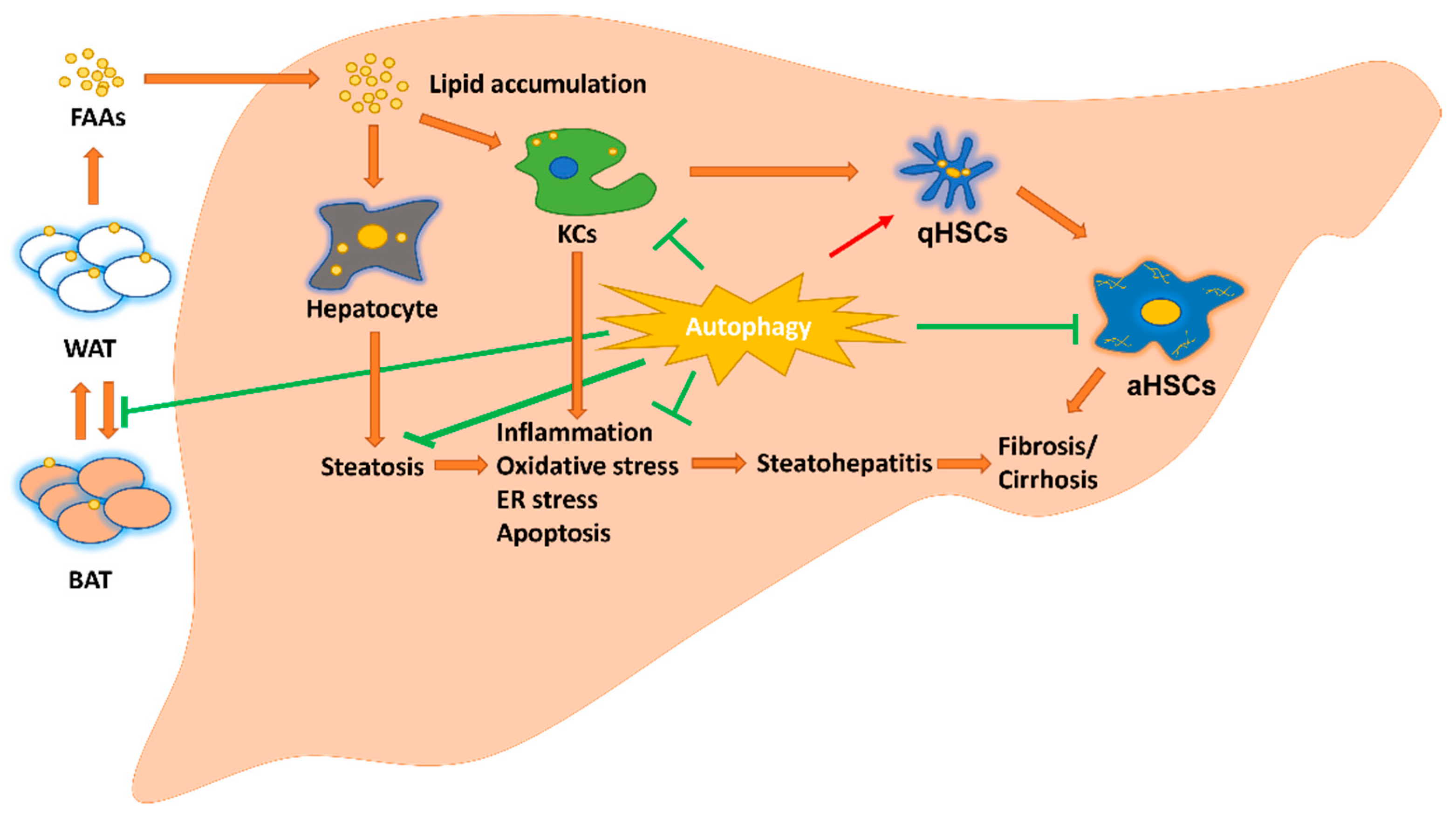
2. Hepatic Parenchymal and Non-Parenchymal Cells and Adipose Tissue in MAFLD
2.1. Hepatocyte in MAFLD
2.2. Macrophage in MAFLD
2.3. Hepatic Stellate Cells (HSCs) in MAFLD
2.4. Adipose Tissue in MAFLD
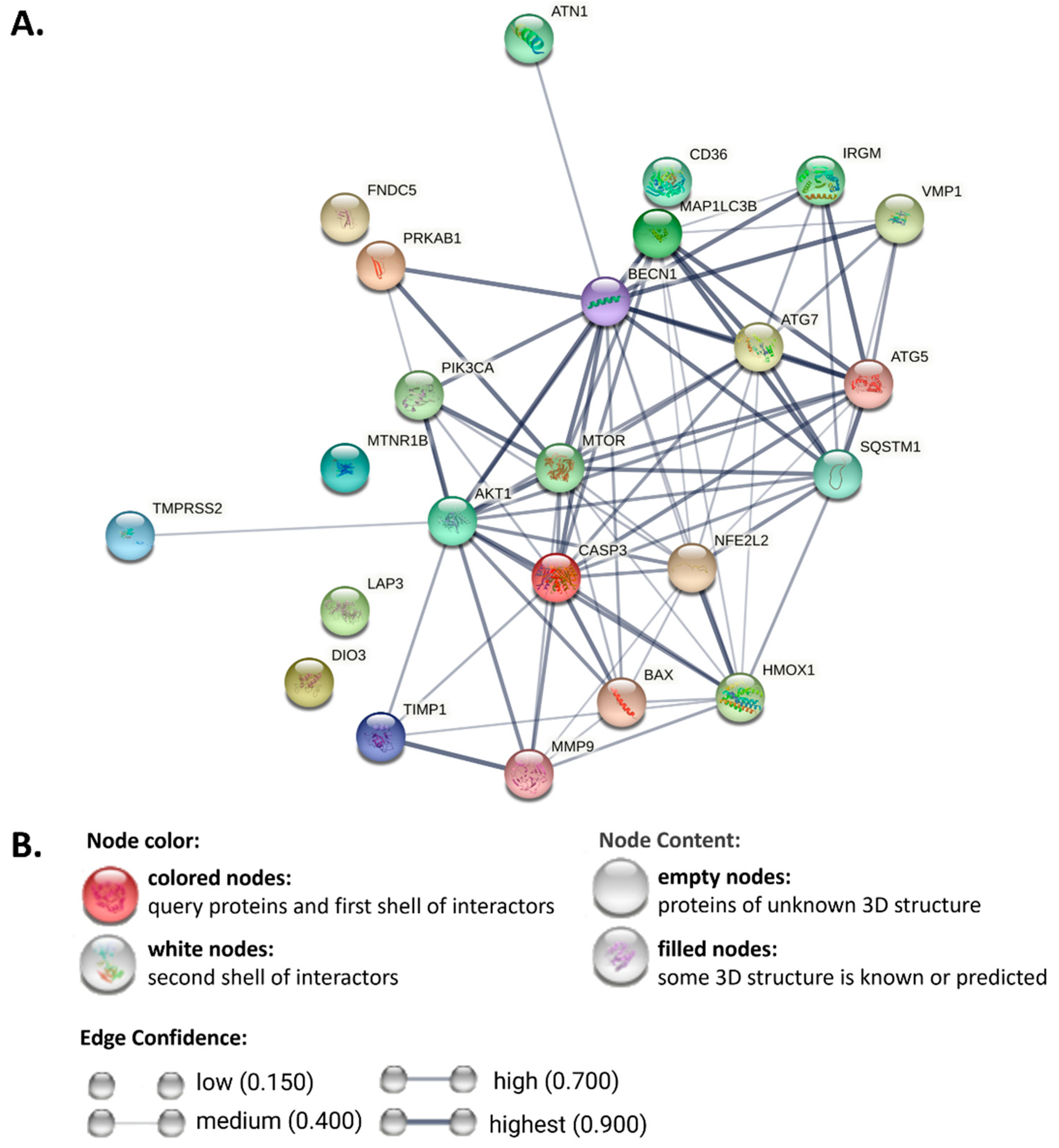
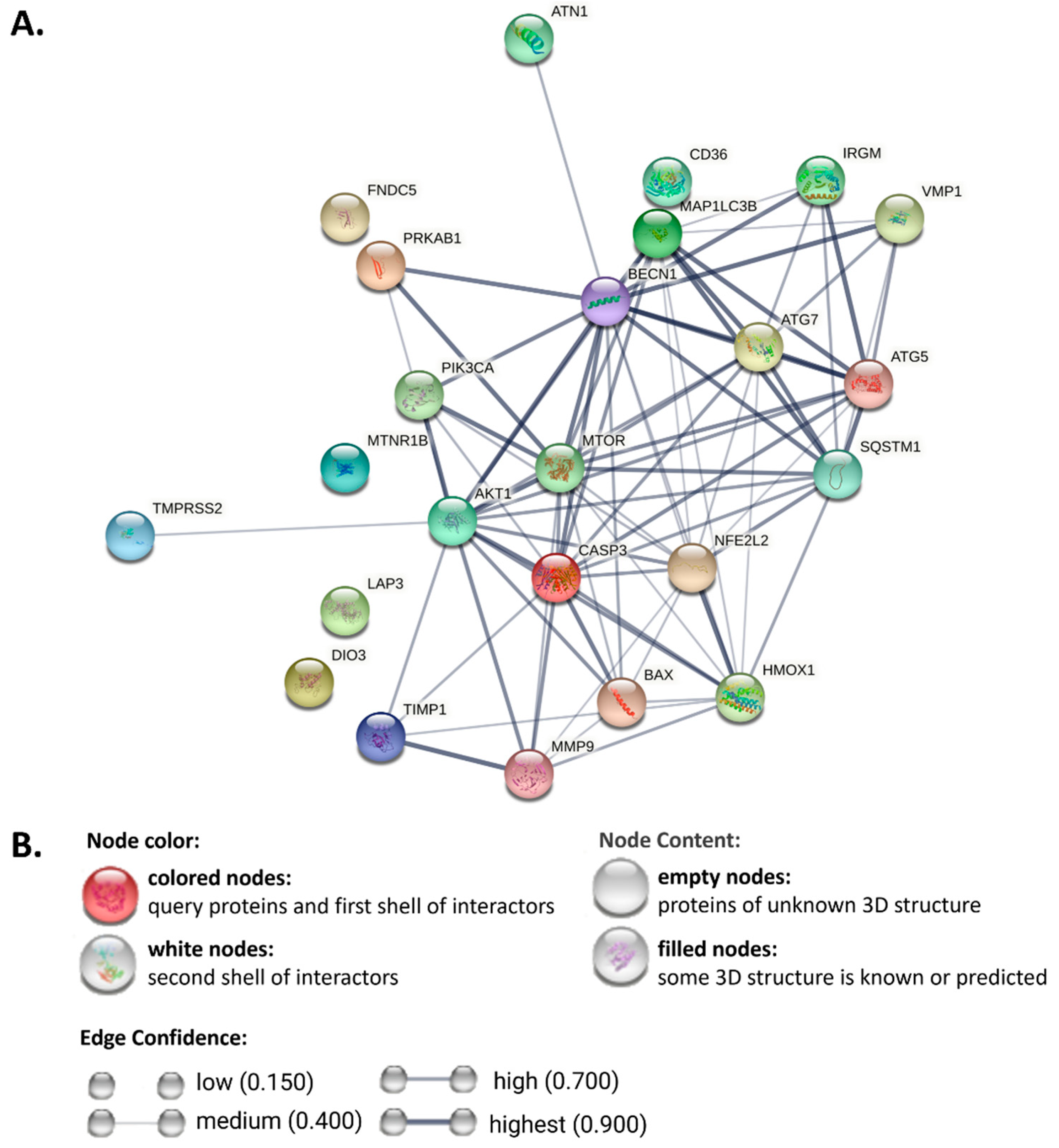
3. Autophagy in MAFLD
3.1. Autophagic Inducer in MAFLD
3.1.1. Thyroid Hormone
3.1.2. Irisin (an Exercise-Derived Myokine from Contractile Muscle)
3.1.3. Hydrogen Sulfide
3.1.4. Melatonin
3.1.5. DA-1241 (a Novel GPR119 Agonist)
3.1.6. ER Transmembrane Protein-Vacuole Membrane Protein 1 (VMP1)
3.1.7. Nuclear Factor Erythroid 2-Related Factor 2 (Nrf2)
3.1.8. Sodium-Glucose Co-Transporter Type-2 Inhibitors (SGLT-2i)
3.1.9. Immunity-Related GTPase M (IRGM)
3.1.10. Autophagy-Related Gene 7 (ATG7)
3.2. Autophagic Repressor in MAFLD
3.2.1. Lipid Receptor CD36
3.2.2. Leucine Aminopeptidase 3 (LAP3)
3.2.3. SARS-CoV-2 Spike Protein

{kind=link}
{kind=link}
| Factors | Effects | Types | Models | Mechanism of Action | Refs. |
|---|---|---|---|---|---|
| TH | induce | protein | C | Increase mitochondrial lipid oxidation and reduce inflammatory response in cultured HepG2-TRb cells | [50,51] |
| M | Decrease serum CHO and ALT levels, hepatic steatosis, inflammation and fibrosis in NASH mice; increase hepatic mitochondrial content and function, hepatic autophagy to enhance b-oxidation of FFAs in NASH mice | ||||
| H | Decreased hepatic steatosis in patients with NAFLD after treatment with low-dose levothyroxine or TRb-selective agonist, resmetirom (MGL-3196) | ||||
| irisin | induce | protein | C | Reduce the PA-dependent increasing of TGF-b1 and type I collagen expression and proinflammatory cytokines production | [55,60] |
| M | Reduce the HFD-induced body weight, liver weight, AST and ALT levels, macrophage infiltration, proinflammatory cytokines production and NAFLD activity score; directly binding with MD2 to compete the binding of MD2 with TLR4 | ||||
| R | Restore the increased apoptotic cells, liver necrosis area and p62 expression level, and the decreased LC3B expression level in aged rats | ||||
| H2S | induce | gas | C | Decrease the lipid level and the apoptosis percentage for OA-treated liver cells and increase the expression levels of Beclin-1 and the LC3 II/I ratio | [69,70] |
| M | Reduce the HFD-induced body weight, liver weight, white fat and brown fat weight, liver TC, TG, AST, ALT, lipid level, TNF-α, IL-1β, and IL-6 levels; inhibit the ROS/PI3K/AKT/mTOR signaling pathway in liver | ||||
| CK | Decrease the ROS/PI3K/AKT/mTOR signaling pathway and increase the expression levels of Beclin1, ATG5 and LC3 II/I ratio | ||||
| melatonin | induce | protein | C | Decrease a-SMA, type 1 collagen, MMP9, TIMP1 and TGF-b expression levels in LX-2 cells and isolated primary HSCs | [80,81,82] |
| M | Decrease serum AST, ALT, liver MDA, histopathological score and expression levels of cleaved caspase-3; increase LC3 II/I ratio and Beclin-1 expression levels | ||||
| DA-1241 | induce | compound | M | Decrease serum CHO and liver TG in HFD mice | [83,84] |
| H | Improve efficacy in the early clinical development for T2D treatment | ||||
| VMP1 | induce | protein | M | Vmp1 deletion in mouse liver impaired mitochondrial b-oxidation and caused the accumulation of neutral lipids in liver, decreased TG levels in serum and the secretion rate for TG; decreased VMP1 expression level in HFD-induced NASH mice | [87,88] |
| Z | vmp1 gene deficiency causes lipoprotein accumulation in the liver and intestine of zebrafish | ||||
| H | Decreased hepatic VMP1 expression level in patients with NASH | ||||
| Nrf2 | induce | protein | M | Nrf2 activator induced the Nrf2/HO-1 dependent pathway to inhibit cleaved caspase-3 and Bcl-2 expression, induce LC3B expression in I/R injury mouse model | [92,93,94] |
| R | Nrf2 activator induced Nrf2/HO-1 axis and autophagy, decreasing inflammation, oxidative stress in I/R injury rat models | ||||
| SGLT-2i | induce | compound | C | Dapagliflozin reduced the lipid droplets contents in HepG2 and L02 cells | [96,97,99] |
| M | Empagliflozin decreased fat mass, body weight, plasma TG and FFA levels, fasting blood glucose levels, NLRP3 inflammasome activity and induced HO-1-adiponectin dependent signaling pathway to prevent obesity | ||||
| R | Dapagliflozin activated AMPK and inhibited mTOR in ZDF rat | ||||
| H | SGLT-2i improved plasma liver enzymes, liver steatosis and NAFLD fibrosis score in T2DM and NAFLD patients | ||||
| IRGM | induce | gene | C | IRGM knockdown in HepG2 and PLC/PRF/5 cells decreased pULK1, Beclin-1 and LC3 II/I ratio and increased lipid droplet | [40,106] |
| H | Obese children with variant IRGM rs10065172 genotype have higher risk of MAFLD | ||||
| ATG7 | induce | gene | C | ATG7 knockdown in hepatocytes increased the lipid droplet amount; ATG7 V471A mutation significantly increased the expression levels of p62, and decreased LC3 II/I ratio in HepG2 cells | [107,108,109] |
| M | HFD-fed Atg7 knockout mice induced the myokines Fgf21 gene expression | ||||
| H | ATG7 rs143545741 variants in severe NAFLD patients | ||||
| CD36 | repress | protein | C | CD36 overexpression in HepG2 or Huh7 cells increased lipid droplets accumulation whereas CD36 knockdown induced lipophagy and fatty acid b-oxidation | [111] |
| M | HFD increased hepatic CD36 expression and decreased LC3II expression in C57BL/6J mice; Cd36 knockout increased LC3II in mouse liver and reconstruction of Cd36 restored the autophagy inhibition | ||||
| LAP3 | repress | protein | C | CHO treatment increased LAP3 expression levels and decreased LC3 II/I ratio in L02 cells | [114] |
| R | HFD increased LAP3 expression level in the E3 rats with NASH; LAP3 knockdown increased LC3 II/I ratio | ||||
| H | Elevated plasm LAP3 levels in patients with NAFLD; positively correlated with fasting blood glucose and TG levels | ||||
| SARS-CoV-2 S | repress | protein | C | Increased lipid deposition in cell membrane, decreased expression levels of LC3I and II in SARS-CoV-2 Spike protein over-expressed HEK293 cells | [120,121] |
| H | SARS-CoV-2 caused hepatocyte death and increased the expression levels of ATG5, LC3, Bax and caspase 3 in the liver of postmortem cases |
3.3. Application of Autophagy-Modulating Agents in Human Diseases
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef]
- Yamamura, S.; Eslam, M.; Kawaguchi, T.; Tsutsumi, T.; Nakano, D.; Yoshinaga, S.; Takahashi, H.; Anzai, K.; George, J.; Torimura, T. MAFLD identifies patients with significant hepatic fibrosis better than NAFLD. Liver Int. 2020, 40, 3018–3030. [Google Scholar] [CrossRef]
- Eslam, M.; Sanyal, A.J.; George, J.; Sanyal, A.; Neuschwander-Tetri, B.; Tiribelli, C.; Kleiner, D.E.; Brunt, E.; Bugianesi, E.; Yki-Järvinen, H. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1. [Google Scholar] [CrossRef]
- Matteoni, C.A.; Younossi, Z.M.; Gramlich, T.; Boparai, N.; Liu, Y.C.; McCullough, A.J. Nonalcoholic fatty liver disease: A spectrum of clinical and pathological severity. Gastroenterology 1999, 116, 1413–1419. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Campbell-Sargent, C.; Mirshahi, F.; Rizzo, W.B.; Contos, M.J.; Sterling, R.K.; Luketic, V.A.; Shiffman, M.L.; Clore, J.N. Nonalcoholic steatohepatitis: Association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001, 120, 1183–1192. [Google Scholar] [CrossRef]
- Schwimmer, J.B.; Deutsch, R.; Rauch, J.B.; Behling, C.; Newbury, R.; Lavine, J.E. Obesity, insulin resistance, and other clinicopathological correlates of pediatric nonalcoholic fatty liver disease. J. Pediatr. 2003, 143, 500–505. [Google Scholar] [CrossRef]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Lebovics, E.; Rubin, J. Non-alcoholic fatty liver disease (NAFLD): Why you should care, when you should worry, what you should do. Diabetes Metab. Res. Rev. 2011, 27, 419–424. [Google Scholar] [CrossRef]
- Duarte, N.; Coelho, I.C.; Patarrao, R.S.; Almeida, J.I.; Penha-Goncalves, C.; Macedo, M.P. How Inflammation Impinges on NAFLD: A Role for Kupffer Cells. Biomed. Res. Int. 2015, 2015, 984578. [Google Scholar] [CrossRef] [PubMed]
- Kazankov, K.; Jorgensen, S.M.D.; Thomsen, K.L.; Moller, H.J.; Vilstrup, H.; George, J.; Schuppan, D.; Gronbaek, H. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 145–159. [Google Scholar] [CrossRef]
- Luo, X.; Luo, S.Z.; Xu, Z.X.; Zhou, C.; Li, Z.H.; Zhou, X.Y.; Xu, M.Y. Lipotoxic hepatocyte-derived exosomal miR-1297 promotes hepatic stellate cell activation through the PTEN signaling pathway in metabolic-associated fatty liver disease. World J. Gastroenterol. 2021, 27, 1419–1434. [Google Scholar] [CrossRef]
- Fianchi, F.; Liguori, A.; Gasbarrini, A.; Grieco, A.; Miele, L. Nonalcoholic Fatty Liver Disease (NAFLD) as Model of Gut-Liver Axis Interaction: From Pathophysiology to Potential Target of Treatment for Personalized Therapy. Int. J. Mol. Sci. 2021, 22, 6485. [Google Scholar] [CrossRef]
- Wynn, T.A.; Barron, L. Macrophages: Master regulators of inflammation and fibrosis. Semin. Liver Dis. 2010, 30, 245–257. [Google Scholar] [CrossRef]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef]
- Weiskirchen, R.; Tacke, F. Relevance of Autophagy in Parenchymal and Non-Parenchymal Liver Cells for Health and Disease. Cells 2019, 8, 16. [Google Scholar] [CrossRef]
- Huda, N.; Zou, H.; Yan, S.; Khambu, B.; Yin, X.M. Analysis of Autophagy for Liver Pathogenesis. Methods Mol. Biol. 2019, 1880, 481–489. [Google Scholar]
- Qin, J.; Xu, Q. Functions and application of exosomes. Acta Pol. Pharm. 2014, 71, 537–543. [Google Scholar]
- Lee, Y.S.; Kim, S.Y.; Ko, E.; Lee, J.H.; Yi, H.S.; Yoo, Y.J.; Je, J.; Suh, S.J.; Jung, Y.K.; Kim, J.H.; et al. Exosomes derived from palmitic acid-treated hepatocytes induce fibrotic activation of hepatic stellate cells. Sci. Rep. 2017, 7, 3710. [Google Scholar] [CrossRef]
- Li, J.; Wang, T.; Liu, P.; Yang, F.; Wang, X.; Zheng, W.; Sun, W. Hesperetin ameliorates hepatic oxidative stress and inflammation via the PI3K/AKT-Nrf2-ARE pathway in oleic acid-induced HepG2 cells and a rat model of high-fat diet-induced NAFLD. Food Funct. 2021, 12, 3898–3918. [Google Scholar] [CrossRef]
- Pedersen, H.K.; Gudmundsdottir, V.; Nielsen, H.B.; Hyotylainen, T.; Nielsen, T.; Jensen, B.A.; Forslund, K.; Hildebrand, F.; Prifti, E.; Falony, G.; et al. Human gut microbes impact host serum metabolome and insulin sensitivity. Nature 2016, 535, 376–381. [Google Scholar] [CrossRef]
- Gadd, V.L.; Skoien, R.; Powell, E.E.; Fagan, K.J.; Winterford, C.; Horsfall, L.; Irvine, K.; Clouston, A.D. The portal inflammatory infiltrate and ductular reaction in human nonalcoholic fatty liver disease. Hepatology 2014, 59, 1393–1405. [Google Scholar] [CrossRef]
- Alharthi, J.; Latchoumanin, O.; George, J.; Eslam, M. Macrophages in metabolic associated fatty liver disease. World J. Gastroenterol. 2020, 26, 1861–1878. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Invest. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Wan, J.; Benkdane, M.; Teixeira-Clerc, F.; Bonnafous, S.; Louvet, A.; Lafdil, F.; Pecker, F.; Tran, A.; Gual, P.; Mallat, A.; et al. M2 Kupffer cells promote M1 Kupffer cell apoptosis: A protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology 2014, 59, 130–142. [Google Scholar] [CrossRef]
- Bleriot, C.; Barreby, E.; Dunsmore, G.; Ballaire, R.; Chakarov, S.; Ficht, X.; De Simone, G.; Andreata, F.; Fumagalli, V.; Guo, W.; et al. A subset of Kupffer cells regulates metabolism through the expression of CD36. Immunity 2021, 54, 2101–2116.e6. [Google Scholar] [CrossRef]
- Friedman, S.L.; Roll, F.J.; Boyles, J.; Bissell, D.M. Hepatic lipocytes: The principal collagen-producing cells of normal rat liver. Proc. Natl. Acad. Sci. USA 1985, 82, 8681–8685. [Google Scholar] [CrossRef] [PubMed]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef]
- Wu, M.H.; Chen, Y.L.; Lee, K.H.; Chang, C.C.; Cheng, T.M.; Wu, S.Y.; Tu, C.C.; Tsui, W.L. Glycosylation-dependent galectin-1/neuropilin-1 interactions promote liver fibrosis through activation of TGF-beta- and PDGF-like signals in hepatic stellate cells. Sci. Rep. 2017, 7, 11006. [Google Scholar] [CrossRef]
- Sakurai, M.; Weber, P.; Wolff, G.; Wieder, A.; Szendroedi, J.; Herzig, S.; Üstünel, B.E. TSC22D4 promotes TGFβ1-induced activation of hepatic stellate cells. Biochem. Biophys. Res. Commun. 2022, 618, 46–53. [Google Scholar] [CrossRef]
- Thoen, L.F.; Guimaraes, E.L.; Grunsven, L.A. Autophagy: A new player in hepatic stellate cell activation. Autophagy 2012, 8, 126–128. [Google Scholar] [CrossRef]
- Zhang, J.; Jiang, N.; Ping, J.; Xu, L. TGFbeta1induced autophagy activates hepatic stellate cells via the ERK and JNK signaling pathways. Int. J. Mol. Med. 2021, 47, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Lucantoni, F.; Martinez-Cerezuela, A.; Gruevska, A.; Moragrega, A.B.; Victor, V.M.; Esplugues, J.V.; Blas-Garcia, A.; Apostolova, N. Understanding the implication of autophagy in the activation of hepatic stellate cells in liver fibrosis: Are we there yet? J. Pathol. 2021, 254, 216–228. [Google Scholar] [CrossRef] [PubMed]
- Schäffler, A.; Schölmerich, J.; Büchler, C. Mechanisms of disease: Adipocytokines and visceral adipose tissue-emerging role in nonalcoholic fatty liver disease. Nat. Clin. Pract. Gastroenterol. Hepatol. 2005, 2, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Takahashi, S.; Higashiura, Y.; Sakai, A.; Koyama, M.; Saitoh, S.; Shimamoto, K.; Ohnishi, H.; Furuhashi, M. Circulating level of fatty acid-binding protein 4 is an independent predictor of metabolic dysfunction-associated fatty liver disease in middle-aged and elderly individuals. J. Diabetes Investig. 2022, 13, 878–888. [Google Scholar] [CrossRef] [PubMed]
- Frayn, K.N. Visceral fat and insulin resistance--causative or correlative? Br. J. Nutr. 2000, 83, S71–S77. [Google Scholar] [CrossRef]
- Meek, S.E.; Nair, K.S.; Jensen, M.D. Insulin regulation of regional free fatty acid metabolism. Diabetes 1999, 48, 10–14. [Google Scholar] [CrossRef]
- Lee, Y.H.; Pratley, R.E. The evolving role of inflammation in obesity and the metabolic syndrome. Curr. Diab. Rep. 2005, 5, 70–75. [Google Scholar] [CrossRef]
- Kim, J.; Lee, S.; Lee, M.S. Suppressive Effect of Autocrine FGF21 on Autophagy-Deficient Hepatic Tumorigenesis. Front. Oncol. 2022, 12, 832804. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cadwell, K.; Cecconi, F.; Choi, A.M.K.; et al. Autophagy in major human diseases. EMBO J. 2021, 40, e108863. [Google Scholar] [CrossRef]
- Goswami, A.B.; Karadarevic, D.; Castano-Rodriguez, N. Immunity-related GTPase IRGM at the intersection of autophagy, inflammation, and tumorigenesis. Inflamm. Res. 2022, 71, 785–795. [Google Scholar] [CrossRef]
- Lin, C.J.; Tsao, Y.N.; Shu, C.W. Autophagy modulation as a potential targeted cancer therapy: From drug repurposing to new drug development. Kaohsiung J. Med. Sci. 2021, 37, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Petibone, D.M.; Majeed, W.; Casciano, D.A. Autophagy function and its relationship to pathology, clinical applications, drug metabolism and toxicity. J. Appl. Toxicol. 2017, 37, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Wirawan, E.; Vande Walle, L.; Kersse, K.; Cornelis, S.; Claerhout, S.; Vanoverberghe, I.; Roelandt, R.; De Rycke, R.; Verspurten, J.; Declercq, W.; et al. Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death Dis. 2010, 1, e18. [Google Scholar] [CrossRef] [PubMed]
- Cairo, M.; Villarroya, J. The role of autophagy in brown and beige adipose tissue plasticity. J. Physiol. Biochem. 2020, 76, 213–226. [Google Scholar] [CrossRef]
- Sinha, R.A.; Bruinstroop, E.; Singh, B.K.; Yen, P.M. Nonalcoholic Fatty Liver Disease and Hypercholesterolemia: Roles of Thyroid Hormones, Metabolites, and Agonists. Thyroid 2019, 29, 1173–1191. [Google Scholar] [CrossRef]
- Feng, X.; Jiang, Y.; Meltzer, P.; Yen, P.M. Thyroid hormone regulation of hepatic genes in vivo detected by complementary DNA microarray. Mol. Endocrinol. 2000, 14, 947–955. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, W.; Yu, X.; Qi, X. Thyroid Function and Risk of Non-Alcoholic Fatty Liver Disease in Euthyroid Subjects. Ann. Hepatol. 2018, 17, 779–788. [Google Scholar] [CrossRef]
- Chi, H.C.; Tsai, C.Y.; Tsai, M.M.; Yeh, C.T.; Lin, K.H. Molecular functions and clinical impact of thyroid hormone-triggered autophagy in liver-related diseases. J. Biomed. Sci. 2019, 26, 24. [Google Scholar] [CrossRef]
- Finan, B.; Clemmensen, C.; Zhu, Z.; Stemmer, K.; Gauthier, K.; Muller, L.; De Angelis, M.; Moreth, K.; Neff, F.; Perez-Tilve, D.; et al. Chemical Hybridization of Glucagon and Thyroid Hormone Optimizes Therapeutic Impact for Metabolic Disease. Cell 2016, 167, 843–857.e14. [Google Scholar] [CrossRef]
- Bruinstroop, E.; Dalan, R.; Cao, Y.; Bee, Y.M.; Chandran, K.; Cho, L.W.; Soh, S.B.; Teo, E.K.; Toh, S.A.; Leow, M.K.S.; et al. Low-Dose Levothyroxine Reduces Intrahepatic Lipid Content in Patients with Type 2 Diabetes Mellitus and NAFLD. J. Clin. Endocrinol. Metab. 2018, 103, 2698–2706. [Google Scholar] [CrossRef]
- Zhou, J.; Tripathi, M.; Ho, J.P.; Widjaja, A.A.; Shekeran, S.G.; Camat, M.D.; James, A.; Wu, Y.; Ching, J.; Kovalik, J.P.; et al. Thyroid Hormone Decreases Hepatic Steatosis, Inflammation, and Fibrosis in a Dietary Mouse Model of Nonalcoholic Steatohepatitis. Thyroid 2022, 32, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Elizondo-Montemayor, L.; Mendoza-Lara, G.; Gutierrez-DelBosque, G.; Peschard-Franco, M.; Nieblas, B.; Garcia-Rivas, G. Relationship of Circulating Irisin with Body Composition, Physical Activity, and Cardiovascular and Metabolic Disorders in the Pediatric Population. Int. J. Mol. Sci. 2018, 19, 3727. [Google Scholar] [CrossRef] [Green Version]
- Arhire, L.I.; Mihalache, L.; Covasa, M. Irisin: A Hope in Understanding and Managing Obesity and Metabolic Syndrome. Front. Endocrinol. 2019, 10, 524. [Google Scholar] [CrossRef]
- Ma, C.; Ding, H.; Deng, Y.; Liu, H.; Xiong, X.; Yang, Y. Irisin: A New Code Uncover the Relationship of Skeletal Muscle and Cardiovascular Health During Exercise. Front. Physiol. 2021, 12, 620608. [Google Scholar] [CrossRef]
- Zhu, W.; Sahar, N.E.; Javaid, H.M.A.; Pak, E.S.; Liang, G.; Wang, Y.; Ha, H.; Huh, J.Y. Exercise-Induced Irisin Decreases Inflammation and Improves NAFLD by Competitive Binding with MD2. Cells 2021, 10, 3306. [Google Scholar] [CrossRef] [PubMed]
- Mazur-Bialy, A.I.; Pochec, E.; Zarawski, M. Anti-Inflammatory Properties of Irisin, Mediator of Physical Activity, Are Connected with TLR4/MyD88 Signaling Pathway Activation. Int. J. Mol. Sci. 2017, 18, 701. [Google Scholar] [CrossRef] [PubMed]
- Mazur-Bialy, A.I.; Bilski, J.; Wojcik, D.; Brzozowski, B.; Surmiak, M.; Hubalewska-Mazgaj, M.; Chmura, A.; Magierowski, M.; Magierowska, K.; Mach, T.; et al. Beneficial Effect of Voluntary Exercise on Experimental Colitis in Mice Fed a High-Fat Diet: The Role of Irisin, Adiponectin and Proinflammatory Biomarkers. Nutrients 2017, 9, 410. [Google Scholar] [CrossRef]
- Mazur-Bialy, A.I.; Bilski, J.; Pochec, E.; Brzozowski, T. New insight into the direct anti-inflammatory activity of a myokine irisin against proinflammatory activation of adipocytes. Implication for exercise in obesity. J. Physiol. Pharmacol. 2017, 68, 243–251. [Google Scholar]
- Mazur-Bialy, A.I. Irisin acts as a regulator of macrophages host defense. Life Sci. 2017, 176, 21–25. [Google Scholar] [CrossRef]
- Bi, J.; Yang, L.; Wang, T.; Zhang, J.; Li, T.; Ren, Y.; Wang, M.; Chen, X.; Lv, Y.; Wu, R. Irisin Improves Autophagy of Aged Hepatocytes via Increasing Telomerase Activity in Liver Injury. Oxid. Med. Cell Longev. 2020, 2020, 6946037. [Google Scholar] [CrossRef]
- Bi, J.; Zhang, J.; Ren, Y.; Du, Z.; Li, T.; Wang, T.; Zhang, L.; Wang, M.; Wu, Z.; Lv, Y.; et al. Irisin reverses intestinal epithelial barrier dysfunction during intestinal injury via binding to the integrin alphaVbeta5 receptor. J. Cell Mol. Med. 2020, 24, 996–1009. [Google Scholar] [CrossRef] [PubMed]
- Shimazu, R.; Akashi, S.; Ogata, H.; Nagai, Y.; Fukudome, K.; Miyake, K.; Kimoto, M. MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J. Exp. Med. 1999, 189, 1777–1782. [Google Scholar] [CrossRef] [PubMed]
- Palsson-McDermott, E.M.; O′Neill, L.A. Signal transduction by the lipopolysaccharide receptor, Toll-like receptor-4. Immunology 2004, 113, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, Y.; Antony, V.; Sun, H.; Liang, G. Metabolism-Associated Molecular Patterns (MAMPs). Trends Endocrinol. Metab. 2020, 31, 712–724. [Google Scholar] [CrossRef] [PubMed]
- Nastos, C.; Kalimeris, K.; Papoutsidakis, N.; Tasoulis, M.K.; Lykoudis, P.M.; Theodoraki, K.; Nastou, D.; Smyrniotis, V.; Arkadopoulos, N. Global consequences of liver ischemia/reperfusion injury. Oxid. Med. Cell Longev. 2014, 2014, 906965. [Google Scholar] [CrossRef] [PubMed]
- Kan, C.; Ungelenk, L.; Lupp, A.; Dirsch, O.; Dahmen, U. Ischemia-Reperfusion Injury in Aged Livers-The Energy Metabolism, Inflammatory Response, and Autophagy. Transplantation 2018, 102, 368–377. [Google Scholar] [CrossRef]
- Kimura, H.; Shibuya, N.; Kimura, Y. Hydrogen sulfide is a signaling molecule and a cytoprotectant. Antioxid. Redox Signal. 2012, 17, 45–57. [Google Scholar]
- Lu, X.; Ding, Y.; Liu, H.; Sun, M.; Chen, C.; Yang, Y.; Wang, H. The Role of Hydrogen Sulfide Regulation of Autophagy in Liver Disorders. Int. J. Mol. Sci. 2022, 23, 4035. [Google Scholar] [CrossRef]
- Wu, D.; Zhong, P.; Wang, Y.; Zhang, Q.; Li, J.; Liu, Z.; Ji, A.; Li, Y. Hydrogen Sulfide Attenuates High-Fat Diet-Induced Non-Alcoholic Fatty Liver Disease by Inhibiting Apoptosis and Promoting Autophagy via Reactive Oxygen Species/Phosphatidylinositol 3-Kinase/AKT/Mammalian Target of Rapamycin Signaling Pathway. Front. Pharmacol. 2020, 11, 585860. [Google Scholar]
- Guo, J.M.; Xing, H.J.; Cai, J.Z.; Zhang, H.F.; Xu, S.W. H2S exposure-induced oxidative stress promotes LPS-mediated hepatocyte autophagy through the PI3K/AKT/TOR pathway. Ecotoxicol. Environ. Saf. 2021, 209, 111801. [Google Scholar] [CrossRef]
- Tan, D.X.; Manchester, L.C.; Esteban-Zubero, E.; Zhou, Z.; Reiter, R.J. Melatonin as a Potent and Inducible Endogenous Antioxidant: Synthesis and Metabolism. Molecules 2015, 20, 18886–18906. [Google Scholar] [PubMed]
- Karasek, M.; Winczyk, K. Melatonin in humans. J. Physiol. Pharmacol. 2006, 57, 19–39. [Google Scholar] [PubMed]
- Mortezaee, K.; Khanlarkhani, N. Melatonin application in targeting oxidative-induced liver injuries: A review. J. Cell Physiol. 2018, 233, 4015–4032. [Google Scholar] [PubMed]
- Carbajo-Pescador, S.; Steinmetz, C.; Kashyap, A.; Lorenz, S.; Mauriz, J.L.; Heise, M.; Galle, P.R.; Gonzalez-Gallego, J.; Strand, S. Melatonin induces transcriptional regulation of Bim by FoxO3a in HepG2 cells. Br. J. Cancer 2013, 108, 442–449. [Google Scholar]
- Hu, W.; Ma, Z.; Jiang, S.; Fan, C.; Deng, C.; Yan, X.; Di, S.; Lv, J.; Reiter, R.J.; Yang, Y. Melatonin: The dawning of a treatment for fibrosis? J. Pineal Res. 2016, 60, 121–131. [Google Scholar]
- Zhang, S.; Chen, S.; Li, Y.; Liu, Y. Melatonin as a promising agent of regulating stem cell biology and its application in disease therapy. Pharmacol. Res. 2017, 117, 252–260. [Google Scholar]
- Yang, Z.; He, Y.; Wang, H.; Zhang, Q. Protective effect of melatonin against chronic cadmium-induced hepatotoxicity by suppressing oxidative stress, inflammation, and apoptosis in mice. Ecotoxicol. Environ. Saf. 2021, 228, 112947. [Google Scholar] [CrossRef]
- Joshi, A.; Upadhyay, K.K.; Vohra, A.; Shirsath, K.; Devkar, R. Melatonin induces Nrf2-HO-1 reprogramming and corrections in hepatic core clock oscillations in Non-alcoholic fatty liver disease. FASEB J. 2021, 35, e21803. [Google Scholar]
- San-Miguel, B.; Fernandez-Palanca, P.; Mauriz, J.L.; Tunon, M.J.; Gonzalez-Gallego, J. Beneficial effects of melatonin on liver fibrosis: A systematic review of current biological evidence. J. Cell Physiol. 2022, 237, 2740–2757. [Google Scholar]
- Das, N.; Mandala, A.; Naaz, S.; Giri, S.; Jain, M.; Bandyopadhyay, D.; Reiter, R.J.; Roy, S.S. Melatonin protects against lipid-induced mitochondrial dysfunction in hepatocytes and inhibits stellate cell activation during hepatic fibrosis in mice. J. Pineal Res. 2017, 62, e12404. [Google Scholar]
- Shajari, S.; Laliena, A.; Heegsma, J.; Tunon, M.J.; Moshage, H.; Faber, K.N. Melatonin suppresses activation of hepatic stellate cells through RORalpha-mediated inhibition of 5-lipoxygenase. J. Pineal Res. 2015, 59, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Barangi, S.; Mehri, S.; Moosavi, Z.; Hayesd, A.W.; Reiter, R.J.; Cardinali, D.P.; Karimi, G. Melatonin inhibits Benzo(a)pyrene-Induced apoptosis through activation of the Mir-34a/Sirt1/autophagy pathway in mouse liver. Ecotoxicol. Environ. Saf. 2020, 196, 110556. [Google Scholar] [CrossRef]
- Kim, M.K.; Cheong, Y.H.; Lee, S.H.; Kim, T.H.; Jung, I.H.; Chae, Y.; Lee, J.H.; Yang, E.K.; Park, H.; Yang, J.S.; et al. A novel GPR119 agonist DA-1241 preserves pancreatic function via the suppression of ER stress and increased PDX1 expression. Biomed. Pharmacother. 2021, 144, 112324. [Google Scholar] [PubMed]
- Kim, Y.; Lee, S.W.; Wang, H.; Kim, R.H.; Park, H.K.; Lee, H.; Kang, E.S. DA-1241, a Novel GPR119 Agonist, Improves Hyperglycaemia by Inhibiting Hepatic Gluconeogenesis and Enhancing Insulin Secretion in Diabetic Mice. Diabetes Metab. J. 2022, 46, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Morita, K.; Hama, Y.; Izume, T.; Tamura, N.; Ueno, T.; Yamashita, Y.; Sakamaki, Y.; Mimura, K.; Morishita, H.; Shihoya, W.; et al. Genome-wide CRISPR screen identifies TMEM41B as a gene required for autophagosome formation. J. Cell Biol. 2018, 217, 3817–3828. [Google Scholar]
- Zhao, Y.G.; Chen, Y.; Miao, G.; Zhao, H.; Qu, W.; Li, D.; Wang, Z.; Liu, N.; Li, L.; Chen, S.; et al. The ER-Localized Transmembrane Protein EPG-3/VMP1 Regulates SERCA Activity to Control ER-Isolation Membrane Contacts for Autophagosome Formation. Mol. Cell 2017, 67, 974–989.e6. [Google Scholar] [CrossRef]
- Morishita, H.; Zhao, Y.G.; Tamura, N.; Nishimura, T.; Kanda, Y.; Sakamaki, Y.; Okazaki, M.; Li, D.; Mizushima, N. A critical role of VMP1 in lipoprotein secretion. Elife 2019, 8, e48834. [Google Scholar]
- Jiang, X.; Fulte, S.; Deng, F.; Chen, S.; Xie, Y.; Chao, X.; He, X.C.; Zhang, Y.; Li, T.; Li, F.; et al. Lack of VMP1 impairs hepatic lipoprotein secretion and promotes non-alcoholic steatohepatitis. J. Hepatol. 2022, 77, 619–631. [Google Scholar] [CrossRef]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Cell biology of ischemia/reperfusion injury. Int. Rev. Cell Mol. Biol. 2012, 298, 229–317. [Google Scholar]
- Sadrkhanloo, M.; Entezari, M.; Orouei, S.; Zabolian, A.; Mirzaie, A.; Maghsoudloo, A.; Raesi, R.; Asadi, N.; Hashemi, M.; Zarrabi, A.; et al. Targeting Nrf2 in ischemia-reperfusion alleviation: From signaling networks to therapeutic targeting. Life Sci. 2022, 300, 120561. [Google Scholar]
- Ashrafizadeh, M.; Paskeh, M.D.A.; Mirzaei, S.; Gholami, M.H.; Zarrabi, A.; Hashemi, F.; Hushmandi, K.; Hashemi, M.; Nabavi, N.; Crea, F.; et al. Targeting autophagy in prostate cancer: Preclinical and clinical evidence for therapeutic response. J. Exp. Clin. Cancer Res. 2022, 41, 105. [Google Scholar]
- Xu, D.; Chen, L.; Chen, X.; Wen, Y.; Yu, C.; Yao, J.; Wu, H.; Wang, X.; Xia, Q.; Kong, X. The triterpenoid CDDO-imidazolide ameliorates mouse liver ischemia-reperfusion injury through activating the Nrf2/HO-1 pathway enhanced autophagy. Cell Death Dis. 2017, 8, e2983. [Google Scholar] [PubMed]
- Yuan, B.; Huang, H.; Qu, S.; Zhang, H.; Lin, J.; Jin, L.; Yang, S.; Zeng, Z. Gastrodin Pretreatment Protects Liver Against Ischemia-Reperfusion Injury via Activation of the Nrf2/HO-1 Pathway. Am. J. Chin. Med. 2020, 48, 1159–1178. [Google Scholar]
- Ma, H.; Yang, B.; Yu, L.; Gao, Y.; Ye, X.; Liu, Y.; Li, Z.; Li, H.; Li, E. Sevoflurane protects the liver from ischemia-reperfusion injury by regulating Nrf2/HO-1 pathway. Eur. J. Pharmacol. 2021, 898, 173932. [Google Scholar] [PubMed]
- Hardman, T.C.; Rutherford, P.; Dubrey, S.W.; Wierzbicki, A.S. Sodium-glucose co-transporter 2 inhibitors: From apple tree to ‘Sweet Pee’. Curr. Pharm. Des. 2010, 16, 3830–3838. [Google Scholar] [PubMed]
- Androutsakos, T.; Nasiri-Ansari, N.; Bakasis, A.D.; Kyrou, I.; Efstathopoulos, E.; Randeva, H.S.; Kassi, E. SGLT-2 Inhibitors in NAFLD: Expanding Their Role beyond Diabetes and Cardioprotection. Int. J. Mol. Sci. 2022, 23, 3107. [Google Scholar]
- Ye, T.; Zhang, J.; Wu, D.; Shi, J.; Kuang, Z.; Ma, Y.; Xu, Q.; Chen, B.; Kan, C.; Sun, X.; et al. Empagliflozin Attenuates Obesity-Related Kidney Dysfunction and NLRP3 Inflammasome Activity Through the HO-1-Adiponectin Axis. Front. Endocrinol. 2022, 13, 907984. [Google Scholar]
- Jojima, T.; Wakamatsu, S.; Kase, M.; Iijima, T.; Maejima, Y.; Shimomura, K.; Kogai, T.; Tomaru, T.; Usui, I.; Aso, Y. The SGLT2 Inhibitor Canagliflozin Prevents Carcinogenesis in a Mouse Model of Diabetes and Non-Alcoholic Steatohepatitis-Related Hepatocarcinogenesis: Association with SGLT2 Expression in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2019, 20, 5237. [Google Scholar]
- Li, L.; Li, Q.; Huang, W.; Han, Y.; Tan, H.; An, M.; Xiang, Q.; Zhou, R.; Yang, L.; Cheng, Y. Dapagliflozin Alleviates Hepatic Steatosis by Restoring Autophagy via the AMPK-mTOR Pathway. Front. Pharmacol. 2021, 12, 589273. [Google Scholar] [CrossRef]
- Chauhan, S.; Mandell, M.A.; Deretic, V. IRGM governs the core autophagy machinery to conduct antimicrobial defense. Mol. Cell 2015, 58, 507–521. [Google Scholar]
- Kim, J.; Kim, Y.C.; Fang, C.; Russell, R.C.; Kim, J.H.; Fan, W.; Liu, R.; Zhong, Q.; Guan, K.L. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell 2013, 152, 290–303. [Google Scholar] [PubMed] [Green Version]
- Egan, D.; Kim, J.; Shaw, R.J.; Guan, K.L. The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy 2011, 7, 643–644. [Google Scholar] [PubMed]
- Schwimmer, J.B.; McGreal, N.; Deutsch, R.; Finegold, M.J.; Lavine, J.E. Influence of gender, race, and ethnicity on suspected fatty liver in obese adolescents. Pediatrics 2005, 115, e561–e565. [Google Scholar] [PubMed]
- Petersen, K.F.; Dufour, S.; Feng, J.; Befroy, D.; Dziura, J.; Dalla Man, C.; Cobelli, C.; Shulman, G.I. Increased prevalence of insulin resistance and nonalcoholic fatty liver disease in Asian-Indian men. Proc. Natl. Acad. Sci. USA 2006, 103, 18273–18277. [Google Scholar] [PubMed]
- Parkes, M.; Barrett, J.C.; Prescott, N.J.; Tremelling, M.; Anderson, C.A.; Fisher, S.A.; Roberts, R.G.; Nimmo, E.R.; Cummings, F.R.; Soars, D.; et al. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn’s disease susceptibility. Nat. Genet. 2007, 39, 830–832. [Google Scholar]
- Lin, Y.C.; Chang, P.F.; Lin, H.F.; Liu, K.; Chang, M.H.; Ni, Y.H. Variants in the autophagy-related gene IRGM confer susceptibility to non-alcoholic fatty liver disease by modulating lipophagy. J. Hepatol. 2016, 65, 1209–1216. [Google Scholar]
- Baselli, G.A.; Jamialahmadi, O.; Pelusi, S.; Ciociola, E.; Malvestiti, F.; Saracino, M.; Santoro, L.; Cherubini, A.; Dongiovanni, P.; Maggioni, M.; et al. Rare ATG7 genetic variants predispose patients to severe fatty liver disease. J. Hepatol. 2022, 77, 596–606. [Google Scholar]
- Kim, K.H.; Kim, S.H.; Min, Y.K.; Yang, H.M.; Lee, J.B.; Lee, M.S. Acute exercise induces FGF21 expression in mice and in healthy humans. PLoS ONE 2013, 8, e63517. [Google Scholar]
- Kim, K.H.; Jeong, Y.T.; Oh, H.; Kim, S.H.; Cho, J.M.; Kim, Y.N.; Kim, S.S.; Kim, D.H.; Hur, K.Y.; Kim, H.K.; et al. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat. Med. 2013, 19, 83–92. [Google Scholar]
- Hajri, T.; Abumrad, N.A. Fatty acid transport across membranes: Relevance to nutrition and metabolic pathology. Annu. Rev. Nutr. 2002, 22, 383–415. [Google Scholar]
- Li, Y.; Yang, P.; Zhao, L.; Chen, Y.; Zhang, X.; Zeng, S.; Wei, L.; Varghese, Z.; Moorhead, J.F.; Chen, Y.; et al. CD36 plays a negative role in the regulation of lipophagy in hepatocytes through an AMPK-dependent pathway. J. Lipid Res. 2019, 60, 844–855. [Google Scholar] [CrossRef] [PubMed]
- Niu, L.; Geyer, P.E.; Wewer Albrechtsen, N.J.; Gluud, L.L.; Santos, A.; Doll, S.; Treit, P.V.; Holst, J.J.; Knop, F.K.; Vilsboll, T.; et al. Plasma proteome profiling discovers novel proteins associated with non-alcoholic fatty liver disease. Mol. Syst. Biol. 2019, 15, e8793. [Google Scholar] [CrossRef]
- Ghorpade, D.S.; Ozcan, L.; Zheng, Z.; Nicoloro, S.M.; Shen, Y.; Chen, E.; Bluher, M.; Czech, M.P.; Tabas, I. Hepatocyte-secreted DPP4 in obesity promotes adipose inflammation and insulin resistance. Nature 2018, 555, 673–677. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Chen, Y.; Xu, K.; Li, Y.; Riaz, F.; Lu, K.; Chen, Q.; Du, X.; Wu, L.; Cao, D.; et al. Cholesterol-induced leucine aminopeptidase 3 (LAP3) upregulation inhibits cell autophagy in pathogenesis of NAFLD. Aging 2022, 14, 3259–3275. [Google Scholar]
- Lu, Y.; Liu, D.X.; Tam, J.P. Lipid rafts are involved in SARS-CoV entry into Vero E6 cells. Biochem. Biophys. Res. Commun. 2008, 369, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Wang, H.; Ye, G.; Cao, X.; Xu, X.; Tan, W.; Zhang, Y. Letter to the Editor: Low-density lipoprotein is a potential predictor of poor prognosis in patients with coronavirus disease 2019. Metabolism 2020, 107, 154243. [Google Scholar] [CrossRef]
- Wei, X.; Zeng, W.; Su, J.; Wan, H.; Yu, X.; Cao, X.; Tan, W.; Wang, H. Hypolipidemia is associated with the severity of COVID-19. J. Clin. Lipidol. 2020, 14, 297–304. [Google Scholar] [CrossRef]
- Cao, X.; Yin, R.; Albrecht, H.; Fan, D.; Tan, W. Cholesterol: A new game player accelerating vasculopathy caused by SARS-CoV-2? Am. J. Physiol. Endocrinol. Metab. 2020, 319, E197–E202. [Google Scholar] [CrossRef]
- Tian, S.; Xiong, Y.; Liu, H.; Niu, L.; Guo, J.; Liao, M.; Xiao, S.Y. Pathological study of the 2019 novel coronavirus disease (COVID-19) through postmortem core biopsies. Mod. Pathol. 2020, 33, 1007–1014. [Google Scholar]
- Shirazi Tehrani, A.; Tabatabaei Mirakabad, F.S.; Abdollahifar, M.A.; Mollazadehghomi, S.; Darabi, S.; Forozesh, M.; Rezaei-Tavirani, M.; Mahmoudiasl, G.R.; Ahrabi, B.; Azimzadeh, Z.; et al. Severe Acute Respiratory Syndrome Coronavirus 2 Induces Hepatocyte Cell Death, Active Autophagosome Formation and Caspase 3 Up-Regulation in Postmortem Cases: Stereological and Molecular Study. Tohoku J. Exp. Med. 2022, 256, 309–319. [Google Scholar] [CrossRef]
- Nguyen, V.; Zhang, Y.; Gao, C.; Cao, X.; Tian, Y.; Carver, W.; Kiaris, H.; Cui, T.; Tan, W. The Spike Protein of SARS-CoV-2 Impairs Lipid Metabolism and Increases Susceptibility to Lipotoxicity: Implication for a Role of Nrf2. Cells 2022, 11, 1916. [Google Scholar] [CrossRef] [PubMed]
- Gasiorkiewicz, B.M.; Koczurkiewicz-Adamczyk, P.; Piska, K.; Pekala, E. Autophagy modulating agents as chemosensitizers for cisplatin therapy in cancer. Invest. New Drugs 2021, 39, 538–563. [Google Scholar] [CrossRef] [PubMed]
- Panda, P.K.; Fahrner, A.; Vats, S.; Seranova, E.; Sharma, V.; Chipara, M.; Desai, P.; Torresi, J.; Rosenstock, T.; Kumar, D.; et al. Chemical Screening Approaches Enabling Drug Discovery of Autophagy Modulators for Biomedical Applications in Human Diseases. Front. Cell Dev. Biol. 2019, 7, 38. [Google Scholar] [CrossRef] [PubMed]
- Martins, W.K.; Silva, M.; Pandey, K.; Maejima, I.; Ramalho, E.; Olivon, V.C.; Diniz, S.N.; Grasso, D. Autophagy-targeted therapy to modulate age-related diseases: Success, pitfalls, and new directions. Curr. Res. Pharmacol. Drug Discov. 2021, 2, 100033. [Google Scholar] [CrossRef]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef]
- Bu, F.; Zhang, J.; Shuai, W.; Liu, J.; Sun, Q.; Ouyang, L. Repurposing drugs in autophagy for the treatment of cancer: From bench to bedside. Drug Discov. Today 2022, 27, 1815–1831. [Google Scholar] [CrossRef]
- Mohsen, S.; Sobash, P.T.; Algwaiz, G.F.; Nasef, N.; Al-Zeidaneen, S.A.; Karim, N.A. Autophagy Agents in Clinical Trials for Cancer Therapy: A Brief Review. Curr. Oncol. 2022, 29, 1695–1708. [Google Scholar] [CrossRef]
- Xu, X.; Sun, Y.; Cen, X.; Shan, B.; Zhao, Q.; Xie, T.; Wang, Z.; Hou, T.; Xue, Y.; Zhang, M.; et al. Metformin activates chaperone-mediated autophagy and improves disease pathologies in an Alzheimer disease mouse model. Protein Cell 2021, 12, 769–787. [Google Scholar] [CrossRef]
- Tiwari, S.; Atluri, V.; Kaushik, A.; Yndart, A.; Nair, M. Alzheimer′s disease: Pathogenesis, diagnostics, and therapeutics. Int. J. Nanomed. 2019, 14, 5541–5554. [Google Scholar] [CrossRef] [Green Version]
- Heckman-Stoddard, B.M.; DeCensi, A.; Sahasrabuddhe, V.V.; Ford, L.G. Repurposing metformin for the prevention of cancer and cancer recurrence. Diabetologia 2017, 60, 1639–1647. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.-L.; Lin, Y.-C. Autophagy Dysregulation in Metabolic Associated Fatty Liver Disease: A New Therapeutic Target. Int. J. Mol. Sci. 2022, 23, 10055. https://doi.org/10.3390/ijms231710055
Chen C-L, Lin Y-C. Autophagy Dysregulation in Metabolic Associated Fatty Liver Disease: A New Therapeutic Target. International Journal of Molecular Sciences. 2022; 23(17):10055. https://doi.org/10.3390/ijms231710055
Chicago/Turabian StyleChen, Chun-Liang, and Yu-Cheng Lin. 2022. "Autophagy Dysregulation in Metabolic Associated Fatty Liver Disease: A New Therapeutic Target" International Journal of Molecular Sciences 23, no. 17: 10055. https://doi.org/10.3390/ijms231710055
APA StyleChen, C.-L., & Lin, Y.-C. (2022). Autophagy Dysregulation in Metabolic Associated Fatty Liver Disease: A New Therapeutic Target. International Journal of Molecular Sciences, 23(17), 10055. https://doi.org/10.3390/ijms231710055