
Reference Genes across Nine Brain Areas of Wild Type and Prader-Willi Syndrome Mice: Assessing Differences in Igfbp7, Pcsk1, Nhlh2 and Nlgn3 Expression

 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Prader–Willi Syndrome
1.2. Reference Genes for Expression Analysis across Brain Regions
2. Results
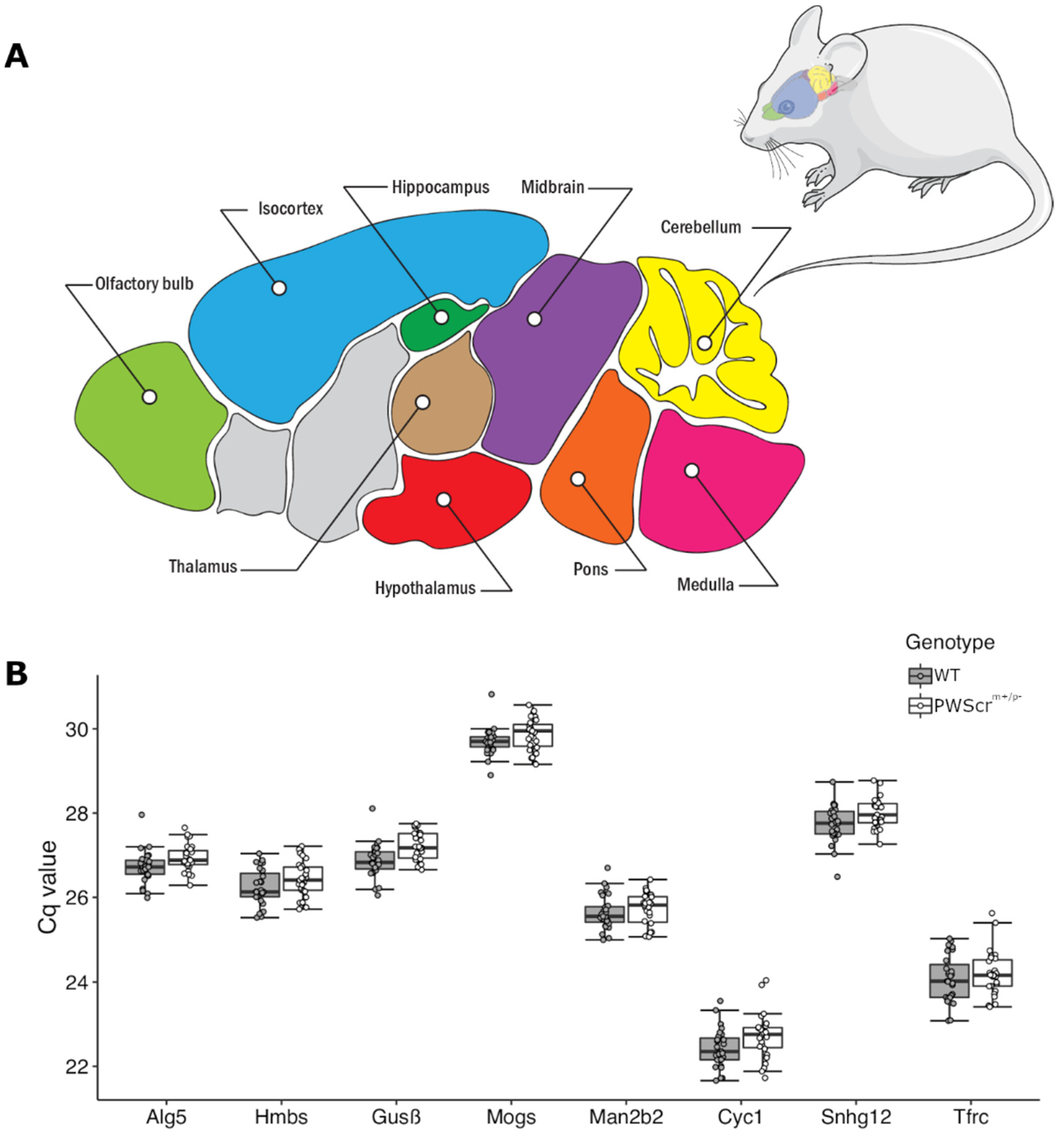
2.1. Partitioning the Mouse Brain into Nine Anatomical District Regions and Reference Gene Candidate Selection
2.2. Compilation of a Reference Gene Panel
2.3. Effect of Snord116 on the Regional Expression of Igfbp7
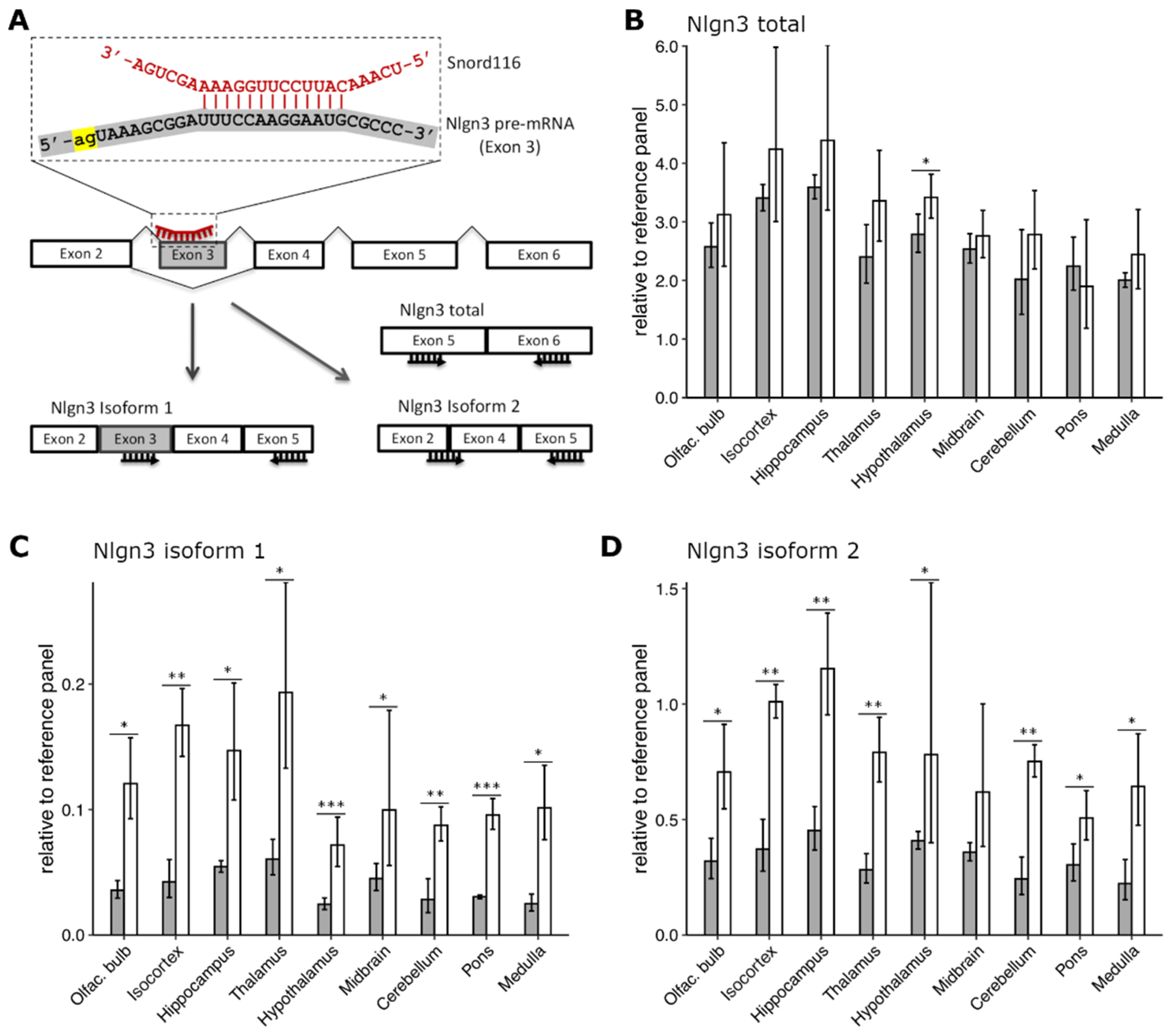
2.4. Lack of Snord116 Influences Abundance but Not Splicing of Nlgn3 mRNA In Vivo
3. Discussion
Potential Interactions of Snord116 with Predicted mRNA Targets
4. Materials and Methods
4.1. Animals and Tissue Collection
4.2. RNA Isolation and cDNA Synthesis
4.3. RT-qPCR
4.4. Ranking of Expression Stability and Statistical Analysis
4.5. Northern Blot Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cassidy, S.B.; Schwartz, S.; Miller, J.L.; Driscoll, D.J. Prader-Willi syndrome. Genet. Med. Off. J. Am. Coll. Med. Genet. 2012, 14, 10–26. [Google Scholar] [CrossRef] [PubMed]
- Cavaillé, J.; Buiting, K.; Kiefmann, M.; Lalande, M.; Brannan, C.I.; Horsthemke, B.; Bachellerie, J.-P.; Brosius, J.; Hüttenhofer, A. Identification of brain-specific and imprinted small nucleolar RNA genes exhibiting an unusual genomic organization. Proc. Natl. Acad. Sci. USA 2000, 97, 14311–14316. [Google Scholar] [CrossRef] [PubMed]
- Runte, M.; Hüttenhofer, A.; Groß, S.; Kiefmann, M.; Horsthemke, B.; Buiting, K. The IC-SNURF-SNRPN transcript serves as a host for multiple small nucleolar RNA species and as an antisense RNA for UBE3A. Hum. Mol. Genet. 2001, 10, 2687–2700. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, T.; Del Gaudio, D.; German, J.R.; Shinawi, M.; Peters, S.U.; Person, R.E.; Garnica, A.; Cheung, S.W.; Beaudet, A.L. Prader-Willi phenotype caused by paternal deficiency for the HBII-85 C/D box small nucleolar RNA cluster. Nat. Genet. 2008, 40, 719–721. [Google Scholar] [CrossRef] [PubMed]
- Skryabin, B.V.; Gubar, L.V.; Seeger, B.; Pfeiffer, J.; Handel, S.; Robeck, T.; Karpova, E.; Rozhdestvensky, T.; Brosius, J. Deletion of the MBII-85 snoRNA Gene Cluster in Mice Results in Postnatal Growth Retardation. PLoS Genet. 2007, 3, e235. [Google Scholar] [CrossRef]
- Kummerfeld, D.-M.; Raabe, C.; Brosius, J.; Mo, D.; Skryabin, B.; Rozhdestvensky, T. A Comprehensive Review of Genetically Engineered Mouse Models for Prader-Willi Syndrome Research. Int. J. Mol. Sci. 2021, 22, 3613. [Google Scholar] [CrossRef]
- Galiveti, C.R.; Raabe, C.A.; Konthur, Z.; Rozhdestvensky, T.S. Differential regulation of non-protein coding RNAs from Prader-Willi Syndrome locus. Sci. Rep. 2014, 4, 6445. [Google Scholar] [CrossRef]
- Butler, M.G.; Miller, J.L.; Forster, J.L. Prader-Willi Syndrome—Clinical Genetics, Diagnosis and Treatment Approaches: An Update. Curr. Pediatr. Rev. 2019, 15, 207–244. [Google Scholar] [CrossRef]
- Diene, G.; Mimoun, E.; Feigerlova, E.; Caula, S.; Molinas, C.; Grandjean, H.; Tauber, M.; French Reference Centre for PWS. Endocrine Disorders in Children with Prader-Willi Syndrome—Data from 142 Children of the French Database. Horm. Res. Paediatr. 2010, 74, 121–128. [Google Scholar] [CrossRef]
- Heksch, R.; Kamboj, M.; Anglin, K.; Obrynba, K. Review of Prader-Willi syndrome: The endocrine approach. Transl. Pediatr. 2017, 6, 274–285. [Google Scholar] [CrossRef]
- Bittel, D.C.; Kibiryeva, N.; Sell, S.M.; Strong, T.; Butler, M.G. Whole genome microarray analysis of gene expression in Prader–Willi syndrome. Am. J. Med Genet. Part A 2007, 143A, 430–442. [Google Scholar] [CrossRef] [PubMed]
- Burnett, L.C.; LeDuc, C.A.; Sulsona, C.R.; Paull, D.; Rausch, R.; Eddiry, S.; Carli, J.F.M.; Morabito, M.V.; Skowronski, A.A.; Hubner, G.; et al. Deficiency in prohormone convertase PC1 impairs prohormone processing in Prader-Willi syndrome. J. Clin. Investig. 2016, 127, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Eddiry, S.; Diene, G.; Molinas, C.; Salles, J.; Auriol, F.C.; Gennero, I.; Bieth, E.; Skryabin, B.V.; Rozhdestvensky, T.S.; Burnett, L.C.; et al. SNORD116 and growth hormone therapy impact IGFBP7 in Prader–Willi syndrome. Genet. Med. 2021, 23, 1664–1672. [Google Scholar] [CrossRef] [PubMed]
- Nillni, E.A. Regulation of Prohormone Convertases in Hypothalamic Neurons: Implications for ProThyrotropin-Releasing Hormone and Proopiomelanocortin. Endocrinology 2007, 148, 4191–4200. [Google Scholar] [CrossRef]
- Seidah, N.G. The Proprotein Convertases, 20 Years Later. In Proprotein Convertases; Mbikay, M., Seidah, N.G., Eds.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2011; pp. 23–57. ISBN 978-1-61779-204-5. [Google Scholar]
- Fox, D.L.; Good, D.J. Nescient Helix-Loop-Helix 2 Interacts with Signal Transducer and Activator of Transcription 3 to Regulate Transcription of Prohormone Convertase 1/3. Mol. Endocrinol. 2008, 22, 1438–1448. [Google Scholar] [CrossRef]
- Coyle, C.A.; Jing, E.; Hosmer, T.; Powers, J.; Wade, G.; Good, D.J. Reduced voluntary activity precedes adult-onset obesity in Nhlh2 knockout mice. Physiol. Behav. 2002, 77, 387–402. [Google Scholar] [CrossRef]
- Cyr, N.E.; Stuart, R.C.; Zhu, X.; Steiner, D.F.; Nillni, E.A. Biosynthesis of proTRH-derived peptides in prohormone convertase 1 and 2 knockout mice. Peptides 2012, 35, 42–48. [Google Scholar] [CrossRef]
- Jing, E.; Nillni, E.A.; Sanchez, V.C.; Stuart, R.C.; Good, D.J. Deletion of the Nhlh2 Transcription Factor Decreases the Levels of the Anorexigenic Peptides? Melanocyte-Stimulating Hormone and Thyrotropin-Releasing Hormone and Implicates Prohormone Convertases I and II in Obesity. Endocrinology 2004, 145, 1503–1513. [Google Scholar] [CrossRef]
- Lloyd, D.J.; Bohan, S.; Gekakis, N. Obesity, hyperphagia and increased metabolic efficiency in Pc1 mutant mice. Hum. Mol. Genet. 2006, 15, 1884–1893. [Google Scholar] [CrossRef]
- Feigerlova, E.; Diene, G.; Oliver, I.; Gennero, I.; Salles, J.-P.; Arnaud, C.; Tauber, M. Elevated Insulin-Like Growth Factor-I Values in Children with Prader-Willi Syndrome Compared with Growth Hormone (GH) Deficiency Children over Two Years of GH Treatment. J. Clin. Endocrinol. Metab. 2010, 95, 4600–4608. [Google Scholar] [CrossRef][Green Version]
- Lein, E.S.; Hawrylycz, M.J.; Ao, N.; Ayres, M.; Bensinger, A.; Bernard, A.; Boe, A.F.; Boguski, M.S.; Brockway, K.S.; Byrnes, E.J.; et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature 2007, 445, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Nestorov, J.; Matić, G.; Elaković, I.; Tanić, N. Gene Expression Studies: How to Obtain Accurate and Reliable Data by Quantitative Real-Time RT PCR. J. Med. Biochem. 2013, 32, 325–338. [Google Scholar] [CrossRef]
- VanGuilder, H.D.; Vrana, K.E.; Freeman, W.M. Twenty-five years of quantitative PCR for gene expression analysis. BioTechniques 2008, 44, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Lowe, R.; Shirley, N.; Bleackley, M.; Dolan, S.; Shafee, T. Transcriptomics technologies. PLoS Comput. Biol. 2017, 13, e1005457. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE Guidelines: Minimum Information for Publication of Quantitative Real-Time PCR Experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef]
- Taylor, S.C.; Mrkusich, E.M. The State of RT-Quantitative PCR: Firsthand Observations of Implementation of Minimum Information for the Publication of Quantitative Real-Time PCR Experiments (MIQE). J. Mol. Microbiol. Biotechnol. 2013, 24, 46–52. [Google Scholar] [CrossRef]
- Chapman, J.R.; Waldenström, J. With Reference to Reference Genes: A Systematic Review of Endogenous Controls in Gene Expression Studies. PLoS ONE 2015, 10, e0141853. [Google Scholar] [CrossRef]
- Riedel, G.; Rüdrich, U.; Fekete-Drimusz, N.; Manns, M.P.; Vondran, F.W.R.; Bock, M. An Extended ΔCT-Method Facilitating Normalisation with Multiple Reference Genes Suited for Quantitative RT-PCR Analyses of Human Hepatocyte-Like Cells. PLoS ONE 2014, 9, e93031. [Google Scholar] [CrossRef] [PubMed]
- Barber, R.D.; Harmer, D.W.; Coleman, R.A.; Clark, B.J. GAPDH as a housekeeping gene: Analysis of GAPDH mRNA expression in a panel of 72 human tissues. Physiol. Genom. 2005, 21, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Selvey, S.; Thompson, E.W.; Matthaei, K.; Lea, R.; Irving, M.; Griffiths, L. β-Actin—An unsuitable internal control for RT-PCR. Mol. Cell. Probes 2001, 15, 307–311. [Google Scholar] [CrossRef]
- Solanas, M.; Moral, R.; Escrich, E. Unsuitability of Using Ribosomal RNA as Loading Control for Northern Blot Analyses Related to the Imbalance between Messenger and Ribosomal RNA Content in Rat Mammary Tumors. Anal. Biochem. 2001, 288, 99–102. [Google Scholar] [CrossRef]
- Thellin, O.; Zorzi, W.; Lakaye, B.; De Borman, B.; Coumans, B.; Hennen, G.; Grisar, T.; Igout, A.; Heinen, E. Housekeeping genes as internal standards: Use and limits. J. Biotechnol. 1999, 75, 291–295. [Google Scholar] [CrossRef]
- Silver, N.; Best, S.; Jiang, J.; Thein, S.L. Selection of housekeeping genes for gene expression studies in human reticulocytes using real-time PCR. BMC Mol. Biol. 2006, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, research0034. [Google Scholar] [CrossRef]
- Pfaffl, M.W.; Tichopad, A.; Prgomet, C.; Neuvians, T.P. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper—Excel-based tool using pair-wise correlations. Biotechnol. Lett. 2004, 26, 509–515. [Google Scholar] [CrossRef]
- Andersen, C.L.; Jensen, J.L.; Ørntoft, T.F. Normalization of Real-Time Quantitative Reverse Transcription-PCR Data: A Model-Based Variance Estimation Approach to Identify Genes Suited for Normalization, Applied to Bladder and Colon Cancer Data Sets. Cancer Res. 2004, 64, 5245–5250. [Google Scholar] [CrossRef]
- Rydbirk, R.; Folke, J.; Winge, K.; Aznar, S.; Pakkenberg, B.; Brudek, T. Assessment of brain reference genes for RT-qPCR studies in neurodegenerative diseases. Sci. Rep. 2016, 6, 37116. [Google Scholar] [CrossRef]
- Xu, D.; Liu, A.; Wang, X.; Zhang, M.; Zhang, Z.; Tan, Z.; Qiu, M. Identifying suitable reference genes for developing and injured mouse CNS tissues. Dev. Neurobiol. 2017, 78, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, A.P.; Kovalenko, A.A.; Malygina, D.A.; Postnikova, T.Y.; Zubareva, O.E.; Zaitsev, A.V. Reference Gene Validation in the Brain Regions of Young Rats after Pentylenetetrazole-Induced Seizures. Biomedicines 2020, 8, 239. [Google Scholar] [CrossRef]
- Ho, K.H.; Patrizi, A. Assessment of common housekeeping genes as reference for gene expression studies using RT-qPCR in mouse choroid plexus. Sci. Rep. 2021, 11, 3278. [Google Scholar] [CrossRef] [PubMed]
- Baldini, L.; Robert, A.; Charpentier, B.; Labialle, S. Phylogenetic and Molecular Analyses Identify SNORD116 Targets Involved in the Prader–Willi Syndrome. Mol. Biol. Evol. 2021, 39, msab348. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.E.; Noel, M.; Lehoux, S.; Cetinbas, M.; Xavier, R.J.; Sadreyev, R.I.; Scolnick, E.M.; Smoller, J.W.; Cummings, R.D.; Mealer, R.G. Mammalian brain glycoproteins exhibit diminished glycan complexity compared to other tissues. Nat. Commun. 2022, 13, 275. [Google Scholar] [CrossRef]
- Galiveti, C.R.; Rozhdestvensky, T.S.; Brosius, J.; Lehrach, H.; Konthur, Z. Application of housekeeping npcRNAs for quantitative expression analysis of human transcriptome by real-time PCR. RNA 2009, 16, 450–461. [Google Scholar] [CrossRef]
- Mathur, D.; Urena-Peralta, J.R.; Lopez-Rodas, G.; Casanova, B.; Coret-Ferrer, F.; Burgal-Marti, M. Bypassing hazard of housekeeping genes: Their evaluation in rat granule neurons treated with cerebrospinal fluid of multiple sclerosis subjects. Front. Cell. Neurosci. 2015, 9, 375. [Google Scholar] [CrossRef]
- Xie, F.; Xiao, P.; Chen, D.; Xu, L.; Zhang, B. miRDeepFinder: A miRNA analysis tool for deep sequencing of plant small RNAs. Plant Mol. Biol. 2012, 80, 75–84. [Google Scholar] [CrossRef]
- Kocher, M.A.; Huang, F.W.; Le, E.; Good, D.J. Snord116 Post-transcriptionally Increases Nhlh2 mRNA Stability: Implications for Human Prader-Willi Syndrome. Hum. Mol. Genet. 2021, 30, 1101–1110. [Google Scholar] [CrossRef]
- Uchigashima, M.; Cheung, A.; Futai, K. Neuroligin-3: A Circuit-Specific Synapse Organizer That Shapes Normal Function and Autism Spectrum Disorder-Associated Dysfunction. Front. Mol. Neurosci. 2021, 14, 749164. [Google Scholar] [CrossRef]
- Uchigashima, M.; Leung, M.; Watanabe, T.; Cheung, A.; Le, T.; Pallat, S.; Dinis, A.; Watanabe, M.; Kawasawa, Y.I.; Futai, K. Neuroligin3 splice isoforms shape inhibitory synaptic function in the mouse hippocampus. J. Biol. Chem. 2020, 295, 8589–8595. [Google Scholar] [CrossRef] [PubMed]
- Bar, C.; Diene, G.; Molinas, C.; Bieth, E.; Casper, C.; Tauber, M. Early diagnosis and care is achieved but should be improved in infants with Prader-Willi syndrome. Orphanet J. Rare Dis. 2017, 12, 118. [Google Scholar] [CrossRef] [PubMed]
- Deal, C.L.; Tony, M.; Höybye, C.; Allen, D.B.; Tauber, M.; Christiansen, J.S.; the 2011 Growth Hormone in Prader-Willi Syndrome Clinical Care Guidelines Workshop Participants. Growth Hormone Research Society Workshop Summary: Consensus Guidelines for Recombinant Human Growth Hormone Therapy in Prader-Willi Syndrome. J. Clin. Endocrinol. Metab. 2013, 98, E1072–E1087. [Google Scholar] [CrossRef] [PubMed]
- Evdokimova, V.; Tognon, C.E.; Benatar, T.; Yang, W.; Krutikov, K.; Pollak, M.; Sorensen, P.H.B.; Seth, A. IGFBP7 Binds to the IGF-1 Receptor and Blocks Its Activation by Insulin-Like Growth Factors. Sci. Signal. 2012, 5, ra92. [Google Scholar] [CrossRef]
- Ahmed, S.; Yamamoto, K.; Sato, Y.; Ogawa, T.; Herrmann, A.; Higashi, S.; Miyazaki, K. Proteolytic processing of IGFBP-related protein-1 (TAF/angiomodulin/mac25) modulates its biological activity. Biochem. Biophys. Res. Commun. 2003, 310, 612–618. [Google Scholar] [CrossRef]
- Sie, C.G.; Hesler, S.; Maas, S.; Kuchka, M. IGFBP7’s susceptibility to proteolysis is altered by A-to-I RNA editing of its transcript. FEBS Lett. 2012, 586, 2313–2317. [Google Scholar] [CrossRef]
- Jin, L.; Shen, F.; Weinfeld, M.; Sergi, C. Insulin Growth Factor Binding Protein 7 (IGFBP7)-Related Cancer and IGFBP3 and IGFBP7 Crosstalk. Front. Oncol. 2020, 10, 727. [Google Scholar] [CrossRef]
- Ding, F.; Li, H.H.; Zhang, S.; Solomon, N.M.; Camper, S.; Cohen, P.; Francke, U. SnoRNA Snord116 (Pwcr1/MBII-85) Deletion Causes Growth Deficiency and Hyperphagia in Mice. PLoS ONE 2008, 3, e1709. [Google Scholar] [CrossRef]
- Stijnen, P.; Ramos-Molina, B.; O’Rahilly, S.; Creemers, J.W.M. PCSK1 Mutations and Human Endocrinopathies: From Obesity to Gastrointestinal Disorders. Endocr. Rev. 2016, 37, 347–371. [Google Scholar] [CrossRef]
- Zhu, X.; Zhou, A.; Dey, A.; Norrbom, C.; Carroll, R.; Zhang, C.; Laurent, V.; Lindberg, I.; Ugleholdt, R.; Holst, J.J.; et al. Disruption of PC1/3 expression in mice causes dwarfism and multiple neuroendocrine peptide processing defects. Proc. Natl. Acad. Sci. USA 2002, 99, 10293–10298. [Google Scholar] [CrossRef]
- Correa-da-Silva, F.; Fliers, E.; Swaab, D.F.; Yi, C. Hypothalamic neuropeptides and neurocircuitries in Prader Willi syndrome. J. Neuroendocr. 2021, 33, e12994. [Google Scholar] [CrossRef]
- Chang, T.-J.; Chiu, Y.-F.; Sheu, W.H.-H.; Shih, K.-C.; Hwu, C.-M.; Quertermous, T.; Jou, Y.-S.; Kuo, S.-S.; Chang, Y.-C.; Chuang, L.-M. Genetic polymorphisms of PCSK2 are associated with glucose homeostasis and progression to type 2 diabetes in a Chinese population. Sci. Rep. 2015, 5, 14380. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, A.; Isomaa, B.; Tuomi, T.; Eriksson, J.G.; Groop, L.; Lyssenko, V. Effect of a common variant of the PCSK2 gene on reduced insulin secretion. Diabetologia 2012, 55, 3245–3251. [Google Scholar] [CrossRef] [PubMed]
- Leak, T.S.; Keene, K.L.; Langefeld, C.D.; Gallagher, C.J.; Mychaleckyj, J.C.; Freedman, B.I.; Bowden, D.W.; Rich, S.S.; Sale, M.M. Association of the proprotein convertase subtilisin/kexin-type 2 (PCSK2) gene with type 2 diabetes in an African American population. Mol. Genet. Metab. 2007, 92, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Clerc, A.; Coupaye, M.; Mosbah, H.; Pinto, G.; Laurier, V.; Mourre, F.; Merrien, C.; Diene, G.; Poitou, C.; Tauber, M. Diabetes Mellitus in Prader-Willi Syndrome: Natural History during the Transition from Childhood to Adulthood in a Cohort of 39 Patients. J. Clin. Med. 2021, 10, 5310. [Google Scholar] [CrossRef] [PubMed]
- Leung, K.N.; Vallero, R.O.; DuBose, A.J.; Resnick, J.L.; LaSalle, J.M. Imprinting regulates mammalian snoRNA-encoding chromatin decondensation and neuronal nucleolar size. Hum. Mol. Genet. 2009, 18, 4227–4238. [Google Scholar] [CrossRef]
- Vitali, P.; Royo, H.; Marty, V.; Bortolin-Cavaillé, M.-L.; Cavaillé, J. Long nuclear-retained non-coding RNAs and allele-specific higher-order chromatin organization at imprinted snoRNA gene arrays. J. Cell Sci. 2010, 123, 70–83. [Google Scholar] [CrossRef]
- Knott, B.; Kocher, M.A.; Paz, H.A.; Hamm, S.E.; Fink, W.; Mason, J.; Grange, R.W.; Wankhade, U.D.; Good, D.J. Dietary Conjugated Linoleic Acid Reduces Body Weight and Fat in Snord116m+/p− and Snord116m−/p− Mouse Models of Prader–Willi Syndrome. Nutrients 2022, 14, 860. [Google Scholar] [CrossRef]
- Polex-Wolf, J.; Lam, B.Y.H.; Larder, R.; Tadross, J.; Rimmington, D.; Bosch, F.; Cenzano, V.J.; Ayuso, E.; Ma, M.K.L.; Rainbow, K.; et al. Hypothalamic loss of Snord116 recapitulates the hyperphagia of Prader-Willi syndrome. J. Clin. Investig. 2018, 128, 960–969. [Google Scholar] [CrossRef]
- Jamain, S.; Quach, H.; Betancur, C.; Råstam, M.; Colineaux, C.; Gillberg, I.C.; Soderstrom, H.; Giros, B.; Leboyer, M.; Gillberg, C.; et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet. 2003, 34, 27–29. [Google Scholar] [CrossRef]
- Tabuchi, K.; Blundell, J.; Etherton, M.R.; Hammer, R.E.; Liu, X.; Powell, C.M.; Südhof, T.C. A Neuroligin-3 Mutation Implicated in Autism Increases Inhibitory Synaptic Transmission in Mice. Science 2007, 318, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Ates, T.; Oncul, M.; Dilsiz, P.; Topcu, I.C.; Civas, C.C.; Alp, M.I.; Aklan, I.; Oz, E.A.; Yavuz, Y.; Yilmaz, B.; et al. Inactivation of Magel2 suppresses oxytocin neurons through synaptic excitation-inhibition imbalance. Neurobiol. Dis. 2018, 121, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Muscatelli, F.; Abrous, D.N.; Massacrier, A.; Boccaccio, I.; Le Moal, M.; Cau, P.; Cremer, H. Disruption of the mouse Necdin gene results in hypothalamic and behavioral alterations reminiscent of the human Prader-Willi syndrome. Hum. Mol. Genet. 2000, 9, 3101–3110. [Google Scholar] [CrossRef] [PubMed]
- Rozhdestvensky, T.S.; Robeck, T.; Galiveti, C.R.; Raabe, C.A.; Seeger, B.; Wolters, A.; Gubar, L.V.; Brosius, J.; Skryabin, B.V. Maternal transcription of non-protein coding RNAs from the PWS-critical region rescues growth retardation in mice. Sci. Rep. 2016, 6, 20398. [Google Scholar] [CrossRef]
- Kishore, S.; Stamm, S. The snoRNA HBII-52 Regulates Alternative Splicing of the Serotonin Receptor 2C. Science 2006, 311, 230–232. [Google Scholar] [CrossRef]
- Brosius, J.; Hüttenhofer, A.; Tiedge, H. Brain-Specific Nonmessenger RNAs. In Non-Coding RNAs: Molecular Biology and Molecular Medicine; Springer Science & Business Media: Berlin, Germany, 2003; pp. 159–169. ISBN 978-0-306-47835-2. [Google Scholar]
- Doe, C.M.; Relkovic, D.; Garfield, A.S.; Dalley, J.; Theobald, D.E.; Humby, T.; Wilkinson, L.S.; Isles, A.R. Loss of the imprinted snoRNA mbii-52 leads to increased 5htr2c pre-RNA editing and altered 5HT2CR-mediated behaviour. Hum. Mol. Genet. 2009, 18, 2140–2148. [Google Scholar] [CrossRef]
- Mo, D.; Raabe, C.A.; Reinhardt, R.; Brosius, J.; Rozhdestvensky, T.S. Alternative Processing as Evolutionary Mechanism for the Origin of Novel Nonprotein Coding RNAs. Genome Biol. Evol. 2013, 5, 2061–2071. [Google Scholar] [CrossRef]
- Bochukova, E.G.; Lawler, K.; Croizier, S.; Keogh, J.M.; Patel, N.; Strohbehn, G.; Lo, K.K.; Humphrey, J.; Hokken-Koelega, A.; Damen, L.; et al. A Transcriptomic Signature of the Hypothalamic Response to Fasting and BDNF Deficiency in Prader-Willi Syndrome. Cell Rep. 2018, 22, 3401–3408. [Google Scholar] [CrossRef]
- Raabe, C.A.; Voss, R.; Kummerfeld, D.-M.; Brosius, J.; Galiveti, C.R.; Wolters, A.; Seggewiss, J.; Huge, A.; Skryabin, B.V.; Rozhdestvensky, T.S. Ectopic expression of Snord115 in choroid plexus interferes with editing but not splicing of 5-Ht2c receptor pre-mRNA in mice. Sci. Rep. 2019, 9, 4300. [Google Scholar] [CrossRef]
- Hebras, J.; Marty, V.; Personnaz, J.; Mercier, P.; Krogh, N.; Nielsen, H.; Aguirrebengoa, M.; Seitz, H.; Pradere, J.-P.; Guiard, B.P.; et al. Reassessment of the involvement of Snord115 in the serotonin 2c receptor pathway in a genetically relevant mouse model. eLife 2020, 9, e60862. [Google Scholar] [CrossRef]
- Bazeley, P.S.; Shepelev, V.; Talebizadeh, Z.; Butler, M.G.; Fedorova, L.; Filatov, V.; Fedorov, A. snoTARGET shows that human orphan snoRNA targets locate close to alternative splice junctions. Gene 2008, 408, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Langhendries, J.-L.; Watzinger, P.; Kötter, P.; Entian, K.-D.; Lafontaine, D.L. Yeast Kre33 and human NAT10 are conserved 18S rRNA cytosine acetyltransferases that modify tRNAs assisted by the adaptor Tan1/THUMPD1. Nucleic Acids Res. 2015, 43, 2242–2258. [Google Scholar] [CrossRef] [PubMed]
- Bortolin-Cavaillé, M.-L.; Quillien, A.; Gamage, S.T.; Thomas, J.M.; Sas-Chen, A.; Sharma, S.; Plisson-Chastang, C.; Vandel, L.; Blader, P.; Lafontaine, D.L.J.; et al. Probing small ribosomal subunit RNA helix 45 acetylation across eukaryotic evolution. Nucleic Acids Res. 2022, 50, 6284–6299. [Google Scholar] [CrossRef] [PubMed]
- Kehr, S.; Bartschat, S.; Tafer, H.; Stadler, P.F.; Hertel, J. Matching of Soulmates: Coevolution of snoRNAs and Their Targets. Mol. Biol. Evol. 2013, 31, 455–467. [Google Scholar] [CrossRef]
- Elliott, B.A.; Ho, H.-T.; Ranganathan, S.V.; Vangaveti, S.; Ilkayeva, O.; Assi, H.A.; Choi, A.K.; Agris, P.F.; Holley, C.L. Modification of messenger RNA by 2′-O-methylation regulates gene expression in vivo. Nat. Commun. 2019, 10, 3401. [Google Scholar] [CrossRef]
- Vitali, P.; Basyuk, E.; Le Meur, E.; Bertrand, E.; Muscatelli, F.; Cavaillé, J.; Huttenhofer, A. ADAR2-mediated editing of RNA substrates in the nucleolus is inhibited by C/D small nucleolar RNAs. J. Cell Biol. 2005, 169, 745–753. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kummerfeld, D.-M.; Skryabin, B.V.; Brosius, J.; Vakhrushev, S.Y.; Rozhdestvensky, T.S. Reference Genes across Nine Brain Areas of Wild Type and Prader-Willi Syndrome Mice: Assessing Differences in Igfbp7, Pcsk1, Nhlh2 and Nlgn3 Expression. Int. J. Mol. Sci. 2022, 23, 8729. https://doi.org/10.3390/ijms23158729
Kummerfeld D-M, Skryabin BV, Brosius J, Vakhrushev SY, Rozhdestvensky TS. Reference Genes across Nine Brain Areas of Wild Type and Prader-Willi Syndrome Mice: Assessing Differences in Igfbp7, Pcsk1, Nhlh2 and Nlgn3 Expression. International Journal of Molecular Sciences. 2022; 23(15):8729. https://doi.org/10.3390/ijms23158729
Chicago/Turabian StyleKummerfeld, Delf-Magnus, Boris V. Skryabin, Juergen Brosius, Sergey Y. Vakhrushev, and Timofey S. Rozhdestvensky. 2022. "Reference Genes across Nine Brain Areas of Wild Type and Prader-Willi Syndrome Mice: Assessing Differences in Igfbp7, Pcsk1, Nhlh2 and Nlgn3 Expression" International Journal of Molecular Sciences 23, no. 15: 8729. https://doi.org/10.3390/ijms23158729
APA StyleKummerfeld, D.-M., Skryabin, B. V., Brosius, J., Vakhrushev, S. Y., & Rozhdestvensky, T. S. (2022). Reference Genes across Nine Brain Areas of Wild Type and Prader-Willi Syndrome Mice: Assessing Differences in Igfbp7, Pcsk1, Nhlh2 and Nlgn3 Expression. International Journal of Molecular Sciences, 23(15), 8729. https://doi.org/10.3390/ijms23158729