
Expression Profiling and MicroRNA Regulatory Networks of Homeobox Family Genes in Sugarcane Saccharum spontaneum L.

 , and
, and
Abstract
:1. Introduction
2. Results
2.1. Genome-Wide Identification of HB Genes in S. spontaneum
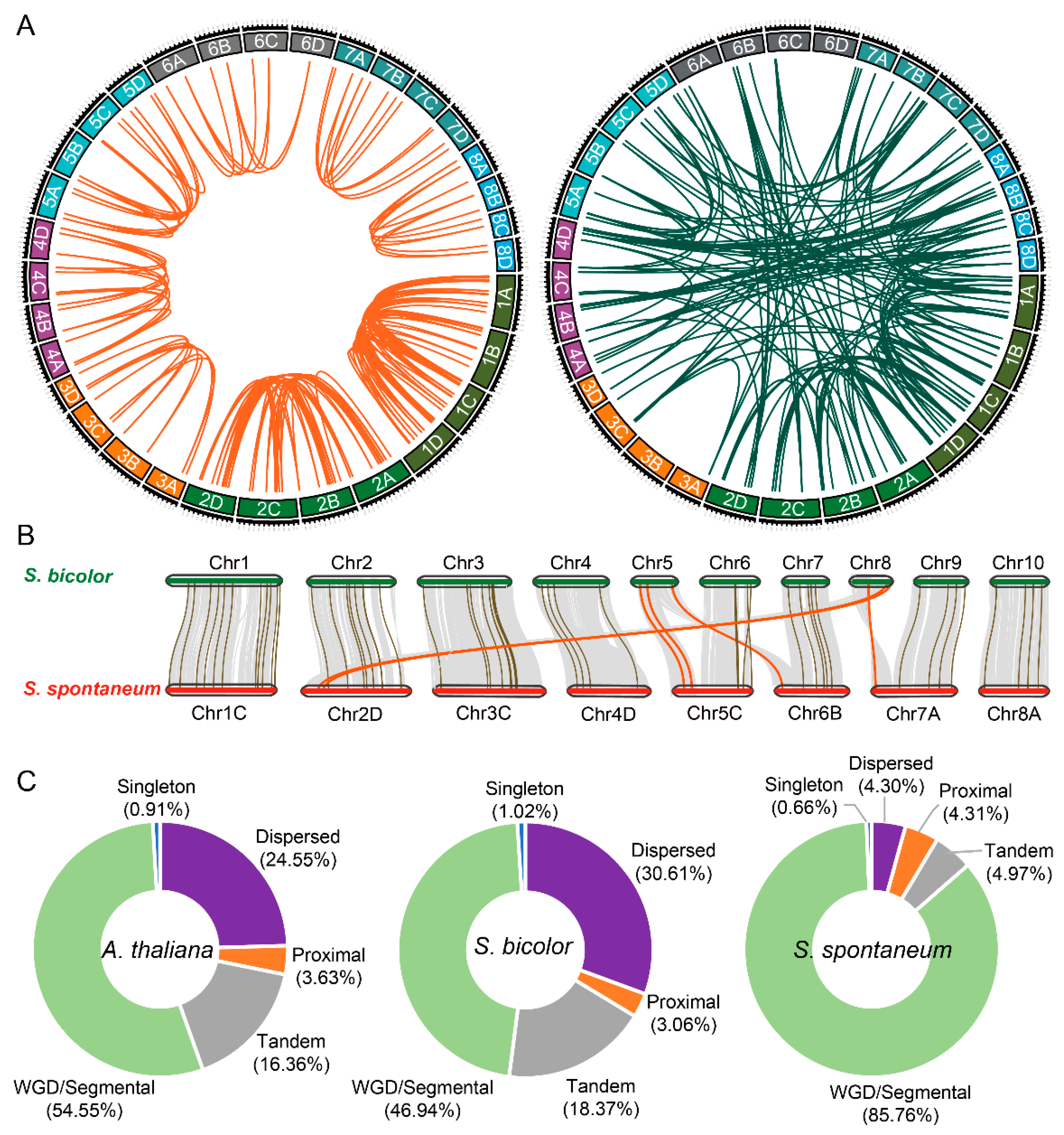
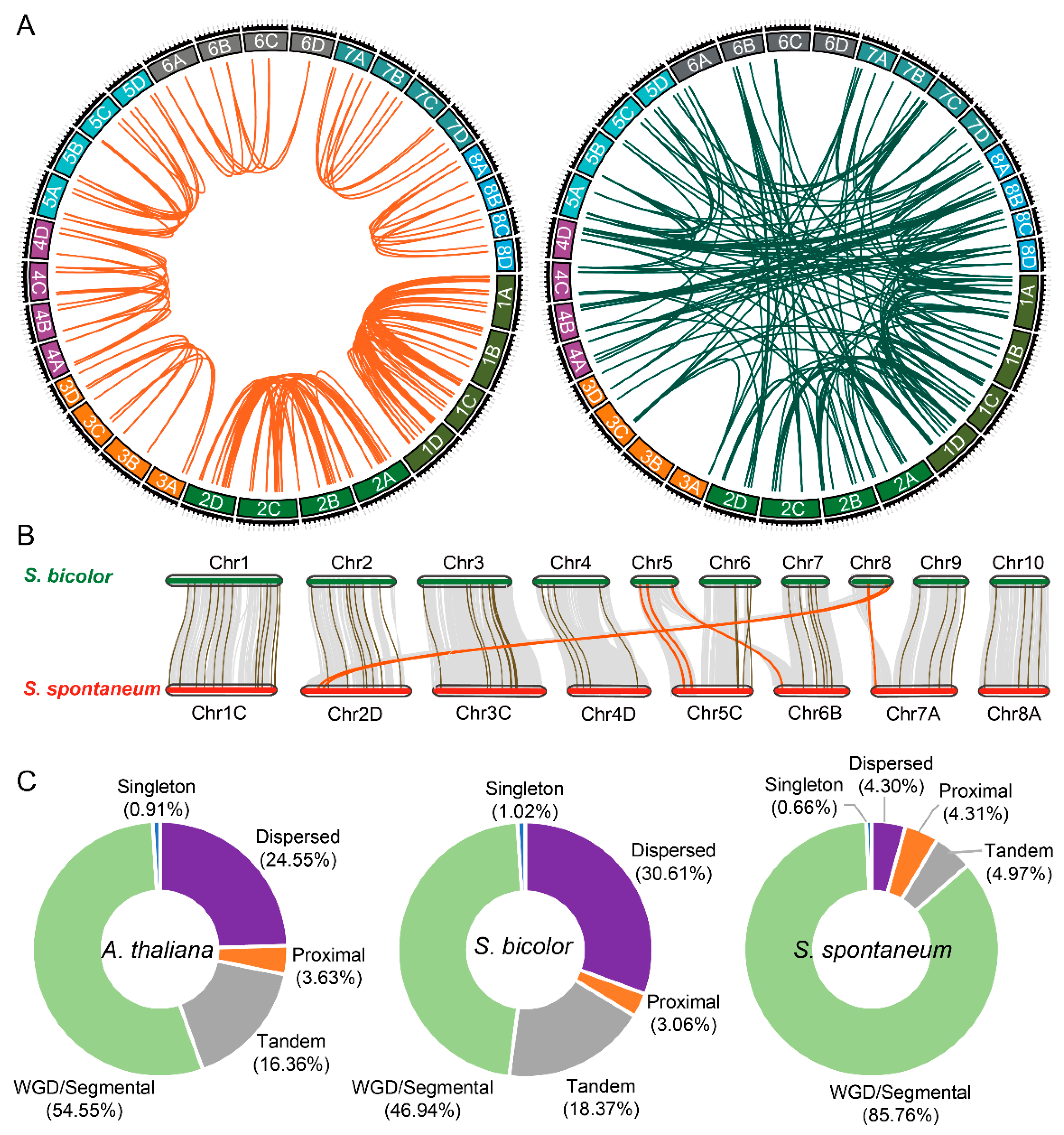
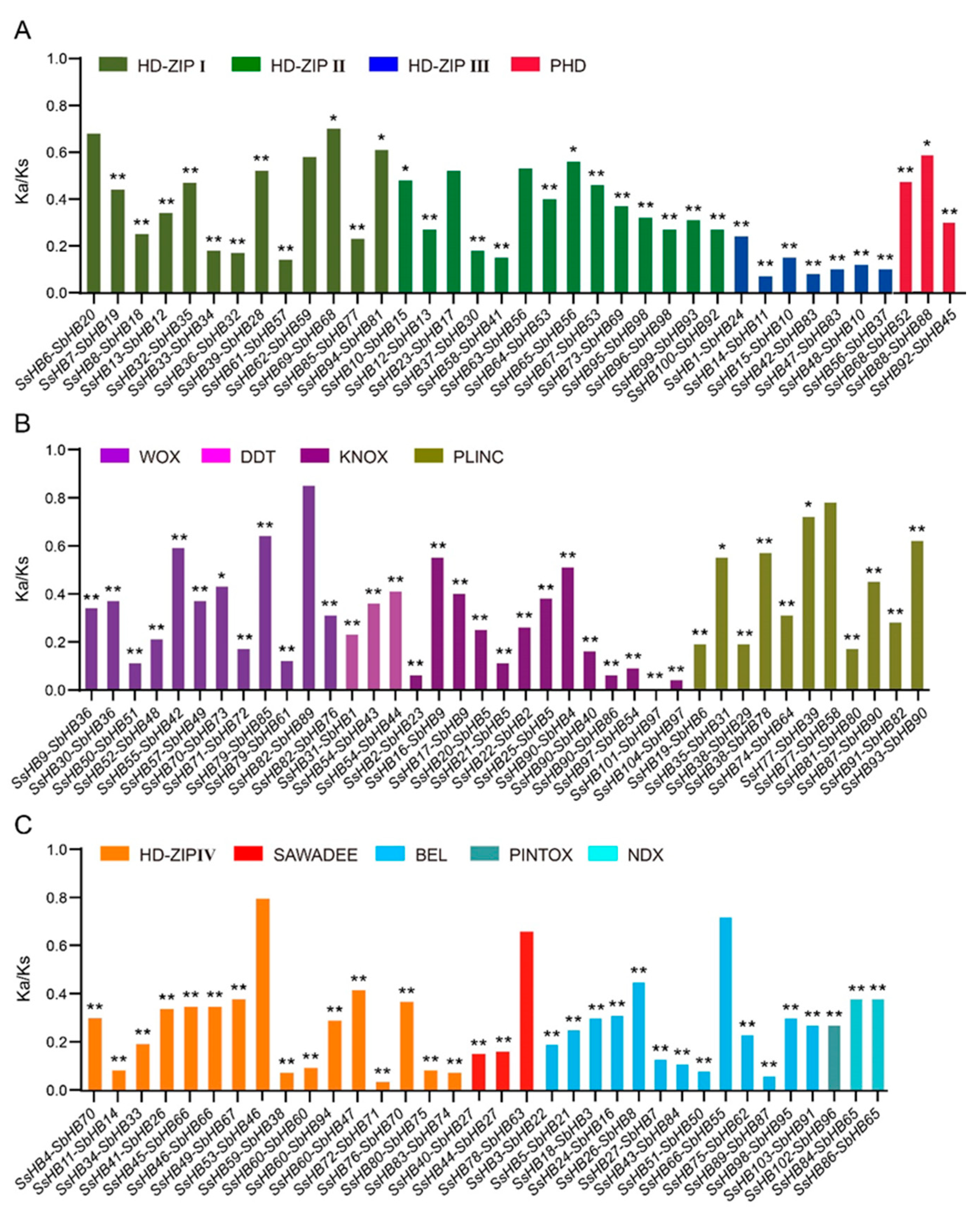
2.2. Chromosome Distribution and Synteny Analysis of SsHBs
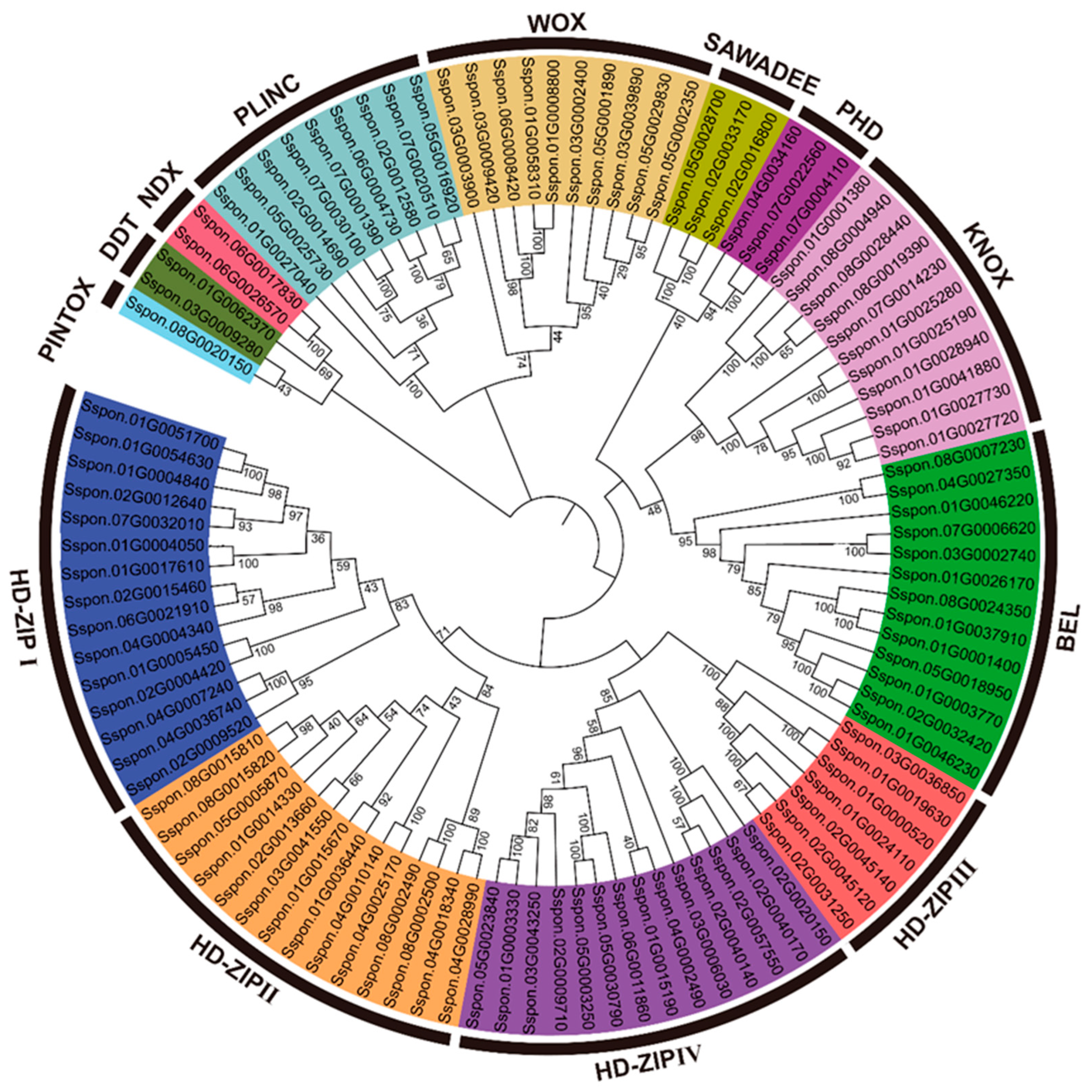
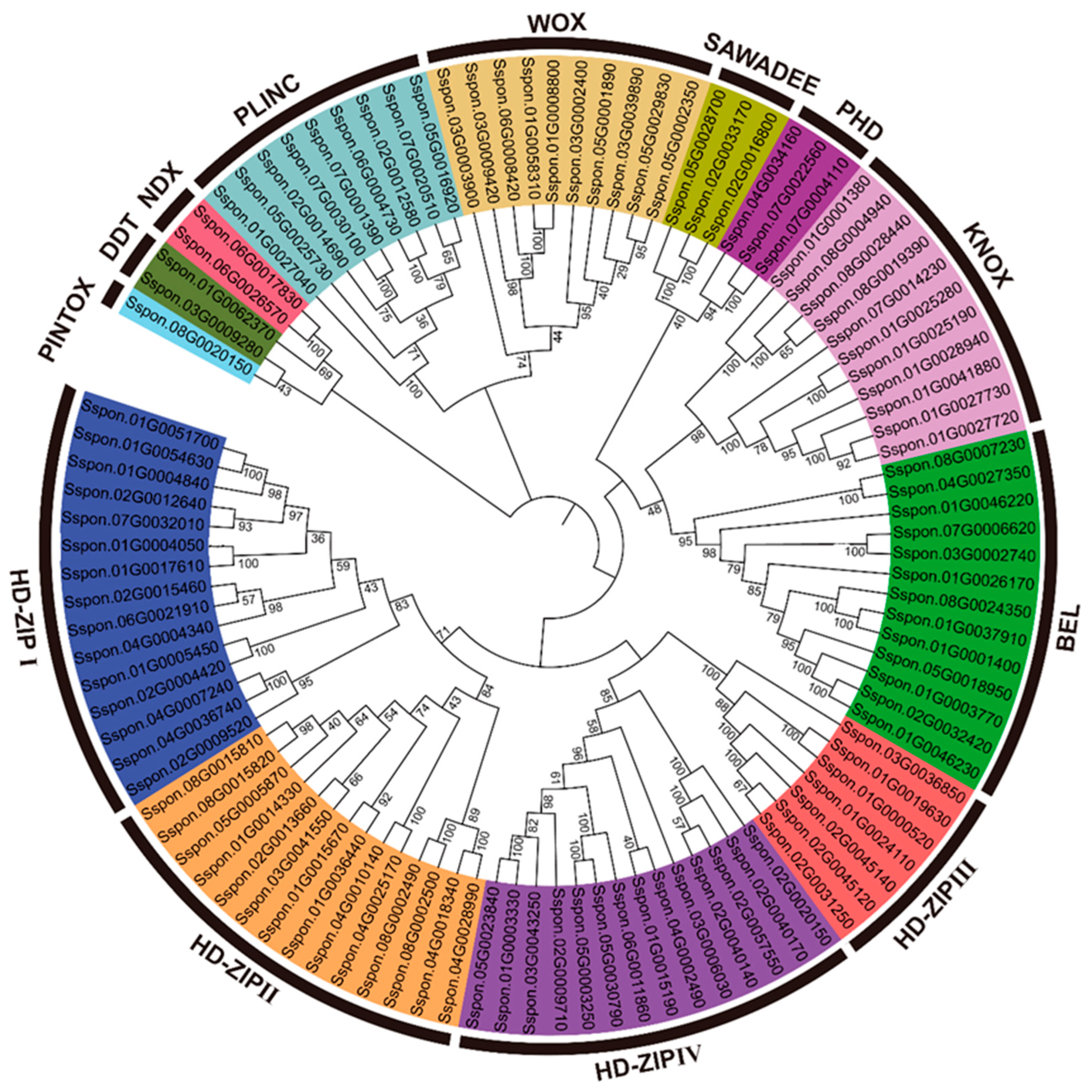
2.3. Classification and Phylogenetic Analysis of SsHBs
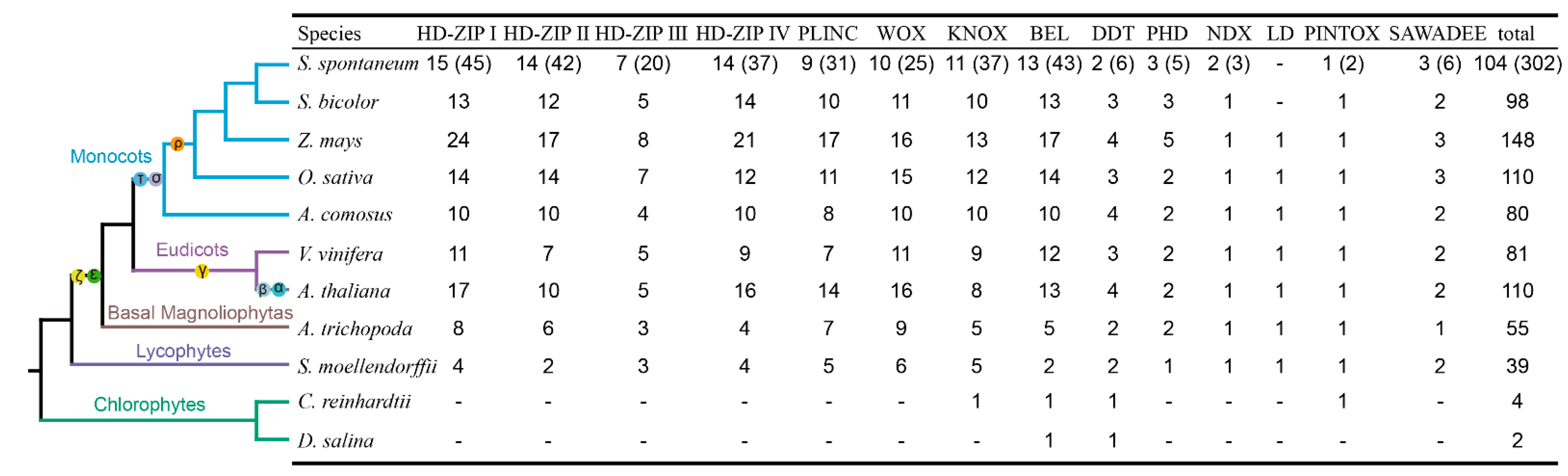
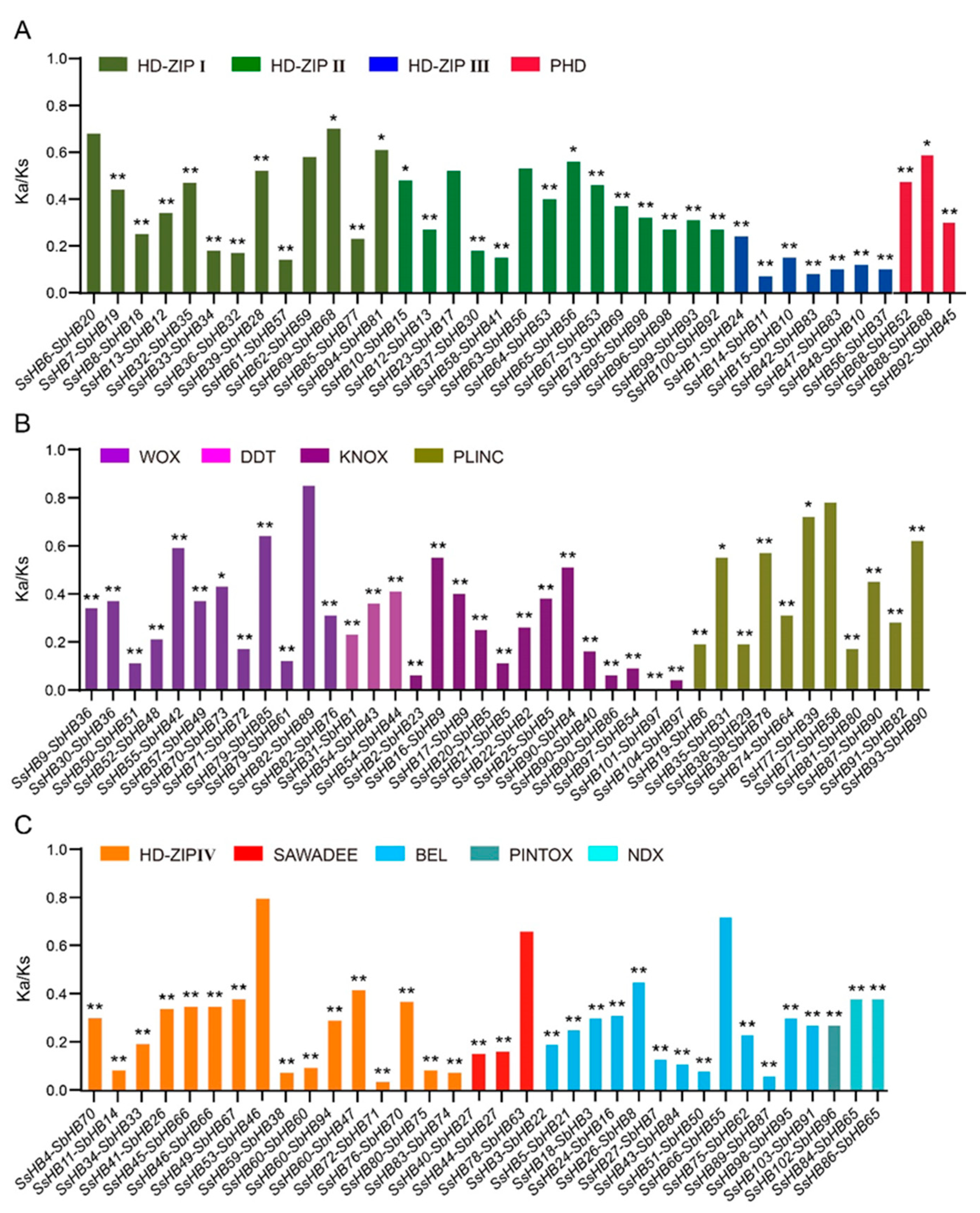
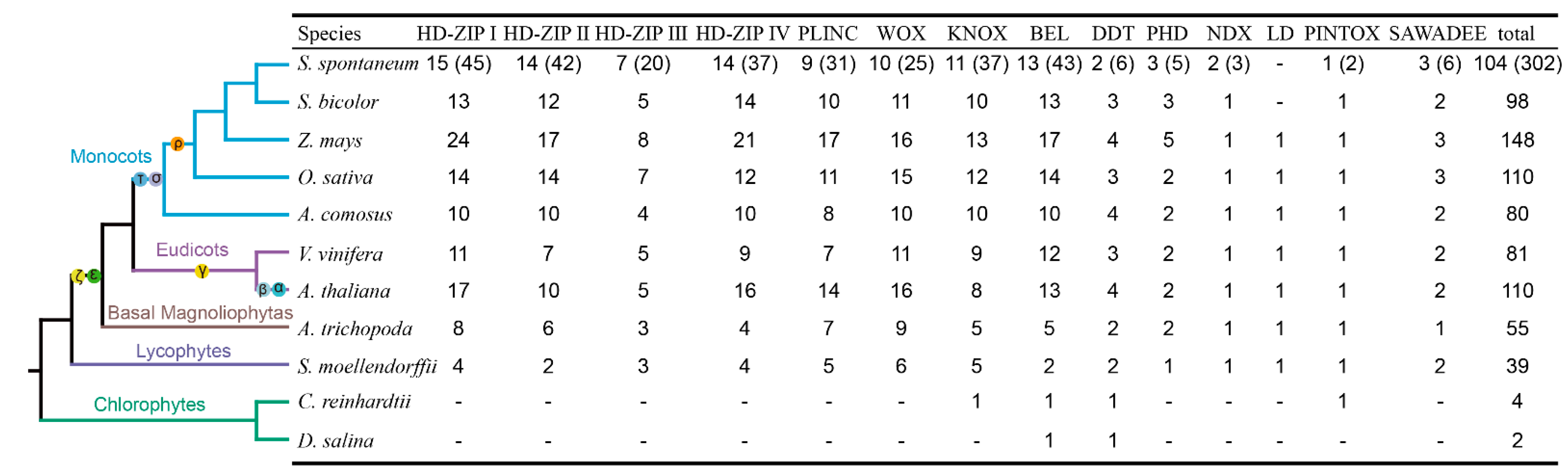
2.4. Comparative Analysis between SsHBs and SbHBs
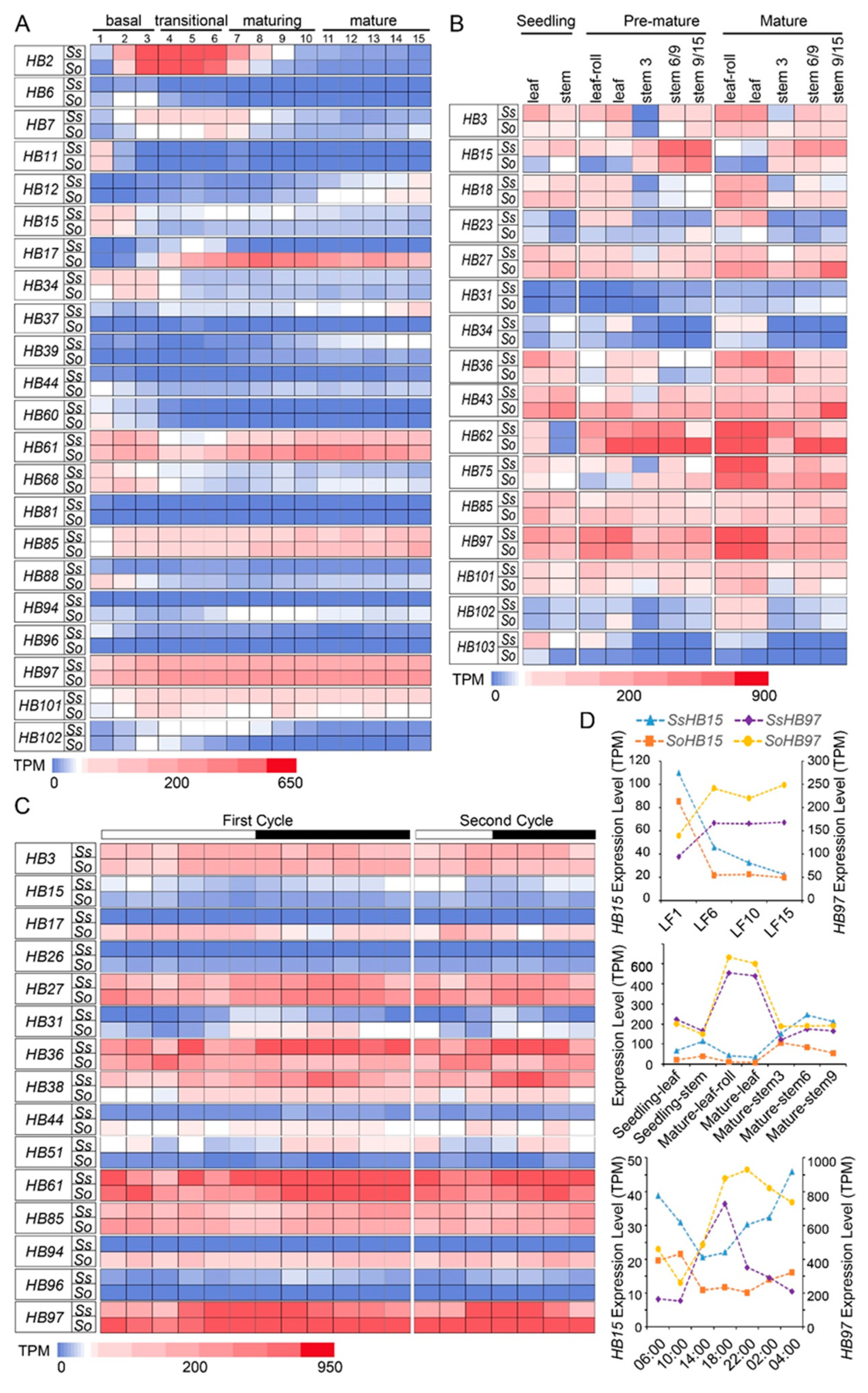
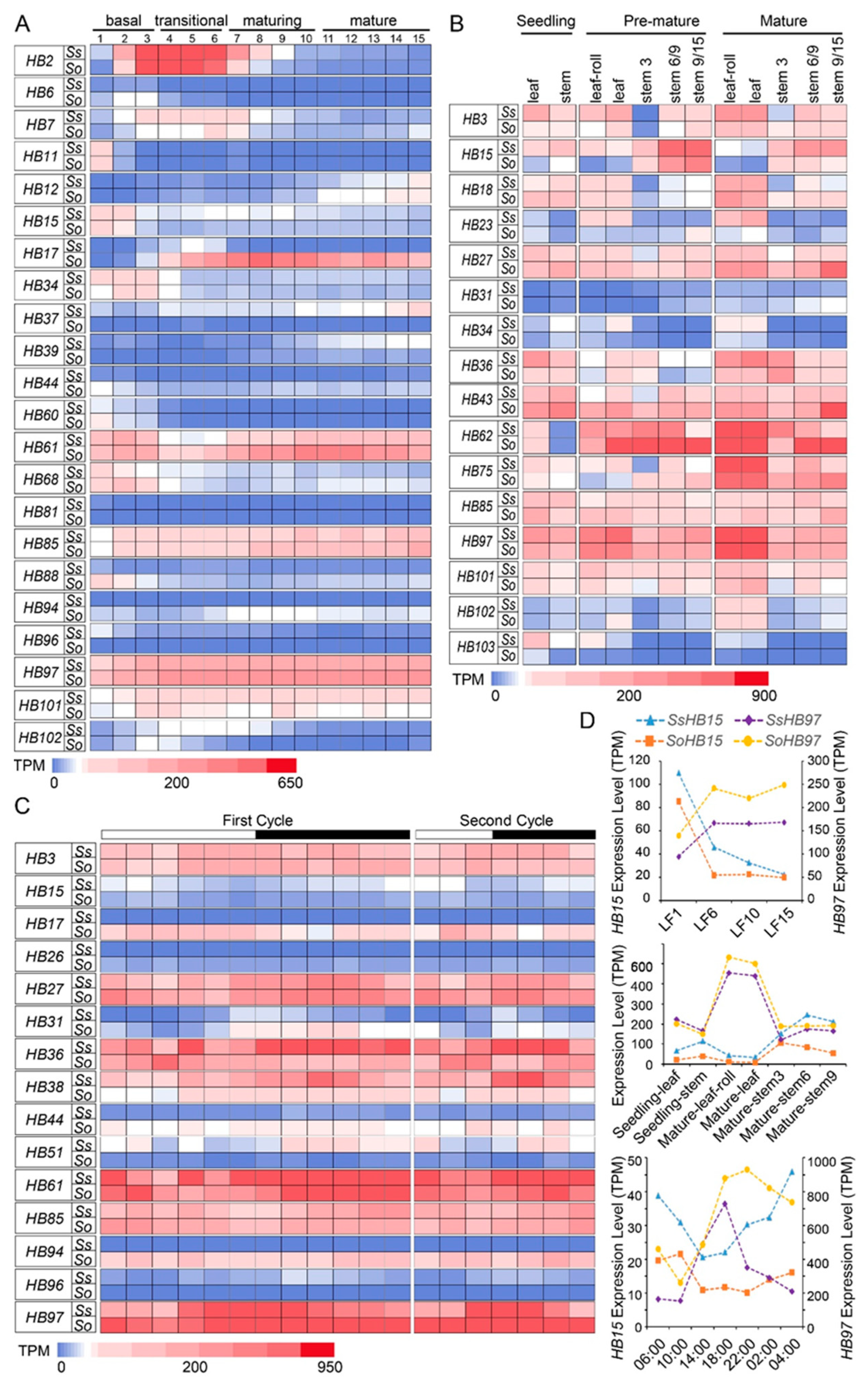
2.5. The Expression Patterns of HB Genes at Different Developmental Stages, Gradient Developmental Leaf Segments, and Circadian Rhythm in S. spontaneum and S. officinarum
2.6. The Regulation of miRNAs in SsHBs Expression
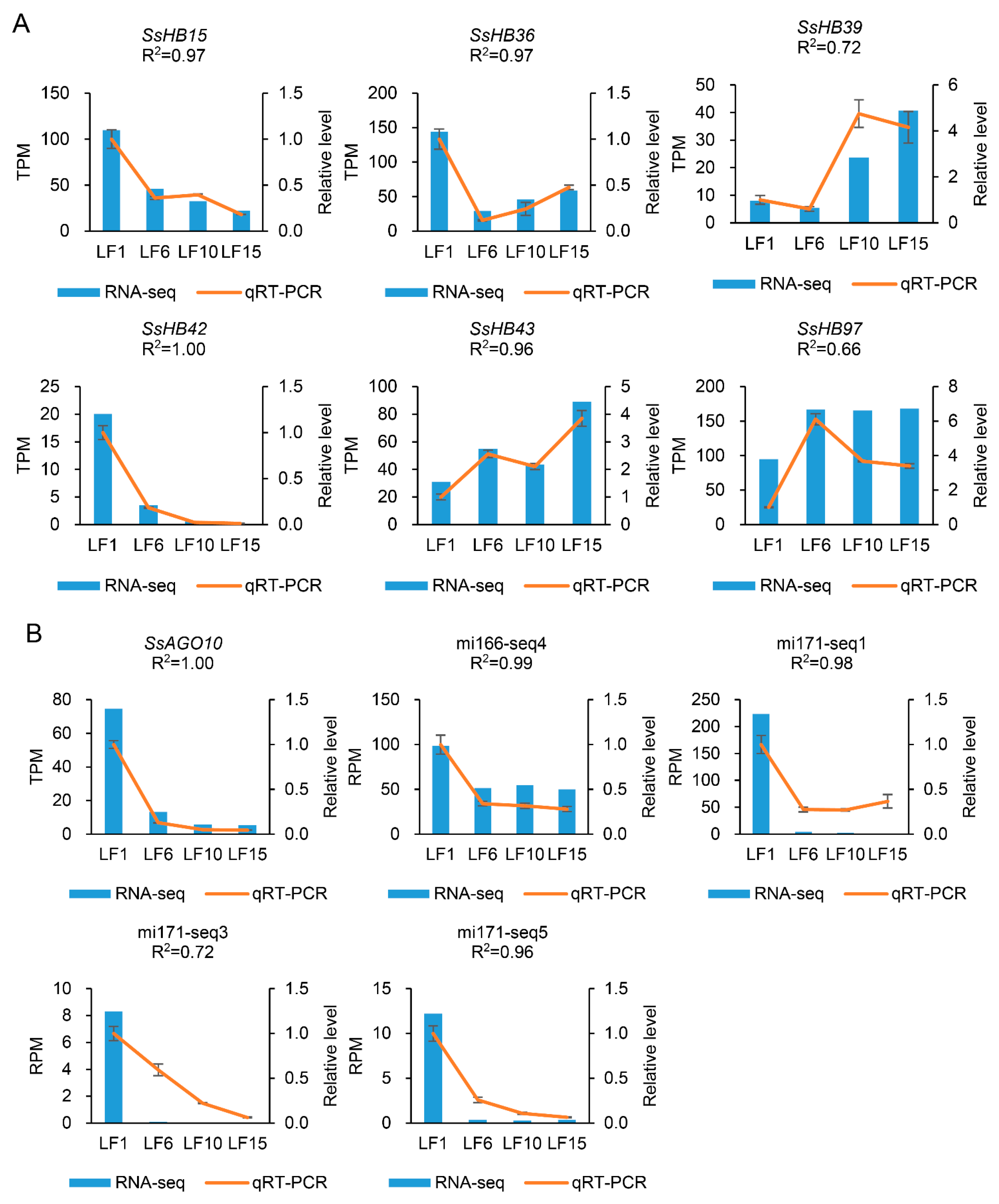
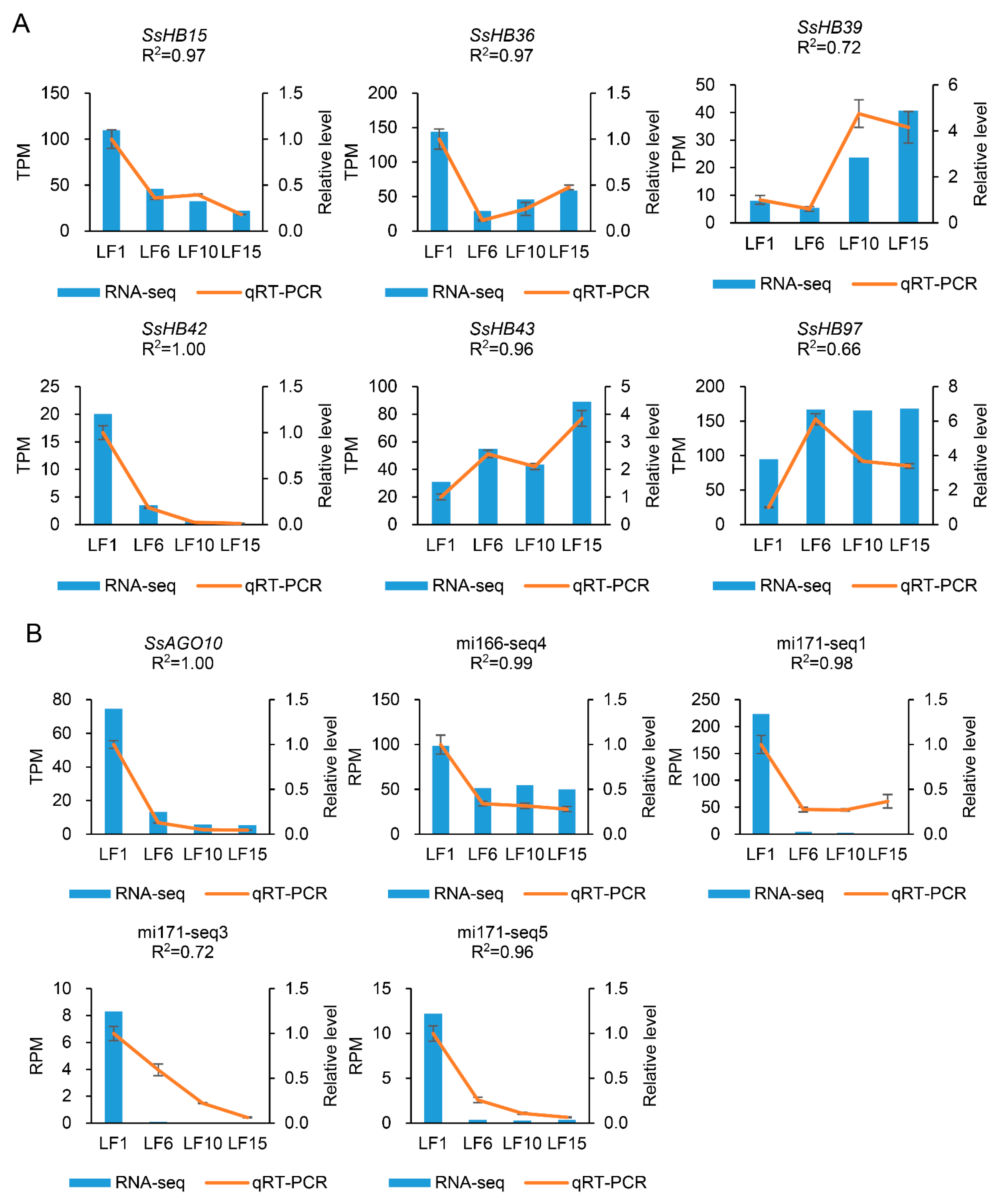
2.7. Verification of Transcriptome Data Using qRT-PCR
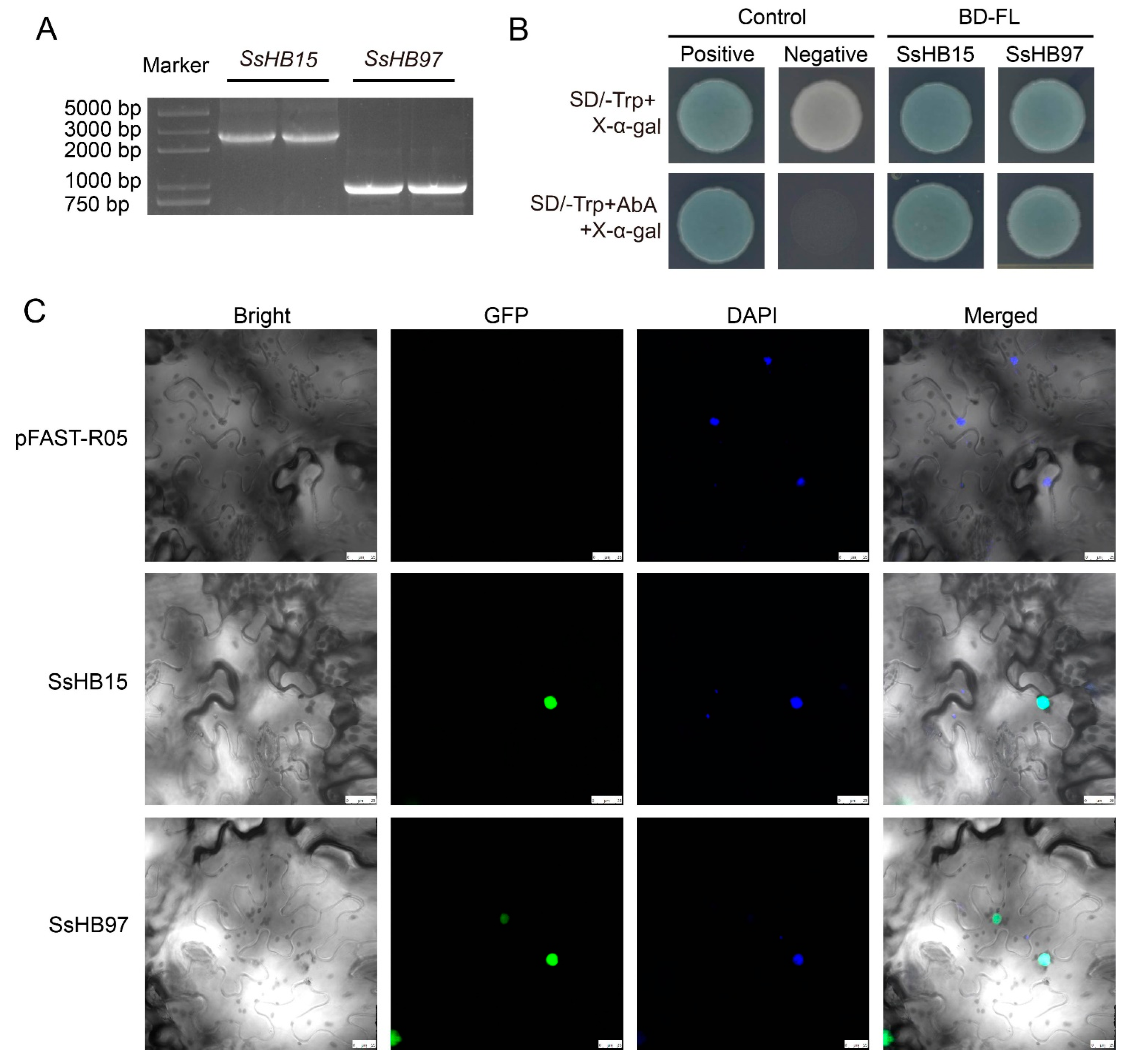
2.8. SsHB15 and SsHB97 Are Located in the Nucleus and Display Transcriptional Activity
3. Discussion
4. Materials and Methods
4.1. Plant Material, RNA Extraction and Sequencing
4.2. The Retrieval of the Homeobox Sequences
4.3. Multiple Sequence Alignments and Phylogenetic Analysis of the Sugarcane Homeobox Genes
4.4. Gene Structure, Protein Conserved Motif Analysis, and Calculation of Ka/Ks
4.5. Gene Duplication and Synteny Analysis
4.6. The miRNA Identification, Quantification, and Identification of Target Genes
4.7. Validation of Gene and miRNA Expression Levels by qRT-PCR
4.8. Subcellular Localization and Transcriptional Activity Detection
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Inukai, S.; Kock, K.H.; Bulyk, M.L. Transcription factor–DNA binding: Beyond binding site motifs. Curr. Opin. Genet. Dev. 2017, 43, 110–119. [Google Scholar] [CrossRef] [Green Version]
- Qu, L.-J.; Zhu, Y.-X. Transcription factor families in Arabidopsis: Major progress and outstanding issues for future research. Curr. Opin. Plant Biol. 2006, 9, 544–549. [Google Scholar] [CrossRef]
- Samad, A.F.A.; Sajad, M.; Nazaruddin, N.; Fauzi, I.A.; Murad, A.M.A.; Zainal, Z.; Ismail, I. MicroRNA and Transcription Factor: Key Players in Plant Regulatory Network. Front. Plant Sci. 2017, 8, 565. [Google Scholar] [CrossRef] [Green Version]
- Derelle, R.; Lopez, P.; Guyader, H.L.; Manuel, M. Homeodomain proteins belong to the ancestral molecular toolkit of eukaryotes. Evol. Dev. 2007, 9, 212–219. [Google Scholar] [CrossRef]
- Bürglin, T.R. Analysis of TALE superclass homeobox genes (MEIS, PBC, KNOX, Iroquois, TGIF) reveals a novel domain conserved between plants and animals. Nucleic Acids Res. 1997, 25, 4173–4180. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, K.; Brocchieri, L.; Bürglin, T.R. A Comprehensive Classification and Evolutionary Analysis of Plant Homeobox Genes. Mol. Biol. Evol. 2009, 26, 2775–2794. [Google Scholar] [CrossRef] [Green Version]
- Desplan, C.; Theis, J.; O’Farrell, P.H. The sequence specificity of homeodomain-DNA interaction. Cell 1988, 54, 1081–1090. [Google Scholar] [CrossRef] [Green Version]
- Chan, R.L.; Gago, G.M.; Palena, C.M.; Gonzalez, D.H. Homeoboxes in plant development. Biochim. Biophys. Acta (BBA)-Gene Struct. Expr. 1998, 1442, 1–19. [Google Scholar] [CrossRef]
- Xu, X.; Lou, Y.; Yang, K.; Shan, X.; Zhu, C.; Gao, Z. Identification of Homeobox Genes Associated with Lignification and Their Expression Patterns in Bamboo Shoots. Biomolecules 2019, 9, 862. [Google Scholar] [CrossRef] [Green Version]
- Hirakawa, Y.; Kondo, Y.; Fukuda, H. TDIF Peptide Signaling Regulates Vascular Stem Cell Proliferation via the WOX4 Homeobox Gene in Arabidopsis. Plant Cell 2010, 22, 2618–2629. [Google Scholar] [CrossRef] [Green Version]
- Ji, J.; Strable, J.; Shimizu, R.; Koenig, D.; Sinha, N.; Scanlon, M.J. WOX4 Promotes Procambial Development. Plant Physiol. 2009, 152, 1346–1356. [Google Scholar] [CrossRef] [Green Version]
- Deveaux, Y.; Toffano-Nioche, C.; Claisse, G.; Thareau, V.; Morin, H.; Laufs, P.; Moreau, H.; Kreis, M.; Lecharny, A. Genes of the most conserved WOX clade in plants affect root and flower development in Arabidopsis. BMC Evol. Biol. 2008, 8, 291. [Google Scholar] [CrossRef] [Green Version]
- Di Giacomo, E.; Laffont, C.; Sciarra, F.; Iannelli, M.A.; Frugier, F.; Frugis, G. KNAT3/4/5-like class 2 KNOX transcription factors are involved in Medicago truncatula symbiotic nodule organ development. New Phytol. 2017, 213, 822–837. [Google Scholar] [CrossRef] [Green Version]
- Haecker, A.; Groß-Hardt, R.; Geiges, B.; Sarkar, A.; Breuninger, H.; Herrmann, M.; Laux, T. Expression dynamics of WOX genes mark cell fate decisions during early embryonic patterning in Arabidopsis thaliana. Development 2004, 131, 657–668. [Google Scholar] [CrossRef] [Green Version]
- Reiser, L.; Modrusan, Z.; Margossian, L.; Samach, A.; Ohad, N.; Haughn, G.W.; Fischer, R.L. The BELL1 gene encodes a homeodomain protein involved in pattern formation in the Arabidopsis ovule primordium. Cell 1995, 83, 735–742. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Hu, X.; Duan, Y.; Li, S.; Wang, Y.; Rehman, A.U.; He, J.; Zhang, J.; Hua, D.; Yang, L.; et al. The Arabidopsis Nodulin Homeobox Factor AtNDX Interacts with AtRING1A/B and Negatively Regulates Abscisic Acid Signaling. Plant Cell 2020, 32, 703–721. [Google Scholar] [CrossRef] [Green Version]
- Shu, Y.; Tao, Y.; Wang, S.; Huang, L.; Yu, X.; Wang, Z.; Chen, M.; Gu, W.; Ma, H. GmSBH1, a homeobox transcription factor gene, relates to growth and development and involves in response to high temperature and humidity stress in soybean. Plant Cell Rep. 2015, 34, 1927–1937. [Google Scholar] [CrossRef]
- Wen, H.; Li, J.; Song, T.; Lu, M.; Kan, P.-Y.; Lee, M.G.; Sha, B.; Shi, X. Recognition of histone H3K4 trimethylation by the plant homeodomain of PHF2 modulates histone demethylation. J. Biol. Chem. 2010, 285, 9322–9326. [Google Scholar] [CrossRef] [Green Version]
- Dolzblasz, A.; Nardmann, J.; Clerici, E.; Causier, B.; van der Graaff, E.; Chen, J.; Davies, B.; Werr, W.; Laux, T. Stem Cell Regulation by Arabidopsis WOX Genes. Mol. Plant 2016, 9, 1028–1039. [Google Scholar] [CrossRef]
- McConnell, J.R.; Emery, J.; Eshed, Y.; Bao, N.; Bowman, J.; Barton, M.K. Role of PHABULOSA and PHAVOLUTA in determining radial patterning in shoots. Nature 2001, 411, 709–713. [Google Scholar] [CrossRef]
- Lincoln, C.; Long, J.; Yamaguchi, J.; Serikawa, K.; Hake, S. A knotted1-like homeobox gene in Arabidopsis is expressed in the vegetative meristem and dramatically alters leaf morphology when overexpressed in transgenic plants. Plant Cell 1994, 6, 1859–1876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hake, S.; Smith, H.M.S.; Holtan, H.; Magnani, E.; Mele, G.; Ramirez, J. The role of knox genes in plant development. Annu. Rev. Cell Dev. Biol. 2004, 20, 125–151. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.K.G.; Irish, V.F. The Arabidopsis zinc finger-homeodomain genes encode proteins with unique biochemical properties that are coordinately expressed during floral development. Plant Physiol. 2006, 140, 1095–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Jiang, T.; Li, L.; Zhang, X.; Yang, T.; Liu, C.; Chu, J.; Zheng, B. The chromatin remodeling complex imitation of switch controls stamen filament elongation by promoting jasmonic acid biosynthesis in Arabidopsis. J. Genet. Genom. 2021, 48, 123–133. [Google Scholar] [CrossRef]
- Lee, I.; Aukerman, M.J.; Gore, S.L.; Lohman, K.N.; Michaels, S.D.; Weaver, L.M.; John, M.C.; Feldmann, K.A.; Amasino, R.M. Isolation of LUMINIDEPENDENS: A gene involved in the control of flowering time in Arabidopsis. Plant Cell 1994, 6, 75–83. [Google Scholar] [CrossRef] [Green Version]
- Byrne, M.E.; Groover, A.T.; Fontana, J.R.; Martienssen, R.A. Phyllotactic pattern and stem cell fate are determined by the Arabidopsis homeobox gene BELLRINGER. Development 2003, 130, 3941–3950. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Lin, T.; Grandellis, C.; Yu, M.; Hannapel, D.J. The BEL1-like family of transcription factors in potato. J. Exp. Bot. 2014, 65, 709–723. [Google Scholar] [CrossRef]
- Steindler, C.; Matteucci, A.; Sessa, G.; Weimar, T.; Ohgishi, M.; Aoyama, T.; Morelli, G.; Ruberti, I. Shade avoidance responses are mediated by the ATHB-2 HD-zip protein, a negative regulator of gene expression. Development 1999, 126, 4235–4245. [Google Scholar] [CrossRef]
- Law, J.A.; Vashisht, A.A.; Wohlschlegel, J.A.; Jacobsen, S.E. SHH1, a Homeodomain Protein Required for DNA Methylation, As Well As RDR2, RDM4, and Chromatin Remodeling Factors, Associate with RNA Polymerase IV. PLoS Genet. 2011, 7, e1002195. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.Y.; Lee, D.; Chung, W.-I.; Kwon, C.S. Arabidopsis ING and Alfin1-like protein families localize to the nucleus and bind to H3K4me3/2 via plant homeodomain fingers. Plant J. 2009, 58, 511–524. [Google Scholar] [CrossRef]
- Pompidor, N.; Charron, C.; Hervouet, C.; Bocs, S.; Droc, G.; Rivallan, R.; Manez, A.; Mitros, T.; Swaminathan, K.; Glaszmann, J.-C.; et al. Three founding ancestral genomes involved in the origin of sugarcane. Ann. Bot. 2021, 127, 827–840. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, Q.; Li, L.; Tang, H.; Zhang, Q.; Chen, Y.; Arrow, J.; Zhang, X.; Wang, A.; Miao, C.; et al. Recent polyploidization events in three Saccharum founding species. Plant Biotechnol. J. 2019, 17, 264–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Zhang, X.; Tang, H.; Zhang, Q.; Hua, X.; Ma, X.; Zhu, F.; Jones, T.; Zhu, X.; Bowers, J.; et al. Allele-defined genome of the autopolyploid sugarcane Saccharum spontaneum L. Nat. Genet. 2018, 50, 1565–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Henriksson, E.; Söderman, E.; Henriksson, K.N.; Sundberg, E.; Engström, P. The arabidopsis homeobox gene, ATHB16, regulates leaf development and the sensitivity to photoperiod in Arabidopsis. Dev. Biol. 2003, 264, 228–239. [Google Scholar] [CrossRef] [Green Version]
- Tsukaya, H. Mechanism of Leaf-Shape Determination. Annu. Rev. Plant Biol. 2006, 57, 477–496. [Google Scholar] [CrossRef]
- Li, Y.; Zhu, Y.; Yao, J.; Zhang, S.; Wang, L.; Guo, C.; van Nocker, S.; Wang, X. Genome-wide identification and expression analyses of the homeobox transcription factor family during ovule development in seedless and seeded grapes. Sci. Rep. 2017, 7, 12638. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Ponnala, L.; Gandotra, N.; Wang, L.; Si, Y.; Tausta, S.L.; Kebrom, T.H.; Provart, N.; Patel, R.; Myers, C.R.; et al. The developmental dynamics of the maize leaf transcriptome. Nat. Genet. 2010, 42, 1060–1067. [Google Scholar] [CrossRef]
- Brodersen, P.; Sakvarelidze-Achard, L.; Bruun-Rasmussen, M.; Dunoyer, P.; Yamamoto Yoshiharu, Y.; Sieburth, L.; Voinnet, O. Widespread Translational Inhibition by Plant miRNAs and siRNAs. Science 2008, 320, 1185–1190. [Google Scholar] [CrossRef]
- Yu, B.; Wang, H. Translational inhibition by microRNAs in plants. Prog. Mol. Subcell. Biol. 2010, 50, 41–57. [Google Scholar] [CrossRef] [Green Version]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef]
- Zhu, H.; Hu, F.; Wang, R.; Zhou, X.; Sze, S.-H.; Liou, L.W.; Barefoot, A.; Dickman, M.; Zhang, X. Arabidopsis Argonaute10 Specifically Sequesters miR166/165 to Regulate Shoot Apical Meristem Development. Cell 2011, 145, 242–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, R.; Chi, X.; Chai, G.; Kong, Y.; He, G.; Wang, X.; Shi, D.; Zhang, D.; Zhou, G. Genome-wide identification, evolutionary expansion, and expression profile of homeodomain-leucine zipper gene family in poplar (Populus trichocarpa). PLoS ONE 2012, 7, e31149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Peer, Y.V. Computational approaches to unveiling ancient genome duplications. Nat. Rev. Genet. 2004, 5, 752–763. [Google Scholar] [CrossRef] [PubMed]
- Cannon, S.B.; Mitra, A.; Baumgarten, A.; Young, N.D.; May, G. The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC Plant Biol. 2004, 4, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Zhou, Y.; Jiang, H.; Li, X.; Gan, D.; Peng, X.; Zhu, S.; Cheng, B. Systematic Analysis of Sequences and Expression Patterns of Drought-Responsive Members of the HD-Zip Gene Family in Maize. PLoS ONE 2011, 6, e28488. [Google Scholar] [CrossRef] [Green Version]
- Ponting, C.P.; Aravind, L. START: A lipid-binding domain in StAR, HD-ZIP and signalling proteins. Trends Biochem. Sci. 1999, 24, 130–132. [Google Scholar] [CrossRef]
- Xu, G.; Guo, C.; Shan, H.; Kong, H. Divergence of duplicate genes in exon–intron structure. Proc. Natl. Acad. Sci. USA 2012, 109, 1187–1192. [Google Scholar] [CrossRef] [Green Version]
- Niehrs, C.; Pollet, N. Synexpression groups in eukaryotes. Nature 1999, 402, 483–487. [Google Scholar] [CrossRef]
- Kong, D.; Hao, Y.; Cui, H. The WUSCHEL Related Homeobox Protein WOX7 Regulates the Sugar Response of Lateral Root Development in Arabidopsis thaliana. Mol. Plant 2016, 9, 261–270. [Google Scholar] [CrossRef] [Green Version]
- Hanson, J.; Johannesson, H.; Engström, P. Sugar-dependent alterations in cotyledon and leaf development in transgenic plants expressing the HDZhdip gene ATHB13. Plant Mol. Biol. 2001, 45, 247–262. [Google Scholar] [CrossRef]
- Leech, R.M.; Rumsby, M.G.; Thomson, W.W. Plastid Differentiation, Acyl Lipid, and Fatty Acid Changes in Developing Green Maize Leaves 1. Plant Physiol. 1973, 52, 240–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evert, R.F.; Russin, W.A.; Bosabalidis, A.M. Anatomical and Ultrastructural Changes Associated with Sink-to-Source Transition in Developing Maize Leaves. Int. J. Plant Sci. 1996, 157, 247–261. [Google Scholar] [CrossRef]
- Ni, Z.; Kim, E.-D.; Ha, M.; Lackey, E.; Liu, J.; Zhang, Y.; Sun, Q.; Chen, Z.J. Altered circadian rhythms regulate growth vigour in hybrids and allopolyploids. Nature 2009, 457, 327–331. [Google Scholar] [CrossRef] [Green Version]
- Green, R.M.; Tingay, S.; Wang, Z.-Y.; Tobin, E.M. Circadian Rhythms Confer a Higher Level of Fitness to Arabidopsis Plants. Plant Physiol. 2002, 129, 576–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michael Todd, P.; Salomé Patrice, A.; Yu Hannah, J.; Spencer Taylor, R.; Sharp Emily, L.; McPeek Mark, A.; Alonso José, M.; Ecker Joseph, R.; McClung, C.R. Enhanced Fitness Conferred by Naturally Occurring Variation in the Circadian Clock. Science 2003, 302, 1049–1053. [Google Scholar] [CrossRef]
- Dodd Antony, N.; Salathia, N.; Hall, A.; Kévei, E.; Tóth, R.; Nagy, F.; Hibberd Julian, M.; Millar Andrew, J.; Webb Alex, A.R. Plant Circadian Clocks Increase Photosynthesis, Growth, Survival, and Competitive Advantage. Science 2005, 309, 630–633. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Castillo-González, C.; Yu, B.; Zhang, X. The functions of plant small RNAs in development and in stress responses. Plant J. 2017, 90, 654–670. [Google Scholar] [CrossRef] [Green Version]
- Shriram, V.; Kumar, V.; Devarumath, R.M.; Khare, T.S.; Wani, S.H. MicroRNAs As Potential Targets for Abiotic Stress Tolerance in Plants. Front. Plant Sci. 2016, 7, 817. [Google Scholar] [CrossRef]
- Li, C.; Zhang, B. MicroRNAs in Control of Plant Development. J. Cell. Physiol. 2016, 231, 303–313. [Google Scholar] [CrossRef]
- Müller, C.J.; Valdés, A.E.; Wang, G.; Ramachandran, P.; Beste, L.; Uddenberg, D.; Carlsbecker, A. PHABULOSA Mediates an Auxin Signaling Loop to Regulate Vascular Patterning in Arabidopsis. Plant Physiol. 2016, 170, 956–970. [Google Scholar] [CrossRef] [Green Version]
- Rong, F.; Chen, F.; Huang, L.; Zhang, J.; Zhang, C.; Hou, D.; Cheng, Z.; Weng, Y.; Chen, P.; Li, Y. A mutation in class III homeodomain-leucine zipper (HD-ZIP III) transcription factor results in curly leaf (cul) in cucumber (Cucumis sativus L.). Theor. Appl. Genet. 2019, 132, 113–123. [Google Scholar] [CrossRef]
- Xu, P.; Ali, A.; Han, B.; Wu, X. Current Advances in Molecular Basis and Mechanisms Regulating Leaf Morphology in Rice. Front. Plant Sci. 2018, 9, 1528. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.-Y.; Shen, A.; Xiong, W.; Sun, Q.-L.; Luo, Q.; Song, T.; Li, Z.-L.; Luan, W.-J. Overexpression of OsHox32 Results in Pleiotropic Effects on Plant Type Architecture and Leaf Development in Rice. Rice 2016, 9, 46. [Google Scholar] [CrossRef] [Green Version]
- Reiser, L.; Sánchez-Baracaldo, P.; Hake, S. Knots in the family tree: Evolutionary relationships and functions of knox homeobox genes. Plant Mol. Biol. 2000, 42, 151–166. [Google Scholar] [CrossRef]
- Li, Z.; Hua, X.; Zhong, W.; Yuan, Y.; Wang, Y.; Wang, Z.; Ming, R.; Zhang, J. Genome-Wide Identification and Expression Profile Analysis of WRKY Family Genes in the Autopolyploid Saccharum spontaneum. Plant Cell Physiol. 2020, 61, 616–630. [Google Scholar] [CrossRef]
- Wang, Y.; Hua, X.; Xu, J.; Chen, Z.; Fan, T.; Zeng, Z.; Wang, H.; Hour, A.-L.; Yu, Q.; Ming, R.; et al. Comparative genomics revealed the gene evolution and functional divergence of magnesium transporter families in Saccharum. BMC Genom. 2019, 20, 83. [Google Scholar] [CrossRef]
- Potter, S.C.; Luciani, A.; Eddy, S.R.; Park, Y.; Lopez, R.; Finn, R.D. HMMER web server: 2018 update. Nucleic Acids Res. 2018, 46, W200–W204. [Google Scholar] [CrossRef] [Green Version]
- Chou, K.-C.; Shen, H.-B. Cell-PLoc: A package of Web servers for predicting subcellular localization of proteins in various organisms. Nat. Protoc. 2008, 3, 153–162. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME Suite: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef]
- Lynch, M.; Conery, J.S. The evolutionary fate and consequences of duplicate genes. Science 2000, 290, 1151–1155. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Tang, H.; DeBarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.-H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Swift, M.L. GraphPad Prism, Data Analysis, and Scientific Graphing. J. Chem. Inf. Comput. Sci. 1997, 37, 411–412. [Google Scholar] [CrossRef]
- Ling, H.; Wu, Q.; Guo, J.; Xu, L.; Que, Y. Comprehensive selection of reference genes for gene expression normalization in sugarcane by real time quantitative RT-PCR. PLoS ONE 2014, 9, e97469. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chromosome in S. bicolor | Number 2 | Corresponding Chromosome in S. spontaneum | Number 2 |
|---|---|---|---|
| Chr1 | 24 | Chr1 | 31 (86) |
| Chr2 | 12 | Chr2 | 14 (46) |
| Chr3 | 15 | Chr3 | 10 (30) |
| Chr4 | 9 | Chr4 | 10 (33) |
| Chr5 | 5 | * Chr5 1 | 4 (11) |
| * Chr6 1 | 2 (3) | ||
| Chr6 | 8 | Chr5 | 7 (21) |
| Chr7 | 8 | Chr6 | 4 (20) |
| Chr8 | 3 | * Chr2 1 | 4 (9) |
| * Chr7 1 | 1 (3) | ||
| Chr9 | 6 | Chr7 | 7 (15) |
| Chr10 | 8 | Chr8 | 10 (25) |
| total | 98 | 104 (302) |
| miRNA | Expression Levels (RPM) | Target Gene | Expression Levels (TPM) | Pearson Correlation | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| LF1 | LF6 | LF10 | LF15 | LF1 | LF6 | LF10 | LF15 | |||
| novel23 | 1.17 | 46.13 | 50.16 | 8.71 | SsHB18 | 30.50 | 117.18 | 118.82 | 89.80 | 0.87 |
| miR166seq1 | 17.08 | 83.22 | 177.36 | 224.85 | SsHB14 | 33.95 | 29.45 | 26.61 | 16.38 | −0.93 |
| miR166seq4 | 98.67 | 51.42 | 54.68 | 50.12 | SsHB15 | 109.86 | 45.91 | 32.53 | 22.35 | 0.97 |
| miR166seq4 | 98.67 | 51.42 | 54.68 | 50.12 | SsHB42 | 20.02 | 3.49 | 0.57 | 0.13 | 0.98 |
| miR166seq4 | 98.67 | 51.42 | 54.68 | 50.12 | SsHB47 | 17.22 | 2.43 | 0.46 | 0.33 | 0.99 |
| miR167seq3 | 6.27 | 49.85 | 93.14 | 109.35 | SsHB27 | 16.69 | 46.23 | 60.82 | 86.03 | 0.97 |
| miR397seq1 | 238.98 | 1209.44 | 974.09 | 1565.64 | SsHB3 | 0.64 | 192.12 | 147.03 | 116.07 | 0.75 |
| miR171seq1 | 223.65 | 4.38 | 3.00 | 0.99 | SsHB43 | 30.97 | 55.00 | 43.61 | 89.00 | −0.64 |
| miR171seq3 | 8.32 | 0.09 | 0.00 | 0.00 | SsHB43 | 30.97 | 55.00 | 43.61 | 89.00 | −0.64 |
| miR171seq5 | 12.22 | 0.39 | 0.30 | 0.36 | SsHB39 | 8.02 | 5.46 | 23.67 | 40.63 | −0.47 |
| miR5168 | 668.85 | 134.94 | 111.77 | 70.96 | SsHB1 | 46.97 | 16.34 | 11.53 | 7.71 | 0.99 |
| miR5168 | 668.85 | 134.94 | 111.77 | 70.96 | SsHB14 | 33.95 | 29.45 | 26.61 | 16.38 | 0.72 |
| miR5168 | 668.85 | 134.94 | 111.77 | 70.96 | SsHB56 | 11.26 | 7.29 | 2.49 | 1.93 | 0.88 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Wang, Y.; Feng, X.; Hua, X.; Dou, M.; Yao, W.; Zhang, M.; Zhang, J. Expression Profiling and MicroRNA Regulatory Networks of Homeobox Family Genes in Sugarcane Saccharum spontaneum L. Int. J. Mol. Sci. 2022, 23, 8724. https://doi.org/10.3390/ijms23158724
Li Y, Wang Y, Feng X, Hua X, Dou M, Yao W, Zhang M, Zhang J. Expression Profiling and MicroRNA Regulatory Networks of Homeobox Family Genes in Sugarcane Saccharum spontaneum L. International Journal of Molecular Sciences. 2022; 23(15):8724. https://doi.org/10.3390/ijms23158724
Chicago/Turabian StyleLi, Yihan, Yongjun Wang, Xiaoxi Feng, Xiuting Hua, Meijie Dou, Wei Yao, Muqing Zhang, and Jisen Zhang. 2022. "Expression Profiling and MicroRNA Regulatory Networks of Homeobox Family Genes in Sugarcane Saccharum spontaneum L." International Journal of Molecular Sciences 23, no. 15: 8724. https://doi.org/10.3390/ijms23158724
APA StyleLi, Y., Wang, Y., Feng, X., Hua, X., Dou, M., Yao, W., Zhang, M., & Zhang, J. (2022). Expression Profiling and MicroRNA Regulatory Networks of Homeobox Family Genes in Sugarcane Saccharum spontaneum L. International Journal of Molecular Sciences, 23(15), 8724. https://doi.org/10.3390/ijms23158724