
On the Computational Design of Azobenzene-Based Multi-State Photoswitches


Abstract
:1. Introduction
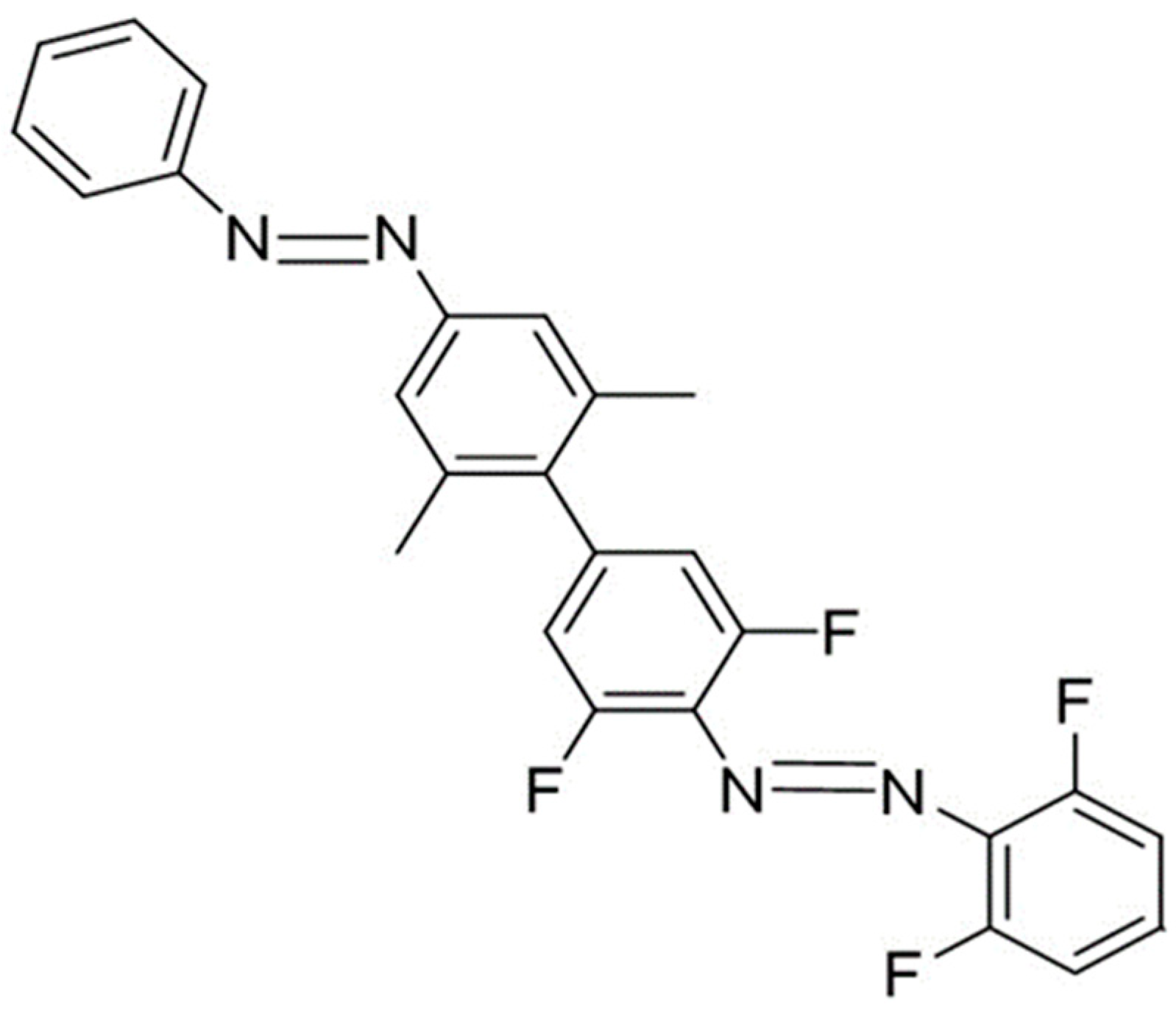
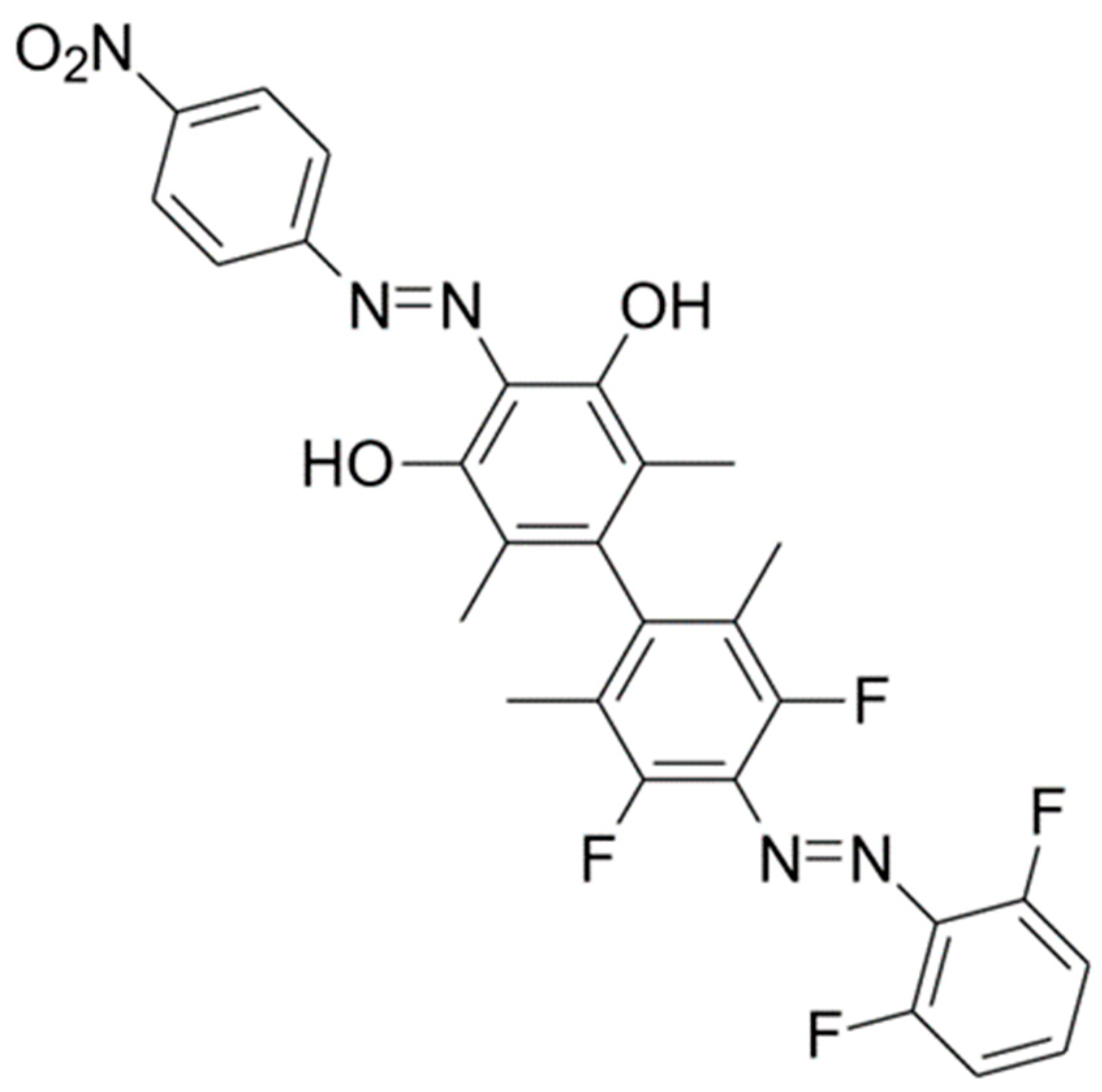
2. Results and Discussion
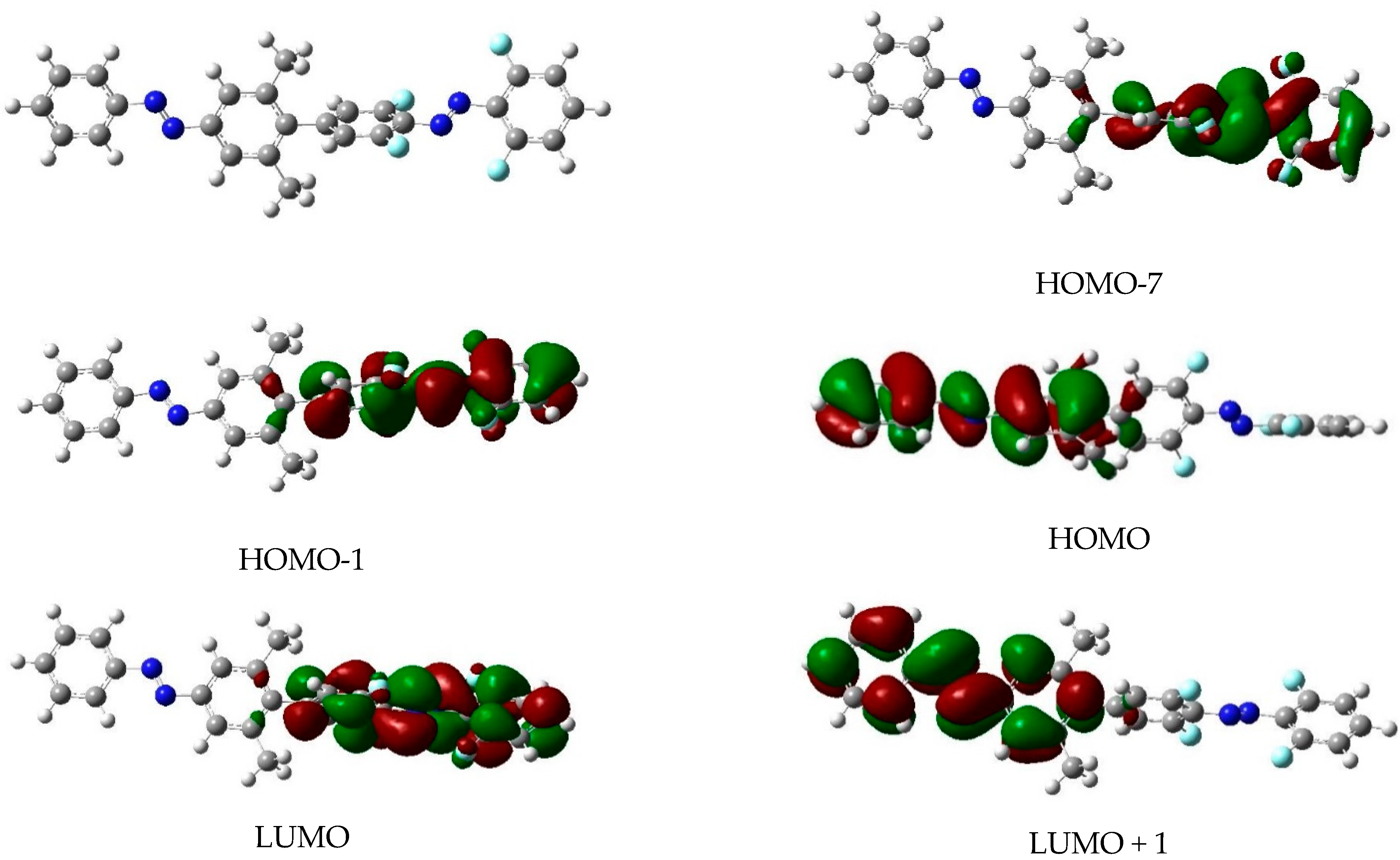
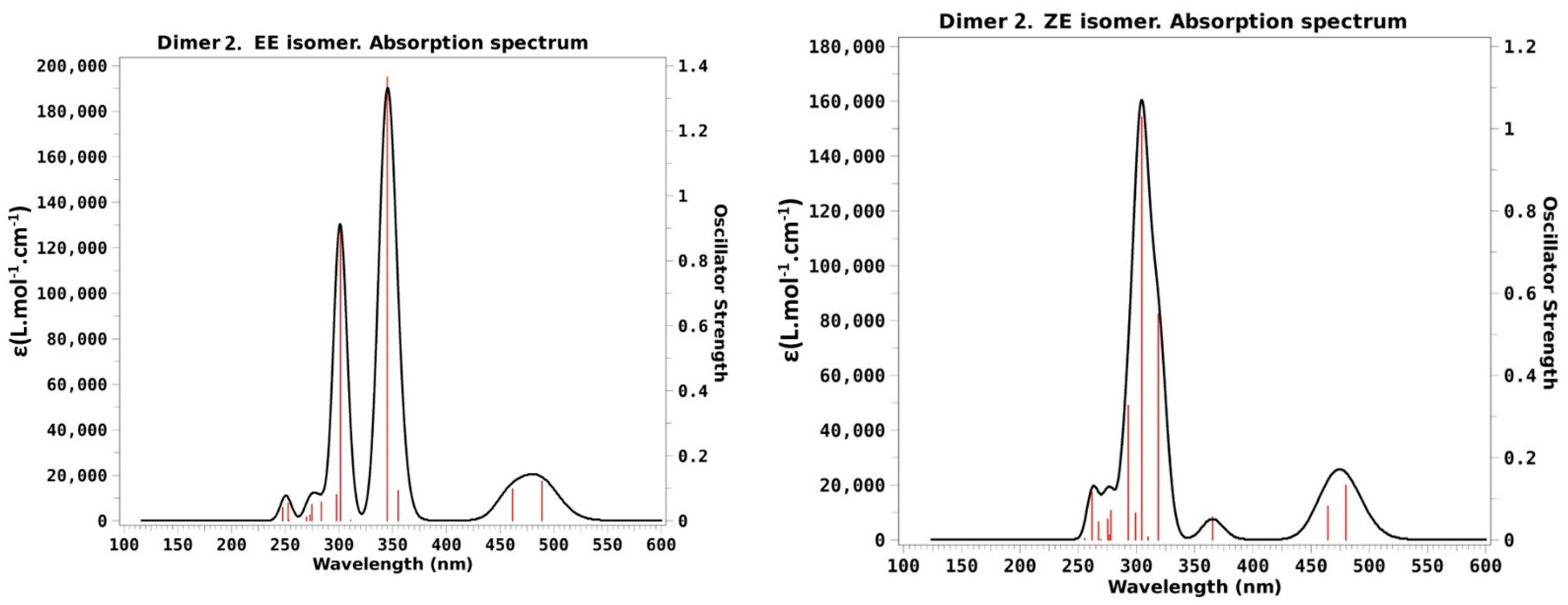
- EE isomer: Experimentally this is the only stable isomer in the absence of irradiation. The main feature is a huge broad band that extends between 280 and 390 nm, with a maximum at approximately 340 nm. An additional, much tidier, but also broad band is seen at 400–500 nm. Our theoretical calculation reveals that the larger band includes both the transitions to S3 and S4 that have large f values 1.61 and 0.78 in our calculation, whereas the less intense band at lower energy will correspond to the transition to S1 calculated at 464 nm that has a small, but not negligible, f value of 0.10. The close transition to S2 calculated at 464 nm has an oscillator strength of 0.10, a small though not negligible value.
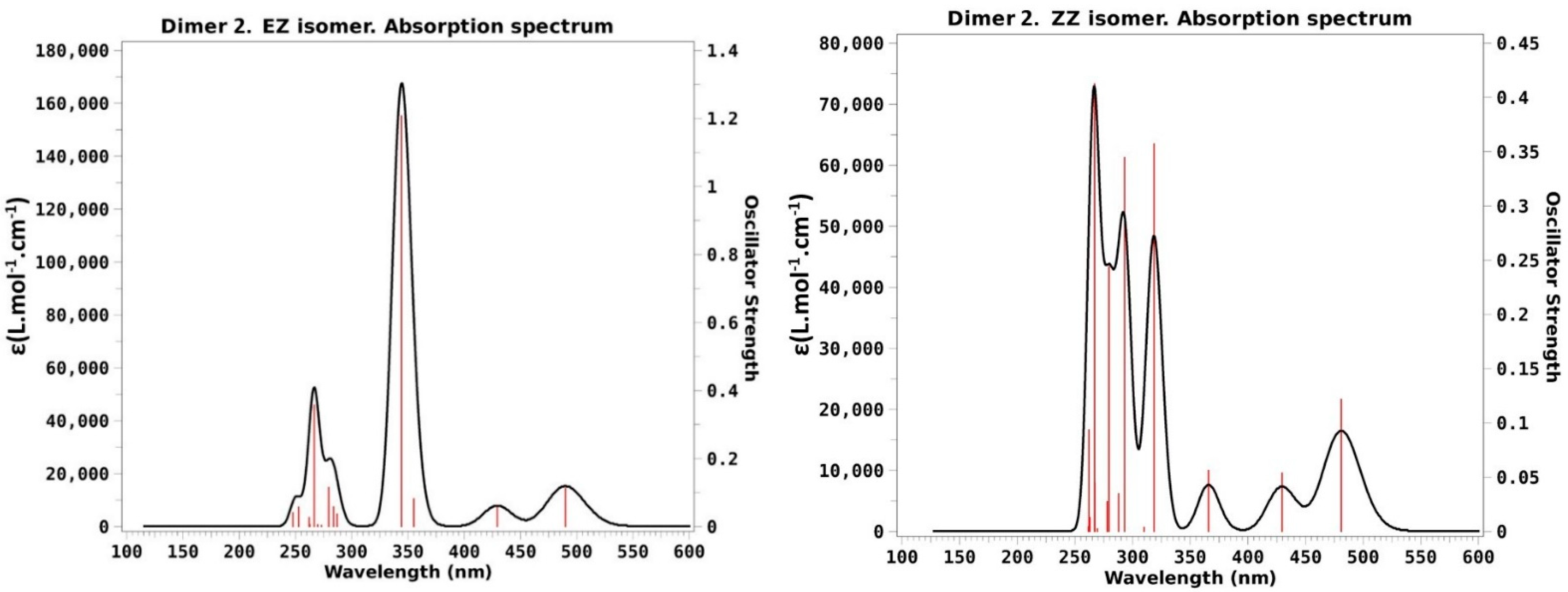
- EZF isomer: Experimentally this isomer is the prevalent one (79%) when the original dimer is irradiated at >500 nm, so we can compare the results in Table 1 with the photostationary state (PSS) spectra after irradiation at this wavelength. In comparison with the EE case just discussed, there are very few differences in the position of the absorption bands. Intensities are different but this may be partially due to the fact that we are now analyzing a mix of different isomers. At any rate the calculations of the excited states of EZF isomer disclose again the presence of three allowed transitions with f > 0.1: The more intense one to S3 at 330 nm (f = 1.29) would make for the higher intensity experimental band with the little help of transition to S4 calculated at 287 nm with f = 0.08. The lower energy will correspond to the S2 transition predicted at 430 nm with a very low but non-zero f value of 0.06.
- ZEF isomer: This structure is not obtained from direct irradiation so that now there is no experimental data to compare with. In any case the calculations predict again rather small differences with respect to the previous discussed cases: The most likely transition is to S3, calculated at 305 nm with f = 1.33, while the lower energy transitions to S1 and S2 are predicted to peak at 465 and 445 nm, respectively, but have, as in the previous cases, quite small intensities (f < 0.1).
- ZZ isomer: This species is dominant (74%) when the original dimer is photoexcited at 350 nm. Now, the obtained spectrum is clearly different from the previous cases just discussed: The larger band is now seen at noticeably shorter wavelengths that almost reach the end of the experimental available range (λ < 300 nm). The less intense band at lower energies is also slightly blue-shifted with a maximum slightly above the 400 nm mark. Our results are in quite nice agreement with these facts as the more intense band is the one to S3 calculated at 278 nm while the two lower transitions to S1 and S2 have very low f values (0.05) and are predicted to appear at 430 and 445 nm. Another noticeable fact of the ZZ absorption spectrum presented in [30] is the lower intensity of the high energy band as compared with the corresponding one of the other two obtained isomers. This is also in nice agreement with our theoretical results as the higher oscillator strength of the ZZ isomer just reaches the 0.36 value for the transition to S3, well below the corresponding maxima of the EE (1.61) and EZF (1.29) structures, as seen in Table 1.
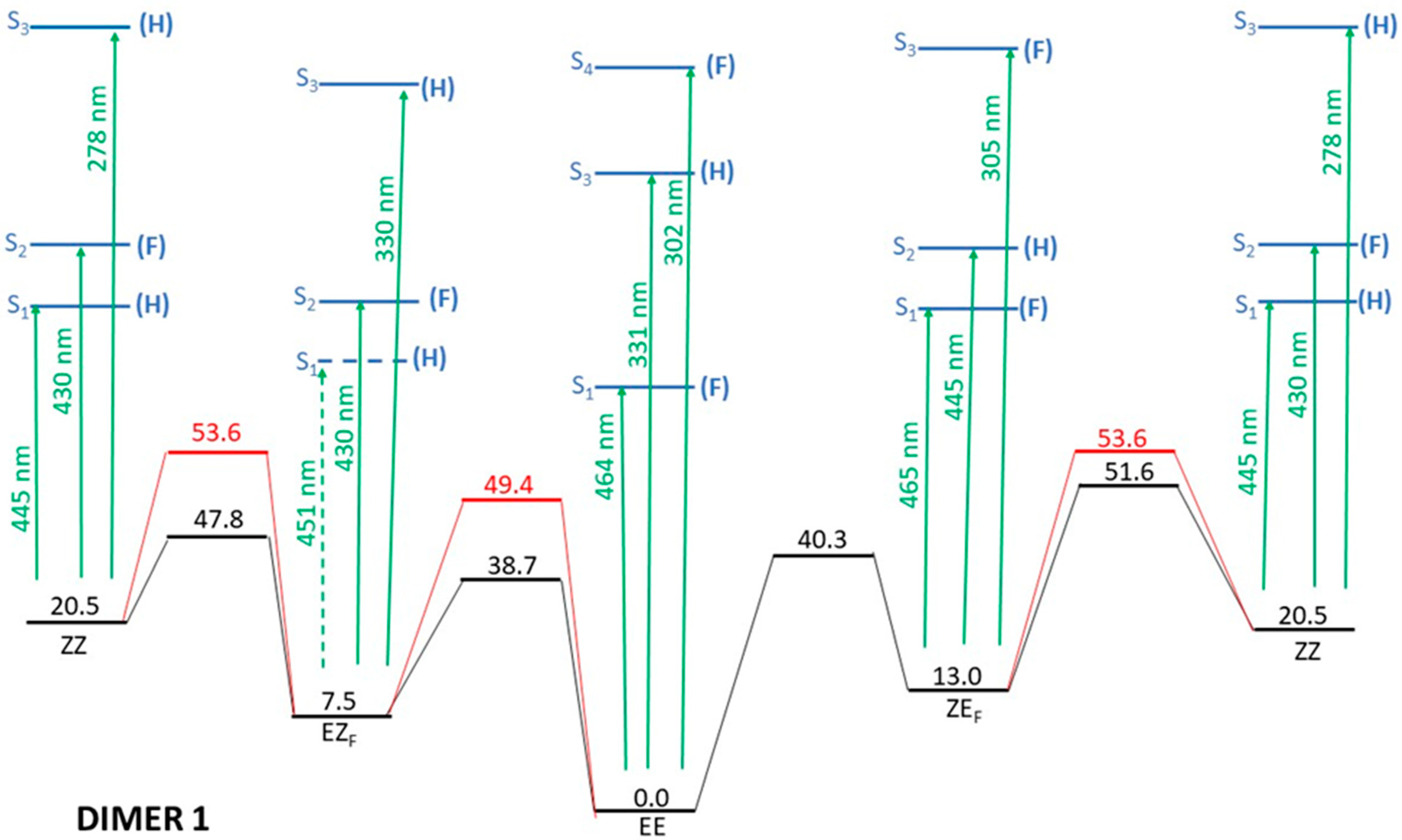
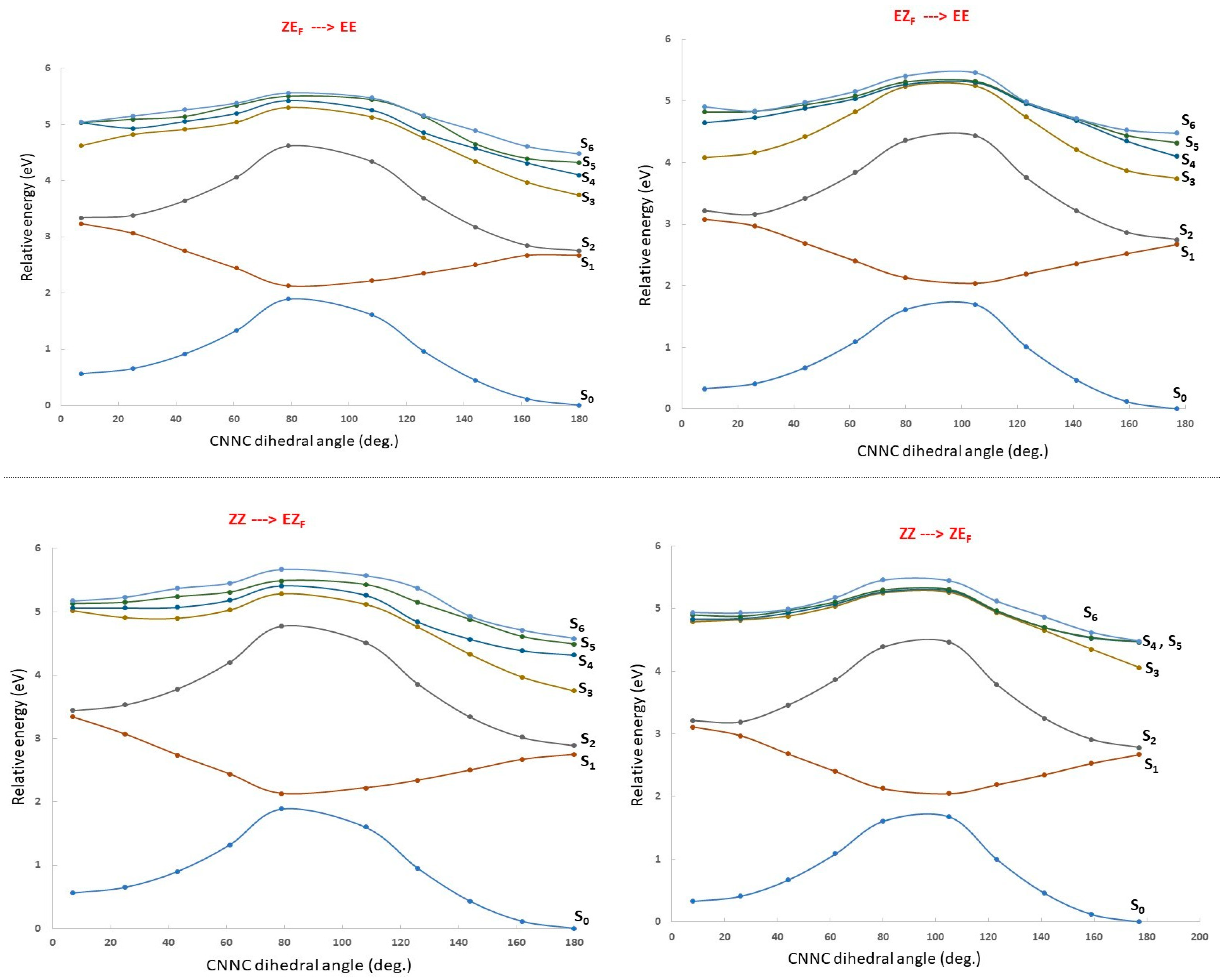
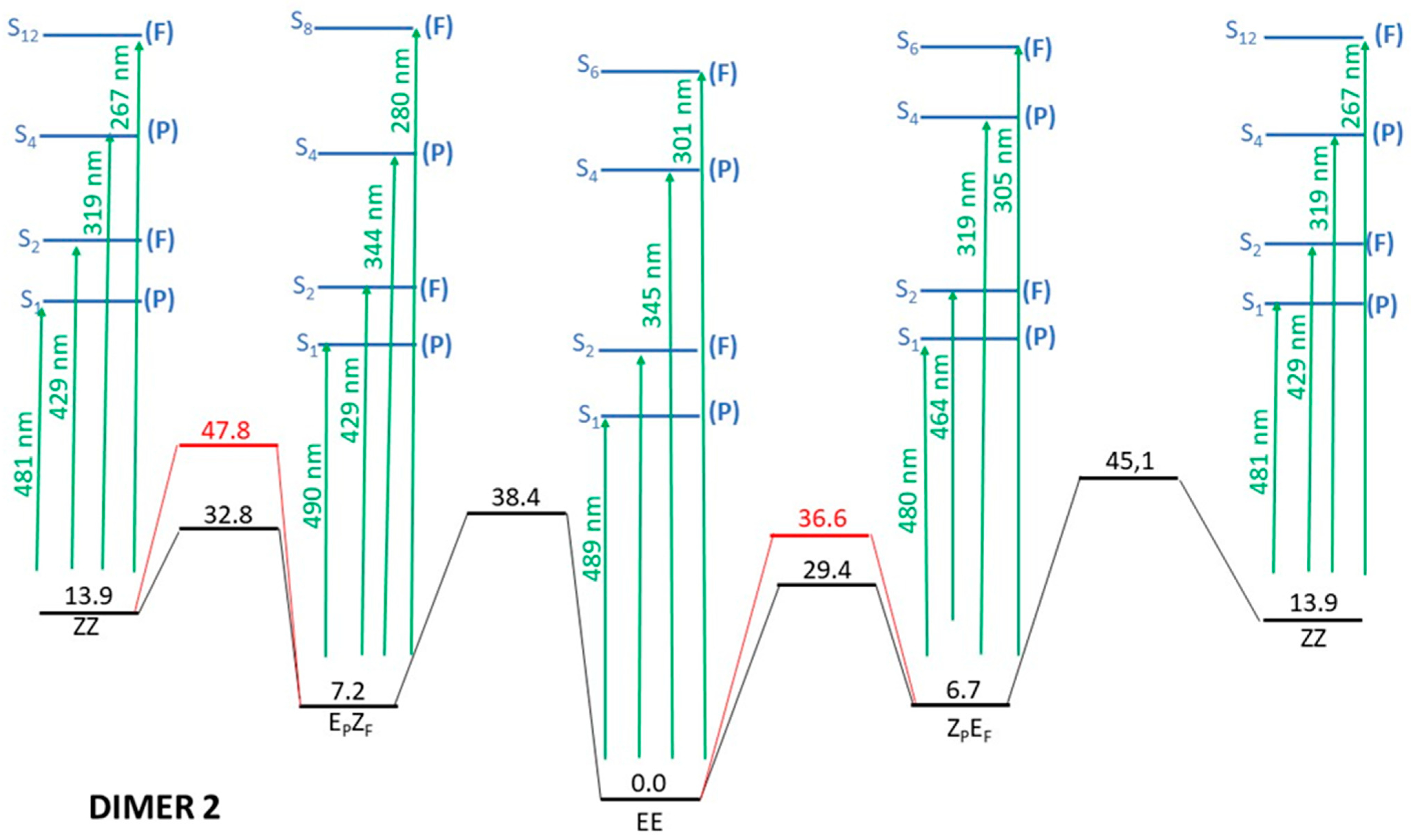
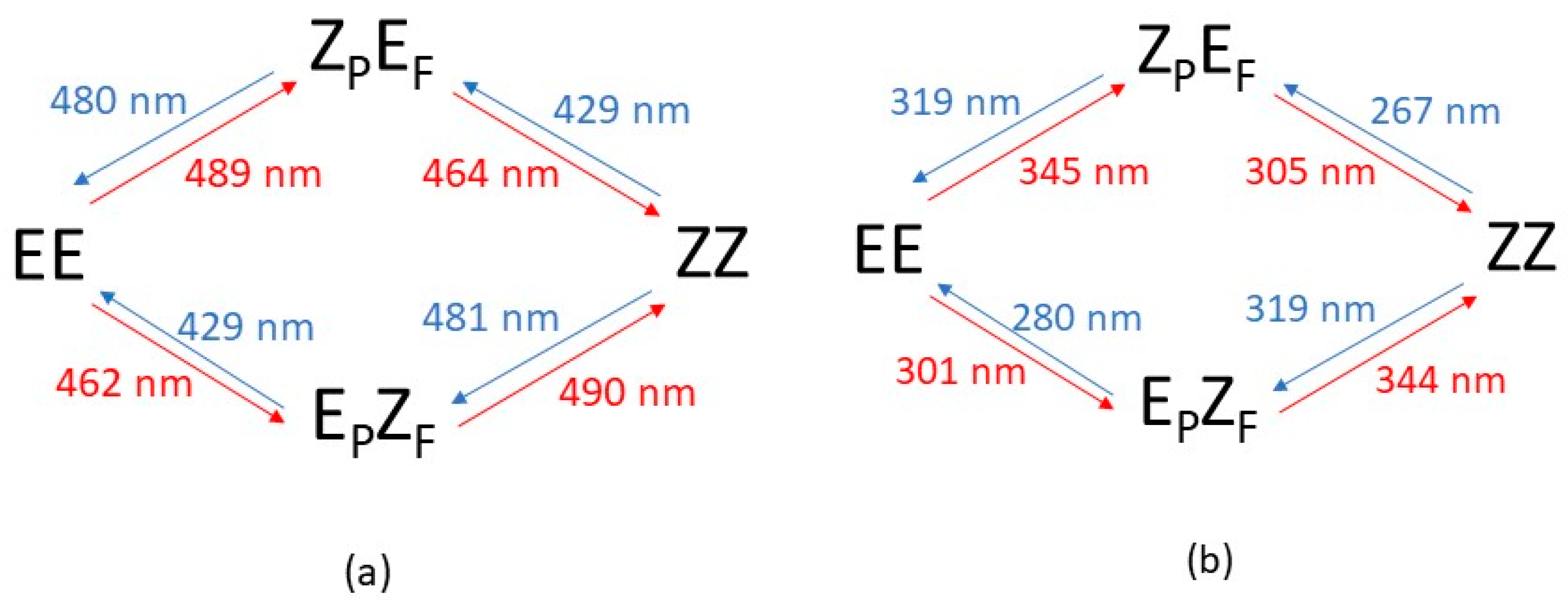
- Irradiation with λ > 500 nm: This low-energy radiation is slightly below the lowest calculated energy excitation of the EE form from S0 to S1 (464 nm), but taking into account the error bars of the calculations and the broadness of the electronic transition bands, we can assume that this radiation allows the excitation of the EE initial form to S1. The oscillator strength of this transition is quite small (0.1), but such a value is usually considered enough to permit the electronic transition. Once in the S1 excited state, the molecule will lose energy and eventually return to the ground electronic state, emitting radiation to return to the original EE structure or else by internal conversion passing through the CI that links EE to EZF. This would explain the predominance of the EZF structure as a result of this irradiation.
- Irradiation with λ = 410 nm: This radiation does not produce any noticeable isomerization reaction as the EE isomer is the dominant species after irradiation. This is easily explained from our results as there are no allowed excitations close to this value for the EE isomer.
- Irradiation with λ = 350 nm: In this case, it is experimentally found that the final majority product is the ZZ isomer. This seems anomalous as in order to obtain the ZZ isomer from the initial EE one, two consecutive isomerizations are needed: The first one from EE to either EZF or ZEF and a second one from each one of these “crossed” isomers to ZZ. According to the results shown in Figure 3, this wavelength should clearly excite the largely allowed transition of EE to S3 and probably also the transition to S4, again with a large f value even if it is slightly above in energy. From our reasoning, this would mean that both EZF and ZEF isomers could be obtained. To explain the formation of the ZZ isomer, a second radiative process should sequentially occur. Our results allow for this to occur given that the 350 nm irradiation can photo-excite both the EZF and ZEF isomers as the two crossed isomers have mainly allowed transition at values slightly above 300 nm. In both cases this second electronic absorption would allow for the isomerization leading to the ZZ isomer. Previous literature extensively reports the feasibility of using such a stepwise, two-photon absorption process to fuel specific photochemical processes of interest [35,36,37]. Parenthetically, we note that at λ > 500 nm, the ZZ adduct is also experimentally obtained as a secondary but noticeable adduct (18%). Its presence should also be explained as the product of two consecutive irradiations of the same energy.
- EZF → EE and ZEF → EE at λ = 410 nm: At this wavelength EZF could access the S2 state. Given that this state implies excitation of the F part it could isomerize to EE. Almost the same pattern can be invoked to explain the isomerization of ZEF back to EE.
- EZF → ZZ and ZEF → ZZ at λ = 350 nm: These two transitions have been already discussed as the second step of the sequential two-photon absorption from EE to ZZ. The high energy irradiation would allow the excitation of the most probable transition (highest value of f) for both crossed structures where isomerization to the ZZ isomer is feasible.
- ZZ → EE at λ = 410 nm: We encounter again a double isomerization process so that, according to our scheme, we will have to claim for a sequential double photo-absorption. For the ZZ isomer the transition to S2 is calculated at 430 nm and at this state the ZEF forms could be obtained. A quite close wavelength of 445 nm would excite the ZEF also to S2, where the isomer EE could be finally obtained. The same process could be envisaged to take place also in the inverted order: ZZ → EZF → EE, implying successive light excitations of 445 and 430 nm (that is: identical values of the former process but also inverted!).
- ZEF → EZF at λ > 500 nm: This seems the most difficult transition to explain as it has to involve a sequential double-electronic excitation by means of two low-energy photons. However, our calculations nicely explain this process assuming that transitions calculated at wavelengths up to 440 nm can be accessed by the considered irradiation. In fact, as in the previous case, we have two possible paths and both are feasible: ZEF → ZZ → EZF with absorptions predicted at 465 and 445 nm, respectively, and ZEF → EE → EZF with two consecutive absorptions now calculated at 445 and 464 nm (curiously enough, again almost the same values but inverted!).
- Irradiation of the EE isomer:
- Irradiation of the ZPEF isomer:
- Irradiation of the EPZF isomer:
- Irradiation of the ZZ isomer:
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Neilson, B.M.; Bielawski, C.W. Illuminating photoswitchable catalysis. ACS Catal. 2013, 3, 1874–1885. [Google Scholar] [CrossRef]
- Beharry, A.A.; Wooley, G.A. Azobenzene photoswitches for biomolecules. Chem. Soc. Rev. 2011, 40, 4422–4437. [Google Scholar] [CrossRef] [PubMed]
- Klajn, R. Spyropyran-based dynamic materials. Chem. Soc. Rev. 2014, 43, 148–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsonis, N.; Lubomska, M.; Pollard, M.M.; Feringa, B.L.; Rudolf, P. Synthetic light-activated molecular switches and motors on surfaces. Prog. Surf. Sci. 2007, 82, 407–434. [Google Scholar] [CrossRef]
- Szymanski, W.; Beierle, J.M.; Kistemaker, H.A.; Velema, W.A.; Feringa, B.L. Reversible photocontrol of biological systems by the incorporation of molecular photoswitches. Chem. Rev. 2013, 113, 6114–6178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hüll, K.; Morstein, J.; Trauner, D. In vivo photopharmacology. Chem. Rev. 2018, 118, 10710–10747. [Google Scholar] [CrossRef]
- Manna, D.; Udayabhaskararao, T.; Zhao, H.; Klajn, R. Orthogonal light-induced self-assembly of nanoparticles using differently substituted azobenzenes. Angew. Chem. Int. Ed. 2015, 54, 12394–12397. [Google Scholar] [CrossRef]
- Borowiak, M.; Nahaboo, W.; Reynders, M.; Nekolla, K.; Jalinot, P.; Hasserodt, J.; Rehberg, M.; Delattre, M.; Zahler, S.; Vollmar, A.; et al. Photoswitchable inhibitors of microtubule dynamics optically control mitosis and cell death. Cell 2015, 162, 403–411. [Google Scholar] [CrossRef] [Green Version]
- Ankenbruck, N.; Courtney, T.; Naro, Y.; Deiters, A. Optochemical control of biological processes in cells and animals. Angew. Chem. Int. Ed. 2018, 57, 2768–2798. [Google Scholar] [CrossRef]
- Dhammika Bandara, H.M.; Burdette, S.C. Photoisomerization in different classes of azobenzene. Chem. Soc. Rev. 2012, 41, 1809–1825. [Google Scholar] [CrossRef]
- Sadovski, O.; Beharry, A.A.; Zhang, F.; Woolley, G.A. Spectral tuning of azobenzene photoswitches for biological applications. Angew. Chem. Int. Ed. 2009, 48, 1484–1486. [Google Scholar] [CrossRef] [PubMed]
- Merino, E.; Ribagorda, M. Control over molecular motion using the cis-trans photoisomerization of the azo group. Beilstein J. Org. Chem. 2012, 8, 1071–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wegener, M.; Hansen, M.J.; Driessen, A.J.M.; Szymanski, W.; Feringa, B.L. Photocontrol of antibacterial activity: Shifting from UV to red light activation. J. Am. Chem. Soc. 2017, 139, 17979–17986. [Google Scholar] [CrossRef]
- Bahrenburg, J.; Sievers, C.M.; Schönbron, J.B.; Hartke, B.; Renth, F.; Temps, F.; Näther, C.; Frank, d.; Sönnichsen, F.D.; Bahrenburg, J. Photochemical properties of multi-azobenzene compounds. Photochem. Photobiol. Sci. 2013, 12, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Vapaavori, J.; Goulet-Hanssens, A.; Heikkinen, I.T.S.; Barrett, C.J.; Priimagi, A. Are two azo groups better than one? Investigating the photoresponse of polimer-bisazobenzene complexes. Chem. Mater. 2014, 26, 5089–5096. [Google Scholar] [CrossRef]
- Fihey, A.; Perrier, A.; Browne, W.R.; Jacquemin, D. Multiphotochromic molecular systems. Chem. Soc. Rev. 2015, 44, 3719–3759. [Google Scholar] [CrossRef]
- Erbas-Cakmak, S.; Leigh, D.A.; McTernan, C.T.; Nussbaumer, A.L. Artificial molecular machines. Chem. Rev. 2015, 115, 10081–10206. [Google Scholar] [CrossRef] [Green Version]
- Coskun, A.; Banaszak, M.; Astumian, R.D.; Stoddart, J.F.; Grzybowski, B.A. Great expectations: Can artificial molecular machines deliver on their promise? Chem. Soc. Rev. 2012, 41, 19–30. [Google Scholar] [CrossRef]
- Hoffmann, K.; Guentner, M.; Mayer, P.; Dube, H. Symmetric and nonsymmetric bis-hemithioindigos precise visible light controlled shape-shifters. Org. Chem. Front. 2019, 6, 1244–1252. [Google Scholar] [CrossRef]
- Cisnetti, F.; Ballardini, R.; Credi, A.; Gandolfi, M.T.; Masiero, S.; Negri, F.; Pieraccini, S.; Spada, G.P. Photochemical and electronic properties of conjugated bis(azo) compounds: An experimental and computational study. Chem. Eur. J. 2004, 10, 2011–2021. [Google Scholar] [CrossRef]
- Robertus, J.; Reker, S.F.; Pijper, T.C.; Dezeman, A.; Browne, W.R.; Feringa, B.L. Kinetic analysis of the thermal isomerization pathways in an asymmetric double azobenzene switch. Phys. Chem. Chem. Phys. 2012, 14, 4374–4382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Floss, G.; Saalfrank, P. The photoinduced E → Z isomerization of bisazobenzenes: A surface hopping molecular dynamics study. J. Phys. Chem. A 2015, 119, 5026–5037. [Google Scholar] [CrossRef] [PubMed]
- Slavov, C.; Yang, C.; Scheighauser, L.; Boumrifak, C.; Drew, A.; Wegner, H.A.; Wachtveitl, J. Connectiviy matters—Ultrafast isomerization dynamics of bisazobenzene photoswitches. Phys. Chem. Chem. Phys. 2016, 18, 14795–14804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heindl, A.H.; Becker, J.; Wegner, H.A. Selective switching of multiple azobenzenes. Chem. Sci. 2019, 10, 7418–7425. [Google Scholar] [CrossRef] [PubMed]
- Reuter, R.; Wegner, H.A. Oligoazobenenophanes-synthesis, photochemistry and properties. Chem. Commun. 2011, 47, 12267–12276. [Google Scholar] [CrossRef]
- Koch, M.; Saphiannikova, M.; Santer, S.; Guskova, O. Photoisomers of azobenzene star with a flat core: Theoretical insights into multiple states from DFT and MD perspective. J. Phys. Chem. B 2017, 121, 8854–8867. [Google Scholar] [CrossRef]
- Yang, C.; Slavov, C.; Wegner, H.A.; Watchtveitl, J.; Drew, A. Computational design of a molecular triple photoswitch for wavelength-selective control. Chem. Sci. 2018, 9, 8665–8672. [Google Scholar] [CrossRef] [Green Version]
- Galanti, A.; Santoro, J.; Mannancherry, R.; Duez, Q.; Diez-Cabanes, V.; Valasek, M.; De Winter, J.; Cornil, J.; Gerbaux, P.; Mayor, M.; et al. A new class of rigid multi(azobenzene) switches featuring electronic decoupling: Unraveling the isomerization in individual photochromes. J. Am. Chem. Soc. 2019, 141, 9273–9283. [Google Scholar] [CrossRef] [Green Version]
- Gupta, D.; Gaur, A.K.; Kumar, P.; Kumar, H.; Mahadevan, A.; Devi, S.; Roy, S.; Venkataramani, S. Tuning of Bistability, Thermal Stability of the Metastable States, and Application Prospects in the C3-Symmetric Designs of Multiple Azo(hetero)arenes Systems. Chem. Eur. J. 2021, 27, 3463–3472. [Google Scholar] [CrossRef]
- Zhao, F.; Grubert, L.; Hecht, S.; Bléger, D. Orthogonal switching in four-state azobenzene mixed-dimers. Chem. Commun. 2017, 53, 3323–3326. [Google Scholar] [CrossRef]
- Bléger, D.; Schwarz, J.; Brouwer, A.M.; Hecht, S. o-Fluoroazobenzenes as readily synthesized photoswitches offering nearly quantitative two-way isomerization with visible light. J. Am. Chem. Soc. 2012, 134, 20597–20600. [Google Scholar] [CrossRef] [PubMed]
- Knie, C.; Utecht, M.; Zhao, F.; Kulla, H.; Kovalenko, S.; Brouwer, A.M.; Saalfrank, P.; Hecht, S.; Bléger, D. ortho-Fluoroazobenzenes: Visible light switches with very long-lived Z isomers. Chem. Eur. J. 2014, 20, 16492–16501. [Google Scholar] [CrossRef] [PubMed]
- Bléger, D.; Dokic, J.; Peters, M.V.; Grubert, L.; Saalfrank, P.; Hecht, S. Electronic decoupling approach to quantitative photoswitching in linear multiazobenzene architectures. J. Phys. Chem. B 2011, 115, 9930–9940. [Google Scholar] [CrossRef] [PubMed]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView, Version 6; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Ward, C.L.; Elles, C.G. Cycloreversion dynamics of a photochromic molecular switch via one-photon and sequential two-photon excitation. J. Phys. Chem. A 2014, 118, 10011–10019. [Google Scholar] [CrossRef] [PubMed]
- Thum, M.D.; Falvey, D.E. Photoreleasable protecting groups triggered by sequential two-photon absorption of visible light: Release of carboxylic acids from a linked anthraquinone-N-alkylpicolinium ester molecule. J. Phys. Chem. A 2018, 122, 3204–3210. [Google Scholar] [CrossRef]
- Ghosh, I.; Ghosh, T.; Bardagi, J.L.; König, B. Reduction of aryl halides by consecutive visible light-induced electron transfer processes. Science 2014, 346, 725–728. [Google Scholar] [CrossRef]
- Zapata, F.; Fernández-González, M.A.; Rivero, D.; Álvarez, A.; Marazzi, M.; Frutos, L.M. Toward an optomechanical control of photoswitches by tuning their spectroscopical properties: Structural and dynamical insights into azobenzene. J. Chem. Theory Comput. 2014, 10, 312–323. [Google Scholar] [CrossRef]
- Bures, F. Fundamental aspects of property tuning in push-pull molecules. RSC Adv. 2014, 4, 58826–58851. [Google Scholar] [CrossRef] [Green Version]
- Silva, D.L.; Krawczyk, P.; Bartkowiak, W.; Mendonça, C.R. Theoretical study of one- and two-photon absorption spectra of azoaromatic compounds. J. Chem. Phys. 2009, 131, 244516. [Google Scholar] [CrossRef]
- Corchado, J.C.; Luz Sánchez, M.; Fdez Galvan, I.; Martín, M.E.; Muñoz-Losa, A.; Barata-Morgado, R.; Aguilar, M.A. Theoretical study of solvent effects on the ground and low-lying excited free energy surfaces of a push-pull substituted azobenzene. J. Phys. Chem. B 2014, 118, 12518–12530. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Miertus, S.; Scrocco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum. A direct utilization of ab initio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Improta, R.; Barone, V.; Scalmani, G.; Frisch, M.J. Using redundant internal coordinates to optimize equilibrium geometries and transition states. J. Comput. Chem. 1996, 17, 49–56. [Google Scholar]
- Handy, N.C. The molecular physics lecture 2004: (i) Density functional theory, (ii) Quantum Monte Carlo, molecular physics. Mol. Phys. 2004, 102, 2399–2409. [Google Scholar] [CrossRef]
- Zhang, M.Y.; Wang, J.Y.; Lin, C.S.; Cheng, W.D. First-principles study of one- and two-photon absorption of the H-bonding complexes from monomeric red fluorescent proteins with large Stokes shifts. J. Phys. Chem. B 2011, 115, 10750–10757. [Google Scholar] [CrossRef] [PubMed]
- List, H.N.; Olsen, J.M.; Rocha-Rinza, T.; Christiansen, O.; Kongsted, J. Performance of popular XC-functionals for the description of excitation energies in GFP-like chromophore models. Int. J. Quantum Chem. 2012, 112, 789–800. [Google Scholar] [CrossRef] [Green Version]
- Randino, C.; Moreno, M.; Gelabert, R.; Lluch, J.M. Peek at the potential energy surfaces of the LSSmkate1 and LSSmKate2 proteins. J. Phys. Chem. B 2012, 116, 14302–14310. [Google Scholar] [CrossRef]
- Moreno, M.; Gelabert, R.; Lluch, J.M. deciphering the grounds of the suitability of acylhydrazones as efficient photoswitches. Phys. Chem. Chem. Phys. 2019, 21, 16075–16082. [Google Scholar] [CrossRef]
- Brémond, E.; Savarese, M.; Adamo, C.; Jacquemin, D. Accuracy of TD-DFT geometries: A fresh look. J. Chem. Theory Comput. 2018, 14, 3715–3727. [Google Scholar] [CrossRef]
- Liang, J.; Feng, X.; Hait, D.; Head-Gordon, M. Revisiting the performance of time-dependent density functional theory for electronic excitations: Assessment of 43 popular and recently developed functionals from rungs one to four. J. Chem. Theory Comput. 2022, 18, 3460–3473. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted gaussian basis set for molecular calculations I. 2nd row atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Raghavachari, K.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods 20. Basis set for correlated wave-functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Domcke, W.; Yarkony, D.R.; Köppel, H. Conical Intersections: Electronic Structure, Dynamics and Spectroscopy; World Scientific Publishing: Singapore, 2004. [Google Scholar]
- Domcke, W.; Yarkony, D.R.; Köppel, H. Conical Intersections: Theory, Computation and Experiment; World Scientific Publishing: Singapore, 2011. [Google Scholar]
- Yarkony, D.R. Nonadiabatic quantum chemistry—Past, present, and future. Chem. Rev. 2012, 112, 481–498. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
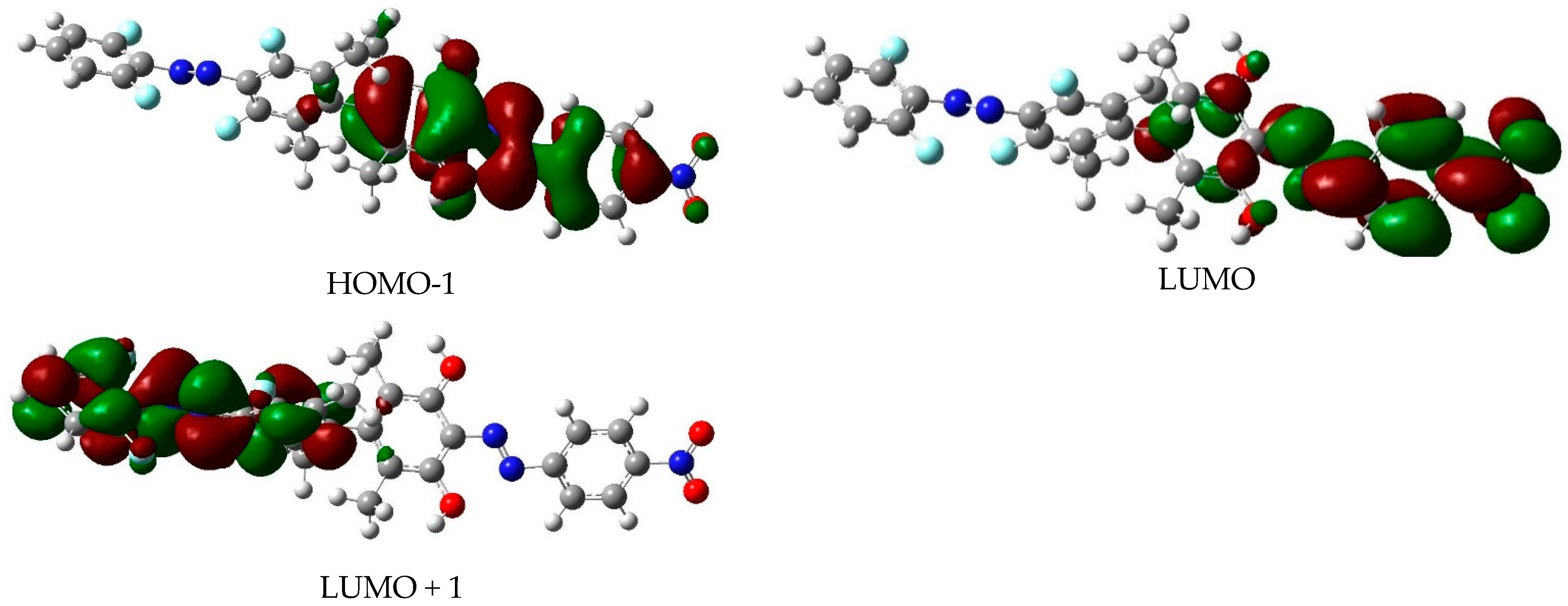
| Isomer | State | E (eV) | λ (nm) | f | Excitation |
|---|---|---|---|---|---|
| EE | S1 | 2.67 | 464 | 0.10 | H−1 → L; H−7 → L (F) |
| S2 | 2.75 | 451 | 0.00 | H−2 → L+1 (H) | |
| S3 | 3.74 | 331 | 1.61 | H → L+1 (H) | |
| S4 | 4.10 | 302 | 0.78 | H−1 → L; H−7 → L (F) | |
| EZF | S1 | 2.75 | 451 | 0.00 | H−2 → L+1 (H) |
| S2 | 2.89 | 430 | 0.06 | H−1 → L (F) | |
| S3 | 3.75 | 330 | 1.29 | H → L+1 (H) | |
| S4 | 4.32 | 287 | 0.08 | H−3 → L+1 (F → H) | |
| ZEF | S1 | 2.67 | 465 | 0.09 | H−1 → L; H−7 → L (F) |
| S2 | 2.78 | 445 | 0.05 | H → L+1 (F) | |
| S3 | 4.06 | 305 | 1.33 | H−1 → L; H−7 → L (F) | |
| S4 | 4.47 | 277 | 0.07 | H−4 → L; H−5 → L (H → F) | |
| S5 | 4.47 | 277 | 0.03 | H−4 → L; H−5 → L (H → F) | |
| S6 | 4.48 | 276 | 0.16 | H−2 → L+1 (H) | |
| ZZ | S1 | 2.78 | 445 | 0.05 | H → L+1 (H) |
| S2 | 2.88 | 430 | 0.05 | H−1 → L (F) | |
| S3 | 4.46 | 278 | 0.36 | H−2 → L+1; H → L+1 (H) | |
| S4 | 4.50 | 275 | 0.02 | H−8 → L (F) | |
| S5 | 4.57 | 271 | 0.12 | H−4 → L (H → F) |
| Isomer | State | E (eV) | λ (nm) | f | Excitation |
|---|---|---|---|---|---|
| EE | S1 | 2.54 | 489 | 0.12 | H−1 → L; H−4 → L (P) |
| S2 | 2.69 | 462 | 0.10 | H−2 → L+1; H−6 → L+1 (F) | |
| S4 | 3.59 | 345 | 1.36 | H−1 → L; H−4 → L (P) | |
| S6 | 4.10 | 301 | 0.9 | H−2 → L+1; H−6 → L+1 (F) | |
| EPZF | S1 | 2.53 | 490 | 0.11 | H−1 → L; H−4 → L (P) |
| S2 | 2.89 | 429 | 0.06 | H−9 → L+1; H−2 → L+1 (F) | |
| S4 | 3.60 | 344 | 1.21 | H−1 → L; H−4 → L (P) | |
| S8 | 4.44 | 280 | 0.11 | H−5 → L+1; H−6 → L+1 (F) | |
| ZPEF | S1 | 2.58 | 480 | 0.13 | H−1 → L; H−1 → L+2 (P) |
| S2 | 2.67 | 464 | 0.08 | H−6 → L+1; H−2 → L+1 (F) | |
| S4 | 3.89 | 319 | 0.55 | H−1 → L; H−5 → L+2 (P) | |
| S6 | 4.07 | 305 | 1.03 | H−6 → L+1; H−2 → L+1 (F) | |
| ZZ | S1 | 2.58 | 481 | 0.12 | H−1 → L; H−1 → L+2 (P) |
| S2 | 2.89 | 429 | 0.05 | H−9 → L+1; H−2 → L+1 (F) | |
| S3 | 3.39 | 366 | 0.06 | H → L; H → L+2 (F → P) | |
| S4 | 3.89 | 319 | 0.36 | H−1 → L; H−5 → L+2 (P) | |
| S12 | 4.64 | 267 | 0.41 | H−9 → L+1 (F) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreno, M.; Lluch, J.M.; Gelabert, R. On the Computational Design of Azobenzene-Based Multi-State Photoswitches. Int. J. Mol. Sci. 2022, 23, 8690. https://doi.org/10.3390/ijms23158690
Moreno M, Lluch JM, Gelabert R. On the Computational Design of Azobenzene-Based Multi-State Photoswitches. International Journal of Molecular Sciences. 2022; 23(15):8690. https://doi.org/10.3390/ijms23158690
Chicago/Turabian StyleMoreno, Miquel, José M. Lluch, and Ricard Gelabert. 2022. "On the Computational Design of Azobenzene-Based Multi-State Photoswitches" International Journal of Molecular Sciences 23, no. 15: 8690. https://doi.org/10.3390/ijms23158690
APA StyleMoreno, M., Lluch, J. M., & Gelabert, R. (2022). On the Computational Design of Azobenzene-Based Multi-State Photoswitches. International Journal of Molecular Sciences, 23(15), 8690. https://doi.org/10.3390/ijms23158690