1. Introduction
Lipid transfer proteins (LTPs) play pivotal roles in the inter-organelle trafficking of lipids in eukaryotes [
1,
2,
3,
4]. The ceramide transport protein, CERT, a typical LTP, mediates the transport of ceramide from the ER to the
trans-Golgi regions, where ceramide is converted to sphingomyelin (SM) [
5,
6]. CERT consists of several functional domains and motifs: an
N-terminal pleckstrin homology (PH) domain, which preferentially binds phosphatidylinositol 4-monophosphate [PtdIns(4)P]; a serine-repeat motif (SRM); an FFAT (two phenylalanines in an acidic tract) motif, which binds vesicle-associated membrane protein-associated protein (VAP); and a
C-terminal steroidogenic acute regulatory-related lipid-transfer (START) domain which effectively encloses and transfers ceramide (
Figure 1A) [
5,
7,
8].
Previous studies have shown that multiple phosphorylation at the SRM down-regulates CERT activity and SM synthesis, while phosphorylation of serine 132 by protein kinase D (PKD) and the following sequential phosphorylation of the downstream serine/threonine residues by casein kinase 1 γ (CK1G) inhibit both PtdIns(4)P-binding and ceramide-transfer activity [
8,
9,
10,
11]. Notably, the phosphoregulatory mechanisms of CERT have recently garnered great attention, because specific missense mutations in the CERT-encoding gene,
CERT1, were found to be associated with a group of autosomal dominant hereditary intellectual disabilities in humans [
12,
13,
14], and some of these mutations were recently shown to compromise CERT SRM hyperphosphorylation [
12,
15].
To explain the simultaneous repression of the activities of the PH and START domains, we previously proposed that the hyperphosphorylation of the SRM triggers a conformational change in CERT, which induces mutual masking between the PH domain and the START domain [
8]. This proposal was supported by co-crystallography of the purified PH and START domains, which suggests that specific surfaces of the two domains of CERT have significant affinity [
16]. In addition, a solution nuclear magnetic resonance (NMR) study suggested that the hyperphosphorylated SRM electrostatically interacts at least partially with the PtdIns(4)P-binding pocket of the CERT PH domain to dissociate the PH domain from the PtdIns(4)P-embedded membrane, thereby possibly facilitating the conformational change in CERT for the PH–START interaction [
17]. However, it is not yet clear how such a dramatic structural alteration to attain the PH/START mutual masking can occur in CERT.
Saus and colleagues have suggested that the predominant form of wild-type CERT [also known as Goodpasture antigen-binding protein (GPBP) Δ26] in cells is a homo-trimer [
18] and recently identified that the five-residue motif SHCIE in CERT/GPBPΔ26 is involved in the self-interaction of the protein [
19] (
Figure 1A). Nonetheless, it remains unclear whether the oligomerization of CERT is relevant to its function and its regulation of CERT.
In this study, we hypothesized that a positively charged region serves as a counterpart of the phosphorylated SRM causing a structural change that suppresses the function of CERT and identified a previously uncharacterized cluster of basic amino acids in CERT as a candidate for this hypothetical counterpart. Then, we found that substitution of the basic amino acids in the cluster with alanine reversed the SRM-dependent repression of CERT activities, i.e., SM synthesis, PtdIns(4)P-binding, VAP-binding, ceramide-transfer activity, and localization to the Golgi, while there was partial recovery of SM synthesis activity. Furthermore, we found that alanine substitution moderately destabilized the trimeric status of CERT. These results suggest that the basic amino acid cluster in the coiled-coil region is involved in the regulation of the CERT function.
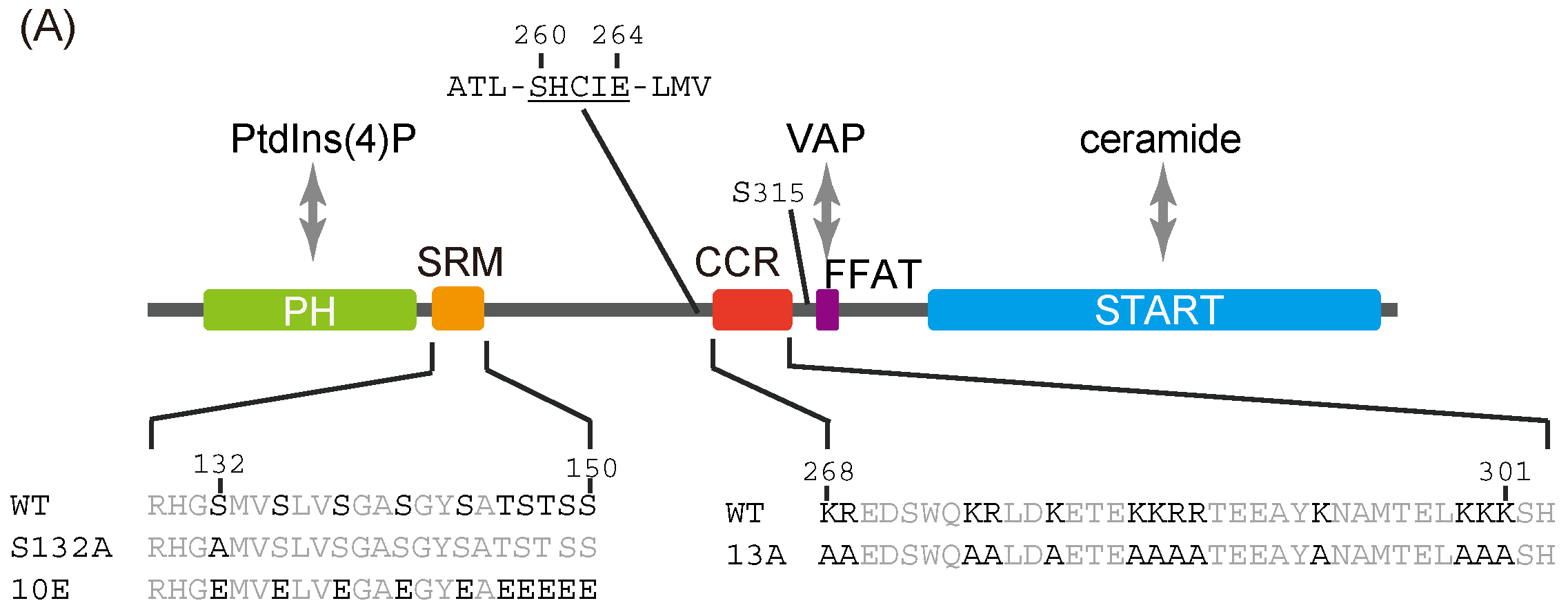
Figure 1.
Identification of a putative coiled-coil region in CERT. (
A) Domains and motifs of CERT. Amino acid sequences are specified for the SRM and CCR in wild-type (WT) and mutants that were used in this study. (
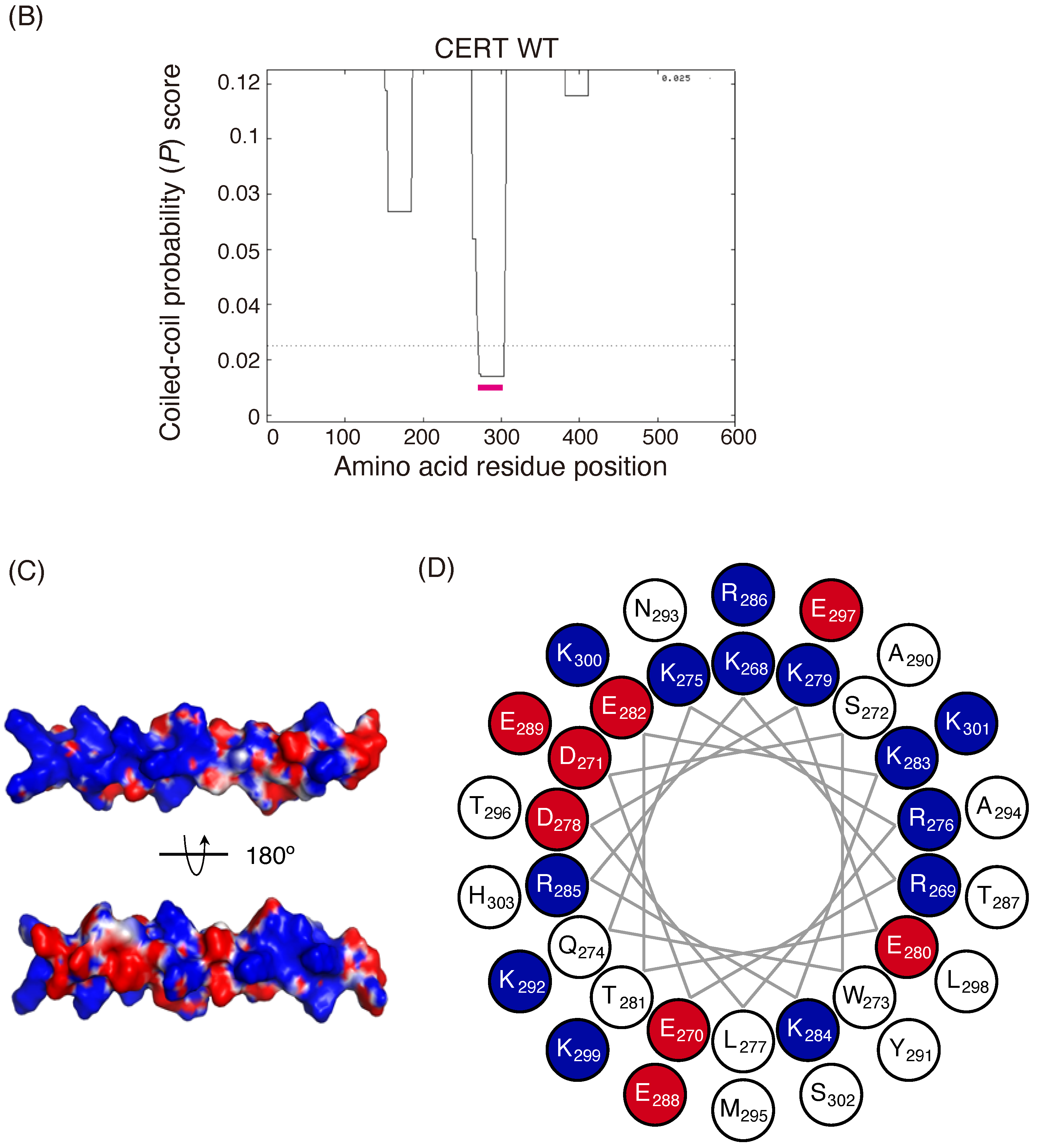
B) Prediction of coiled-coil regions (CCRs) in CERT. The amino acid residues 268–303 of human CERT (highlighted with a magenta line) were predicted to be in a coiled-coil structure. The Paircoil2 program [
20] was used to make the predictions. The decision threshold was set to 0.025 (dashed line), and a sliding window of 21 was used. (
C) Three-dimensional protein modeling of the CCR in human CERT. CCBuilder 2.0 [
21] was used for protein modeling, and PyMol (
https://pymol.org/2/ accessed on 14 July 2018) was used to visualize charged surfaces. Basic and acidic amino acid residues are colored in blue and red, respectively. Other residues are colored in white. (
D) Wheel model of the CCR from wild-type CERT. Lysine/arginine and glutamic acid/aspartic acid are colored in blue and red, respectively. ORIGAMI program [
22] was used for wheel model projection.
Figure 1.
Identification of a putative coiled-coil region in CERT. (
A) Domains and motifs of CERT. Amino acid sequences are specified for the SRM and CCR in wild-type (WT) and mutants that were used in this study. (
B) Prediction of coiled-coil regions (CCRs) in CERT. The amino acid residues 268–303 of human CERT (highlighted with a magenta line) were predicted to be in a coiled-coil structure. The Paircoil2 program [
20] was used to make the predictions. The decision threshold was set to 0.025 (dashed line), and a sliding window of 21 was used. (
C) Three-dimensional protein modeling of the CCR in human CERT. CCBuilder 2.0 [
21] was used for protein modeling, and PyMol (
https://pymol.org/2/ accessed on 14 July 2018) was used to visualize charged surfaces. Basic and acidic amino acid residues are colored in blue and red, respectively. Other residues are colored in white. (
D) Wheel model of the CCR from wild-type CERT. Lysine/arginine and glutamic acid/aspartic acid are colored in blue and red, respectively. ORIGAMI program [
22] was used for wheel model projection.


3. Discussion
Multiple phosphorylation of the CERT SRM results in simultaneous repression of the activities of the
N-terminal PH domain and
C-terminal START domain of CERT [
8,
9]. Interestingly, the repression of the PH domain activity requires the START domain in CERT, while the repression of the START domain activity requires the PH domain, providing a model of SRM phosphorylation-dependent conformational change in CERT [
8]. The model is supported by co-crystallography evidence of the purified PH and START domains, which suggests that specific surfaces of the two domains of CERT have significant affinity [
16]. However, it is poorly understood how the SRM phosphorylation-dependent conformational change occurs.
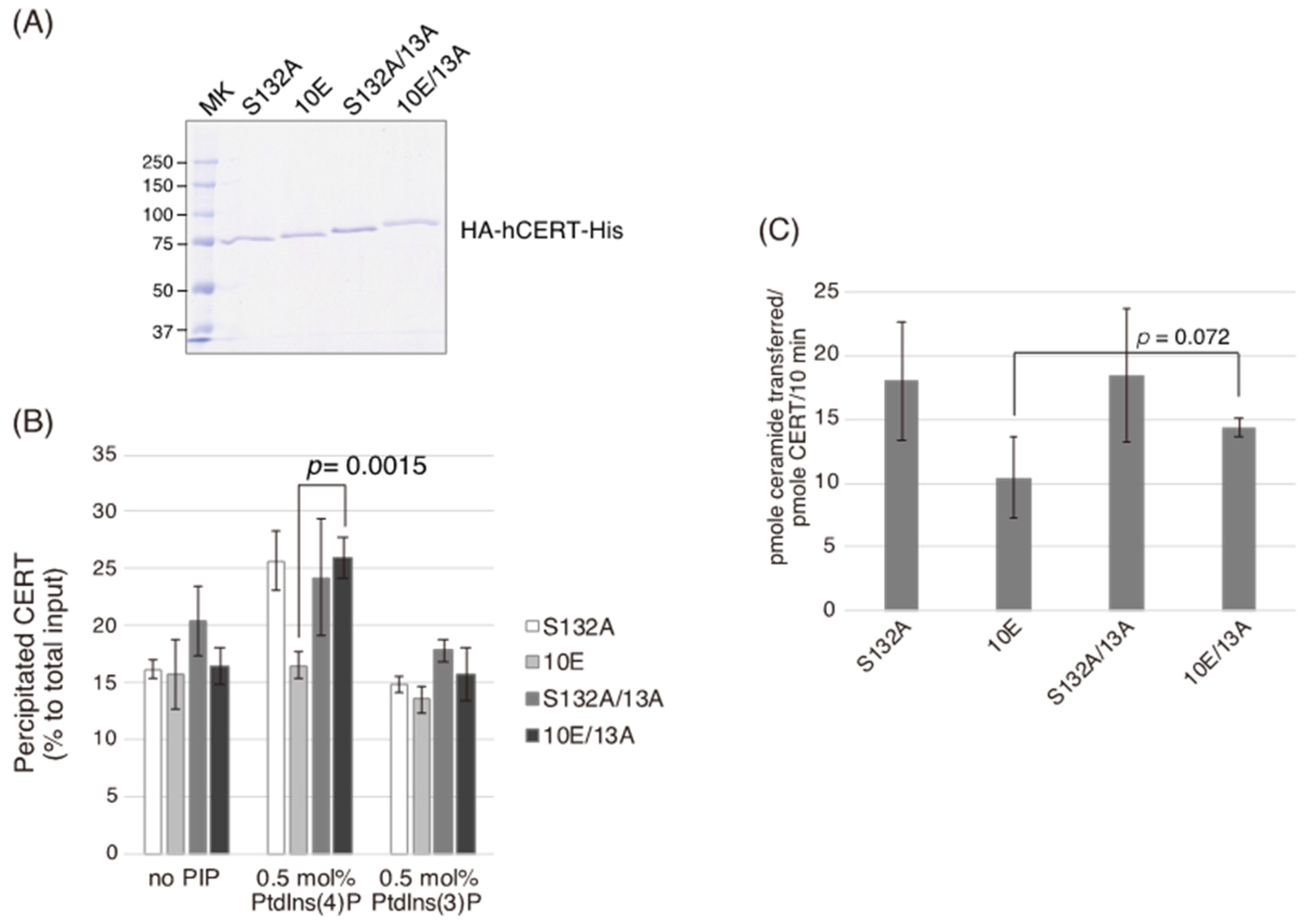
In the current study, we identified a previously uncharacterized cluster of lysine/arginine residues, which were predicted to be located on the outer surface of a probable coiled-coil fold (
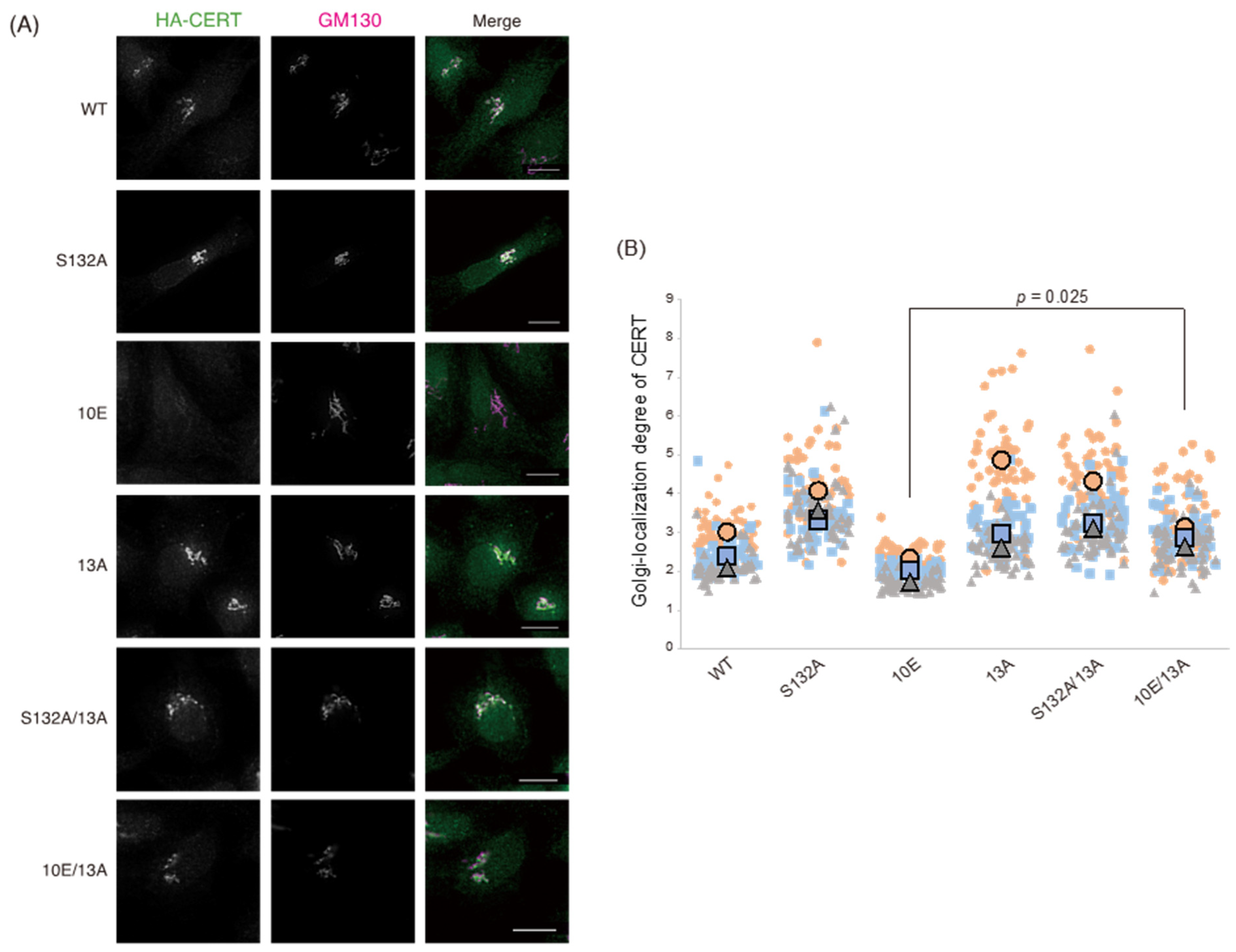
Figure 1). Analysis of PtdIns(4)P-binding activity, VAP-binding activity, S315 phosphorylation, and Golgi localization (
Figure 3,
Figure 4 and
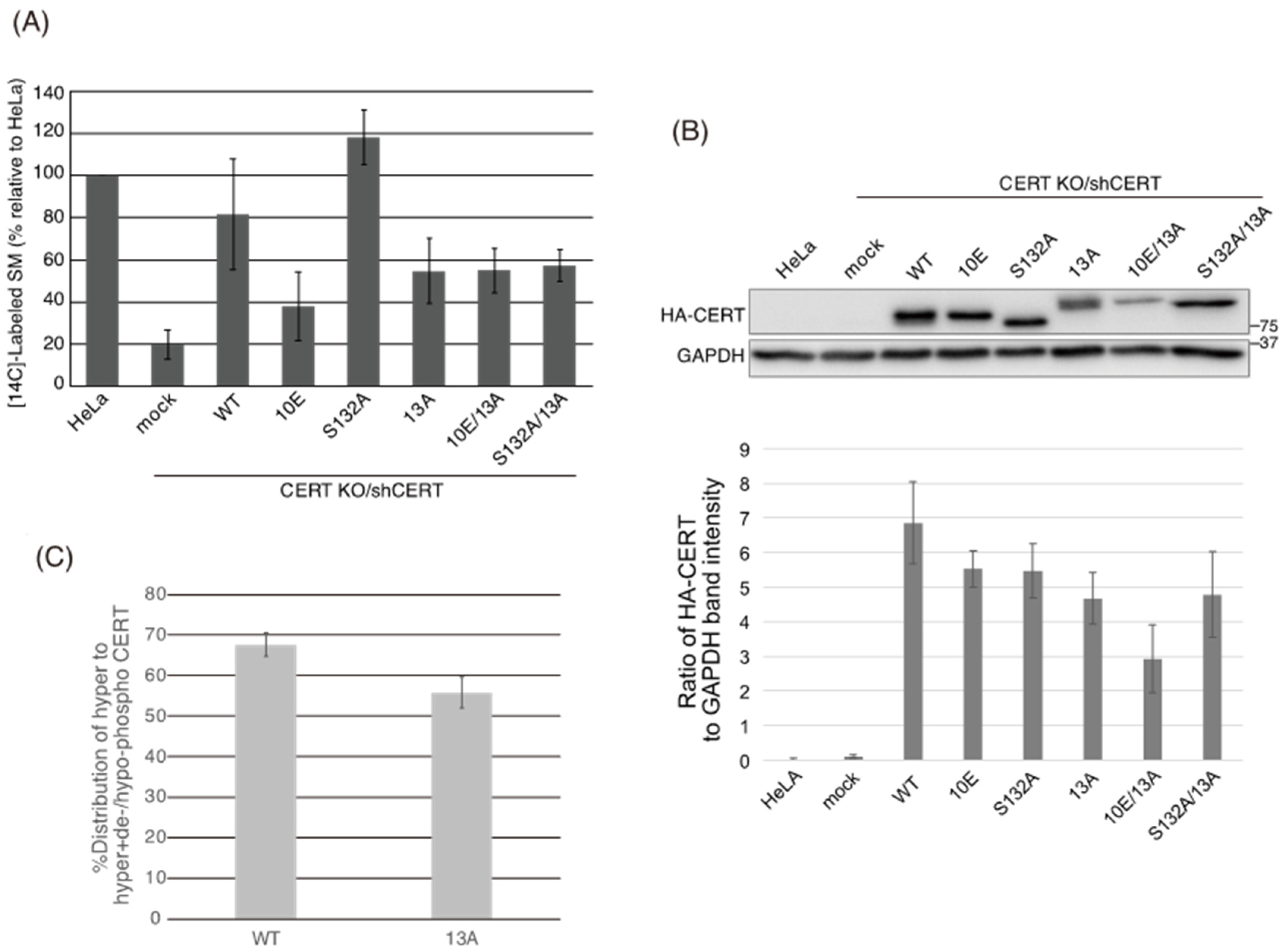
Figure 5B) demonstrated that the negative effects of the 10E mutation on these activities were reversed near to the positive control level (the level of CERT S132A) by alanine substitutions (i.e., the 13A mutation) of the basic amino acids in the cluster. On the other hand, the effects of the 13A mutation on the de novo synthesis of SM in intact cells and the ceramide-transfer activity in a cell-free assay system were complicated: The de novo synthesis of SM activity in CERT 10E/13A cells was similar to (but tended to be higher than) that in CERT 10E cells but never reached the level seen in CERT S132A cells (
Figure 2A), and the level of the ceramide-transfer activity of CERT 10E/13A did not reach the level of CERT S132A (
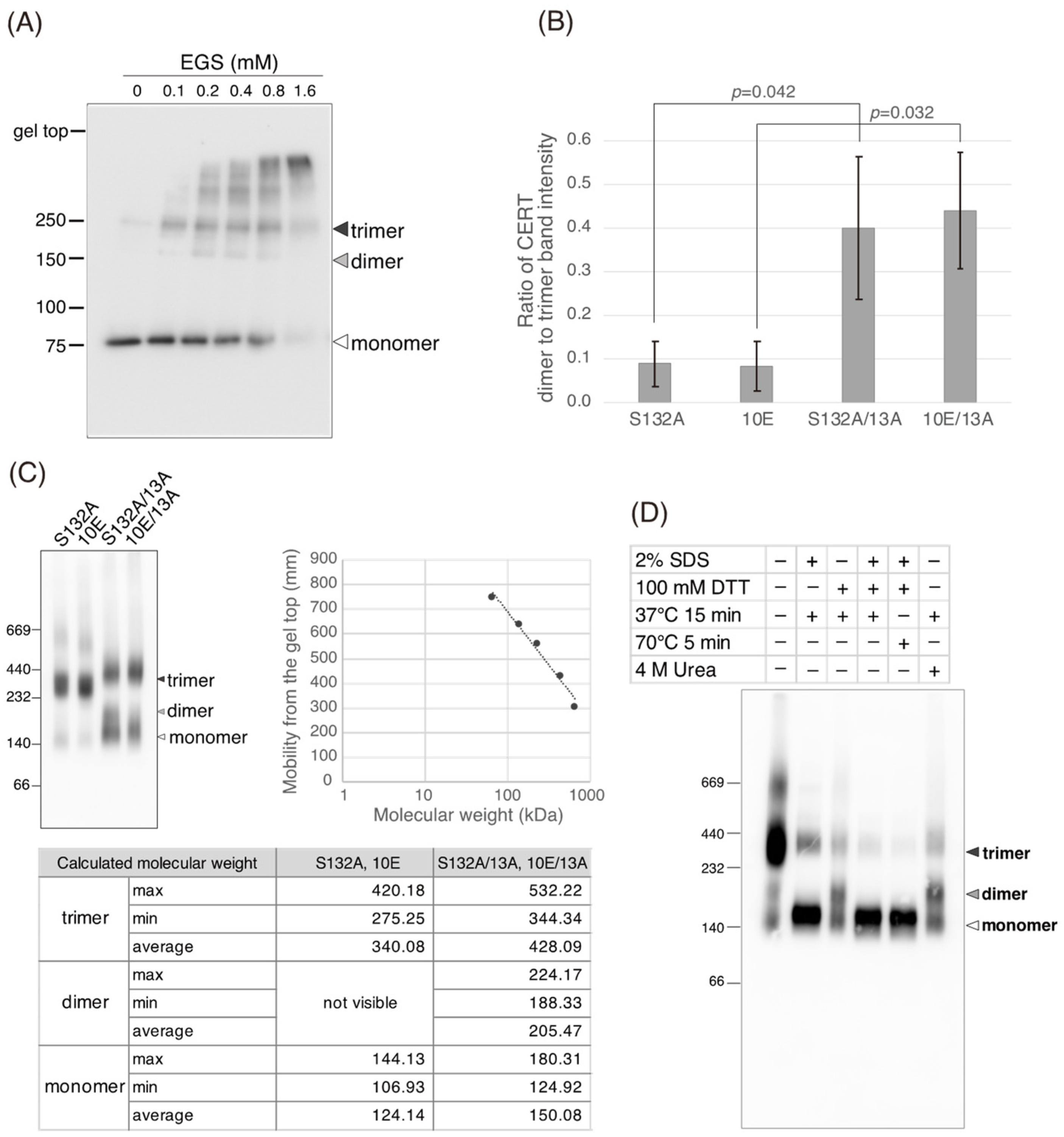
Figure 5C). Saus and colleagues showed that the predominant form of wild-type CERT (also known as GPBPΔ26) in cells is a homo-trimer [
18,
19], which is supported by our present study (
Figure 6). The five-residue motif SHCIE, which is located in the vicinity of the CCR (
Figure 1A), was shown to be crucial for the trimerization of CERT [
19], and our present study suggested that the 13A mutation partially destabilized the trimer organization of CERT (
Figure 6). The 13A mutation itself may have a marginally negative impact on intermembrane ceramide-transfer activity via partially destabilizing the trimeric state of CERT, and, therefore, the CERT 10E/13A construct could not fully reverse its SM synthesis and ceramide-transfer activities to the CERT S132A level. Taking these findings together, we conclude that the negative impact of SRM hyperphosphorylation on the function of CERT is reversed by the 13A mutation, albeit not perfectly, and that the basic amino acid cluster of the CCR is involved in the SRM phosphorylation-dependent repression of CERT.
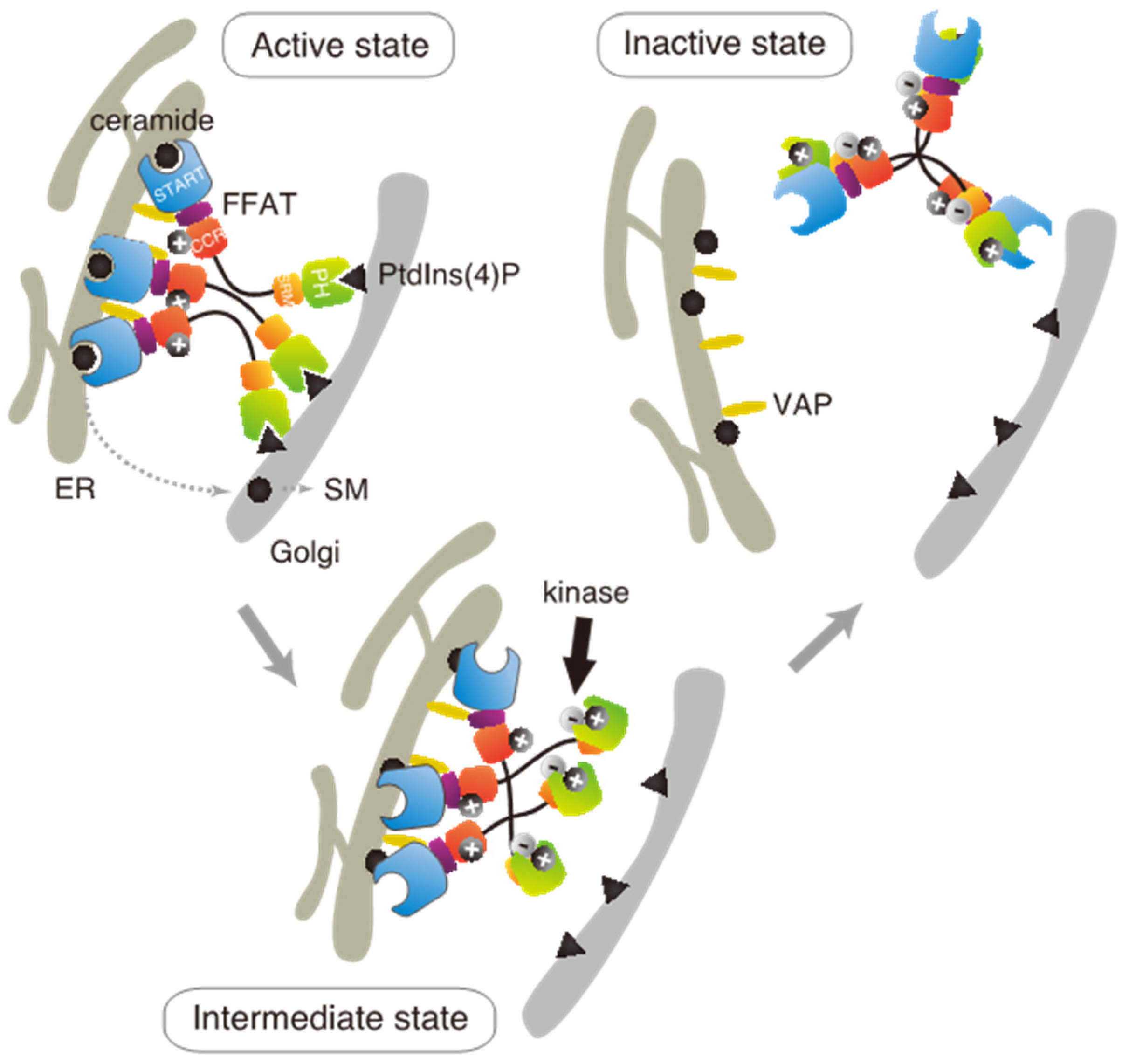
Based on these results in the present and previous studies, we propose an updated model to explain how multiple phosphorylation of the SRM represses the function of CERT (
Figure 7). This model suggests that the positively charged surface of the CCR interacts with the highly phosphorylated SRM and that the 13A mutation loses this electrostatic interaction. Consequently, CERT 10E/13A, similar to the constitutively activated CERT S132A, may form an “open structure” in which the PH and START domains are more accessible to their lipid ligands, i.e., PtdIns(4)P and ceramide, respectively (
Figure 7). In addition, the open structure facilitates the FFAT motif to interact with VAP, which is an important step to exert CERT-mediated transport of ceramide at the ER–Golgi contact sites [
7,
27]. However, it should be pointed out that our model does not determine whether the basic amino acid cluster in the CCR and the phosphorylated SRM interact intramolecularly (i.e., in the same CERT molecule) or in an intermolecular manner (between different CERT molecules in one trimer, analogous to “braided hair”). Structural biological studies of the native complex of the full-size CERT will be needed to further elucidate how mutual masking between the PH and START domains in CERT occurs. It should also be emphasized that we do not deny the possibility that the oligomerizing nature of CERT in intact cells may be more dynamic than the trimeric model, which is derived mainly from static “snapshot” data. Although we putatively depicted that all FFAT motifs and PH domains of one CERT trimer are simultaneously interacting with VAP and PtdIns(4)P, respectively, in the model (
Figure 7), the binding of one or two FFAT motifs and PH domains in the CERT trimer to their partners may be enough for CERT to act at the ER–Golgi contact site.
Several missense mutations in human
CERT1 have been reported to be associated with inherited intellectual disorders (IDs). ID-associated amino acid substitutions are enriched in the SRM region [
12,
13,
14,
28,
29], and these substitutions likely abrogate the SRM phosphorylation-dependent inactivation of CERT [
12]. In addition, some ID-associated substitutions occur outside the SRM, i.e., T166A, F182L, and G243R substitutions in CERT [
30,
31]. A recent study demonstrated that CERT G243R exhibits a constitutively activated phenotype [
15]. Although none of T166A, F182L, or G243R are constituents of the CCR (which is defined from K268 to K301), they may also abrogate the SRM-dependent regulatory system in CERT. If ID-associated mutations in
CERT1 are mapped in the CCR in the future, it should provide strong evidence for the involvement of the CCR in human health-relevant functional regulation of CERT.
4. Materials and Methods
4.1. Materials
Phosphatidylcholine (more specifically, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine), phosphatidylethanolamine (more specifically, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine), and lactosylceramide were obtained from Avanti Polar Lipids. Phosphatidylinositol 3-monophosphate [PtdIns(3)P] and PtdIns(4)P were obtained from Cayman Chemicals. Ricinus communis agglutinin was obtained from Vector Laboratories, Inc. Phosphate-buffered saline (PBS) was obtained from Fujifilm Wako Pure Chemical Co.
4.2. Antibodies and Plasmids
Anti-HA antibody (#3F10) was obtained from Roche Diagnostics; anti-His (#D291-3S) from Medical & Biological Laboratories Co., Ltd.; anti-CERT from Abcam (#ab72536); anti-GM130 (#610823) from BD Biosciences; and anti-GAPDH (#016-25523) from Fujifilm Wako Pure Chemical Co. The production of anti-VAP-A chicken polyclonal antibody and anti-phospho-serine 315 antiserum was described previously [
27].
The construction of the CERT S132A and 10E mutants was described previously [
8]. The
N-terminal HA-tagged WT CERT cDNA in pBlueScript II SK (+) (pBS/nHAcFL-CERT WT) was used as a template to mutagenize the thirteen lysine/arginine residues in the CCR (amino acids 268–303) to alanine (CERT 13A). The CCR was divided into two overlapping sub-regions, CC1 (amino acids 268–291, CERT 9A) and CC2 (287–303, CERT 4A), and the following two sets of oligonucleotides were used for mutagenesis, respectively. CC1: 5′-GAACTAATGGTTGCAGCTGAGGACAGCTGGCAGGCGGCCCTGGATGCGGAAACTGAGGCGGCAGCAGCAACAGAGGAAGCATAT-3′ and 5′-TGCTTCCTCTGTTGCTGCTGCCGCCTCAGTTTCCGCATCCAGGGCCGCCTGCCAGCTGTCCTCAGCTGCAACCATTAGTTCAAT-3′, CC2: 5′-ACAGAGGAAGCATATGCAAATGCAATGACAGAACTTGCGGCAGCATCCCACTTTGGAGGACCAGAT-3′ and 5′-TGGTCCTCCAAAGTGGGATGCTGCCGCAAGTTCTGTCATTGCATTTGCATATGCTTCCTCTG-3′. WT and mutant CERT cDNAs were transferred to pMXs-IRES-Bsd vector [
32] or pENTR/D-TOPO vector (Thermo Fisher Scientific) for expression in mammal cells or insect cells, respectively.
A retrovirus-based CERT shRNA expression vector (shCERTv1) was constructed by annealing the following two oligonucleotides and cloning the annealed nucleotides into pSilencer v 5.1 vector (Thermofisher). shCERT-pSilencer v1 sense: 5′-GATCCGCGAGAGTATCCTAAATTTAAGTTCTCTAAATTTAGGATACTCTCGCTTTTTTGGAAA-3′ and shCERT-pSilencer v1 antisense: 5′-AGCTTTTCCAAAAAAGCGAGAGTATCCTAAATTTAGAGAACTTAAATTTAGGATACTCTCGCG-3′.
4.3. Cell Culture and Transfections
HeLa cells expressing a mouse ecotropic retroviral receptor, HeLa-mCAT#8 cells [
33], were cultured in DMEM with 10% (
v/v) FBS at 37 °C with 5% CO
2. A
CERT-disrupted mutant HeLa cell line (clone name, TAL-CE#14 clone) that was previously established in our laboratory [
34] was used as CERT KO HeLa cells. For the establishment of CERT KO/shCERT, the CERT KO cells were transduced with virus particles produced using the shCERTv1 plasmid. An isolated clone (TAL-CE#14-shCE#1 clone) was transduced with virus particles produced using pMXs-IRES-Bsd expression vectors [
32] encoding
N-terminal HA-tagged human CERT S132A, CERT 10E, CERT 13A, CERT S132A/13A, or CERT 10E/13A. Sf21 cells (Thermo Fisher Scientific, Waltham, MA, USA) were cultured in SF900-II medium in monolayer or suspension at 27 °C. Cell transfection was performed as described below.
4.4. Baculovirus Expression and Purification
WT and mutant CERT cDNAs cloned in pENTR/D-TOPO vector (Thermo Fisher Scientific) were transferred into BaculoDirect linear DNA (Thermo Fisher Scientific, Waltham, MA, USA) by recombination, and the resulting ligation mixtures were transfected to Sf21 cells in monolayer using Cellfectin II (Thermo Fisher Scientific, Waltham, MA, USA). High-titer viral stocks were generated under selection with 100 μM ganciclovir (Tokyo Chemical Industry, Tokyo, Japan), transduced into Sf21 cells in suspension, and cultured for 72 h at 27 °C. Cells were harvested, lysed, and purified using an anti-His-tag and an anti-HA-tag affinity resin. Briefly, cells were resuspended in TALON buffer [50 mM HEPES-NaOH (pH 7.5), 300 mM NaCl] containing 30 mM imidazole and protease inhibitor cocktail (complete EDTA-free, Roche, Basel, Switzerland) and disrupted by passing through 18-gauge and 25-gauge needles followed by sonication with a probe-type sonicator (UP50H, Dr. Hielscher GmbH, Chamerau, Germany). Cell lysates were collected by centrifugation at 100,000× g for 1 h at 4 °C, then loaded on a TALON metal affinity column (Clontech Laboratories, Inc., Mountain View, CA, USA). The N-terminal His-tagged CERT proteins were on-column treated with λ protein phosphatase (NEB) for 1 h at 4 °C, then eluted with TALON buffer containing 150 mM imidazole. The eluent was subjected to concentration and buffer exchange [50 mM HEPES-NaOH (pH 7.5), 500 mM NaCl] using a 30-kDa cutoff Amicon Ultra column (Merck Millipore, Burlington, MA, USA), bound to anti-HA affinity resin (Merck Millipore, Burlington, MA, USA) o/n at 4 °C, and eluted with a buffer [50 mM HEPES-NaOH (pH 7.5), 500 mM NaCl] containing 1 μM HA peptide. Aliquots of the eluent were stored at −80 °C.
4.5. In Vitro PtdIns(4)-Binding and Ceramide-Transfer Assays
PtdIns(4)P-binding by purified recombinant CERT proteins was measured using liposomes without or with 0.5 mol% of PtdIns(3)P or PtdIns(4)P, as previously described but with some modifications [
8]. Liposomes composed of phosphatidylcholine:phosphatidylethanolamine:lactosylceramide:phosphoinositide (15.87:4:6:0.13, mol/mol) were prepared by sonication using a probe-type sonicator (UP50H, Dr. Hielscher GmbH, Chamerau, Germany) and pre-centrifuged at 20,400×
g for 5 min at 4 °C. Purified CERT was pre-centrifuged at 100,000×
g for 30 min at 4 °C. Liposomes containing in total 26 nmol of lipids were incubated with 200 fmol of recombinant CERT in 50 μL of buffer C containing 0.3 mg/mL BSA and 0.1 (
v/v) CHAPS for 30 min at 4 °C.
Ricinus communis agglutinin (18.75 μg) was added to terminate the reaction, incubated on ice for 10 min to aggregate the liposomes, and centrifuged at 20,400×
g for 1 min at 4 °C. PtdIns(4)P-bound (pellet fraction) or non-bound (supernatant fraction) CERT was quantified by immunoblotting analysis using an anti-HA antibody.
Ceramide-transfer between artificial donor and acceptor liposomes was measured as described elsewhere, with some modifications [
5,
35]. Briefly, donor and acceptor liposomes composed of phosphatidylcholine:phosphatidylethanolamine:lactosylceramide:[
14C]ceramide (64:16:40:1, mol/mol), and phosphatidylcholine: phosphatidylethanolamine (320:80, mol/mol), respectively, were prepared by sonication using a probe-type sonicator (UP50H, Dr. Hielscher Gmb) and pre-centrifuged at 20,400×
g for 1 min at 4 °C to remove lipid aggregates. Purified CERT was pre-centrifuged at 100,000×
g for 30 min at 4 °C to exclude aggregated protein. Acceptor liposomes containing a total 400 nmol of lipids were incubated with 2 pmol of recombinant CERT in 80 μL of buffer C [20 mM HEPES-NaOH (pH 7.5), 50 mM NaCl, 1 mM EDTA] on ice. Donor liposomes (20 μL) were added to the assay mixtures to start the reactions and incubated for 10 min at 37 °C. The reactions were terminated by adding 37.5 μg of
Ricinus communis agglutinin, incubated for 15 min on ice, and the donor liposomes were precipitated by centrifugation at 20,400×
g for 3 min at 4 °C. The radioactivity of the supernatant including the acceptor liposomes was measured using liquid scintillation counting.
4.6. Chemical Cross-Linking
Recombinant CERT proteins [10.8 ng of protein in 30 μL of 50 mM HEPES-NaOH (pH 7.4)] were incubated with or without various concentrations of ethylene glycol bis (succinimidyl succinate) (EGS, Thermo Fisher Scientific, Waltham, MA, USA) on ice for 2 h. After addition of Tris-HCl (pH 7.4) to the mixture at a final concentration of 47.6 mM, the resultant mixture was incubated for 15 min on ice to terminate the cross-linking reaction. The terminated mixture was diluted in SDS-PAGE loading buffer and subjected to western blotting analysis.
4.7. Blue Native PAGE
Samples were prepared using the NativePAGETM sample preparation kit (Thermo Fisher Scientific, Waltham, MA, USA), according to the manufacturer’s instructions. Briefly, purified recombinant CERT samples and HMW Native Marker Kit (GE Healthcare, Chicago, IL, USA) were diluted in native PAGE buffer supplemented with 1% digitonin, and CBB G-250 was added to a final concentration of 0.25%. Samples were separated on a NativePAGE™ Novex® 4–16% Bis-Tris gel (Thermo Fisher Scientific, Waltham, MA, USA), and the gel was incubated in 25 mM Tris-glycine buffer (pH 8.3) with 0.1% SDS for 15 min at room temperature with shaking. Proteins were transferred to a PVDF membrane (Bio-Rad, Hercules, CA, USA), and the membrane was fixed in 10% acetic acid for 15 min, rinsed with distilled water, and air-dried. The membrane was de-stained in methanol, rinsed with distilled water, and subjected to western blotting analysis.
4.8. Immunoblotting and Co-Immunoprecipitation
Protein samples were prepared in NuPAGE lithium dodecyl sulfate sample buffer (Thermo Fisher Scientific) with 100 mM DTT and heated for 5 min at 70 °C. Samples were resolved by SDS-PAGE and transferred to PVDF membranes (Bio-Rad). Proteins were visualized by incubation with primary antibodies, which was followed by incubation with secondary HRP-conjugated antibodies for detection using a LuminoGraph II system (ATTO Corporation). Precision Plus Protein All Blue Standards (Bio-Rad) were used as molecular standards for SDS-PAGE.
Co-immunoprecipitation of CERT and VAP-A was carried out as previously described, with some modifications [
27]. Briefly, CERT KO cells were transiently co-transfected with pcDNAneo/nHA-hCERT and pcDNAhyg/nFL-hVAP-A in 6-well plates and cultured for 24 h at 37 °C. Cells were washed twice with PBS and incubated on ice in PBS containing a cross-linker [2 mM dithiobis (succinimidyl propionate)] (Thermo Fisher Scientific, Waltham, MA, USA) for 30 min. The cross-linking reaction was terminated by adding 50 mM Tris-HCl (pH 7.4) buffer, followed by 10 min incubation on ice. Cells were washed twice with PBS and lysed on ice in buffer A [50 mM Tris-HCl (pH 7.4), 1 mM EDTA, 1 mM EGTA, 100 mM NaCl, 50 mM NaF, 5 mM Na-pyrophosphate, 10 mM Na-β glycerophosphate] containing 1% (
v/v) Triton X-100 and supplemented with a protease inhibitor cocktail (cOmplete EDTA-free, Roche) of PPI-2 and PPI-3 phosphatase inhibitors (Merck Millipore). Lysates were precleared by centrifugation at 14,000×
g for 10 min at 4 °C, and the protein concentrations of the precleared lysate fractions were determined using the BCA protein assay kit (Thermo Fisher Scientific, Waltham, MA, USA). HA-CERT or FLAG-VAP was immunoprecipitated from supernatants containing equivalent amounts of total protein using an anti-HA agarose gel (Merck Millipore, Burlington, MA, USA) or an anti-FLAG M2 agarose gel (Sigma-Aldrich, St. Louis, MO, USA), washed twice with buffer A containing 0.1% (
v/v) Triton X-100, and resuspended in NuPAGE lithium dodecyl sulfate sample buffer (Thermo Fisher Scientific, Waltham, MA, USA). Immunoprecipitates were subjected to SDS-PAGE and immunoblotting analysis using appropriate antibodies.
4.9. Immunofluorescence Microscopy
Cells cultured on glass coverslips (Matsunami, Tokyo, Japan) were fixed in Mildform® 10N (Fujifilm Wako Pure Chemical Co., Osaka, Japan) for 10 min at room temperature, permeabilized in 0.1% (v/v) Triton X-100/PBS for 10 min at room temperature, blocked in 3% (v/v) BSA/PBS, and incubated with antibodies diluted in 0.1% (v/v) BSA/PBS. The cover slips were rinsed with distilled water and mounted with Fluoromount (Diagnostic BioSystems, Pleasanton, CA, USA). Fluorescence images were obtained with a fluorescence microscope (BZ-X710 with a 60× Plan-Apochromat V NA 1.20 objective lens, Keyence).
4.10. Quantification of the Golgi-Localization Degree of CERT Fluorescence Signals
To quantify the Golgi-localization degree of each CERT mutant, the mean ratio of the fluorescence intensity of the CERT signal between the Golgi-related region and the whole cell region was determined using Fiji/ImageJ software (NIH) [
36]. For segmentation of the Golgi-related regions, raw fluorescence images of GM130 were processed by the automatic thresholding Otsu method, and the binary images obtained were further processed by “Dilate” processing. The whole cell regions were segmented manually. The mean ratio of the fluorescence intensity of CERT in the two segmented regions in each cell was calculated.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







